


Abstract
A complex disease generally results not from malfunction of individual molecules but from dysfunction of the relevant system or network, which dynamically changes with time and conditions. Thus, estimating a condition-specific network from a single sample is crucial to elucidating the molecular mechanisms of complex diseases at the system level. However, there is currently no effective way to construct such an individual-specific network by expression profiling of a single sample because of the requirement of multiple samples for computing correlations. We developed here with a statistical method, i.e. a sample-specific network (SSN) method, which allows us to construct individual-specific networks based on molecular expressions of a single sample. Using this method, we can characterize various human diseases at a network level. In particular, such SSNs can lead to the identification of individual-specific disease modules as well as driver genes, even without gene sequencing information. Extensive analysis by using the Cancer Genome Atlas data not only demonstrated the effectiveness of the method, but also found new individual-specific driver genes and network patterns for various types of cancer. Biological experiments on drug resistance further validated one important advantage of our method over the traditional methods, i.e. we can even identify such drug resistance genes that actually have no clear differential expression between samples with and without the resistance, due to the additional network information.
INTRODUCTION
One key to achieving personalized medicine is to elucidate molecular mechanisms of individual-specific diseases, which generally result from the dysfunction of individual-specific networks/systems rather than the malfunction of single molecules (1–4). In fact, it has been recognized that the phenotypic change of a living organism can seldom be fully understood by merely analyzing single molecules, and it is the relevant system or specific network that is ultimately responsible for such a phenomenon (3,4). With rapid advances in high-throughput technologies, applying molecular networks to the analysis of human diseases is attracting increasingly wide attention (2). A molecular network, e.g. a gene regulatory network, or a co-expression network, can be generally estimated by correlation coefficients of molecule pairs from expression or sequence data of multiple samples. Based on biological and clinical data, a number of network-based methods were proposed not only to identify disease modules and pathways but also to elucidate molecular mechanisms of disease development at the network level (5–7). To determine a person's state of health, many studies have shown that network-based biomarkers, e.g. subnetwork markers (5,6), network biomarkers (8) and edge biomarkers (9,10), are superior to traditional single-molecule biomarkers for accurately characterizing disease states due to their additional information on interactions and networks. In particular, an individual-specific network is considered to be reliable for accurately characterizing the specific disease state of an individual. It can be directly used to identify the biomarkers and disordered pathways, and further elucidate the molecular mechanisms of a disease for individual patients. However, it is generally difficult to obtain individual-specific networks (i.e. networks on an individual basis) because constructing an individual-specific network from expression data by traditional approaches requires multiple samples so as to evaluate correlations or other quantitative measures (6,11–13) between molecules for each individual, which are usually not available in clinical practice, and thus this requirement seriously limits their application in personalized medicine. In other words, although we can now obtain information of individual-specific differentially expressed genes or somatic mutations from expression or sequence data (14–16) of a single sample, there is still no effective methodology to construct the individual-specific network from such data of the single sample, which is the key personalized feature of each individual at a system level.
In this study, we developed a statistical method to construct an individual-specific network solely based on expression data of a single sample, i.e. a single-sample network or sample-specific network (SSN), rather than the aggregated network for a group of samples, based on statistical perturbation analysis of a single sample against a group of given control samples. In particular, we derived the SSN method to quantify the individual-specific network of each sample in terms of statistical significance in an accurate manner, which is the theoretical foundation of this method. Analyses of the Cancer Genome Atlas (TCGA) data with nine different cancers not only validated the effectiveness of our method, but also led to the following discoveries: (i) we found that there are several common network patterns in the same types of cancer, which, however, are not shared by other types of cancer; (ii) personalized features of various types of cancer were characterized by SSNs, which in turn also revealed important regulatory patterns of driver genes in the cancer; (iii) individual somatic mutations for a sample were strongly correlated with its SSN on a single-sample basis, which was also validated by the pathway enrichment and functional analysis; and (iv) in contrast to the mutational driver genes, the functional driver genes, which functionally affect the occurrence and development of cancer, can be predicted from the hub genes of an SSN for an individual sample. As further applications of TCGA to big data, SSNs were used to predict individual driver mutations for various types of cancer solely based on gene expressions without DNA sequence information, classify cancer phenotypes and identify cancer subtypes by network biomarkers for accurate diagnosis and prediction of diseases in individuals, which all agrees well with the experimental data. Although some previous methods can do the personalized analysis of single samples based on networks or pathways (17,18), there is no reported method to construct an SSN and predict driver genes only based on the expression profile of a single sample as far as the authors know.
Moreover, knockdown experiments validated our prediction of drug-resistant genes in the lung cancer cell line PC9. In contrast to traditional methods that are based on the differential gene expression between the samples with and without drug resistance, we further identified such drug-resistant genes that actually have no differential expression and thus are generally missed by traditional methods.
MATERIALS AND METHODS
In this paper, an SSN also implies an individual-specific network.
Constructing an SSN from a single sample
The SSN for each sample or individual is constructed based on statistical perturbation analysis of this sample against a group of given control samples. For this, we need to have expression data for a group of samples, which serve as the control or reference samples. As shown in Figure 1, by using this group of samples, we can construct the reference network by Pearson correlation coefficients (PCCs) (Figure 1A), i.e. compute the PCC of each pair of molecules as an edge with or without a template or background network. Generally, the reference network would have the common attributes of these reference samples. We then add the single test sample d to this group and construct another network by PCCs; this new network is called the perturbed network (Figure 1B). Thus, we can obtain the differential network between the reference and perturbed networks, which can clearly characterize the specific features of the additional sample d against this group. This differential network is referred to as the SSN of this new sample (Figure 1C). The difference between the reference and perturbed networks is due to sample d. If the single sample d is similar to the reference samples in terms of the gene expression pattern, even after adding the sample d to the reference samples, the change or perturbation of the PCC on any edge would be insignificant. In contrast, if there were obvious differences between the single sample d and the reference samples in terms of expression patterns, adding the single sample to the reference samples would cause significant changes of the PCC on some edges in the perturbed network. Thus, if the differential PCC (ΔPCC) of an edge is statistically significant based on the evaluation of our SSN method, the edge would be kept on the SSN for the individual sample d (Figure 1C). After the ΔPCC is calculated on every edge of the STRING network (http://string-db.org) and filtered by a significant value of ΔPCC, the SSN for sample d is constructed from this single sample's expression data against the reference network. The key is how to quantify the statistical significance of each differential edge (i.e. ΔPCC) in the differential network. Based on the analysis of perturbation statistics, we derived SSN theory (see section ‘Theoretical foundation of SSN’ and Figure 2A) to accurately quantify each differential edge in the network for a single sample in terms of statistical significance, which is the theoretical foundation for this method (Figure 1C). All of the edges with significant differential correlations were used to constitute the SSN for the single sample d (Figure 1B and C). In this study, the functional association network with high confidence (confidence score ≥ 0.9) was used as the template or background network from the STRING database version 9.1 (http://string-db.org) that includes physical interactions, regulatory interactions and the co-expression network of molecules, and all edges in the template network were measured by the PCC, which was calculated by the ‘SciPy’ extension module (http://www.scipy.org/) of the Python programming language.
Figure 1.

Flowchart for constructing an individual-specific network. (A) For a group of reference samples (n samples), a reference network can be constructed by the correlations between molecules based on expression data of this group of samples (multisamples), i.e. computing the PCCn (the Pearson correlation coefficient (PCC) of an edge in the reference network with n samples) of each pair of molecules as an edge in the network. Generally, the reference network has the common attributes of these reference samples. (B) A new sample d is added to the group, and the perturbed network with this additional sample is constructed in the same way by the correlation PCCn + 1 of the combined data. The difference between the reference and perturbed networks is due to sample d. (C) The differential network is constructed by the difference of the corresponding edge between the reference and perturbed networks in terms of PCC, i.e. ΔPCCn = PCCn + 1–PCCn for each edge. Based on sample-specific network (SSN) theory, we can quantify the significance of each edge, i.e. ΔPCCn in the network. The SSN for sample d is constituted by those edges with significant ΔPCCn.
Figure 2.

The significance of a differential ΔPCC or an edge. (A) The theoretical result to evaluate the significance of ΔPCCn by Equation 2, (B) the distribution of ΔPCCn numerically obtained by random simulation (n = 100), (C) the significant value of ΔPCCn evaluated by the numerical simulation (i.e. from the distribution of the random simulation) and the theoretical result (i.e. from Equation (2)). ΔPCCn in the area above the curve is statistically significant with a P-value of < 0.05. Clearly, the simulated curve (the red color) and theoretical curve (the blue color), i.e. the values of ΔPCCn for the random simulation and the theoretical calculation of Equation 2 with a P-value of 0.05 are almost identical with little difference, which well validates Equation 2 of the SSN method.
Topological distance between genes in the SSN and somatic mutation genes (SMGs) in the same sample
Let the genes in the SSN be a set S, and the somatic mutation genes (SMGs) in this single sample be a set D. The topological distance is based on the shortest distance between these two sets S and D in the background network. Specifically, for a single sample, the average shortest distance was calculated by averaging the shortest distances between each gene on the SSN and each SMG of this sample based on the connection of the background network, where the ‘igraph’ extension module (http://igraph.sourceforge.net/) of the Python programming language was used to obtain the shortest distance between two genes in the network. If two genes cannot be linked based on the background network, then the shortest distance between the two genes is assigned 100, which is a sufficiently large value for the shortest distance in the network. Then, the same number of genes of the SSN were randomly chosen from a background network for this single sample, and the average shortest distance between these randomly chosen genes and the SMGs of this sample were again computed by the ‘igraph’ and the average shortest distance for the random genes was compared with the average shortest distance for the genes in the SSN. This permutation was repeated 100 times. The proportion, in which the average shortest distance for random genes is less than the average shortest distance for the genes in the SSN, is defined as the topological distance between the SSN and the SMGs for this single sample. If the proportion is <0.05, we consider the topological distance to be significant for this sample. Otherwise, the topological distance is not significant. The significant samples of the topological distance were identified by testing the topological distance between SSN and mutation genes (SMGs and driver mutation genes (DMGs)) for every sample in various cancers against the background network.
Functional distance between the SSN and the SMGs in the same sample
For a single sample, the pathway or functional enrichment of the genes in the SSN based on the KEGG (Kyoto Encyclopedia of Genes and Genomes) pathways or GO (Gene Ontology) items was calculated by the hypergeometric test (19) as follows:
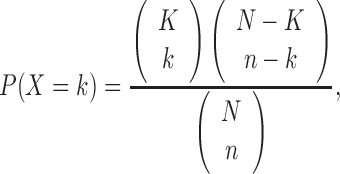 |
where, N is the number of genes on the whole background network, K is the number of genes sharing a pathway or a GO term on the background network, n is the number of genes in the SSN of a single sample and k is the number of genes sharing the same pathway or GO term in the SSN, P(X = k) is the probability observing exactly k shared genes in the hypergeometric distribution. The P-value of the hypergeometric distribution is calculated by the cumulative probability P(X ≥ k) (http://en.wikipedia.org/wiki/Hypergeometric_distribution). If the P-value of the enrichment for a pathway or a GO term is <0.05, then we regard that this pathway or GO function is significantly enriched in the SSN of this single sample. Otherwise, we regard that the pathway or GO function is not enriched in the SSN. Subsequently, the functional association of the SSN and somatic mutations in the same sample can be defined as the number of shared pathways in KEGG or functions of the GO terms between the enriched pathways or GO terms of the SSN and each SMG. The genes with the same number as that of the SSN were then randomly chosen from this single sample, and the enriched pathways or GO terms were again identified by the hypergeometric test. The functional association between these randomly chosen genes and the SMGs of this sample was separately obtained based on KEGG and GO, and the random functional association was compared with the actual functional association between the SSN and somatic mutations of this sample. This permutation was repeated 1000 times. The proportion, in which the random functional association is more than the actual functional association, is defined as the functional distance between the SSN and SMGs for this single sample. If the proportion is <0.05, we consider the functional distance to be significant for this sample; otherwise, the functional distance is not significant for this sample. The significant samples of the functional distance were identified by testing the functional distance between SSN and mutation genes (SMGs and DMGs) on KEGG pathways and GO terms for every sample in various types of cancer.
Classification of phenotypes and subtypes of cancer
A 5-fold cross-validation was conducted for the classification of phenotypes by the ‘ksvm’ package in Bioconductor (http://www.bioconductor.org/) for the R language to implement the function of SVM (supporting vector machine), and the ROC (receiver operating characteristic) curve is drawn by the ‘ROCR’ package in Bioconductor for the R language. The hierarchical clustering was also used to classify the phenotype in the R language.
For the subtype of cancer, the top 100 nodes (differentially expressed genes) or edges (ΔPCCs) with standard deviations were chosen for the consensus clustering and the ‘ConsensusClusterPlus’ package in Bioconductor for the R language was used to perform the consensus clustering. The ‘survival’ package in Bioconductor was used to calculate the log-rank value of the survival curve.
Cell culture and siRNA transfection
The NSCLC (non-small cell lung cancer) cell line PC9 expressing the EGFR (epidermal growth factor receptor) exon 19 deletion mutation was purchased from ATCC (Manassas, VA, USA) and was grown in RPMI 1640 medium Supplementary with 10% fetal bovine serum. The gefitinib-resistant cell line derived from PC9 (PC9-DR) was established by treating PC9 cells with gefitinib continuously for 3 months. siRNA (small interfering RNA) transfection was performed using RNAimax (Life Technologies, Carlsbad, CA, USA) following the manufacturer's protocol. Two siRNAs were used for each gene.
Constructing SSNs for PC9 and PC9-DR
The expression profiles of PC9 and PC9-DR were detected by a Human Genome U219 Array (Affymetrix, Santa Clara, CA, USA) and two repeats for both PC9 and PC9-DR were performed in this study. Sixty normal samples were chosen from the GSE19804 dataset (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE19804) as the reference samples for constructing the reference network. Two PC9 SSNs and two PC9-DR SSNs were then separately constructed based on the reference network and the expression profiles of PC9 and PC9-DR. We only chose the correlation-gained edges (Supplementary Data Note S9) from these SSNs for the functional validation experiments (due to gene knockdown experiments). After filtering the correlation-lost edges (Supplementary Data Note S9), the overlapped network of two PC9 SSNs was taken as the SSN of PC9 and the overlapped network of two PC9-DR SSNs was taken as the SSN of PC9-DR. Subsequently, the differential network (6) between the SSNs of PC9 and PC9-DR was constructed by subtracting the SSN of PC9 from that of PC9-DR (i.e. removing the common SSN edges of PC9 and PC9-DR from the SSN of PC9-DR). The high-degree (>10) genes in the differential network were then selected as potential genes for drug resistance. Clearly, all edges in this differential network are the upregulated edges (correlation-gained edges) from PC9 to PC9-DR.
Drug treatment and cell growth assay
Cells were plated in triplicate at a density of 3000 cells/well in 96-well plates. The cells were then treated with 1 μM gefitinib for 72 hours before 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide (MTT) staining. And the cell growth assay was performed as previously described (20).
shRNA experiments
The shRNAs (short hairpin RNA) against the genes were subcloned into the pLKO.1 vector (Addgene, Cambridge, MA, USA). The shRNA against the luciferase gene was used as the control. The shRNAs were packaged in lentiviral particles by co-transfecting with packaging plasmids into 293T cells and the filtered cell culture supernatant was further used to infect PC9-DR cells as previously reported (21). 293T cells were cultured in DMEM with 8% FBS. The shRNA sequences used are listed in Supplementary Table S8.
Reverse transcription PCR and quantitative real-time PCR (qPCR)
RNA was extracted using Trizol reagent (Invitrogen, Carlsbad, CA, USA) and phenol/chloroform methods and then reverse-transcribed into first-strand complementary DNA with a RevertAid First Strand cDNA Synthesis Kit (Fermentas, Waltham, MA, USA). Gene overexpression and knockdown efficiency were detected by qPCR with gene-specific primers using a 7500 Fast Real-Time PCR System (Applied Biosystems, Foster City, CA, USA) and SYBR Green Master PCR Mix (Invitrogen). Glyceraldehyde 3-phosphate dehydrogenase (human) served as an internal control. The primers used for PCR are listed in Supplementary Table S8.
Statistical analysis
Statistical analysis was performed using a two-tailed Student's t-test.
RESULTS
Theoretical foundation of SSN
For the differential network, each edge is a ΔPCC and we provide a quantitative measure to evaluate its statistical significance. Assuming that there are n samples for the group of the given reference samples, we refer to the PCCs of an edge in the reference network with n samples and the perturbed network with n + 1 samples (due to one additional test sample) as PCCn and PCCn
+ 1, respectively. Then, the ΔPCC of the edge between the reference and perturbed networks is ΔPCCn = PCCn
+ 1–PCCn. We can show that the mean and standard deviation of ΔPCCn for the population are 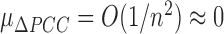 and
and 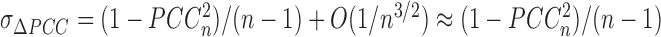 with a large n, where O(1/n) implies the term with the order of 1/n. It is well known that the p value of PCCn can be evaluated by Student's t test with n − 2 degrees of freedom (Supplementary Data Note S2), i.e.,
with a large n, where O(1/n) implies the term with the order of 1/n. It is well known that the p value of PCCn can be evaluated by Student's t test with n − 2 degrees of freedom (Supplementary Data Note S2), i.e.,
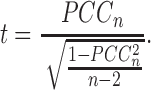 |
(1) |
In this work, we can theoretically further show that the P-value of such ΔPCC follows a new type of symmetrical distribution defined as ‘volcano distribution’ in this paper (Supplementary Data Note S3), whose tail regions are similar to those of the normal distribution. Hence, the statistical hypothesis test Z-test (or U-test) can be used to evaluate the significance level of each ΔPCC because of the central limit theorem (22). The null hypothesis is that the ΔPCCn is equal to the population mean of ΔPCCn, and thus we have,
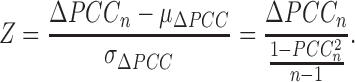 |
(2) |
From Equation 2, the P-value for the edge can be obtained from the statistical Z-value (Figure 2A). If the P-value of the Z-test is <0.05, then the ΔPCC or this edge is significant and there is such an edge in the SSN. Clearly, different numbers of control samples and the correlation of the reference edge will yield a different significance of the edge in the SSN, even with the same value of the ΔPCCn.
For validation of Equation 2, we randomly generated two series of reference numbers (i.e. the expression of two molecules) to estimate their correlation as an edge based on multivariate normal distribution with the different correlation PCCn = 0, 0.1 to 0.9 of the two series of numbers by the ‘Numpy’ package (http://www.numpy.org/). The number or length n of the two series was changed from 5 to 200 (i.e. the number of the reference samples). For every pair of n and PCCn, the random digital simulation was repeated 2 000 000 times, where the value of ΔPCCn with a P-value of 0.05 in the two-tails area was selected from every distribution of simulation, i.e. the significant value (Figure 2C, the red line and Supplementary Figure S2). As shown in Figure 2B (n = 100), the distribution of ΔPCCn follows a new type of distribution defined as volcano distribution, whose tail areas are similar to those of a normal distribution in a random condition. At the same time, the significant value of ΔPCCn with the P-value of 0.05 can also be obtained from the theoretical calculation (Figure 2C, the blue line and Supplementary Figure S3), i.e. Equation 2, where ΔPCCn in the area above the curve is statistically significant with a P-value of < 0.05 (Figure 2C). The simulated and theoretical curves, i.e. the values of ΔPCCn for the random simulation and the theoretical calculation of Equation 2 with the P-value of 0.05 are almost identical (Figure 2C, and in particular, Supplementary Figures S2 and 3 for all n and PCCn) with little difference, which well validates Equation 2 of the SSN method. Note that we can also directly use the volcano distribution for the significance test in an accurate manner.
Theoretical relations between differential correlations and differential expression
We first define three types of edges in a network when there is a sample in addition to the reference samples; (i) the correlation-gained edge is the edge whose correlation or PCC in the absolute value is increased from the reference samples to the single sample, (ii) the correlation-lost edge is the edge whose correlation or PCC in the absolute value is decreased from the reference network to the single sample and (iii) the correlation-invariant edge is the edge whose correlation or PCC in the absolute value exhibits little change from the reference network to the single sample.
We assume that there are two genes X and Y; the expressions of X and Y in the reference samples are Xr and Yr with r = 1,…, n; the expressions of X and Y in the single sample is XS and YS; the differential expression of X is ΔX = XS −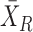 and the differential expression of Y is ΔY = YS −
and the differential expression of Y is ΔY = YS −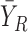 , where
, where 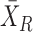 and
and 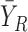 are average values of X and Y in the reference samples, respectively. Based on the analysis in Supplementary Data Note S9, when the number of the reference samples, i.e. n, is sufficiently large, we can theoretically derive the following relations between differential correlation ΔPCCn and differential expression of X and Y:
are average values of X and Y in the reference samples, respectively. Based on the analysis in Supplementary Data Note S9, when the number of the reference samples, i.e. n, is sufficiently large, we can theoretically derive the following relations between differential correlation ΔPCCn and differential expression of X and Y:
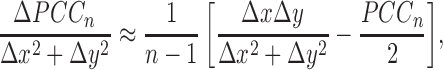 |
(3) |
or
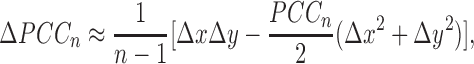 |
(4) |
where 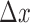 and
and 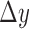 are
are 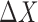 and
and 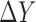 normalized by the reference samples, defined as follows:
normalized by the reference samples, defined as follows:
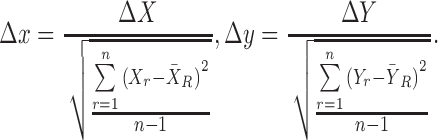 |
From the above Equations 3 and 4, we can obtain Supplementary Tables S6 and 7, which describe the various cases between ΔPCC and differential expression of X and Y, and a graphical explanation is also given in Supplementary Figure S14. The details of the different gene expression levels for a single sample affecting the correlation are given in Supplementary Data Notes S9 and 10 with Supplementary Tables S6 and 7.
SSNs reveal common network patterns for cancers at molecular level
We chose the datasets of nine different types of cancer from the TCGA (http://cancergenome.nih.gov/) database (Supplementary Table S1 and Supplementary Data Note S1) with gene expression profiling and matched clinic information. For each type of cancer, 8–17 normal samples were selected as the reference samples (Supplementary Table S1) and every single cancer sample was used to construct its SSN (Figure 1 and Supplementary Table S2). We collected the expression data and clinic information of the nine types of cancer, and detailed information for the numbers of control and cancer samples is given in Supplementary Table S1. The SSN for each sample was constructed for all cancer datasets, and only the correlation-lost edges (see Supplementary Data Note S9), whose correlations in absolute values decrease from the reference to this sample, were chosen to perform the following analysis. We focused here on the analysis of the sample-specific subnetworks with correlation-lost edges related to tumor protein p53 (TP53), which is a crucial gene in cancer, to demonstrate the power of this analysis for characterizing the personalized features (Figure 3). The subnetwork of TP53 is composed of genes directly connected with TP53, or its first-order neighboring genes. Figure 3A shows the subnetworks of TP53 from four breast invasive carcinoma (BRCA) samples, which clearly characterize their individual features at the network level. Although most edges in the four subnetworks of TP53 are different from each other, the associations between TP53 and both MUC1 and CCNB1 exist in all four subnetworks (Figure 3A). In fact, by analyzing subnetworks of TP53 for all 761 samples in breast cancer, we found that there is an association between TP53 and MUC1 in 65.18% of breast cancer samples, and an association between TP53 and CCNB1 in 64.52% of breast cancer samples (Supplementary File S1). In other words, for 65.18% of individuals with breast cancer, the correlation between TP53 and MUC1 has a significant loss in the cancer status and the correlation between TP53 and CCNB1 has been lost in the cancer status for 64.52% of individuals with breast cancer. MUC1 is a known marker for breast cancer (23), and it associates with TP53 (24,25) and plays an important role in breast cancer (26,27). CCNB1 is an important known marker for breast cancer, and it was reported to be related with TP53 (28,29) and have important implications for cancer prognosis (30,31).
Figure 3.

Individual-specific networks characterize personalized features and also reveal common network patterns for cancer. (A) The four individual-specific subnetworks of tumor protein p53 (TP53) from four samples for breast invasive carcinoma (BRCA). The numbers of the connections with TP53 for the four samples are respectively 8, 7, 6 and 7, and the genes linked to TP53 are also different in the four samples, i.e. DDB2 for BRCA_A0B0, CHEK1 for BRCA_A25A, IGF1R for BRCA_A0HO and ESR1 for BRCA_A0DP are unique genes for the respective subnetworks of the four samples. However, MUC1 and CCNB1 (the yellow color) are common genes appearing in the four subnetworks. Actually, we found that 65.18% of BRCA samples include a significant connection (the bold lines) between TP53 and MUC1, and 64.52% of BRCA samples include a significant connection (the bold lines) between TP53 and CCNB1. i.e. these connections have a significant loss of correlation between BRCA and normal samples, and are the common network pattern related to TP53. (B) The individual-specific subnetworks of TP53 from four samples for kidney renal clear cell carcinoma (KIRC). There are different targets and numbers connected with TP53 in the four samples of KIRC. Three genes, VEGFA, IGFBP3 and MUC1 (the yellow color) appeared in all four samples (the bold lines). Actually, we found that 94.74% of KIRC samples have a significant loss of correlation for connection between VEGFA and TP53, 92.34% of KIRC samples have a significant loss of correlation for connection between IGFBP3 and TP53, and 87.32% of KIRC samples have a significant loss of correlation for connection between MUC1 and TP53, which are the common network patterns related to TP53.
By analyzing the gene expression of the four breast cancer samples, we found that TP53 is significantly upregulated only in two (BRCA_A0B0 and BRCA_A25A) of the four samples, as compared with its expression in the reference samples and the gene expression of TP53 was not significantly changed in the other two samples (BRCA_A0HO and BCRA_A0DP) compared with the reference samples, but MUC1 was significantly upregulated in all four samples. The correlation between TP53 and MUC1 takes a positive value relative to the reference samples, and the correlation coefficient is significantly decreased in the four samples. Thus, we called the relationship between TP53 and MUC1 in breast cancer ±UU (Positive Correlation Decreases due to X Upregulation and Y Upregulation, TP53 as Y, see Class-5 in Supplementary Data Note S10 and Supplementary Table S6) for sample BRCA_A0B0 and BRCA_A25A and ±*N (Positive Correlation Decreases due to X Upregulation and Y No change, TP53 as Y, see Supplementary Table S6) for sample BRCA_A0HO and BRCA_A0DP (Supplementary Data Note S10). The gene CCNB1 is also significantly upregulated in all four samples. The correlation between TP53 and CCNB1 takes a positive value based on the reference samples, and the correlation coefficient is significantly decreased in the four samples. Thus, we called the relationship between TP53 and CCNB1 ±UU for samples BRCA_A0B0 and BRCA_A25A, and ±*N for samples BRCA_A0HO and BRCA_A0DP (TP53 as Y, Supplementary Data Note S10 and Supplementary Table S6).
On the other hand, in the subnetworks of TP53 for kidney renal clear cell carcinoma (KIRC), the network patterns or targets of TP53 are also diverse and individual dependent (Figure 3B), but there are three consistent edges connected with VEGFA, IGFBP3 and MUC1 in all four subnetworks of TP53. By analyzing all individual-specific subnetworks of TP53 for 418 kidney cancer samples, we found the edge with VEGFA in 94.74% of SSNs, the edge with IGFBP3 in 92.34% of SSNs and the edge with MUC1 in 87.32% of SSNs (Supplementary File S1). This means that the edge between TP53 and VEGFA among 94.74% of samples, the edge between TP53 and IGFBP3 among 92.34% of samples and the edge between TP53 and MUC1 among 87.32% of samples have significant loss of correlation, or these associations in most kidney cancer samples suffer from significant loss compared with normal samples, i.e. they are the common network patterns related to TP53 in kidney cancer. The VEGFA gene is an important growth factor acting in kidney cancer (32,33), the IGFBP3 gene is a cell growth factor and an important marker in kidney cancer (34,35), and the MUC1 gene affects invasive and migratory properties of kidney cancer cells and is a potential therapeutic target (36). From the above analysis, the abnormal interactions of TP53 with VEGFA, IGFBP3 and MUC1 are considered to be potential factors contributing to kidney cancer.
By analyzing the gene expression of the four kidney cancer samples, we found that in three (KIRC_2444, KIRC_3362 and KIRC_5457) of the four samples TP53 is significantly upregulated relative to the reference samples and the gene expression of TP53 did not significantly change only in sample KIRC_4807, and the VEGFA gene is significantly upregulated in all four samples. The correlation between TP53 and VEGFA takes a negative value based on the reference samples, and the correlation coefficient is increased (loss of negative correlation) in the four samples. Thus, we called the relationship between TP53 and VEGFA ±UU (Negative Correlation Increases due to X Upregulation and Y Upregulation, TP53 as Y) for samples KIRC_2444, KIRC_3362 and KIRC_5457, and ±*N (Negative Correlation Increases due to X Upregulation and Y No change, TP53 as Y) for sample KIRC_4807. The IGFBP3 gene is significantly upregulated in all four samples, the correlation between TP53 and IGFBP3 takes a positive value based on the reference samples, and the correlation coefficient is decreased (loss of a positive correlation) in all four samples. Thus, we called the relationship between TP53 and IGFBP3 ±UU for samples KIRC_2444, KIRC_3362 and KIRC_5457 and ±*N for sample KIRC_4807. The gene MUC1 is downregulated in all four samples, the correlation between TP53 and MUC1 takes a positive value based on the reference samples, and the correlation coefficient is decreased (loss of positive correlation) in all four samples. Thus, we called the relationship between TP53 and MUC1 in kidney cancer ±DU (Positive Correlation Decreases due to X Downregulation and Y Upregulation, TP53 as Y) for samples KIRC_2444, KIRC_3362 and KIRC_5457 and ±*N (Positive Correlation Decreases due to X Downregulation and Y No change, TP53 as Y) for sample KIRC_4807 (Supplementary Data Notes S9 and 10 and Supplementary Table S6).
Here, the loss of correlation means that the correlation in terms of the absolute value decreases from the reference samples.
SSNs characterize personalized features and also reveal different regulatory patterns of driver genes in cancer
Each cancer sample has its own individual-specific pathogenesis. We used the individual-specific subnetworks related to the DMG TP53 as an example to show the personalized features and regulatory patterns for various types of cancer.
The samples were selected from stomach adenocarcinoma (STAD), BRCA and glioblastoma multiforme (GBM), and the subnetworks of TP53 are shown in Figure 4 and Supplementary Figures S4–6, where each network is the individual-specific subnetwork for one sample. We classified the edges into two types, i.e. one is the correlation-gained edges (the red lines) whose PCCs are increased from the reference network with positive correlation coefficients or decreased from the reference network with negative correlation coefficients and another is the correlation-lost edges (the green lines) whose PCCs are decreased from the reference network with positive correlation coefficients or increased from the reference network with negative correlation coefficients. For STAD, there are almost equal numbers of the correlation-gained and correlation-lost edges in the TP53 subnetwork (one sample in Figure 4A and four samples in Supplementary Figure S4). Actually, on average from all 183 STAD samples, there are 28.37 increasing edges and 28.89 decreasing edges, which means that TP53 as a driver gene affects both the correlation-gained and correlation-lost edges in STAD. For BRCA, the number of the correlation-lost edges is obviously greater than that of the correlation-gained edges in the TP53 subnetwork (one sample in Figure 4B and four samples in Supplementary Figure S5). On average from all 761 BRCA samples, there are 2.22 correlation-gained edges and 26.34 correlation-lost edges; this implies that the interaction partners of TP53 are significantly reduced in BRCA, which is a general feature of BRCA. In contrast, for GBM, the number of the correlation-gained edges is much greater than that of the correlation-lost edges for TP53 subnetworks (one sample in Figure 4C and four samples in Supplementary Figure S6). On average from 333 GBM samples, there are 120.24 correlation-gained edges and 24.02 correlation-lost edges for the driver gene TP53, which characterizes the regulatory patterns of GBM and means that interaction partners of TP53 are significantly increased. TP53 may promote tumor onset by mainly inducing the correlation-gained edges in GBM. It is consistent with the report that TP53 is gain-of-function in GBM (37,38).
Figure 4.

Individual-specific networks related to driver gene tumor protein p53 (TP53) reveal the regulatory patterns in different types of cancer. (A) The individual-specific subnetwork of TP53 in sample 4255 from stomach adenocarcinoma (STAD). (B) The individual-specific subnetwork of TP53 in sample A0CV from BRCA. (C) The individual-specific subnetwork of TP53 in sample 2574 from glioblastoma multiforme. The network analyses related to the driver gene TP53 not only reveal the personalized features of each sample but also indicate that each cancer type has a specific regulatory pattern. The red and green lines represent the correlation-gained and correlation-lost edges, respectively. The green node indicates an oncogene from NCBI (http://www.ncbi.nlm.nih.gov/).
SSNs validated by literature
In addition to the descriptions in the preceding section, our findings for many other differential associations or regulations between normal and cancer samples are also consistent with previous reports. ESR1 (estrogen receptor 1) is an important factor for the pathological process of breast cancer. We found a significant loss of correlation between TP53 and ESR1 in 42.97% of breast cancer samples compared with the normal reference samples. Indeed, the ESR1 gene was found to be upregulated by TP53 in the breast cancer cell line MCF-7 (39,40). The overexpression of ESR1 could cause the observed loss of correlation in breast cancer samples.
TP63 (tumor protein p63) is a member of the p53 family of transcription factors, and the correlation between TP63 and TP53 is usually low in normal lungs. A recent study showed a high frequency of co-expression between TP53 and TP63 in lung squamous cell carcinoma, but not in lung adenocarcinoma (LUAD) (41). Coincidentally, our analysis showed that despite low or even no correlation between TP53 and TP63 in the reference and normal samples, 74.2% of lung squamous cell carcinoma samples showed a significant gain in correlation compared with the reference samples, whereas only 6.5% of LUAD samples showed a significant correlation variation compared with the reference samples.
The CDC25C (cell division cycle 25C) gene is involved in the regulation of cell division. It is well known that CDC25C regulates cellular entry into mitosis in the G2/M phase (42), and its expression is also regulated by TP53 (43). Hence, the abnormal regulation between TP53 and CDC25C may disrupt the regulation of CDC25C by TP53, and render the cell cycle and cell division out of control. The correlation between TP53 and CDC25C is 0.52 (the P-value of significant PCC is 0.04) in the reference samples, which is significantly high. However, 52.04% of breast cancer samples showed a significant loss of correlation (decreasing correlation coefficient) between these two genes compared with the reference samples. This result is consistent with those of previous studies, i.e. it was found that CDC25C expression is regulated by TP53 (43) in normal samples, but in breast cancer its expression and that of its splice variants are not dependent on TP53 (44).
SSNs validated as personalized features by disease gene enrichment, individual somatic mutations, functional analysis and pathway enrichment
SMGs of cancer that provide individual-specific information for each sample (45) can be used to validate SSNs and our method (45). Specifically, we measured the relationship between the SSN and the SMGs (Supplementary Data Note S4) in the same sample to validate the sample specificity of each SSN. The topological distance (‘Materials and Methods’ section and Supplementary Data Note S5) and functional distance (‘Materials and Methods’ section and Supplementary Data Note S6) between an SSN and a set of SMGs were used to measure their relationships for every sample in various cancer datasets. As shown in Figure 5A, the topological distance between the SSN and corresponding SMGs is significantly small in more than 99% of samples on average in each cancer, i.e. they are significantly related to each other, which implies that the SSN is indeed sample specific and reflects the personalized features. From a functional viewpoint, there are more than 72% of samples on average in all cancers in which each SSN and the corresponding somatic mutations are significantly related in terms of the functional distance of GO annotations (Figure 5B and Supplementary Table S3), and more than 63% of samples on average of all cancers, in which each SSN and the corresponding somatic mutations are significantly related in terms of the functional distance in KEGG pathways (Figure 5B and Supplementary Table S3). In most of the samples, the SSN and somatic mutations are also significantly related in terms of the functional distance in the Biological Biochemical Image Database (http://bbid.grc.nia.nih.gov/) and BIOCARTA (http://www.biocarta.com/) pathways (Supplementary Table S3). In addition to the somatic mutations, the driver mutations of cancer (Supplementary Data Note S4) are also individual-specific mutations, and currently, 125 DMGs have been determined for cancer (46). Then, instead of using SMGs, we similarly performed another analysis using the overlapped genes between the 125 DMGs and SMGs as a set of the driver genes in each sample for measuring the relationships. Figure 5A–C show that the results for the driver genes are similar to those for the SMGs, but are more significant in the functional distance analysis (Figure 5C). In particular, comparing with 63% of samples on average as shown in Figure 5B, there are more than 94% of samples on average in which each SSN and the DMGs are significantly related in terms of the KEGG pathways (Figure 5C and Supplementary Table S3). These results indicate that the SSN is indeed sample specific and characterizes the personalized features of each sample at the network level. Note that we also performed the multiple-test correction, i.e. the Bonferroni correction, and the results have no significant differences from the above analyses.
Figure 5.

Validating individual-specific networks and predicting driver mutation genes (DMG) in different types of cancer. (A) The proportion of significant samples in terms of the topological distance between a sample's SSN and its mutation genes (SMGs and DMGs). The x-axis is the nine types of cancer and the y-axis is the percentage of significant samples in terms of the topological distance for somatic mutation genes (SMGs) (the blue color) and DMGs (the red color) in various types of cancer. (B) The proportion of significant samples in terms of the functional distance between a sample's SSN and its SMGs. The x-axis is the nine types of cancer and the y-axis is the percentage of significant samples in terms of the functional distance for SMGs on GO terms (the blue color) and KEGG pathways (the red color) in various types of cancer. (C) The proportion of significant samples in terms of the functional distance between one sample's SSN and its DMGs. The x-axis is the nine types of cancer and the y-axis is the percentage of significant samples in terms of the functional distance for DMGs on GO terms (the blue color) and KEGG pathways (the red color) in various types of cancer. (D) The proportion of significant samples in the enrichment analysis of known cancer genes for each SSN in the database of Cancer Gene Census (CGC) and Genetic Association Database (GAD). The x-axis is the nine types of cancer and the y-axis is the percentage of significant samples in the enrichment analysis of known cancer genes in the database of CGC (the blue color) and GAD (the red color). The results in A–D show that each SSN is significantly related to the mutations of the same sample in terms of the topological and functional distances, and indeed characterizes the personalized features of the individual. (E) The proportion of SMGs to be DMGs in top 30, 20, 10 and 5 highest degree genes of SSN and random genes. Predicting individual DMGs by each SSN in various types of cancer. The rate that a SMG with a high degree is also a DMG increases in each SSN, and thus the accuracy of the prediction increases with the degree. The x-axis is the nine types of cancer and the y-axis is the percentage of SMGs to be DMGs in top 30 (the blue color), 20 (the red color), 10 (the green color) and 5 (the purple color) highest degree genes of SSN and random genes (the cyan color) in various types of cancer.
The known cancer genes can be downloaded from the CGC (Cancer Gene Census, http://cancer.sanger.ac.uk/cosmic/census) database and the GAD (Genetic Association Database, http://geneticassociationdb.nih.gov/), and the enrichment of an SSN relative to the known cancer genes can be used to validate its functional specificity (Supplementary Data Note S7). The result shows that the known cancer genes in the CGC database are significantly enriched in all of the SSNs in the cancer datasets (Figure 5D). For genes in GAD, we chose the cancer-associated genes and then evaluated the significance of enrichment for each SSN (Figure 5D and Supplementary Data Note S7); our results clearly show that the corresponding cancer genes are significantly enriched in most SSNs.
SSNs predict individual driver mutations for cancer solely based on gene expression without DNA sequence information
A hub node in an SSN is a gene that is highly connected with other genes (a gene with a high degree or with many links). Generally, the higher the gene degree in an SSN, the greater the variations or changes in the regulation related to this gene from normal to tumor samples, i.e. a gene with a high degree is a gene with large variations in interactions on a network level. Thus, a high-degree gene in the SSN is more likely to be a DMG for cancer. Based on this hypothesis, we predicted the DMGs for the genes with different degrees in SSNs (Supplementary Data Note S8), and found that the higher the gene degree, the more likely this gene is a DMG (Figure 5E). We conducted the computation for the top 30, 20, 10 and 5 highest degree genes for every SSN, and the rate that a gene with the high degree is also a DMG was calculated for every cancer by further checking the somatic mutation data of the genes. Clearly, from the top 30 highest degree genes the rate is monotonically increased (Figure 5E). For example, within the top 30 highest degree genes, about a half of those SMGs (51.23% rate) are DMGs for stomach cancer (STAD) in Figure 5E. With the top five highest degree genes, 84.44% of those SMGs are DMGs. However, in the random condition, only 2.71% of SMGs are DMGs in STAD (Figure 5E). Clearly, these results indicate that the hub genes in an SSN are strongly related to the driver mutations in the same sample, and thus can be used to predict the potential driver genes (including driver mutation and driver non-mutation genes, also called functional driver genes) on an individual basis for each sample, even without its DNA sequence information. As shown in Figure 5E, the accuracy of the prediction of the DMGs increases with the degree of the hubs in SSNs. These results also imply that high-degree genes in the network are high-risk genes that are more likely to be related to the tumor onset than other genes. The potential driver genes for each sample (Supplementary File S2) were obtained from the top 10 high-degree genes in the SSN of each sample. The functional enrichment analysis was performed for the potential driver genes of each sample using known DMGs and oncogenes from NCBI (http://www.ncbi.nlm.nih.gov), and we found that the known DMGs (46) and oncogenes were significantly enriched (Supplementary Figure S7). The significant enrichment of the known DMGs and oncogenes in the potential driver genes of most samples for every cancer clearly indicates that the SSN method is valid for predicting driver genes of cancer solely based on gene expression without DNA sequence information. In other words, the SSN method provides an effective strategy for personalized medicine, enabling the prediction of potential driver genes or potential oncogenes for a specific patient based on single-sample data.
SSNs classify phenotypes of cancer and identify subtypes of cancer by network biomarkers for accurate diagnosis
Molecular networks are reliable forms to accurately characterize complex diseases, in contrast to individual molecules. Many network-based approaches have been proposed to extract a discriminative gene set as a biomarker for the classification of samples with distinct phenotypes by considering network information, but when diagnosing a new sample, such a gene set is simply used in a similar way to traditional molecular biomarkers without effectively exploiting the network information of that sample. In this sense, those biomarkers are not network biomarkers but essentially molecule biomarkers. In contrast, our method can construct the SSN for each sample and therefore open a new way for diagnosing a single sample by network biomarkers, i.e. diagnose or classify each sample by the SSN/subnetwork/edges.
We first used hierarchical clustering to classify the normal and tumor samples, by using the top five differentially expressed genes (i.e. node biomarkers, by the traditional method) and the top five differential edges or ΔPCCs for SSNs (i.e. edge biomarkers, by our method) as the biomarkers. The edge biomarkers are clearly superior to node markers in terms of the accuracy of the classification of normal and tumor samples (STAD) by hierarchical clustering, i.e. there are only four samples for edge biomarkers but 29 samples for node markers, which were wrongly classified (top five edge biomarkers with accuracy of 98.1% in Figure 6A, and top five node biomarkers with accuracy of 86.5% in Figure 6B).
Figure 6.

Classifying phenotypes and cancer subtypes. (A) The classification of cancer (183 samples, the green bar) and normal (33 samples, the red bar) samples by hierarchical clustering of the edge biomarkers (top five differential edges or differential ΔPCCs by our method) for STAD with 98.1% accuracy. (B) The classification of normal and cancer samples by hierarchical clustering of the node biomarkers (top five differential genes by the traditional method) for STAD with 86.5% accuracy. (C) The classification result of using the top five differential genes and edges for lung adenocarcinoma (LUAD). (D) The classification using the bottom five differential genes and edges for LUAD. (E) The log-rank P-value of the survival curve for the subtyping in BRCA, uterine corpus endometrial carcinoma (UCEC), KIRC, LUAD and ovarian serous cystadenocarcinoma (OV). We used the top 100 variable genes as node biomarkers for subtyping cancer (the traditional method), and top 100 variable edges (or ΔPCCs) as edge biomarkers for subtyping cancer (our method). All results show that individual-specific subnetworks or edge biomarkers are superior to the traditional node or molecular biomarkers in terms of classification and subtyping.
We then used the SVM model with 5-fold cross-validation to classify the phenotypes or samples for various types of cancer. When we chose the top five differentially expressed genes and edges as the biomarkers to classify the normal and tumor samples, we found that the area under the curves (AUCs) for edge biomarkers and node biomarkers are respectively 99.66 and 99.65% for LUAD, and thus the accuracies of both are very similar and high (Figure 6C LUAD and Supplementary Figure S8 for the other eight types of cancer). However, for the same data of LUAD, when we chose the bottom five differential-expression genes and edges as the biomarkers to classify the two phenotypes, the AUC for edge biomarkers is still as high as 95.5% but the AUC for node biomarkers is significantly reduced to 53.6%, which implies that the edge biomarkers are robust and have synergetic power in the classification, compared with the node biomarkers (Figure 6D for LUAD and Supplementary Figure S8 for the other eight cancers).
Subtyping cancer is an important topic in recent cancer research, and most subtyping methods mine the information based on expression of genes, rather than individual networks. Here, although a sophisticated algorithm may considerably improve the accuracy, we used a simple subtyping method (47) to identify the potential subtypes in different types of cancer separately based on gene expression data (i.e. node biomarkers, top 100 variable genes) and SSN data (i.e. network/edge biomarkers, top 100 variable edges) by package ‘ConsensusClusterPlus’ in Bioconductor. The log-rank P-value of the survival curve was then used to evaluate the effect of the cancer subtyping. The numbers of subtypes for various types of cancer were referred to the following publications for BRCA (48), KIRC (49), LUAD (50), ovarian serous cystadenocarcinoma (50) and uterine corpus endometrial carcinoma (50). As shown in Figure 6E, the accuracy (the P-value) of subtype identification by our edge biomarkers (i.e. differential ΔPCCs) is superior to the traditional node biomarkers (i.e. differential genes), which demonstrates that the information of associations on an individual basis is useful and powerful in subtype identification of complex diseases.
The functional driver genes from SSN
A mutation driver is a mutation that can confer growth advantage to the cell and be positively selected on cancer occurrence (51). However, known DMGs are mutated in many cancer samples and the existing driver mutations cannot explain all cancer onsets and typically have low coverage in cancer samples. Thus, in this work, we developed the concept of functional driver genes, whose dysfunction will benefit the formation or maintenance of the cancer occurrence with or without somatic mutations. The dysfunction can be measured by the changes in associations or a network between the reference samples and the single sample, and thus the functional driver genes can be obtained by analyzing the SSN.
The high-degree genes in the SSN are important features for a cancer sample, and characterize their importance in the dysfunctional network or SSN of the single sample. Actually, they are also strongly related to the mutation driver genes in the individual sample (Figure 5E). Thus, the high-degree genes in the SSN can be considered as a measurement of the functional driver genes from the network viewpoint, and they may play an important functional role in the SSN for cancer occurrence and have the ability to drive the normal cell to a cancerous phenotype, similarly to mutation driver genes.
SSNs can be used to detect the functional drivers solely based on the expression data, even without the sequence information (i.e. without mutation information). We collected all of the top five highest degree genes, which are regarded as the potential functional driver genes of each single sample, for every sample in various types of cancer. The genes that appeared in at least five samples were chosen as the functional driver genes of this kind of cancer (Supplementary Table S4), and were further validated as the potential disease genes by their mutation ratio in the cancer genomic data of TCGA with cBioPortal (52). That is, we compared the ratio of the number of samples with mutations in these potential disease genes against the number of total samples (the blue color, Supplementary Figure S9), and the ratio of the number of samples with mutations in random genes (with the same number as the potential disease genes) against the number of total samples (the orange color, Supplementary Figure S9). The result shows that there is an obviously higher mutation ratio for these functional driver genes than random genes in cancer samples of TCGA. This result implies that the functional driver genes tend toward mutation or dysfunction in cancer samples.
Experiments validated that SSNs identified functional driver genes contributing to drug resistance
Lung cancer is the leading cause of cancer-related deaths worldwide, with NSCLC being the predominant form of the disease (53). The EGFR signaling pathway, essential for normal epithelial cell proliferation, is frequently deregulated in lung cancer (54). EGFR kinase inhibitors, including gefitinib and erlotinib, are clinically effective therapeutics of NSCLCs with EGFR kinase domain mutations (55,56). However, the clinical efficacy of gefitinib is limited by the development of acquired drug resistance. Using the human lung cancer cell line PC9 that harbors an EGFR kinase domain mutation and is sensitive to tyrosine kinase inhibitor (TKI) treatment, and the TKI-resistant cell line (PC9-DR) derived through long-term exposure to TKI, we performed microarray analyses of gene expression. The expression profiles of PC9 and PC9-DR were obtained separately, and then the two SSNs for PC9 and PC9-DR were both constructed based on the expression profiles and the reference dataset from GSE19804 of the Gene Expression Omnibus database (see ‘material and methods’ section), and only the correlation-gained edges in the SSN were retained and used for the following analysis. Their differential network (6) was constructed by removing the common SSN edges of PC9-DR and PC9 from the SSN of PC9-DR, and 59 candidate genes as the potential functional driver genes were identified from the differential network based on the degree distribution (i.e. genes with degree >10 in the differential network between the SSNs of PC9-DR and PC9) (Figure 7A, Supplementary Figure S10 and Supplementary Table S5). Interestingly, most of these genes did not show significant expression changes between PC9-DR and PC9 and thus might not be identified by classical methods that are generally based on differential gene expression (Supplementary Table S5). To test the power of our methods, we then performed individual gene knockdown of the top 15 candidates with siRNA and analyzed their influence on cell growth and drug response (Supplementary Figure S11). Our data show that knockdown of these genes did not impact cell growth in both PC9 and PC9-DR cells (Supplementary Figure S12). However, 73% of these genes (11 out of 15) were identified as important regulators of drug resistance since knockdown of any one of these 11 genes significantly conferred the PC9-DR cells with sensitivity to gefitinib (Figure 7B and C). Among them, five genes (BRCA1, GRB2, TGFBR1, CASP3 and CSNK2A1) showed the most significant effects when knocked down in PC9-DR cells (Figure 7B and C). Moreover, to compare with the analytical strategy that focuses on expression changes, we further knocked down the top five upregulated genes (FAM171A1, COL13A1, vimentin, BMP5 and CYB5R2) with shRNA (The shRNA sequences used are listed in Supplementary Table S8). However, none of these genes could overcome drug resistance when knocked down (Supplementary Figure S13a). We also randomly chose 29 other genes to perform knockdown screening with siRNA, and PC9-DR cells showed no significant difference in drug response after their knockdown (Supplementary Figure S13b). Taken together, our experimental data validate the effectiveness of the SSN method, and demonstrated the superiority of SSNs in the identification of genes important for drug resistance by considering individual networks. Our method is more powerful than the traditional differential expression methods for identifying the functional driver genes.
Figure 7.

Experimental identification of functional driver genes contributing to drug resistance in the lung cancer cell line PC9-DR. (A) The subnetwork for the top 15 candidate genes. The node size indicates the degree of individual genes. The 15 candidate genes are highlighted as yellow, and other nodes are the first-order neighbors (genes) with the top 15 candidate genes in the differential network between PC9 and PC9-DR. The detailed differential network is provided in Supplementary Figure S10. (B) The basic information for the top 15 candidate genes. Gene symbols: the official symbol for each gene; Degree: the degree of each gene in the differential network (or the number of neighbors for each gene); Fold change: the expression change of each gene from PC9 to PC9-DR; Significance to drug resistance: the significance of the result of drug resistance after gene knockdown. Clearly, none of the 15 genes exhibit significant differential expression (0.66 < fold change < 1.5) and thus may not be identified by traditional statistical analyses, although most of them actually show significant effects on drug resistance (11 genes among 15 candidate genes) in knockdown experiments. (C) The growth inhibition percentage when either PC9 or PC9-DR cells with indicated gene knockdown were treated with 1 μM gefitinib for 72 h. PC9 siScramble was set as the positive control and PC9-DR siScramble was set as the negative control. Data are shown as means ± SEM. **P < 0.01 and *P < 0.05. Note that all edges shown in (A) are the upregulated edges (correlation-gained edges) from PC9 to PC9-DR.
DISCUSSION
The ΔPCC is the interferential degree of correlation of a single sample perturbing the reference samples, and it depicts the changed degree of correlation by adding the single sample to the reference samples. Thus, it describes the difference in associations from a network viewpoint. If the single sample can affect the correlation of two genes in the reference samples with a significant change, the regulation of two genes in the single sample is considered to be inconsistent with the regulation in the reference samples. This inconsistency of regulation may be due to the differential gene expression in either or both of the two genes, or caused by a functional alteration, e.g. a mutation that cannot be identified by traditional testing of differential expression of the genes. Therefore, ΔPCC testing is a more sensitive method than the traditional differential expression testing, and can identify the potential disease genes that even display no differential expression from normal/control samples. In such a sense, the SSN is complementary to the traditional methods from the network perspective. In other words, those non-differential expressed genes which are usually removed from the traditional analyses may have rich information on diseases, not at the gene expression level but at the network level. Thus, similar to non-coding RNAs (or non-coding regions of DNA) that are now considered as the ‘dark matter’ in sequence (57), our analysis shows that those non-differential genes may play important roles in disease progression (or biological processes) and are actually the ‘dark matter’ in expression (57).
A biological function is generally facilitated not by individual molecules but by their regulations or molecular networks, which dynamically change with time and conditions. Thus, identifying the condition-specific network or SSN is crucial to elucidate molecular mechanisms of complex biological processes at a system level. However, although expression data or sequencing data provide information about the profiles of molecules on a single-sample basis, there is no effective method to construct a molecular network on a single-sample basis. In this work we proposed a statistical method to construct the SSN for a single sample, which opens a new way for both characterizing personalized features and analyzing biological systems at a network level. The analyses of TCGA data not only validated the effectiveness of our method but also demonstrated that SSNs can characterize the network patterns on a single-sample basis. We also reported new discoveries for regulatory patterns, personalized networks and edge biomarkers in several cancer types.
Although a group of reference samples is required in our method, it is generally available even in clinical practice and also there is no strict condition on the reference samples. Theoretically, the reference samples can be composed of any type of samples, but choosing those reference samples with distinct expression profiles from the test samples certainly increases the discriminatory power of its SSN. Actually, to check the robustness of the results against the different choices of the reference samples, we tested breast cancer data from TCGA. There are 99 normal samples from TCGA, and we randomly chose 17 normal samples from these as a group of reference data. With these 17 randomly chosen reference samples, we could then construct the SSN for each cancer sample by our method and compared the new SSNs with the old SSNs. We repeated the process 100 times, and obtained the average recurrence ratio of the edges in the SSN from different reference samples. The comparison results show that the average recurrence ratio of the edges in the SSN from the different reference samples is as high as 81.01%, which indicates that the method is stable and robust with respect to the choice of the reference (or normal) samples. Another test was also performed to check the robustness of the SSN from different reference sample sizes based on a breast cancer dataset. We randomly chose 15, 20, 30 and 50 normal samples from 99 normal samples as reference samples, and calculated the SSN for all tumor samples. The new SSNs were then compared with the old SSNs from 17 control samples, and this test was repeated 100 times. The average recurrence rate of edges in the new SSNs relative to the old SSNs from 17 control samples is 80.34, 82.38, 83.8 and 85.17% for reference sample sizes 15, 20, 30 and 50, respectively. As the number of reference samples increases, the standard deviation of these percentages slowly decreases from 7.9% for 15 samples and 6.9% for 20 samples to 6.2% for 30 samples and 5.9% for 50 samples. These results indicate that the method is robust and stable for the different reference sample sizes. In addition, we studied the basic statistics of SSNs for BRCA with normal samples as reference samples. The average size of the 99 SSNs is 167.9 edges per SSN, and in contrast, that of SSNs for 761 disease samples is 2466.3 edges per SSN as shown in Supplementary Table S2. Clearly, the sizes of the SSNs are significantly different between normal and cancer samples. The average number of the connected components for the 99 SSNs from normal samples is 62.11, while that for the 761 SSNs from disease samples is 209.94. Hence, the SSNs of cancer samples have bigger network sizes and more connected components than the SSNs of normal samples by using the normal samples as reference samples.
The high-degree genes in the SSN represent important features for a cancer sample, and are strongly related to the DMGs in the individual sample, which may be beneficial to personalized diagnosis and individualized treatment. We show that the high-degree genes of an SSN can be used to predict the DMGs for each sample, and the accuracy of the prediction for the DMGs increases with the degree of the gene in SSNs. This property implies that SSNs can be used to detect the functional drivers solely based on the expression data even without the sequence information.
Generally, there are three types of perturbations for a hub in an SSN, i.e. a hub in an SSN could be the result of (i) a perturbation in the hub gene itself, (ii) a perturbation of its interaction partners or (iii) a combination of both, although there are few hub genes for the type ‘(i)’ among TCGA samples. Because a hub gene is generally connected with other downstream and upstream genes through a feedback network and it is impossible for a perturbation of the hub gene not to affect downstream genes by regulation or upstream genes by feedback, so there is few type ‘(i)’ in biological systems. On the other hand, types ‘(ii)’ and ‘(iii)’ are widely observed in TCGA samples. Even for one hub gene in the same disease, there are both types ‘(ii)’ and ‘(iii)’. For example, TP53 has no differential expression in samples A0HO and A0DP in breast cancer (Figure 3A), i.e. TP53 is the type ‘(ii)’ in these two samples, but in samples A0B0 and A25A, TP53 is overexpressed relative to the control samples (Figure 3A), i.e. TP53 is the type ‘(iii)’ in these two samples. In particular, there are 37531 hub genes with high degrees (i.e. at least 10 neighbors) in SSNs of 761 breast cancer samples, where no sample is of type ‘(i)’, 93 of 37 531 hub genes belong to type ‘(ii)’ and most of the hub genes are of type ‘(iii)’.
It should be noted that an SSN in this work is not a real molecular network for each sample but a perturbation network for a single sample against the reference network. It reflects the variation between normal and disease samples in terms of interactions, regulations or a network, similarly to differential expression of a gene, which is not the real gene expression level for each sample but the variation of the gene expression between normal and disease samples. In contrast to a molecular network inferred by traditional methods, which is actually an aggregated network for multiple samples, our method can construct an SSN on a single-sample basis and thus can be directly applied to the data analysis of single samples, in particular with potential applications to precision medicine and personalized medicine.
This method is based on the PCC, and thus the correlation network is the desired choice to construct the SSN. However, humans have more than 20 000 genes, which implies that the full-correlation network for humans has more than 200 000 000 edges. Hence, construction of such a network is computationally intense; in addition, the correlation network includes indirect associations, which are false-positive connections in a molecular network. In this work, we adopted the background or reference network to reduce the false-positive connections, which also significantly alleviates the computational and storage requirements.
Some recent studies (58) developed a method to decompose the aggregated associations of a group of samples into those of individual samples. This method has some similarities with ours but is notably different. In particular, this method approximately decomposes the association or PCC into a group of networks corresponding to individual samples. There are three major differences between those two methods. First, an SSN in this work is actually a perturbed network of an individual sample from a group of reference samples, which characterizes the individual sample at the network level in an accurate manner. In contrast, this method uses an approximation scheme to decompose an association (or aggregated) network of a group of samples approximately into individual networks corresponding to individual samples in a linear manner. Second, our method evaluates a new single sample based on a group of reference samples, whereas this method evaluates a single sample in the group. Third, an SSN is constructed using a statistic based on a new type of distribution, the volcano distribution, which can be proven mathematically and ensured by statistical theory. However, this method uses no such statistic, and furthermore the correlation in each individual network can be >1 or <−1 due to its heuristic scheme (see Equation 31 in (58)).
Biological experiments on drug resistance validated not only the effectiveness of our method for constructing SSNs by single samples, but also one advantage of our method, i.e. identifying non-differentially expressed disease genes or factors as functional drivers, which are generally missed by traditional methods. As shown in Figure 7, although there are no differentially expressed genes between PC9-DR and PC9 in the top 15 candidate functional driver genes, the expression of some genes was actually reduced from PC9 to PC9-DR (Figure 7B) (not significant in terms of fold change). After knocking down these genes, i.e. BRCA1, TGFBR1 and CASP3, the drug resistance of PC9-DR showed very significant changes (Figures 7B and C). This result cannot be explained by traditional analysis based on differential expression, but in our work, we show that the correlations of these genes with their neighbors were increased from PC9 to PC9-DR, i.e. the regulation between these genes and their neighbors increased with the drug resistance, even though their gene expression decreased. Thus, deeply knocking down those genes reduced the regulation with their neighbors and therefore changed the drug resistance. For TCGA data, we found a number of representative or significant edges in each cancer stage, but it is an interesting problem to identify the consistent network patterns formed by those edges in different cancer stages. In addition, we used both the background network and the FDR correction to reduce the noise and false-positive ratio, but how to further remove the inherent noise is our future topic.
In this work, we constructed an association network by correlation, which includes both the effect of direct and indirect regulation between two genes. Actually, instead of the correlation or PCC, we can similarly use partial correlation, conditional mutual information or part mutual information to construct a direct association network (13,59,60), which will be addressed in future studies.
Supplementary Material
Acknowledgments
The authors also appreciate the valuable suggestions of Dr Dangsheng Li on both the experiments and the manuscript.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Strategic Priority Research Program of the Chinese Academy of Sciences [XDB13040700]; National Program on Key Basic Research Projects [2014CB910504, 2012CB910800]; National Natural Science Foundation of China (NSFC) [91529303, 61134013, 91439103, 61403363, 81430066, 81402276, 81402371, 81401898, 81402498, 81325015, 31370747, 81101583, 81372509, 81471047]; Science and Technology Commission of Shanghai Municipality [15XD1504000]; Key projects of natural science of Anhui Provincial Education Department (KJ2016A002); JSPS KAKENHI [15H05707]; JST's “Super Highway", the accelerated research program to bridge university IPs and practical use. Funding for open access charge: Strategic Priority Research Program of the Chinese Academy of Sciences [XDB13040700].
Conflict of interest statement. None declared.
REFERENCES
- 1.Hood L., Flores M. A personal view on systems medicine and the emergence of proactive P4 medicine: predictive, preventive, personalized and participatory. N. Biotechnol. 2012;29:613–624. doi: 10.1016/j.nbt.2012.03.004. [DOI] [PubMed] [Google Scholar]
- 2.Barabasi A.L., Gulbahce N., Loscalzo J. Network medicine: a network-based approach to human disease. Nat. Rev. Genet. 2011;12:56–68. doi: 10.1038/nrg2918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen L., Wang R.-S., Zhang X.-S. Biomolecular Networks: Methods and Applications in Systems Biology. Hoboken: John Wiley & Sons Inc; 2009. [Google Scholar]
- 4.Chen L., Wang R., Li C., Aihara K. Modeling Biomolecular Networks in Cells: Structures and Dynamics. London: Springer-Verlag; 2010. [Google Scholar]
- 5.Ideker T., Krogan N.J. Differential network biology. Mol. Syst. Biol. 2012;8:565. doi: 10.1038/msb.2011.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu X., Liu Z.P., Zhao X.M., Chen L. Identifying disease genes and module biomarkers by differential interactions. J. Am. Med. Inform. Assoc. 2012;19:241–248. doi: 10.1136/amiajnl-2011-000658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fang Z., Tian W., Ji H. A network-based gene-weighting approach for pathway analysis. Cell Res. 2012;22:565–580. doi: 10.1038/cr.2011.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu R., Wang X., Aihara K., Chen L. Early diagnosis of complex diseases by molecular biomarkers, network biomarkers, and dynamical network biomarkers. Med. Res. Rev. 2014;34:455–478. doi: 10.1002/med.21293. [DOI] [PubMed] [Google Scholar]
- 9.Zhang W., Zeng T., Liu X., Chen L. Diagnosing phenotypes of single-sample individuals by edge biomarkers. J. Mol. Cell Biol. 2015;7:231–241. doi: 10.1093/jmcb/mjv025. [DOI] [PubMed] [Google Scholar]
- 10.Zhang W., Zeng T., Chen L. EdgeMarker: Identifying differentially correlated molecule pairs as edge-biomarkers. J. Theor. Biol. 2014;362:35–43. doi: 10.1016/j.jtbi.2014.05.041. [DOI] [PubMed] [Google Scholar]
- 11.De Bodt S., Proost S., Vandepoele K., Rouze P., Van de Peer Y. Predicting protein-protein interactions in Arabidopsis thaliana through integration of orthology, gene ontology and co-expression. BMC Genomics. 2009;10:288. doi: 10.1186/1471-2164-10-288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bhardwaj N., Lu H. Correlation between gene expression profiles and protein-protein interactions within and across genomes. Bioinformatics. 2005;21:2730–2738. doi: 10.1093/bioinformatics/bti398. [DOI] [PubMed] [Google Scholar]
- 13.Zhang X., Liu K., Liu Z.P., Duval B., Richer J.M., Zhao X.M., Hao J.K., Chen L. NARROMI: a noise and redundancy reduction technique improves accuracy of gene regulatory network inference. Bioinformatics. 2013;29:106–113. doi: 10.1093/bioinformatics/bts619. [DOI] [PubMed] [Google Scholar]
- 14.Meric-Bernstam F., Farhangfar C., Mendelsohn J., Mills G.B. Building a personalized medicine infrastructure at a major cancer center. J. Clin. Oncol. 2013;31:1849–1857. doi: 10.1200/JCO.2012.45.3043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mendelsohn J. Personalizing oncology: perspectives and prospects. J. Clin. Oncol. 2013;31:1904–1911. doi: 10.1200/JCO.2012.45.3605. [DOI] [PubMed] [Google Scholar]
- 16.Liu X., Wang J., Chen L. Whole-exome sequencing reveals recurrent somatic mutation networks in cancer. Cancer Lett. 2013;340:270–276. doi: 10.1016/j.canlet.2012.11.002. [DOI] [PubMed] [Google Scholar]
- 17.Ahn T., Lee E., Huh N., Park T. Personalized identification of altered pathways in cancer using accumulated normal tissue data. Bioinformatics. 2014;30:i422–i429. doi: 10.1093/bioinformatics/btu449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Drier Y., Sheffer M., Domany E. Pathway-based personalized analysis of cancer. Proc. Natl. Acad. Sci. U.S.A. 2013;110:6388–6393. doi: 10.1073/pnas.1219651110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rivals I., Personnaz L., Taing L., Potier M.C. Enrichment or depletion of a GO category within a class of genes: which test. Bioinformatics. 2007;23:401–407. doi: 10.1093/bioinformatics/btl633. [DOI] [PubMed] [Google Scholar]
- 20.Li F., Han X., Li F., Wang R., Wang H., Gao Y., Wang X., Fang Z., Zhang W., Yao S., et al. LKB1 inactivation elicits a Redox imbalance to modulate non-small cell lung cancer plasticity and therapeutic response. Cancer Cell. 2015;27:698–711. doi: 10.1016/j.ccell.2015.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gao Y., Zhang W., Han X., Li F., Wang X., Wang R., Fang Z., Tong X., Yao S., Li F., et al. YAP inhibits squamous transdifferentiation of Lkb1-deficient lung adenocarcinoma through ZEB2-dependent DNp63 repression. Nat. Commun. 2014;5:4629. doi: 10.1038/ncomms5629. [DOI] [PubMed] [Google Scholar]
- 22.Rice J.A. Mathematical statistics and data analysis. 3rd edn. Belmont: Thomson/Brooks/Cole; 2007. [Google Scholar]
- 23.Wreschner D.H., Zrihan-Licht S., Baruch A., Sagiv D., Hartman M.L., Smorodinsky N., Keydar I. Does a novel form of the breast cancer marker protein, MUC1, act as a receptor molecule that modulates signal transduction. Adv. Exp. Med. Biol. 1994;353:17–26. doi: 10.1007/978-1-4615-2443-4_3. [DOI] [PubMed] [Google Scholar]
- 24.Wei X., Xu H., Kufe D. Human MUC1 oncoprotein regulates p53-responsive gene transcription in the genotoxic stress response. Cancer Cell. 2005;7:167–178. doi: 10.1016/j.ccr.2005.01.008. [DOI] [PubMed] [Google Scholar]
- 25.Wei X., Xu H., Kufe D. Human mucin 1 oncoprotein represses transcription of the p53 tumor suppressor gene. Cancer Res. 2007;67:1853–1858. doi: 10.1158/0008-5472.CAN-06-3063. [DOI] [PubMed] [Google Scholar]
- 26.Gimmi C.D., Morrison B.W., Mainprice B.A., Gribben J.G., Boussiotis V.A., Freeman G.J., Park S.Y., Watanabe M., Gong J., Hayes D.F., et al. Breast cancer-associated antigen, DF3/MUC1, induces apoptosis of activated human T cells. Nat. Med. 1996;2:1367–1370. doi: 10.1038/nm1296-1367. [DOI] [PubMed] [Google Scholar]
- 27.Kufe D.W. MUC1-C oncoprotein as a target in breast cancer: activation of signaling pathways and therapeutic approaches. Oncogene. 2013;32:1073–1081. doi: 10.1038/onc.2012.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yu M., Zhan Q., Finn O.J. Immune recognition of cyclin B1 as a tumor antigen is a result of its overexpression in human tumors that is caused by non-functional p53. Mol. Immunol. 2002;38:981–987. doi: 10.1016/s0161-5890(02)00026-3. [DOI] [PubMed] [Google Scholar]
- 29.Innocente S.A., Abrahamson J.L., Cogswell J.P., Lee J.M. p53 regulates a G2 checkpoint through cyclin B1. Proc. Natl. Acad. Sci. U.S.A. 1999;96:2147–2152. doi: 10.1073/pnas.96.5.2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ding K., Li W., Zou Z., Zou X., Wang C. CCNB1 is a prognostic biomarker for ER+ breast cancer. Med. Hypotheses. 2014;83:359–364. doi: 10.1016/j.mehy.2014.06.013. [DOI] [PubMed] [Google Scholar]
- 31.Suzuki T., Urano T., Miki Y., Moriya T., Akahira J., Ishida T., Horie K., Inoue S., Sasano H. Nuclear cyclin B1 in human breast carcinoma as a potent prognostic factor. Cancer Sci. 2007;98:644–651. doi: 10.1111/j.1349-7006.2007.00444.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Luan F.L., Ding R., Sharma V.K., Chon W.J., Lagman M., Suthanthiran M. Rapamycin is an effective inhibitor of human renal cancer metastasis. Kidney Int. 2003;63:917–926. doi: 10.1046/j.1523-1755.2003.00805.x. [DOI] [PubMed] [Google Scholar]
- 33.Rennel E., Mellberg S., Dimberg A., Petersson L., Botling J., Ameur A., Westholm J.O., Komorowski J., Lassalle P., Cross M.J., et al. Endocan is a VEGF-A and PI3K regulated gene with increased expression in human renal cancer. Exp. Cell Res. 2007;313:1285–1294. doi: 10.1016/j.yexcr.2007.01.021. [DOI] [PubMed] [Google Scholar]
- 34.Chuang S.T., Patton K.T., Schafernak K.T., Papavero V., Lin F., Baxter R.C., Teh B.T., Yang X.J. Over expression of insulin-like growth factor binding protein 3 in clear cell renal cell carcinoma. J. Urol. 2008;179:445–449. doi: 10.1016/j.juro.2007.09.106. [DOI] [PubMed] [Google Scholar]
- 35.Cheung C.W., Vesey D.A., Nicol D.L., Johnson D.W. The roles of IGF-I and IGFBP-3 in the regulation of proximal tubule, and renal cell carcinoma cell proliferation. Kidney Int. 2004;65:1272–1279. doi: 10.1111/j.1523-1755.2004.00535.x. [DOI] [PubMed] [Google Scholar]
- 36.Aubert S., Fauquette V., Hemon B., Lepoivre R., Briez N., Bernard D., Van Seuningen I., Leroy X., Perrais M. MUC1, a new hypoxia inducible factor target gene, is an actor in clear renal cell carcinoma tumor progression. Cancer Res. 2009;69:5707–5715. doi: 10.1158/0008-5472.CAN-08-4905. [DOI] [PubMed] [Google Scholar]
- 37.Wang X., Chen J.X., Liu J.P., You C., Liu Y.H., Mao Q. Gain of function of mutant TP53 in glioblastoma: prognosis and response to temozolomide. Ann. Surg. Oncol. 2014;21:1337–1344. doi: 10.1245/s10434-013-3380-0. [DOI] [PubMed] [Google Scholar]
- 38.Li Y., Guessous F., Kwon S., Kumar M., Ibidapo O., Fuller L., Johnson E., Lal B., Hussaini I., Bao Y., et al. PTEN has tumor-promoting properties in the setting of gain-of-function p53 mutations. Cancer Res. 2008;68:1723–1731. doi: 10.1158/0008-5472.CAN-07-1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Angeloni S.V., Martin M.B., Garcia-Morales P., Castro-Galache M.D., Ferragut J.A., Saceda M. Regulation of estrogen receptor-alpha expression by the tumor suppressor gene p53 in MCF-7 cells. J. Endocrinol. 2004;180:497–504. doi: 10.1677/joe.0.1800497. [DOI] [PubMed] [Google Scholar]
- 40.Rasti M., Arabsolghar R., Khatooni Z., Mostafavi-Pour Z. p53 binds to estrogen receptor 1 promoter in human breast cancer cells. Pathol. Oncol. Res. 2012;18:169–175. doi: 10.1007/s12253-011-9423-6. [DOI] [PubMed] [Google Scholar]
- 41.Choy B., Findeis-Hosey J.J., Li F., McMahon L.A., Yang Q., Xu H. High frequency of coexpression of maspin with p63 and p53 in squamous cell carcinoma but not in adenocarcinoma of the lung. Int. J. Clin. Exp. Pathol. 2013;6:2542–2547. [PMC free article] [PubMed] [Google Scholar]
- 42.Moon D.O., Kim M.O., Nam T.J., Kim S.K., Choi Y.H., Kim G.Y. Pectenotoxin-2 induces G2/M phase cell cycle arrest in human breast cancer cells via ATM and Chk1/2-mediated phosphorylation of cdc25C. Oncol. Rep. 2010;24:271–276. doi: 10.3892/or_00000856. [DOI] [PubMed] [Google Scholar]
- 43.St Clair S., Giono L., Varmeh-Ziaie S., Resnick-Silverman L., Liu W.J., Padi A., Dastidar J., DaCosta A., Mattia M., Manfredi J.J. DNA damage-induced downregulation of Cdc25C is mediated by p53 via two independent mechanisms: one involves direct binding to the cdc25C promoter. Mol. Cell. 2004;16:725–736. doi: 10.1016/j.molcel.2004.11.002. [DOI] [PubMed] [Google Scholar]
- 44.Albert H., Battaglia E., Monteiro C., Bagrel D. Genotoxic stress modulates CDC25C phosphatase alternative splicing in human breast cancer cell lines. Mol. Oncol. 2012;6:542–552. doi: 10.1016/j.molonc.2012.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Blau C.A., Liakopoulou E. Can we deconstruct cancer, one patient at a time. Trends Genet. 2013;29:6–10. doi: 10.1016/j.tig.2012.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vogelstein B., Papadopoulos N., Velculescu V.E., Zhou S., Diaz L.A., Jr, Kinzler K.W. Cancer genome landscapes. Science. 2013;339:1546–1558. doi: 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tothill R.W., Tinker A.V., George J., Brown R., Fox S.B., Lade S., Johnson D.S., Trivett M.K., Etemadmoghadam D., Locandro B., et al. Novel molecular subtypes of serous and endometrioid ovarian cancer linked to clinical outcome. Clin. Cancer Res. 2008;14:5198–5208. doi: 10.1158/1078-0432.CCR-08-0196. [DOI] [PubMed] [Google Scholar]
- 48.Reis-Filho J.S., Pusztai L. Gene expression profiling in breast cancer: classification, prognostication, and prediction. Lancet. 2011;378:1812–1823. doi: 10.1016/S0140-6736(11)61539-0. [DOI] [PubMed] [Google Scholar]
- 49.Kim J.K., Kim T.K., Ahn H.J., Kim C.S., Kim K.R., Cho K.S. Differentiation of subtypes of renal cell carcinoma on helical CT scans. AJR. Am. J. Roentgenol. 2002;178:1499–1506. doi: 10.2214/ajr.178.6.1781499. [DOI] [PubMed] [Google Scholar]
- 50.Hofree M., Shen J.P., Carter H., Gross A., Ideker T. Network-based stratification of tumor mutations. Nat. Methods. 2013;10:1108–1115. doi: 10.1038/nmeth.2651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Stratton M.R., Campbell P.J., Futreal P.A. The cancer genome. Nature. 2009;458:719–724. doi: 10.1038/nature07943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cerami E., Gao J., Dogrusoz U., Gross B.E., Sumer S.O., Aksoy B.A., Jacobsen A., Byrne C.J., Heuer M.L., Larsson E., et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–404. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Siegel R., Ma J., Zou Z., Jemal A. Cancer statistics, 2014. CA Cancer J. Clin. 2014;64:9–29. doi: 10.3322/caac.21208. [DOI] [PubMed] [Google Scholar]
- 54.Sordella R., Bell D.W., Haber D.A., Settleman J. Gefitinib-sensitizing EGFR mutations in lung cancer activate anti-apoptotic pathways. Science. 2004;305:1163–1167. doi: 10.1126/science.1101637. [DOI] [PubMed] [Google Scholar]
- 55.Mok T.S., Wu Y.L., Thongprasert S., Yang C.H., Chu D.T., Saijo N., Sunpaweravong P., Han B., Margono B., Ichinose Y., et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N. Eng. J. Med. 2009;361:947–957. doi: 10.1056/NEJMoa0810699. [DOI] [PubMed] [Google Scholar]
- 56.Ji H., Li D., Chen L., Shimamura T., Kobayashi S., McNamara K., Mahmood U., Mitchell A., Sun Y., Al-Hashem R., et al. The impact of human EGFR kinase domain mutations on lung tumorigenesis and in vivo sensitivity to EGFR-targeted therapies. Cancer Cell. 2006;9:485–495. doi: 10.1016/j.ccr.2006.04.022. [DOI] [PubMed] [Google Scholar]
- 57.Jiang J. The ‘dark matter’ in the plant genomes: non-coding and unannotated DNA sequences associated with open chromatin. Curr. Opin. Plant Biol. 2015;24:17–23. doi: 10.1016/j.pbi.2015.01.005. [DOI] [PubMed] [Google Scholar]
- 58.Kuijjer M.L., Tung M., Yuan G., Quackenbush J., Glass K. Estimating sample-specific regulatory networks. ArXiv e-prints. 2015 doi: 10.1016/j.isci.2019.03.021. http://arxiv.org/abs/1505.06440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang X., Zhao J., Hao J.K., Zhao X.M., Chen L. Conditional mutual inclusive information enables accurate quantification of associations in gene regulatory networks. Nucleic Acids Res. 2015;43:e31. doi: 10.1093/nar/gku1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhao J., Zhou Y., Zhang X., Chen L. Part mutual information for quantifying direct associations in networks. Proc. Natl. Acad. Sci. U.S.A. 2016;113:5130–5135. doi: 10.1073/pnas.1522586113. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.