


Abstract
Gene 32 protein (gp32) is the single-stranded (ss) DNA binding protein of the bacteriophage T4. It binds transiently and cooperatively to ssDNA sequences exposed during the DNA replication process and regulates the interactions of the other sub-assemblies of the replication complex during the replication cycle. We here use single-molecule FRET techniques to build on previous thermodynamic studies of gp32 binding to initiate studies of the dynamics of the isolated and cooperative binding of gp32 molecules within the replication complex. DNA primer/template (p/t) constructs are used as models to determine the effects of ssDNA lattice length, gp32 concentration, salt concentration, binding cooperativity and binding polarity at p/t junctions. Hidden Markov models (HMMs) and transition density plots (TDPs) are used to characterize the dynamics of the multi-step assembly pathway of gp32 at p/t junctions of differing polarity, and show that isolated gp32 molecules bind to their ssDNA targets weakly and dissociate quickly, while cooperatively bound dimeric or trimeric clusters of gp32 bind much more tightly, can ‘slide’ on ssDNA sequences, and exhibit binding dynamics that depend on p/t junction polarities. The potential relationships of these binding dynamics to interactions with other components of the T4 DNA replication complex are discussed.
INTRODUCTION
The DNA replication system of T4 bacteriophage has been studied for many years by our groups and others (1–6), and is clearly established as the simplest model for understanding many aspects of the more complex systems of higher organisms. This follows in part because T4 is the simplest creature that utilizes the three major replication sub-assemblies characteristic of higher organisms, including a hexameric helicase–primase complex that ‘opens’ the duplex DNA genome, exposes the single-stranded (ss) DNA templates and primes (with short RNA sequences) DNA synthesis on the lagging strand template; a pair of DNA polymerases that engage in ‘coupled’ leading and lagging strand DNA synthesis (and initial ‘editing’ to assure high replication fidelity) within the ‘trombone-shaped’ DNA framework of the replication complex (7,8); and circular trimeric (or dimeric) replication clamps that are opened and closed and properly positioned on the polymerases by a pentameric complex that hydrolyses ATP and serves as a clamp-loader (and in many cases clamp-‘remover’) to control the processivity of DNA synthesis (9–11).
The activities of these three sub-assemblies are regulated and integrated by a ssDNA binding protein (called gene product 32 or gp32 in the T4 system) that binds cooperatively to the ssDNA sequences, and which are transiently formed during the replication process. This binding serves to protect the exposed strands against attack by ssDNA nucleases, to prevent the formation of disruptive secondary structures within these ssDNA sequences, and to extend the ssDNA into an optimal templating conformation for the homologous polymerases. Slightly weaker, yet equally cooperative binding of gp32 to single-stranded sequences within its cognate messenger RNA also controls the translational synthesis of gp32, and thus its concentration within the phage-infected E. coli system (12,13). The binding and removal of cooperatively bound clusters of gp32 molecules from ssDNA sequences, which is necessary for the proper function of the helicases, polymerases and clamp-loaders within the overall replication cycle, is controlled by additional T4-coded protein factors. All these factors and complexes must interact within the replication complex with appropriate binding affinities and association–dissociation kinetics to permit—for the T4 bacteriophage—successful infection of the host E. coli, and subsequent DNA replication to produce progeny phage.
In earlier cycles of research on the mechanisms and interactions of gp32 (and also on the ssDNA binding proteins of higher organisms), bulk solution studies were used to define the overall binding thermodynamics of this protein to ssDNA as a function of lattice length and base composition. However, significant high-resolution work is only now getting underway to establish the detailed structure and interactions of gp32 with ssDNA and other regulatory proteins during the replication cycle. Initial bulk solution studies involving the site-specific placement of fluorescent (and CD-active) base analogues within the DNA of replication fork model complexes have permitted the ‘mapping,’ to single nucleotide resolution, of the interactions within the gp32 binding site that are responsible for ssDNA binding and cooperativity (14,15). These bulk solution studies have thus far been confined to investigations of equilibrium and steady-state binding questions.
The advent of single-molecule spectroscopic techniques now permits us to use optical probes to study the dynamics of gp32–ssDNA interactions, and to begin to learn how these molecules function within the DNA replication cycle. In this work, we present an initial approach to these issues that is based on the analysis of single-molecule Förster resonance energy transfer (smFRET) studies of ssDNA backbone fluctuations at ∼100 ms time resolution. These experiments, using appropriately positioned fluorescent cyanine dyes as smFRET pairs on DNA replication-fork DNA constructs, have led to an increased understanding of the assembly dynamics of cooperatively bound clusters of gp32 molecules on the ssDNA ‘tails’ of model replication constructs as a function of ssDNA length, strand polarity and protein interactions with the primer-template (p/t) DNA junction.
MATERIALS AND METHODS
DNA constructs
All DNA oligonucleotides were purchased from Integrated DNA Technologies (Coralville, IA, USA) (Table 1). The DNA substrates were annealed by adding the biotinylated strand and the non-biotinylated strand in a 1:1.5 ratio for single-molecule or in a 1:1 ratio for bulk FRET experiments at 100 nM concentration of DNA strands to a 1.5 ml siliconized Eppendorf tube with a standard imaging buffer (10 mM Tris at pH 8.0, 100 mM NaCl and 6 mM MgCl2), as described in detail elsewhere (6,22).
Table 1. Nucleotide base sequences and nomenclature for the p/t DNA constructs used in these studies.
| DNA construct | Nucleotide base sequence |
|---|---|
| 3′-tail-pdT7-Cy3/Cy5 | 5′-GTCGCCAGCCTCGCAGCCTTTTTTT/Cy3/-3′ |
| 3′-/biotin/CAGCGGTCGGAGCGTCGG-Cy5/-5′ | |
| 3′-tail-pdT14-Cy3/Cy5 | 5′-GTCGCCAGCCTCGCAGCCTTTTTTTTTTTTTT/Cy3/-3′ |
| 3′-/biotin/CAGCGGTCGGAGCGTCGG-Cy5/-5′ | |
| 3′-tail-pdT15-Cy3/Cy5 | 5′-GTCGCCAGCCTCGCAGCCTTTTTTTTTTTTTTT/Cy3/-3′ |
| 3′-/biotin/CAGCGGTCGGAGCGTCGG-Cy5/-5′ | |
| 3′-tail-pd(mixed-base-seq)15-Cy3/Cy5 | 5′-GTCGCCAGCCTCGCAGCCCAAGAGTAATAGACG/Cy3/-3′ |
| 3′-/biotin/CAGCGGTCGGAGCGTCGG-Cy5/-5′ | |
| 3′-tail-pdT22-Cy3/Cy5 | 5′-GTCGCCAGCCTCGCAGCCTTTTTTTTTTTTTTTTTTTTTT/Cy3/-3′ |
| 3′-/biotin/CAGCGGTCGGAGCGTCGG-Cy5/-5′ | |
| 5′-tail-pdT7-Cy3/Cy5 | 5′-/Cy3/TTTTTTTCCGACGCTCCGACCGCTG-3′ |
| 3′-/Cy5/GGCTGCGAGGCTGGCGAC/biotin/-5′ | |
| 5′-tail-pdT15-Cy3/Cy5 | 5′-/Cy3/TTTTTTTTTTTTTTTCCGACGCTCCGACCGCTG-3′ |
| 3′-/Cy5/GGCTGCGAGGCTGGCGAC/biotin/-5′ | |
| 5′-tail-pdT22-Cy3/Cy5 | 5′-/Cy3/TTTTTTTTTTTTTTTTTTTTTTCCGACGCTCCGACCGCTG-3′ |
| 3′-/Cy5/GGCTGCGAGGCTGGCGAC/biotin/-5′ |
Preparation of gp32 and determination of protein concentration
Psy6/AR120 cells were grown to an optical density (OD600) of 0.9–1.0 at 37°C in Luria–Bertani liquid medium (LB broth) containing 50 μg/ml ampicillin. Cells were induced by adding nalidixic acid to a final concentration of 40 μg/ml, grown for an additional 8 to 10 h at 37°C and harvested. The gp32 protein was then purified according to the procedure of Bittner et al. (41) and the concentrations were determined by absorbance at 280 nm, assuming a molar extinction coefficient of 37 × l03 M−1 cm-1 (16,17). The gp32 protein (∼260 μM) was kept in storage buffer (20 mM Tris-OAc buffer, pH 8.1 containing 0.5 mM DTT, 1 mM EDTA, 50 mM KOAc, 50% glycerol), and properly diluted to make a specified concentration with a standard imaging buffer for bulk and smFRET experiments.
Bulk measurements
Absorbance spectra of our samples were obtained using a Cary UV-Vis 3E spectrophotometer equipped with a Peltier temperature controller. Fluorescence spectra were measured using a Horiba FluoroMax 4 spectrophotometer. For our bulk fluorescence FRET measurements, DNA oligomers were annealed at 100 nM concentrations in standard imaging buffer in 1.5 ml Eppendorf tubes using a heating block, and then transferred to a quartz 1-cm path length cuvette for measurement. The samples were excited at 532 nm while maintaining the temperature of the cuvette at 20°C using a Neslab RTE-210 thermostat. Fluorescence spectra were recorded over the range 545–750 nm, as shown in Figure 1. The bulk FRET efficiency was calculated using the formula 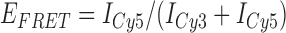 , with
, with 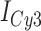 and
and 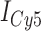 representing the peak fluorescence intensities of the donor and acceptor chromophores, respectively. For our protein titration experiments, the proper amount of protein (gp32) solution was added sequentially to the cuvette to vary the gp32 concentration from 0.1 to 10 μM, while accounting for the dilution of the Cy3/Cy5 p/t DNA construct. At each of the specified gp32 concentrations, the solution was incubated for 2 min before recording fluorescence spectra.
representing the peak fluorescence intensities of the donor and acceptor chromophores, respectively. For our protein titration experiments, the proper amount of protein (gp32) solution was added sequentially to the cuvette to vary the gp32 concentration from 0.1 to 10 μM, while accounting for the dilution of the Cy3/Cy5 p/t DNA construct. At each of the specified gp32 concentrations, the solution was incubated for 2 min before recording fluorescence spectra.
Figure 1.

Schematic illustrations of the non-cooperative and cooperative binding of gp32 monomers to: (A) the 3′-tail-pdT7-Cy3/Cy5 p/t DNA construct; (B) the 3′-tail-pdT15-Cy3/Cy5 p/t DNA construct; and (C) the 3′-tail-pdT22-Cy3/Cy5 p/t DNA construct. The single-stranded regions of these DNA constructs can bind (A) 0 or 1, (B) 0, 1 or 2 or (C) 0, 1, 2 or 3 gp32 proteins, respectively. (D) Bulk fluorescence measurements of the 3′-tail-pdT15-Cy3/Cy5 construct at 100 nM oligomer concentration exhibited changes in the FRET efficiency upon titration with gp32. Red, blue and green curves are partially overlapping. The inset shows the values of 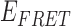 calculated from the peak Cy3/Cy5 fluorescence intensities. (E) The results of bulk fluorescence gp32 titration measurements (at 100 mM NaCl, 6 mM MgCl2 and 10 mM Tris (pH 8.0) concentrations) for initial 100 nM concentrations of six of the p/t DNA constructs used in our studies (for both 5′ and 3′ oliogo-dT tails of length N = 7, 15 and 22).
calculated from the peak Cy3/Cy5 fluorescence intensities. (E) The results of bulk fluorescence gp32 titration measurements (at 100 mM NaCl, 6 mM MgCl2 and 10 mM Tris (pH 8.0) concentrations) for initial 100 nM concentrations of six of the p/t DNA constructs used in our studies (for both 5′ and 3′ oliogo-dT tails of length N = 7, 15 and 22).
smFRET experiments
All of our single-molecule experiments were performed using a prism-based TIRF microscope that was equipped with a 532 nm laser and 100x NA, 1.4 oil-immersion objective (Plan Apo, Nikon), as previously described (6,22). Experiments were carried out at room temperature (22°C) in a temperature-controlled laboratory that is stabilized to within ±0.5°F. Sample solutions were prepared using the standard imaging buffer unless otherwise specified. These solutions contained the oxygen scavenging and triplet quenching system used only for smFRET measurements: 165 U/ml glucose oxidase (Sigma), 0.8% (w/v) D-glucose (Sigma), 2170 U/ml Catalase (Sigma) in a Trolox solution (≥1 mM, Sigma). Protein (gp32) solutions at various concentrations containing these oxygen scavenging and triplet quenching system were incubated in a microfluidic sample chamber for 2–3 min before data acquisition. Unless otherwise specified, data were acquired without flushing the unbound proteins from the sample chamber. Data sets were generally collected on the same day for a given experiment (e.g. salt concentration dependence of a particular substrate, gp32 concentration dependence), and the results of these experiments were reproduced on separate days.
Data analysis
Individual image frames of a typical data movie contained hundreds of single molecule features, which we analyzed using software packages (IDL and MatLab) that were generously provided by Prof. Taekjip Ha (Johns Hopkins). The program identifies single-molecule features in sequence that simultaneously exhibited donor (Cy3) and acceptor (Cy5) signals, and plots their respective time-dependent intensities as individual trajectories (see, for example, the left-hand column of Figure 2). These donor and acceptor intensities were used to calculate the smFRET efficiencies according to the same formula that we used for bulk experiments: 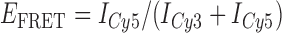 . Histograms of the smFRET efficiencies (e.g. those shown in the right-hand column of Figure 2) were each constructed from ∼10 single-molecule data sets, each of which monitored ∼200 single-molecule features over a duration of 120 s (1200 frames per data set at 100 ms per frame). The protocol computes the average
. Histograms of the smFRET efficiencies (e.g. those shown in the right-hand column of Figure 2) were each constructed from ∼10 single-molecule data sets, each of which monitored ∼200 single-molecule features over a duration of 120 s (1200 frames per data set at 100 ms per frame). The protocol computes the average 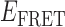 value for each single-molecule feature detected within the first 12 frames of the data set (while discarding frames 1 and 2). For each of the ∼100 molecules detected within an image frame, its average
value for each single-molecule feature detected within the first 12 frames of the data set (while discarding frames 1 and 2). For each of the ∼100 molecules detected within an image frame, its average 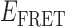 value was recorded as a single ‘count’ that was entered into the histogram. For all of the
value was recorded as a single ‘count’ that was entered into the histogram. For all of the 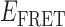 histograms shown in this work, an artifact feature appears with maximum
histograms shown in this work, an artifact feature appears with maximum 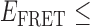 0.1, which is due to the inclusion of signals from inactive (i.e. photo-bleached) acceptor molecules and some minor leakage of donor fluorescence into the acceptor channel (see Supplementary Figure S8). The software we used to perform the Hidden Markov Model (HMM) analysis was also provided by Taekjip Ha, which we used to determine peak-to-peak persistence times, and transition density plots (TDPs). We confirmed the validity of our HMM analysis by testing it against simulated smFRET trajectories for two-state systems with known initial and final
0.1, which is due to the inclusion of signals from inactive (i.e. photo-bleached) acceptor molecules and some minor leakage of donor fluorescence into the acceptor channel (see Supplementary Figure S8). The software we used to perform the Hidden Markov Model (HMM) analysis was also provided by Taekjip Ha, which we used to determine peak-to-peak persistence times, and transition density plots (TDPs). We confirmed the validity of our HMM analysis by testing it against simulated smFRET trajectories for two-state systems with known initial and final 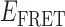 values (see Supplementary Figure S9). We varied the initial and final
values (see Supplementary Figure S9). We varied the initial and final 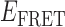 values by 0.05 increments over the range 0.0–1.0, thus generating an array of 20
values by 0.05 increments over the range 0.0–1.0, thus generating an array of 20  20 = 400 smFRET trajectories to uniformly sample the space of possible transitions. For all our simulated trajectories, we assumed a noise level of ∼4%, which closely resembles the data we have presented in the current work. Our results indicate that we can reliably identify transitions between initial and final FRET values with magnitudes
20 = 400 smFRET trajectories to uniformly sample the space of possible transitions. For all our simulated trajectories, we assumed a noise level of ∼4%, which closely resembles the data we have presented in the current work. Our results indicate that we can reliably identify transitions between initial and final FRET values with magnitudes 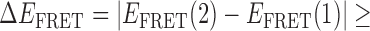 0.1.
0.1.
Figure 2.

Representative smFRET trajectories of the 3′-tail-pdT15-Cy3/Cy5 p/t DNA substrate at various gp32 concentrations. (A) p/t DNA substrates alone; (B) in the presence of 0.1 μM gp32; (C) 1.0 μM gp32; and (D) 10 μM gp32. Histograms of the smFRET efficiencies [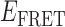 = IA/(ID + IA)], based on compilation of thousands of individual smFRET values as indicated by the vertical axes and by the number of entries, N, shown at the right of each histogram. For the trajectories shown in panels (B) and (C), the Cy5 fluorescence levels have been vertically displaced to clearly display the anti-correlated signal fluctuations. The histogram feature centered at
= IA/(ID + IA)], based on compilation of thousands of individual smFRET values as indicated by the vertical axes and by the number of entries, N, shown at the right of each histogram. For the trajectories shown in panels (B) and (C), the Cy5 fluorescence levels have been vertically displaced to clearly display the anti-correlated signal fluctuations. The histogram feature centered at 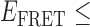 0.1 is due to the presence of inactive Cy5 acceptors on some detected molecules, as discussed in Materials and Methods. The red-shaded regions show the probability distribution of background signals, which was determined from control experiments using biotin-Cy3 samples (see Supplementary Figure S8). The buffer solution used in this and the subsequent figures, unless otherwise noted, contained 100 mM NaCl, 6 mM MgCl2 and 10 mM Tris (pH 8.0).
0.1 is due to the presence of inactive Cy5 acceptors on some detected molecules, as discussed in Materials and Methods. The red-shaded regions show the probability distribution of background signals, which was determined from control experiments using biotin-Cy3 samples (see Supplementary Figure S8). The buffer solution used in this and the subsequent figures, unless otherwise noted, contained 100 mM NaCl, 6 mM MgCl2 and 10 mM Tris (pH 8.0).
RESULTS AND DISCUSSION
Equilibrium gp32 binding to short ssDNA lattices
The functional activity of the gp32 protein in DNA replication is known to involve its preferential and cooperative binding to ssDNA at and near replication fork junctions (16). The binding site size (n) of a gp32 monomer on ssDNA is 7 nucleotide residues (17), and it binds in cooperative clusters to long ssDNA lattices at approximately physiological salt concentrations with an (association) equilibrium constant K of ∼105 M−1 per gp32 monomer and a cooperativity factor 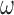 of ∼103 per gp32 monomer (18). In the absence of cooperativity, the direct binding of individual gp32 molecules to ssDNA lattices is primarily stabilized by electrostatic interactions between positively charged amino acid side chains within the protein binding site and the negatively charged phosphates of the ssDNA backbone. This direct affinity of isolated gp32 molecules for ssDNA is strongly dependent on salt concentration, reflecting the significant free energy contributions of the displacement of condensed counter-ions (Na+ and Mg2+) from the phosphates by the multivalent positively charged gp32 binding site (15,19).
of ∼103 per gp32 monomer (18). In the absence of cooperativity, the direct binding of individual gp32 molecules to ssDNA lattices is primarily stabilized by electrostatic interactions between positively charged amino acid side chains within the protein binding site and the negatively charged phosphates of the ssDNA backbone. This direct affinity of isolated gp32 molecules for ssDNA is strongly dependent on salt concentration, reflecting the significant free energy contributions of the displacement of condensed counter-ions (Na+ and Mg2+) from the phosphates by the multivalent positively charged gp32 binding site (15,19).
For ssDNA lattices that are sufficiently long to permit cooperative binding, the net binding affinity per cooperatively bound gp32 cluster is equal to 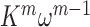 , where
, where 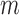 represents the number of cooperatively bound protein monomers that can fit onto the ssDNA lattice at protein saturation. The exponent to which
represents the number of cooperatively bound protein monomers that can fit onto the ssDNA lattice at protein saturation. The exponent to which 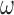 is raised is equal to the number (
is raised is equal to the number (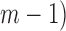 of monomer–monomer interfaces that exist within a bound cluster of
of monomer–monomer interfaces that exist within a bound cluster of 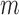 molecules. The binding affinity per gp32 monomer within the cluster is thus equal to
molecules. The binding affinity per gp32 monomer within the cluster is thus equal to 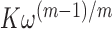 . If the cooperativity factor
. If the cooperativity factor 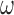 is large, as it is for gp32 binding to ssDNA, this parameter provides a significant contribution to the stability of a cooperatively bound protein–ssDNA complex, with the exponent of the cooperativity parameter per gp32 monomer
is large, as it is for gp32 binding to ssDNA, this parameter provides a significant contribution to the stability of a cooperatively bound protein–ssDNA complex, with the exponent of the cooperativity parameter per gp32 monomer 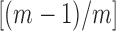 increasing toward unity as the length of the ssDNA binding lattice increases. Thus, the net affinity of a cooperatively bound gp32 cluster to ssDNA increases with increasing cluster size, and the extent of binding saturation at a given solution concentration of gp32 also increases with increasing ssDNA lattice length. The dependence of the overall binding affinity of gp32 on ssDNA lattice length is a very significant consideration for the experiments that follow.
increasing toward unity as the length of the ssDNA binding lattice increases. Thus, the net affinity of a cooperatively bound gp32 cluster to ssDNA increases with increasing cluster size, and the extent of binding saturation at a given solution concentration of gp32 also increases with increasing ssDNA lattice length. The dependence of the overall binding affinity of gp32 on ssDNA lattice length is a very significant consideration for the experiments that follow.
Bulk FRET techniques can be used to monitor the binding equilibria of gp32 to ssDNA lattices
To monitor the binding of gp32 to ssDNA lattices associated with physiologically relevant DNA constructs, we performed bulk FRET experiments on appropriately designed Cy3/Cy5-labeled primer-template (p/t) DNA substrates. Unless otherwise stated, all of our measurements were performed in solutions containing 100 mM NaCl, 6 mM MgCl2 and 10 mM Tris buffer at pH 8.0. In Table 1, we list the nomenclature and sequences of the eight different p/t DNA substrates we investigated. The majority of these substrates consisted of an 18 bp dsDNA sequence with a polydeoxythymine [p(dT)N] N-mer ‘tail’ of variable length (N = 7, 14, 15, 22 nucleotide residues) and polarity (3′- or 5′-), thus forming p/t junction constructs. In addition, we performed some control measurements on a 3′- 14-mer-tail p/t construct in which the ssDNA tail comprised a mixed base sequence, as described further below.
The length of the p(dT)N tail determines the number of gp32 monomers that can bind to a ssDNA sequence at saturating gp32 concentrations. For example, the 3′-p(dT)7 construct shown schematically in Figure 1A can only bind to a single gp32 monomer, while the 3′-p(dT)15 construct shown in Figure 1B can either bind a single gp32 monomer randomly at nine (= N − n + 1 = 15 − 7 + 1) different ssDNA lattice positions, or two gp32 monomers in a cooperative binding mode at 2 ssDNA lattice positions (1–14 or 2–15), with each gp32 monomer of the dimeric cluster spanning 7 dT residues. The 3′-p(dT)22 construct shown in Figure 1C can bind up to three gp32 monomers, with the cooperatively bound gp32 trimer occurring at either of two lattice positions (1–21 or 2–22). A 3′-p(dT)14 construct was also studied to remove these latter binding ambiguities for the 15-mer case – and look for cluster ‘sliding’ (see below). In all of these constructs, the dsDNA sections of the p/t substrate were labeled at their 5′-positions with a Cy5 acceptor fluorophore, and the 3′ (or 5′)-ends of the p(dT)N tail with a Cy3 donor fluorophore, thus comprising an ‘intra-molecular’ Cy3 / Cy5 donor–acceptor FRET p/t DNA construct. We denote the particular p/t constructs shown in Figure 1A–C as 3′-tail-pdTN-Cy3/Cy5, and analogous constructs with opposite polarity as 5′-tail-pdTN-Cy3/Cy5.
In the absence of protein, the p(dT)15 and p(dT)22 p/t DNA constructs exhibited a high bulk FRET signal. We assume that this reflects the relatively high random coil ‘flexibility’ of the unbound ssDNA segment, which results in a small average end-to-end distance between the FRET probes located at the ends of the ssDNA tail. Binding one, and then cooperatively binding two (or three) gp32 monomers to the p(dT)15(22) tail of the p/t construct would then be expected to progressively ‘stiffen’ (decrease the flexibility) of the gp32-complexed ssDNA sequence, and thus to increase the average separation between the dye chromophores to yield a concomitant reduction in the FRET efficiency. [The FRET efficiency is defined as 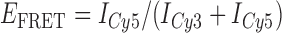 , with
, with 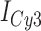 and
and 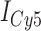 representing the peak fluorescence intensities of the donor and acceptor chromophores, respectively (20,21).] We confirmed this anticipated behavior for the indicated substrates by performing bulk fluorescence experiments, which showed that the FRET signal changed as stoichiometric, and then excess, concentrations of gp32 were added to the p/t DNA for the p(dT)15 and p(dT)22 constructs. As an example, a bulk fluorescence experiment is shown in Figure 1D for the 3′-tail-pdT15-Cy3/Cy5 construct, and the results for these six substrates are summarized in Figure 1E.
representing the peak fluorescence intensities of the donor and acceptor chromophores, respectively (20,21).] We confirmed this anticipated behavior for the indicated substrates by performing bulk fluorescence experiments, which showed that the FRET signal changed as stoichiometric, and then excess, concentrations of gp32 were added to the p/t DNA for the p(dT)15 and p(dT)22 constructs. As an example, a bulk fluorescence experiment is shown in Figure 1D for the 3′-tail-pdT15-Cy3/Cy5 construct, and the results for these six substrates are summarized in Figure 1E.
We performed control measurements to determine whether gp32 binding to the dsDNA segment of our p/t constructs might contribute some background signal, and thereby interfere with measurements of the smFRET efficiency (see Supplementary Figure S1). These control substrates contained Cy3/Cy5 FRET chromophore pairs positioned on opposing DNA strands at the p/t junction (Supplementary Figure S1A) or in the middle of the duplex region (Supplementary Figure S1B). We observed no significant changes to the fluorescence spectra or the FRET signals in bulk gp32 titrations with these DNA substrates (Supplementary Figure S1C and S1D). This is in contrast to the systematic FRET signal changes we observed in analogous bulk titrations of the p(dT)15 and p(dT)22 substrates (Figure 1E). The results of our bulk FRET experiments confirm that gp32 binds preferentially to the ssDNA region of the p/t DNA constructs that have sufficiently long tails to involve the cooperative binding mode of the protein, and that the FRET changes we observed reflect this binding. The p(dT)7-containing constructs showed only marginal changes in the FRET signal, even at elevated protein concentrations. Moreover, our smFRET experiments with the p(dT)7 constructs revealed no direct indication of gp32-induced state-to-state transitions, such as those we observed for the p(dT)15 substrates that are described further below. These results are consistent with previous findings that the gp32 interaction with the p(dT)7 substrates, which involves no cooperative binding, is weak and unstable at the protein and DNA concentrations used in these studies (14,15).
The results of our bulk fluorescence experiments reflect the expectation that each gp32 monomer binds to the ssDNA tails of the various p/t DNA substrates with a binding affinity that depends sensitively on the cooperativity factor per gp32 monomer 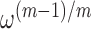 , where
, where 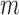 is the number of cooperatively bound gp32 monomers within a cluster at binding saturation (14,15). For the shortest length [p(dT)7] substrates, the association binding constant per gp32 monomer at physiological salt concentrations is
is the number of cooperatively bound gp32 monomers within a cluster at binding saturation (14,15). For the shortest length [p(dT)7] substrates, the association binding constant per gp32 monomer at physiological salt concentrations is 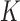 ≈ 105 M−1, while for the intermediate length [p(dT)15] construct tails that can bind two gp32 molecules cooperatively, the net association constant per gp32 molecule in a dimer cluster at saturation is
≈ 105 M−1, while for the intermediate length [p(dT)15] construct tails that can bind two gp32 molecules cooperatively, the net association constant per gp32 molecule in a dimer cluster at saturation is 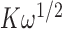 . This corresponds to an apparent association constant per gp32 monomer of 105 ·103/2 M−1 or ∼3.3 × 106 M−1 (for K = 105 M−1 and
. This corresponds to an apparent association constant per gp32 monomer of 105 ·103/2 M−1 or ∼3.3 × 106 M−1 (for K = 105 M−1 and 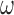 = 103). For the longest p(dT)22 constructs, which can bind three gp32 molecules at lattice saturation, the net association constant per gp32 monomer within the trimer cluster is
= 103). For the longest p(dT)22 constructs, which can bind three gp32 molecules at lattice saturation, the net association constant per gp32 monomer within the trimer cluster is 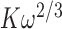 , which corresponds to a value of 105 ·102 M−1 ∼107 M−1 under these solution conditions. Our bulk FRET experiments (Figure 1E) show that the concentrations of gp32 required to achieve saturation of longer ssDNA p(dT)N substrates are somewhat lower than those necessary to achieve saturation for shorter strands, as expected for the cooperative binding of gp32 clusters to short lattices because of the dependence of the cooperativity parameter per gp32 monomer on lattice length. Finally, our bulk FRET titrations suggest that the binding affinity of cooperatively bound gp32 clusters depends also on the polarity of the p(dT)N-tail, with apparent binding saturation being achieved at slightly lower gp32 concentrations for the 5′- than for the 3′-end labeled p(dT)15 and p(dT)22 constructs (see Figure 1E).
, which corresponds to a value of 105 ·102 M−1 ∼107 M−1 under these solution conditions. Our bulk FRET experiments (Figure 1E) show that the concentrations of gp32 required to achieve saturation of longer ssDNA p(dT)N substrates are somewhat lower than those necessary to achieve saturation for shorter strands, as expected for the cooperative binding of gp32 clusters to short lattices because of the dependence of the cooperativity parameter per gp32 monomer on lattice length. Finally, our bulk FRET titrations suggest that the binding affinity of cooperatively bound gp32 clusters depends also on the polarity of the p(dT)N-tail, with apparent binding saturation being achieved at slightly lower gp32 concentrations for the 5′- than for the 3′-end labeled p(dT)15 and p(dT)22 constructs (see Figure 1E).
gp32 binds cooperatively to the 3′-tail-pdT15-Cy3/Cy5 p/t DNA constructs, and induces continuous smFRET fluctuations
Our bulk FRET experiments on the three classes of p/t constructs described above showed that gp32 binding affinities to the p(dT)N-tails differs significantly as a function of tail length N (as well as slightly as a function of binding polarity, see below). Binding saturation occurred at very low input gp32 protein concentrations (at ∼0.5 μM, or close to ∼3 gp32 monomers added per ssDNA tail) for 3′-end labeled p(dT)22 constructs, which are long enough to allow three gp32 proteins to bind cooperatively, while slightly higher gp32 concentrations appeared to be required to approach binding saturation on the p(dT)15-tailed constructs at the construct concentrations used. In contrast, for the p(dT)7-tailed constructs, which can only bind to one gp32 monomer, binding was very weak and saturation was not achieved (see Figure 1D and E). These results clearly reflect the increased binding stability induced by the increasing contribution (per gp32 monomer) of the exponent of the cooperativity parameter as the length of the bound clusters of gp32 molecules increased.
We studied the kinetic properties and FRET distributions of the above three classes of constructs using smFRET techniques with 100-ms time resolution, as described previously and briefly reviewed in the Materials and Methods section (6,22). While the results of our single-molecule experiments were consistent with the equilibrium findings of our bulk studies, they also revealed very different dynamic behavior of the constructs as a function of ssDNA [p(dT)N] lattice length. We found that smFRET measurements of gp32-dependent binding to constructs with p(dT)15 tails showed very pronounced fluctuations between bound and unbound states on the resolution time scale of our experiments (100-ms, see Figure 2), while the smFRET changes seen with the p(dT)22 constructs were quite stable over the same time scales. In contrast, p(dT)7 constructs showed only minimal smFRET changes in the presence of gp32, suggesting that these fluctuations were too fast to be clearly resolved at the time resolution of our instrument. Typical fluctuation patterns for the p(dT)7- and the p(dT)22-tailed constructs are shown in Supplementary Figure S2. From the above observations, we concluded that the most useful kinetic information for this series of constructs should be obtainable from the p(dT)15-tailed constructs for the salt and protein concentration conditions (and time resolution window) used here. In most of the remainder of this paper, we focus on the kinetic behavior of these p(dT)15-tailed constructs as a model system to initiate our study of the dynamic properties of gp32 molecules, both singly and cooperatively bound to ssDNA.
In the absence of the gp32 protein, the majority of smFRET trajectories of the 3′-tail-pdT15-Cy3/Cy5 p/t DNA substrates exhibited relatively high, quasi-stable smFRET signals (see Figure 2A, left). From thousands of such trajectories (with each trajectory contributing a single data point, see Materials and Methods), we constructed histograms of the smFRET efficiency that exhibited a prominent feature peaked at 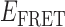 ≈ 0.65 (see Figure 2A, right). We note here that an artifact feature, which peaks at
≈ 0.65 (see Figure 2A, right). We note here that an artifact feature, which peaks at 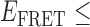 0.1, is present for all of the
0.1, is present for all of the 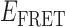 histograms presented in this work. The occurrence of this feature is due to the contributions of single-molecules in which the Cy5 acceptor chromophore has photo-bleached, and to a small amount of Cy3 donor fluorescence that leaks into the acceptor detector channel (see Materials and Methods, and Supplementary Figure S7). The red shaded region indicates the probability distribution associated with this ‘background’ signal.
histograms presented in this work. The occurrence of this feature is due to the contributions of single-molecules in which the Cy5 acceptor chromophore has photo-bleached, and to a small amount of Cy3 donor fluorescence that leaks into the acceptor detector channel (see Materials and Methods, and Supplementary Figure S7). The red shaded region indicates the probability distribution associated with this ‘background’ signal.
Upon addition of 0.1 μM gp32 to the sample chamber, we observed that the majority of smFRET trajectories (>70% of the molecules within the sample imaging area) exhibited a continuous pattern of anti-correlated Cy3/Cy5 fluorescence fluctuations occurring on sub-second time scales (see Figure 2B, left). This pattern of smFRET fluctuations suggested that individual molecules undergo multiple transitions between discrete states during the time window of the experiments, although the specific values of 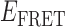 appeared to vary somewhat from one molecule to another. The time period over which we could monitor these trajectories was limited by the onset of photo-degradation after 1 to 2 min. Histograms of the value of
appeared to vary somewhat from one molecule to another. The time period over which we could monitor these trajectories was limited by the onset of photo-degradation after 1 to 2 min. Histograms of the value of 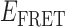 obtained from trajectories recorded in the presence of this low gp32 concentration exhibited a prominent feature that peaked at
obtained from trajectories recorded in the presence of this low gp32 concentration exhibited a prominent feature that peaked at 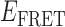 ≈ 0.4, which appeared to be broader than the feature obtained from DNA constructs alone. This shift and broadening of the
≈ 0.4, which appeared to be broader than the feature obtained from DNA constructs alone. This shift and broadening of the 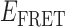 distributions at low protein levels could reflect a binding equilibrium in which populations of 0-bound, 1-bound and 2-bound states coexist.
distributions at low protein levels could reflect a binding equilibrium in which populations of 0-bound, 1-bound and 2-bound states coexist.
We note that our observations of gp32-dependent smFRET fluctuations are reminiscent of those observed by others in previous studies of DNA–protein interactions using similar p/t DNA substrates with different proteins. These include studies of the Escherichia coli ssDNA-binding protein ssb (23–25), dsRNA binding protein (26) and helicases that bind to ss-ds DNA fork junctions (27–31). In all of these cases, repetitive smFRET fluctuations were interpreted in terms of the assembly of protein–DNA filaments, the diffusion or sliding of protein along ssDNA, or the unwinding of duplex DNA (32).
A number of possible mechanisms could, in principle, be responsible for the fluctuations we observed with the gp32-bound 3′-tail-pdT15-Cy3/Cy5 construct. These include: (i) rapid association/dissociation of gp32 molecules to/from the ssDNA sequences of the p/t construct; (ii) diffusive translocation (e.g. hopping or sliding) of gp32 protein along the ssDNA tails; (iii) conformational fluctuations of the ssDNA backbone of the p(dT)15 tails induced by the binding and release of gp32 monomers; and (iv) conformational fluctuations of the gp32 molecules themselves, which are associated with their isolated or cooperative binding to the ssDNA tails of the construct. These mechanisms can be divided into two groups, based on their responses to changing gp32 concentrations. Fluctuations reflecting the sliding or hopping of single gp32 monomers or of cooperatively-bound gp32 dimer clusters along the ssDNA component of the p/t constructs should not be gp32 concentration sensitive, while those that involve association and dissociation of gp32 proteins to and from the ssDNA tails of the constructs should change with changing gp32 concentration.
To this end, the smFRET fluctuations of the p/t DNA substrate in the presence of 0.1 μM gp32 were initially monitored, followed by the injection of a flush buffer to remove unbound protein. After excess protein had thus been removed from the sample cell, we observed an immediate cessation of the quasi-continuous smFRET fluctuations, suggesting that the gp32 must have dissociated rapidly from the ssDNA on the time scale of our measurements. These results suggest that the smFRET fluctuations we observed at these low gp32 concentrations cannot be attributed to conformational fluctuations of proteins that remain bound to the p/t DNA substrate (29–33), but rather reflect the association-dissociation of (likely single) gp32 molecules to and from the ssDNA p(dT)15 tails of the constructs.
Changing gp32 concentration influences the pattern of smFRET fluctuations and the equilibrium distribution of protein–ssDNA association complex sub-states
We next studied the influence of increasing gp32 concentration on the pattern of smFRET fluctuations. It was previously shown that, over the concentration range ∼0.1 to ∼1 μM, gp32 exists primarily as monomers in solution and that gp32-oligomers form at concentrations greater than 1 μM (15,33). However, these solution phase oligomers differ in their specific interactions and conformation from those bound cooperatively to ssDNA lattices, and therefore these gp32 oligomers formed in the absence of ssDNA must first be dissociated to monomers before they can bind to ssDNA (15). As discussed in previous sections, cooperative interactions between gp32 monomers bound to ssDNA result in enhanced binding affinity to ssDNA at elevated protein concentrations (18), and this enhancement is less pronounced for short lattices (13,34). We therefore performed a series of measurements by introducing, in succession, 100–250 μl aliquots of gp32 into the sample flow chamber to achieve the final gp32 concentrations of 0.1 μM, 1 μM and 10 μM. After each change in gp32 concentration, the samples were incubated for 2 to 3 min before data acquisition.
As suggested above, we found that the pattern of smFRET fluctuations of the p/t DNA substrates to be quite sensitive to the concentration of the gp32 protein used. In the presence of 0.1 μM gp32, the distribution of the observed values of the smFRET efficiency 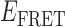 for the 3′-tail-pdT15-Cy3/Cy5 p/t DNA substrate was shifted to smaller values and broadened relative to that of the p/t DNA substrate alone (see Figure 2A and B). Moreover, individual smFRET trajectories recorded at these low protein concentrations exhibited intermittent fluctuations between a dominant ‘high-
for the 3′-tail-pdT15-Cy3/Cy5 p/t DNA substrate was shifted to smaller values and broadened relative to that of the p/t DNA substrate alone (see Figure 2A and B). Moreover, individual smFRET trajectories recorded at these low protein concentrations exhibited intermittent fluctuations between a dominant ‘high-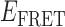 ’ state (centered at ∼0.65) and a short-lived ‘low-
’ state (centered at ∼0.65) and a short-lived ‘low-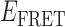 ’ state (centered at ∼0.4). This pattern changed when the concentration of gp32 was increased to 1 μM. At this higher protein concentration, the smFRET signal fluctuated between a dominant low-
’ state (centered at ∼0.4). This pattern changed when the concentration of gp32 was increased to 1 μM. At this higher protein concentration, the smFRET signal fluctuated between a dominant low-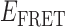 and a minority high-
and a minority high-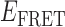 state (see Figure 2C). The inversion of population between high- and low-
state (see Figure 2C). The inversion of population between high- and low-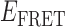 states at this gp32 concentration was reflected by the appearance of two modes in the
states at this gp32 concentration was reflected by the appearance of two modes in the 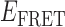 distribution, a dominant mode that peaked at
distribution, a dominant mode that peaked at 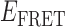 ≈ 0.35 and a weaker mode with peak at
≈ 0.35 and a weaker mode with peak at 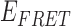 ≈ 0.6.
≈ 0.6.
We note that smFRET trajectories obtained from samples incubated with 0.1 μM and 1 μM gp32 yielded a majority of molecules (>70%) exhibiting quasi-continuous fluctuations between high- and low-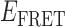 states. Such fluctuations were not observed for samples incubated with 10-μM gp32 (see Figure 2D, left). At this very high protein concentration, single-molecule trajectories exhibited a bimodal distribution of quasi-stationary
states. Such fluctuations were not observed for samples incubated with 10-μM gp32 (see Figure 2D, left). At this very high protein concentration, single-molecule trajectories exhibited a bimodal distribution of quasi-stationary 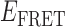 values, which occurred either within the
values, which occurred either within the 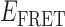 ≈ 0.2–0.4 range, or within the
≈ 0.2–0.4 range, or within the 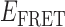 ≈ 0.4–0.7 range (see Figure 2D, right). As pointed out above, we note that at 10 μM concentrations, gp32 in solution exists as an equilibrium distribution of monomeric and largely di- and trimeric states, and that these oligomeric gp32 species must dissociate prior to binding to ssDNA (15,33). The observed bimodal distribution of
≈ 0.4–0.7 range (see Figure 2D, right). As pointed out above, we note that at 10 μM concentrations, gp32 in solution exists as an equilibrium distribution of monomeric and largely di- and trimeric states, and that these oligomeric gp32 species must dissociate prior to binding to ssDNA (15,33). The observed bimodal distribution of 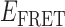 values is consistent with such a coupled equilibrium of gp32-ssDNA complexes (with
values is consistent with such a coupled equilibrium of gp32-ssDNA complexes (with 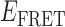 ≈ 0.2–0.4), unbound ssDNA (with
≈ 0.2–0.4), unbound ssDNA (with 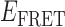 ≈ 0.4–0.7) and solution phase monomeric and oligomeric species of gp32.
≈ 0.4–0.7) and solution phase monomeric and oligomeric species of gp32.
The quasi-stationary trajectories we observed from samples incubated with 10 μM gp32 suggest that, under these conditions, a saturation point is reached for the loading of protein onto the p/t DNA substrates that prohibits the rapid conformational fluctuations of the ssDNA end-to-end (intra-FRET dye pair) distance that we observed at lower protein concentrations. This saturation condition could reflect a variety of molecular mechanisms, including, the presence of two tightly (and cooperatively) bound gp32 molecules associated with the ssDNA region of the p/t DNA substrate; the presence of additional gp32 proteins bound to the duplex region near the p/t junction; and/or a dynamic exchange between proteins free in solution and those bound to the ssDNA on a time scale much faster than the resolution of our experiments. If the smFRET fluctuations we observed at lower protein concentrations (0.1 μM) had been due to one-dimensional sliding/hopping of single gp32 monomers, or cooperatively bound dimeric gp32 clusters along the ssDNA, or to a protein-induced ssDNA conformational change, we might expect this pattern of fluctuations to persist at the 1 μM gp32 concentrations. On the contrary, we observed that this pattern depended sensitively on protein concentration.
The above considerations suggest that the signal fluctuations we observe at lower (0.1 μM) gp32 concentrations likely reflect gp32 monomer binding to, and dissociating from, the ssDNA portions of the p/t constructs. At 10 μM gp32 concentrations, the low-FRET state centered at 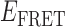 ≈ 0.3 can then be interpreted to represent cooperatively bound gp32 dimers that dissociate too slowly (presumably as gp32 monomers from the ends of cooperatively-bound dimeric clusters) to induce signal fluctuations. Furthermore, if these dimers were to slide (as clusters between positions 1–14 and 2–15) this process must be either too fast or too slow under these conditions to induce observable signal fluctuations. An alternative interpretation would be that these two cooperatively bound dimer states do not exhibit distinct smFRET values.
≈ 0.3 can then be interpreted to represent cooperatively bound gp32 dimers that dissociate too slowly (presumably as gp32 monomers from the ends of cooperatively-bound dimeric clusters) to induce signal fluctuations. Furthermore, if these dimers were to slide (as clusters between positions 1–14 and 2–15) this process must be either too fast or too slow under these conditions to induce observable signal fluctuations. An alternative interpretation would be that these two cooperatively bound dimer states do not exhibit distinct smFRET values.
Given the above considerations, we tentatively make the following state assignments to the 3′-tail-pdT15-Cy3/Cy5 construct. The quasi-stationary signal observed in the absence of gp32 (with peak value 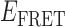 = 0.65) is assigned to the 0-bound state, since this state appears to be present at all protein concentrations. As the concentration of gp32 was increased, we observed that the relative occupancy of the 0-bound state was reduced. Similarly, we assign the quasi-stationary signal we observed at the 10 μM saturation conditions with peak value
= 0.65) is assigned to the 0-bound state, since this state appears to be present at all protein concentrations. As the concentration of gp32 was increased, we observed that the relative occupancy of the 0-bound state was reduced. Similarly, we assign the quasi-stationary signal we observed at the 10 μM saturation conditions with peak value 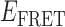 ≈ 0.3 to the 2-bound state. This state also appears to be present within our smFRET trajectories as a transient intermediate at both 0.1 and 1.0 μM gp32 concentrations. We expect 1-bound states to show
≈ 0.3 to the 2-bound state. This state also appears to be present within our smFRET trajectories as a transient intermediate at both 0.1 and 1.0 μM gp32 concentrations. We expect 1-bound states to show 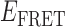 values intermediate to these limits, and we anticipate that such states might persist for relatively short times in comparison to the average lifetimes of the 0-bound and 2-bound states.
values intermediate to these limits, and we anticipate that such states might persist for relatively short times in comparison to the average lifetimes of the 0-bound and 2-bound states.
To determine the time scales associated with the state-to-state inter-conversions within the gp32 – 3′-tail-pdT15-Cy3/Cy5 system, we constructed histograms of the 0-bound and 2-bound state persistence times at the various gp32 concentrations investigated (see Figure 3). To this end, we implemented a HMM analysis of our smFRET trajectories to determine the most likely number of discrete states and associated lifetimes that could be resolved on the 100-ms time scale of our measurements (35). The HMM analysis assumes that each single-molecule fluorescence trajectory is representative of a system undergoing stochastic fluctuations between a fixed number of discrete states with well-defined 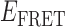 values. Since the experimental single-molecule trajectories contain noise, the HMM analysis provides a systematic procedure to compare the agreement between these data with models in which the number of states, and their associated
values. Since the experimental single-molecule trajectories contain noise, the HMM analysis provides a systematic procedure to compare the agreement between these data with models in which the number of states, and their associated 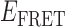 values, is varied. We tested the accuracy of our HMM software using simulated single molecule trajectories with known numbers of states and associated
values, is varied. We tested the accuracy of our HMM software using simulated single molecule trajectories with known numbers of states and associated 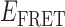 values (see Supplementary Figure S9).
values (see Supplementary Figure S9).
Figure 3.

smFRET trajectories with hidden Markov model (HMM) analysis. (A) Representative smFRET trajectory (shown in black) of the p/t DNA construct 3′-tail-pdT15-Cy3/Cy5 in the presence of 0.1 μM gp32 superimposed onto an HMM derived idealized FRET trajectory (shown in red). The HMM analysis shows that these data are consistent with a two-state interpretation in which the system fluctuates between a doubly bound state (2-bound) and an unbound state (0-bound). (B) Representative histograms are shown for the persistence times, 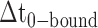 and
and 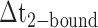 , which were obtained from the HMM analysis. Histograms were constructed from ∼5000 transitions from ∼50 individual single molecule trajectories of 120-s duration. Average lifetimes
, which were obtained from the HMM analysis. Histograms were constructed from ∼5000 transitions from ∼50 individual single molecule trajectories of 120-s duration. Average lifetimes 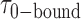 = 1.31 s and
= 1.31 s and 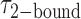 = 0.32 s were calculated from single exponential fits to these histograms, which correspond to the apparent equilibrium constant
= 0.32 s were calculated from single exponential fits to these histograms, which correspond to the apparent equilibrium constant 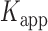 = [2-bound]/[0-bound] = 0.24. (C and D) Same as for panels (A) and (B), except that the gp32 protein concentration was 1.0 μM. Under these conditions, the values of the average lifetimes and apparent equilibrium constant are
= [2-bound]/[0-bound] = 0.24. (C and D) Same as for panels (A) and (B), except that the gp32 protein concentration was 1.0 μM. Under these conditions, the values of the average lifetimes and apparent equilibrium constant are 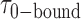 = 0.16 s,
= 0.16 s, 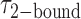 = 0.51 s, and
= 0.51 s, and 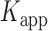 = 3.2.
= 3.2.
Our data are well suited to an HMM analysis, since we observed similar, relatively simple and well-defined fluctuations for the majority of the 120 s trajectories that we obtained, and each trajectory exhibited over 100 transitions. This suggests that for each set of conditions we should indeed be observing equilibrium fluctuations between a discrete number of well-defined states during the 120 s observation period. Figure 3A shows an example of an optimized HMM-derived smFRET trajectory (red) superimposed upon an experimental trajectory (black) that had been recorded in the presence of 0.1 μM gp32. Our HMM analysis reveals that at this protein concentration, 85% of the smFRET trajectories were well fit using a two-state model, and that the remaining 15% were well fit using a three-state model. Similarly, for the 1.0 μM gp32 sample, 92% of the trajectories were well fit using a two-state model, and the remaining 8% were well fit using a three-state model. We used the HMM analysis to identify the presence of distinct smFRET states, their associated smFRET efficiencies, and their persistence times. Histograms of the resulting series of persistence times provided the necessary information to determine the average lifetime of the presumed 0-bound [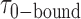 ] and 2-bound [
] and 2-bound [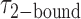 ] states (see Figure 3B). These histograms were calculated from ∼5000 transitions that were identified from 50 individual molecular trajectories. For the 0.1 μM gp32 concentration condition, we obtained average lifetime values
] states (see Figure 3B). These histograms were calculated from ∼5000 transitions that were identified from 50 individual molecular trajectories. For the 0.1 μM gp32 concentration condition, we obtained average lifetime values 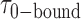 = 1.31 s and
= 1.31 s and 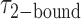 = 0.32 s, corresponding to an apparent equilibrium constant
= 0.32 s, corresponding to an apparent equilibrium constant 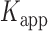 ([2-bound]/[0-bound]) of 0.24. For the 1.0 μM gp32 condition,
([2-bound]/[0-bound]) of 0.24. For the 1.0 μM gp32 condition, 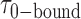 = 0.16 s and
= 0.16 s and 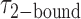 = 0.51 s, corresponding to
= 0.51 s, corresponding to 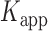 = 3.2. We note that these parameters are not real rate or equilibrium constants; rather these times represent the average forward and backward rates of the reactions, and the resulting apparent equilibrium constants can be considered to represent the ‘occupancy’ of the p(dT)15-tails of the p/t DNA constructs (presumably by cooperatively-bound dimer clusters of gp32 molecules) under the conditions of the experiment.
= 3.2. We note that these parameters are not real rate or equilibrium constants; rather these times represent the average forward and backward rates of the reactions, and the resulting apparent equilibrium constants can be considered to represent the ‘occupancy’ of the p(dT)15-tails of the p/t DNA constructs (presumably by cooperatively-bound dimer clusters of gp32 molecules) under the conditions of the experiment.
Although the above analysis suggests that we can detect the presence of both 0- and 2-bound states of the (gp32)2-ssDNA assembly reaction, there must also exist one or more 1-bound state(s) that function as intermediates. The detection and assignment of 1-bound states is complicated by the fact that they may exist on a time scale that is much shorter than the resolution of our experiments, or that they might result in 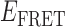 values that cannot be distinguished from those of the 0- and 2-bound states. However, since the binding of a single gp32 molecule to the p(dT)15 lattice is expected to stiffen the chain locally, the 1-bound state—if it exists for a long enough time to be detected—should produce
values that cannot be distinguished from those of the 0- and 2-bound states. However, since the binding of a single gp32 molecule to the p(dT)15 lattice is expected to stiffen the chain locally, the 1-bound state—if it exists for a long enough time to be detected—should produce 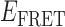 values that lie intermediate to those of the 0- and 2-bound states. Based on the persistence times of the 0- and 2-bound states that we determined above, we expect that the 1-bound states must have lifetimes on the order of tens-of-milliseconds. Further single molecule experiments—currently underway—that are performed with much higher time resolution should help to further define the equilibrium and kinetic roles of the 1-bound state in the gp32 binding and assembly process.
values that lie intermediate to those of the 0- and 2-bound states. Based on the persistence times of the 0- and 2-bound states that we determined above, we expect that the 1-bound states must have lifetimes on the order of tens-of-milliseconds. Further single molecule experiments—currently underway—that are performed with much higher time resolution should help to further define the equilibrium and kinetic roles of the 1-bound state in the gp32 binding and assembly process.
The density of smFRET state-to-state transitions reveals the presence of ssDNA conformational sub-states that are associated with gp32 ssDNA lattice binding and dissociation processes
We examined the distribution of 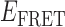 values associated with individual state-to-state transitions by using the results of our HMM analyses to construct TDPs (35). A TDP is a two-dimensional histogram that indicates the occurrences of transitions between distinct states with unique
values associated with individual state-to-state transitions by using the results of our HMM analyses to construct TDPs (35). A TDP is a two-dimensional histogram that indicates the occurrences of transitions between distinct states with unique 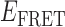 values. In the context of this work, each point on the TDP represents a transition between distinct bound and unbound gp32-ssDNA states. In Figure 4, we present examples of TDPs constructed from individual single molecule trajectories obtained with the 3′-tail-pdT15-Cy3/Cy5 DNA substrates in the presence of 0.1 μM gp32. Each plot is a two-dimensional contour diagram, with horizontal and vertical axes representing initial [
values. In the context of this work, each point on the TDP represents a transition between distinct bound and unbound gp32-ssDNA states. In Figure 4, we present examples of TDPs constructed from individual single molecule trajectories obtained with the 3′-tail-pdT15-Cy3/Cy5 DNA substrates in the presence of 0.1 μM gp32. Each plot is a two-dimensional contour diagram, with horizontal and vertical axes representing initial [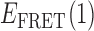 ] and final [
] and final [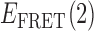 ] values, respectively. TDPs are useful to resolve state-to-state inter-conversion events that occur during an individual single-molecule trajectory (here of 120 s duration), and to ascertain the degree of non-degeneracy (or dispersion) within a sample containing hundreds of molecules (27). In general, a feature appearing below the yellow-dashed diagonal line corresponds to an association process (indicated by an orange arrow), converting the 0- (or a 1-) bound state to a 2- (or to a 1-) bound state. Conversely, a feature appearing above the diagonal line corresponds to a dissociation process (indicated by a red arrow), converting a 2- (or a 1-) bound state to a 1- or 0-bound state. The mirror symmetry of the TDP along the diagonal line is consistent with a reversible equilibrium system obeying detailed balance conditions, since the probability of high-to-low
] values, respectively. TDPs are useful to resolve state-to-state inter-conversion events that occur during an individual single-molecule trajectory (here of 120 s duration), and to ascertain the degree of non-degeneracy (or dispersion) within a sample containing hundreds of molecules (27). In general, a feature appearing below the yellow-dashed diagonal line corresponds to an association process (indicated by an orange arrow), converting the 0- (or a 1-) bound state to a 2- (or to a 1-) bound state. Conversely, a feature appearing above the diagonal line corresponds to a dissociation process (indicated by a red arrow), converting a 2- (or a 1-) bound state to a 1- or 0-bound state. The mirror symmetry of the TDP along the diagonal line is consistent with a reversible equilibrium system obeying detailed balance conditions, since the probability of high-to-low 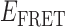 transitions must be equal to the probability of low-to-high transitions. This assumes that the duration of the single-molecule trajectory is long enough to fully sample the equilibrium distribution of fully reversible association and disassociation processes.
transitions must be equal to the probability of low-to-high transitions. This assumes that the duration of the single-molecule trajectory is long enough to fully sample the equilibrium distribution of fully reversible association and disassociation processes.
Figure 4.

Transition density plots (TDPs) from smFRET trajectories of 3′-tail-pdT15-Cy3/Cy5 in the presence of 0.1 μM gp32. Panels (A) and (B) show TDPs corresponding to two-state behavior. Peaks below the dashed yellow diagonal line indicate a protein–DNA association event (from a high-to-low 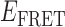 value), while peaks above the diagonal line indicate a dissociation event (from a low-to-high
value), while peaks above the diagonal line indicate a dissociation event (from a low-to-high 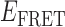 value). Individual molecular trajectories exhibit some variation in the
value). Individual molecular trajectories exhibit some variation in the 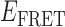 values, as evident from comparison between panels (A) and (B). Red and orange arrows connecting initial and final
values, as evident from comparison between panels (A) and (B). Red and orange arrows connecting initial and final 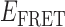 values indicate dissociative and associative ‘transition pathways,’ respectively, which are determined by the analysis. Panel (C) shows the TDP resulting from a trajectory that was well fit to a three-state model. In this example, there is indication of a short-lived intermediate with
values indicate dissociative and associative ‘transition pathways,’ respectively, which are determined by the analysis. Panel (C) shows the TDP resulting from a trajectory that was well fit to a three-state model. In this example, there is indication of a short-lived intermediate with 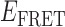 ≈ 0.4, which is coupled to longer-lived end-states with
≈ 0.4, which is coupled to longer-lived end-states with 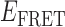 ≈ 0.55 and ≈ 0.65. The low intensity blue-shaded feature appears vertically displaced from the high intensity red feature above the diagonal line, and its symmetry related counterpart appears horizontally displaced from the high intensity feature below the diagonal line. Thus, the presence of multiple features vertically (horizontally) displaced above (below) the diagonal line of the TDP indicates the occurrence of multiple coupled states along a transition pathway.
≈ 0.55 and ≈ 0.65. The low intensity blue-shaded feature appears vertically displaced from the high intensity red feature above the diagonal line, and its symmetry related counterpart appears horizontally displaced from the high intensity feature below the diagonal line. Thus, the presence of multiple features vertically (horizontally) displaced above (below) the diagonal line of the TDP indicates the occurrence of multiple coupled states along a transition pathway.
When we compared the TDPs constructed from randomly selected trajectories taken under identical experimental conditions, we found that there was variation in the specific 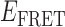 values we obtained for the distinct 0-, 1- and 2-bound states defined by our assembly models (see Figure 4A and B). As mentioned previously, ∼85% of the trajectories obtained at 0.1 μM gp32 were well fit to a two-state model, while only ∼15% were well fit to a three-state model. For the majority of trajectories, the TDPs indicate that state-to-state inter-conversions occurred primarily along distinct pathways. For example, the pathway corresponding to the TDP shown in Figure 4A has
values we obtained for the distinct 0-, 1- and 2-bound states defined by our assembly models (see Figure 4A and B). As mentioned previously, ∼85% of the trajectories obtained at 0.1 μM gp32 were well fit to a two-state model, while only ∼15% were well fit to a three-state model. For the majority of trajectories, the TDPs indicate that state-to-state inter-conversions occurred primarily along distinct pathways. For example, the pathway corresponding to the TDP shown in Figure 4A has 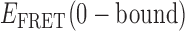 ≈ 0.5 and
≈ 0.5 and 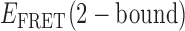 ≈ 0.27, while the pathway for the TDP shown in Figure 4B has
≈ 0.27, while the pathway for the TDP shown in Figure 4B has 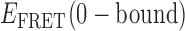 = 0.61 and
= 0.61 and 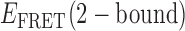 = 0.35. For the minority population of trajectories where the three-state model was operative, we observed the presence of an additional short-lived intermediate that was coupled to two other states. An example of a TDP for this case is shown in Figure 4C. If we assign the short-lived intermediate to a 1-bound state, we note that its average
= 0.35. For the minority population of trajectories where the three-state model was operative, we observed the presence of an additional short-lived intermediate that was coupled to two other states. An example of a TDP for this case is shown in Figure 4C. If we assign the short-lived intermediate to a 1-bound state, we note that its average 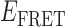 value of ∼0.4 lies (as expected) intermediate to the range of values we have assigned to the 0- and 2-bound states. The inter-conversion pathway corresponding to this TDP has two 0-bound states with
value of ∼0.4 lies (as expected) intermediate to the range of values we have assigned to the 0- and 2-bound states. The inter-conversion pathway corresponding to this TDP has two 0-bound states with 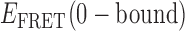 ≈ 0.55 and ≈ 0.65. Each of these 0-bound states undergo association to a 1-bound state with
≈ 0.55 and ≈ 0.65. Each of these 0-bound states undergo association to a 1-bound state with 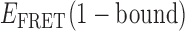 ≈ 0.4, as indicated by solid and dashed orange arrows. Dashed and solid red arrows indicate the reverse dissociation processes, respectively.
≈ 0.4, as indicated by solid and dashed orange arrows. Dashed and solid red arrows indicate the reverse dissociation processes, respectively.
To obtain further insights into the relationship between the observed signal fluctuations and the underlying state-to-state transitions, we examined cumulative TDPs constructed from ∼50 single molecule trajectories (total of ∼5000 state-to-state transitions). Such TDPs corresponding to the 3′-tail-pdT15-Cy3/Cy5 substrate in the presence of 0.1 μM gp32 and 1 μM gp32 are shown, respectively, in Figure 5A and B. Projected along the horizontal and vertical axes of each plot are the associated integrated transition density distributions (solid gray). We use the information contained in the TDPs to identify association/dissociation transitions between bound and unbound states that can be effectively resolved on the 100-ms time scale of the measurements. To accomplish this, we draw horizontal and vertical lines (dashed gray) through the most prominent features (those with intensity > 40% of the peak value in Figure 5A, and with intensity > 50% of the peak value for all other examples) of the TDPs. These cumulative TDPs show that at low gp32 concentration (Figure 5A), the majority of transitions occur between 0- and 2-bound states (i.e, between high- and low-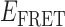 values) via four distinguishable pathways, which each appear to occur independently of one another. Each of the four pathways occurs with varying intensity; there is a singular (red) feature with dissociation coordinate (0.24, 0.47), and a less intense (orange) feature with dissociation coordinate (0.29, 0.54). Two relatively weak (green) features are also present with dissociation coordinates (0.21, 0.43) and (0.33, 0.56).
values) via four distinguishable pathways, which each appear to occur independently of one another. Each of the four pathways occurs with varying intensity; there is a singular (red) feature with dissociation coordinate (0.24, 0.47), and a less intense (orange) feature with dissociation coordinate (0.29, 0.54). Two relatively weak (green) features are also present with dissociation coordinates (0.21, 0.43) and (0.33, 0.56).
Figure 5.

Cumulative TDPs constructed from the sum of ∼50 molecular trajectories (corresponding to ∼5000 state-to-state transitions) from 3′-tail-pdT15-Cy3/Cy5 DNA in the presence of (A) 0.1 μM gp32; and (B) 1 μM gp32. TDPs for individual single molecule trajectories taken under identical conditions are shown in Figure 4. Integrated transition density distributions are shown (in gray) projected along the horizontal and vertical axes. Horizontal and vertical line segments connecting low- and high-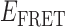 values indicate the most prominent state-to-state transition pathways. For the 0.1 μM gp32 sample shown in Panel A, there are four prominent state-to-state transitions (indicated by purple Gaussians). We note that the cumulative transition density distributions shown in panels A and B contain only a subset of the
values indicate the most prominent state-to-state transition pathways. For the 0.1 μM gp32 sample shown in Panel A, there are four prominent state-to-state transitions (indicated by purple Gaussians). We note that the cumulative transition density distributions shown in panels A and B contain only a subset of the 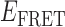 values encompassed by the histograms shown in Figure 2B and C, respectively. This suggests that only a discrete set of conformational states participate in gp32 association and dissociation events.
values encompassed by the histograms shown in Figure 2B and C, respectively. This suggests that only a discrete set of conformational states participate in gp32 association and dissociation events.
The association/dissociation pathways we observe at 0.1 μM concentrations of gp32 appear to connect one of four 2-bound states (each with a distinct 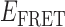 value within the range 0.15 <
value within the range 0.15 < 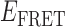 < 0.37) to one of four 0-bound states (each with a distinct
< 0.37) to one of four 0-bound states (each with a distinct 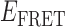 value within the range 0.4 <
value within the range 0.4 < 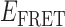 < 0.7). The existence of four 2-bound states is plausible, given that there are two different ways that a cooperatively bound gp32 dimer can occupy the ssDNA lattice (at positions 1–14 versus 2–15), and for each of these two conformations the cooperative interaction between the proteins of the gp32 dimer must be disrupted (or formed) during the dissociation (or association) pathway. The four 0-bound states presumably represent the four unbound ssDNA positions that can productively bind one gp32 monomer, and then get ‘trapped’ by the binding of an adjacent gp32 monomer to form a cooperatively bound gp32 dimer. The other five possible association positions for a 1-bound gp32 molecule cannot lead to dimer formation and thus likely dissociate rapidly (see further comments on this point in the next section). The presumed existence of distinguishable 0-bound states suggests that certain unbound conformations may persist for the 3′-tail-pdT15-Cy3/Cy5 substrate on time scales longer than the 100-ms time resolution of our experiments. Further evidence for the existence of ‘long-lived’ 0-bound states is presented in subsequent sections.
< 0.7). The existence of four 2-bound states is plausible, given that there are two different ways that a cooperatively bound gp32 dimer can occupy the ssDNA lattice (at positions 1–14 versus 2–15), and for each of these two conformations the cooperative interaction between the proteins of the gp32 dimer must be disrupted (or formed) during the dissociation (or association) pathway. The four 0-bound states presumably represent the four unbound ssDNA positions that can productively bind one gp32 monomer, and then get ‘trapped’ by the binding of an adjacent gp32 monomer to form a cooperatively bound gp32 dimer. The other five possible association positions for a 1-bound gp32 molecule cannot lead to dimer formation and thus likely dissociate rapidly (see further comments on this point in the next section). The presumed existence of distinguishable 0-bound states suggests that certain unbound conformations may persist for the 3′-tail-pdT15-Cy3/Cy5 substrate on time scales longer than the 100-ms time resolution of our experiments. Further evidence for the existence of ‘long-lived’ 0-bound states is presented in subsequent sections.
Although the cumulative TDP at lower gp32 concentration (0.1 μM) does not exhibit significant coupling between a short-lived 1-bound intermediate and the 0- and 2-bound end-states, we note that a ‘gap’ in the transition density distribution appears near the value 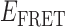 ≈ 0.4, which is the same efficiency we have assigned to the 1-bound state. The absence of a feature with
≈ 0.4, which is the same efficiency we have assigned to the 1-bound state. The absence of a feature with 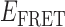 ≈ 0.4 at this low protein concentration, a value consistent with that of a 1-bound state, is consistent with our conjecture that the average lifetime of this intermediate (at least at low protein concentrations) must be short in comparison to the 100-ms time scale of our measurements.
≈ 0.4 at this low protein concentration, a value consistent with that of a 1-bound state, is consistent with our conjecture that the average lifetime of this intermediate (at least at low protein concentrations) must be short in comparison to the 100-ms time scale of our measurements.
Deriving detailed assembly-disassembly pathways from transition density plots
The observations (inferred from Figure 5A) of four distinct assembly/disassembly pathways at relatively low protein concentration (0.1 μM gp32) could be a reflection of the four possible ways that the (gp32)2-p(dT)15 complex bound to the 3′-tail-pdT15-Cy3/Cy5 substrate might undergo inter-conversion with a 0-bound state. As noted previously, a cooperatively bound gp32 dimer can occupy the ss-p(dT)15 lattice in two ways: at positions 1–14, or at positions 2–15, with each gp32 monomer spanning 7 dT residues. For each of these cases, the (gp32)2-p(dT)15 complex can undergo disassembly in two different ways: (i) by a pathway in which the gp32 monomer at the 3′ distal end of the dimer cluster disassociates from the template first; or (ii) by a pathway in which the gp32 monomer that lies closest to the 5′ distal end (the p/t junction) dissociates first. Both pathways (i) and (ii) must involve disruption of the cooperative interaction between gp32 molecules, which would presumably occur by a different pathway for each case. The differences in the intensities of the four TDP features would thus represent the varying likelihoods that the cooperative 2-bound state can undergo a lattice-conformation-dependent dissociation step. It is therefore conceivable that each of these four pathways might involve 0- and 2-bound end-states that are distinct from one another, and from unbound lattice conformations that do not participate in gp32 binding and dissociation events.
At higher gp32 concentrations (1 μM gp32), bound states are expected to occur with higher probability than they do at lower concentrations. Cumulative TDPs constructed from the sum of ∼50 single molecule trajectories exhibited multiple features, suggesting a pattern of coupled transitions of even higher complexity than seen at the lower protein concentration (see Figure 5B). Unlike the pattern we observed at 0.1 μM gp32, which shows four individual transitions between distinct sub-states that occur independently of one another, the pattern at 1 μM gp32 indicates the presence of one prominent feature occurring at the dissociation coordinate (0.26, 0.42), in addition to multiple minor features. These minor features might be assigned to transitions between 2- and 0-bound ‘families’ of end-states. However, we suggest that the single most prominent feature in the transition density distribution at 1 μM gp32 represents a transition between a 2-bound state (with 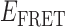 ≈ 0.27) and a 1-bound state (with
≈ 0.27) and a 1-bound state (with 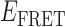 ≈ 0.42, indicated by a blue Gaussian curve in Figure 5B). The presence of transitions between this 1-bound state and only one of the multiple observed 2-bound states suggests that this represents the most likely association/dissociation pathway at elevated protein concentrations. The fact that we do not observe transitions between this 1-bound state and any of the 0-bound states may indicate that the formation and disassociation of 1-bound states occurs too quickly to be resolved at the 100-ms resolution of our measurements.
≈ 0.42, indicated by a blue Gaussian curve in Figure 5B). The presence of transitions between this 1-bound state and only one of the multiple observed 2-bound states suggests that this represents the most likely association/dissociation pathway at elevated protein concentrations. The fact that we do not observe transitions between this 1-bound state and any of the 0-bound states may indicate that the formation and disassociation of 1-bound states occurs too quickly to be resolved at the 100-ms resolution of our measurements.
To test our conjecture that the four association/dissociation pathways we observed at low gp32 concentration (0.1 μM) are related to the two ways that a cooperatively bound dimer can occupy the p(dT)15 lattice, we performed similar experiments on a 3′-tail-pdT14-Cy3/Cy5 substrate. For this p(dT)14 substrate, the behavior of individual single-molecule trajectories and histograms of 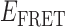 values as a function of gp32 concentration was qualitatively very similar to that we observed for the corresponding p(dT)15 substrate (see Supplementary Figure S3). However, the cumulative TDPs constructed from ∼50 single-molecule trajectories (corresponding to ∼5000 state-to-state transitions) taken from the p(dT)14 substrates at low protein concentration (0.1 μM gp32) exhibited only a single prominent transition pathway connecting a 2-bound state with
values as a function of gp32 concentration was qualitatively very similar to that we observed for the corresponding p(dT)15 substrate (see Supplementary Figure S3). However, the cumulative TDPs constructed from ∼50 single-molecule trajectories (corresponding to ∼5000 state-to-state transitions) taken from the p(dT)14 substrates at low protein concentration (0.1 μM gp32) exhibited only a single prominent transition pathway connecting a 2-bound state with 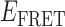 ≈ 0.3 to a 0- or 1-bound state with
≈ 0.3 to a 0- or 1-bound state with 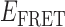 ≈ 0.46 (see Figure 6A). Less pronounced features in the cumulative TDP can also be observed, which appear to connect a 1-bound state to 0-bound states. At elevated protein concentration (1.0 μM gp32), the most prominent transition pathway occurs between a 2-bound state with
≈ 0.46 (see Figure 6A). Less pronounced features in the cumulative TDP can also be observed, which appear to connect a 1-bound state to 0-bound states. At elevated protein concentration (1.0 μM gp32), the most prominent transition pathway occurs between a 2-bound state with 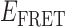 ≈ 0.17 and a 1-bound state with
≈ 0.17 and a 1-bound state with 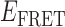 ≈ 0.37 (see Supplementary Figure S4A). The simplification of the pattern of association/dissociation pathways that we observe for the p(dT)14 substrate supports our conjecture that the more complex pattern we see for the 3′-tail-pdT15-Cy3/Cy5 substrate reflects the four possible ways that a cooperatively bound dimer may undergo association/dissociation transitions.
≈ 0.37 (see Supplementary Figure S4A). The simplification of the pattern of association/dissociation pathways that we observe for the p(dT)14 substrate supports our conjecture that the more complex pattern we see for the 3′-tail-pdT15-Cy3/Cy5 substrate reflects the four possible ways that a cooperatively bound dimer may undergo association/dissociation transitions.
Figure 6.

Cumulative TDPs constructed from the sum of ∼50 molecular trajectories from (A) the 3′-tail-pdT14-Cy3/Cy5 DNA in the presence of 0.1 μM gp32 and 100 mM NaCl; (B) the 3′-tail-pdT15-Cy3/Cy5 DNA in the presence of 0.1 μM gp32 and 10 mM NaCl; and (C) the 5′-tail-pdT15-Cy3/Cy5 DNA in the presence of 0.1 μM gp32 and 100 mM NaCl. Integrated transition density distributions are shown (in gray) projected along the horizontal and vertical axes. Horizontal and vertical gray dashed line segments connecting low- and high-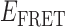 values indicate the most prominent state-to-state transition pathways.
values indicate the most prominent state-to-state transition pathways.
The above observations are consistent with the notion that end-dissociation (or association) is a favored pathway for the p(dT)15 lattices, which is facilitated in different ways for each of the two cooperatively bound positions. For example, end-dissociation/association may occur more freely from the 3′-end when the cluster is positioned against the p/t junction (occupying sites 1–14). Alternatively, end-dissociation/association might occur more freely at the 5′-end when the cluster is positioned at sites 2–15, such that there is a vacant dT ‘space’ at the p/t junction. As indicated above, the observation of only four association pathways—when there are nine positions at which a single gp32 molecule can potentially bind—suggests that only binding at productive positions 1–7, 2–8, 8–14 or 9–15 can lead to dimer formation, while binding at unproductive positions 3–9, 4–10, 5–11, 6–12 or 7–13 only leads to dissociation. This also implies that occupation of the four productive intermediate positions for single gp32 molecules by sliding of isolated gp32 molecules from unproductive positions is very unlikely, and effectively does not occur. (If sliding were to indirectly populate 1-bound productive intermediate states, we would expect to see more than four association pathways.) The distribution of intensities of the four disassociation/association pathways indicated by the TDP of Figure 5A is consistent with this scenario. The fact that only a single prominent feature occurs in the TDPs for the case of the p(dT)14 substrate (see Figure 6A and Supplementary Figure S4A), suggests that dissociation (or association) can occur only in one way—from the 3′-end—which is also consistent with our earlier work and biological requirements (14,15).
The above results suggest that association/dissociation events occur between distinct 2-bound and 0-bound sub-states, each of which is a member of a ‘family’ with discrete (and heterogeneous or disperse) 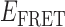 values. Inter-conversion between sub-states within a family is not observed, suggesting that if such transitions occur they do so more slowly than the 120 s duration of an individual single-molecule trajectory, or more quickly than the 100-ms instrumental resolution. At low protein concentration (0.1 μM gp32), the 1-bound state appears to have too short a lifetime to observe directly. However, at the higher 1 μM gp32 concentration, the 1-bound state appears to be directly coupled to at least one 2-bound state. Direct formation of the 1-bound state appears to be too fast to observe with the 100-ms resolution of these experiments. Alternative explanations for the relatively broad transition density distributions include the possibility of variations in the local environments of different molecules (e.g. transient surface attachment). However, we believe that such random sources of dispersion are unlikely to explain the well-defined and structured features of the TDPs.
values. Inter-conversion between sub-states within a family is not observed, suggesting that if such transitions occur they do so more slowly than the 120 s duration of an individual single-molecule trajectory, or more quickly than the 100-ms instrumental resolution. At low protein concentration (0.1 μM gp32), the 1-bound state appears to have too short a lifetime to observe directly. However, at the higher 1 μM gp32 concentration, the 1-bound state appears to be directly coupled to at least one 2-bound state. Direct formation of the 1-bound state appears to be too fast to observe with the 100-ms resolution of these experiments. Alternative explanations for the relatively broad transition density distributions include the possibility of variations in the local environments of different molecules (e.g. transient surface attachment). However, we believe that such random sources of dispersion are unlikely to explain the well-defined and structured features of the TDPs.
The effects of salt concentration changes on gp32–ssDNA association/dissociation dynamics
As mentioned in the previous section, our results suggest the presence of multiple distinguishable and relatively long-lived ( 100-ms) conformations of the 3′-tail-pdT15-Cy3/Cy5 substrate, even in the absence of protein. The existence of such conformations would imply either that multiple low activation barriers must be overcome for their inter-conversion, or that the intervening activation barriers are high in comparison to thermal energies. Furthermore, the distribution of the various substrate conformations is expected to affect the available transition pathways for protein–DNA complex assembly, since these represent potentially different binding targets for gp32 association and possibly different template conformations from which the cooperatively bound gp32 dimers can dissociate. In nearly all the experiments discussed in this paper to this point, we studied gp32-ssDNA-binding interactions with the 3′-tail-pdT15-Cy3/Cy5 construct in 100 mM NaCl, 6 mM MgCl2 and 10 mM Tris (pH 8.0). However, varying the monovalent cation concentration provides an additional approach to further examine these interactions using smFRET techniques.
100-ms) conformations of the 3′-tail-pdT15-Cy3/Cy5 substrate, even in the absence of protein. The existence of such conformations would imply either that multiple low activation barriers must be overcome for their inter-conversion, or that the intervening activation barriers are high in comparison to thermal energies. Furthermore, the distribution of the various substrate conformations is expected to affect the available transition pathways for protein–DNA complex assembly, since these represent potentially different binding targets for gp32 association and possibly different template conformations from which the cooperatively bound gp32 dimers can dissociate. In nearly all the experiments discussed in this paper to this point, we studied gp32-ssDNA-binding interactions with the 3′-tail-pdT15-Cy3/Cy5 construct in 100 mM NaCl, 6 mM MgCl2 and 10 mM Tris (pH 8.0). However, varying the monovalent cation concentration provides an additional approach to further examine these interactions using smFRET techniques.
To investigate the interplay between gp32 binding affinity, and the distribution of 3′-tail-pdT15-Cy3/Cy5 substrate target conformations, we performed smFRET experiments at NaCl concentrations ranging from 10 to 400 mM, while retaining buffer and divalent ion concentrations at their previous levels. Previous bulk solution studies have shown that the value of the association constant 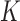 for both isolated gp32 monomer binding and cooperative (at monovalent salt concentration greater than 0.2 M) binding to ssDNA is salt concentration-dependent, with low salt concentrations favoring tighter binding, while the inter-gp32-subunit cooperativity parameter
for both isolated gp32 monomer binding and cooperative (at monovalent salt concentration greater than 0.2 M) binding to ssDNA is salt concentration-dependent, with low salt concentrations favoring tighter binding, while the inter-gp32-subunit cooperativity parameter 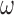 is not salt concentration dependent (36–39).
is not salt concentration dependent (36–39).
We also carried out bulk solution FRET experiments using the 3′-tail-pdT15-Cy3/Cy5 substrate as a function of NaCl and gp32 concentrations – the results are summarized in Supplementary Figure S5. For all of the protein concentrations examined (ranging from 0 to 10 μM gp32), we observed that the bulk FRET values generally increased as the NaCl concentration was increased (over the range 10 to 400 mM NaCl), which is consistent with the view that elevated salt concentrations lead to weaker protein–DNA interactions. However, we also observed some notable exceptions to this general behavior. Thus, at 0.1 μM gp32 concentrations, we observed that the lowest FRET value, which presumably corresponds to the tightest binding condition, occurs at physiological salt concentrations (∼100 mM NaCl). At salt concentrations lower and higher than physiological, the FRET value was seen to increase, indicating weaker binding. These observations suggest that at low protein concentrations another salt-dependent factor, such as substrate conformation, may influence protein-binding affinity. We therefore turned to smFRET experiments to investigate the effects of changing salt concentration on gp32 binding to ssDNA lattices.
In Figure 7, we compare histograms of 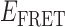 values obtained from single-molecule trajectories as a function of NaCl concentration (varied by row), and as a function of gp32 concentration (varied by column). For the DNA construct alone (far-left column), we found that the
values obtained from single-molecule trajectories as a function of NaCl concentration (varied by row), and as a function of gp32 concentration (varied by column). For the DNA construct alone (far-left column), we found that the 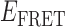 histogram at low NaCl concentration (10 mM) was very similar to that obtained at approximately physiological monovalent cation concentrations (100 mM). In both cases, the histograms showed a single broad feature centered at
histogram at low NaCl concentration (10 mM) was very similar to that obtained at approximately physiological monovalent cation concentrations (100 mM). In both cases, the histograms showed a single broad feature centered at 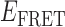 ≈ 0.6. As the NaCl concentration was raised above physiological levels (to ∼200 mM NaCl), we observed this feature to shift to the slightly lower value
≈ 0.6. As the NaCl concentration was raised above physiological levels (to ∼200 mM NaCl), we observed this feature to shift to the slightly lower value 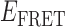 ≈ 0.57. At still higher salt concentrations (300 and 400 mM NaCl) this FRET distribution bifurcated into two separate broad peaks, one centered at
≈ 0.57. At still higher salt concentrations (300 and 400 mM NaCl) this FRET distribution bifurcated into two separate broad peaks, one centered at 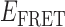 ≈ 0.35 and the other at
≈ 0.35 and the other at 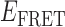 ≈ 0.64. We conjecture that the feature with peak value
≈ 0.64. We conjecture that the feature with peak value 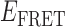 ≈ 0.35 represents a family of locally-stiffened p(dT)15 conformations of the ssDNA tail that are stabilized at high salt, while the feature with peak value
≈ 0.35 represents a family of locally-stiffened p(dT)15 conformations of the ssDNA tail that are stabilized at high salt, while the feature with peak value 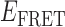 ≈ 0.65 represents a family of more flexible conformations that are similar to those populated at low salt. We note that an alternative explanation for the appearance of the low-FRET feature at ∼0.35 could be a chromophore-induced effect. We address this possibility further below.
≈ 0.65 represents a family of more flexible conformations that are similar to those populated at low salt. We note that an alternative explanation for the appearance of the low-FRET feature at ∼0.35 could be a chromophore-induced effect. We address this possibility further below.
Figure 7.

Histograms of the smFRET efficiencies for the 3′-tail-pdT15-Cy3/Cy5 substrate are shown as a function of NaCl concentration (10, 100, 200, 300 and 400 mM, varied by row), and as a function of gp32 concentration (0, 0.1, 1 and 10 μM, varied by column). The feature centered at 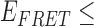 0.1 is an artifact (indicated by red-shaded distribution), as discussed in Materials and Methods. Each histogram is based on the compilation of thousands of individual smFRET data points. All the experiments were performed at 6 mM MgCl2 and 10 mM Tris (pH 8.0).
0.1 is an artifact (indicated by red-shaded distribution), as discussed in Materials and Methods. Each histogram is based on the compilation of thousands of individual smFRET data points. All the experiments were performed at 6 mM MgCl2 and 10 mM Tris (pH 8.0).
The remaining three columns of Figure 7 show how changing monovalent salt concentration affects the binding of gp32. For reference, the second row illustrates the gp32 concentration dependence at physiological salt, as previously discussed (see Figure 2). For samples incubated under low salt conditions (10 mM NaCl, top row), the peak feature at ∼0.4 appears to broaden and to shift to a slightly higher 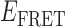 value (∼0.45), indicating the weakening of gp32 binding affinity. This could be a consequence of altered base stacking interactions at low salt, which favor the extended strand conformations that provide preferred substrates for gp32 binding. At salt concentrations above physiological (200, 300 and 400 mM NaCl, bottom three rows), gp32 binding interactions are greatly reduced. Such reduction in apparent gp32 binding might be attributed to increased electrostatic shielding and reduced ion-condensation-dependent mixing entropy effects as the monovalent salt concentration is increased, but the observed effects are unexpectedly large, and also interpretations of this sort offer no explanation for the second FRET peak (centered at ∼0.35) that appears in these high salt histograms in both the absence and the presence of gp32. We consider that this new peak in the high salt FRET histograms likely represents a new class of ssDNA conformations that are stabilized by high salt, and that also disfavor gp32 binding.
value (∼0.45), indicating the weakening of gp32 binding affinity. This could be a consequence of altered base stacking interactions at low salt, which favor the extended strand conformations that provide preferred substrates for gp32 binding. At salt concentrations above physiological (200, 300 and 400 mM NaCl, bottom three rows), gp32 binding interactions are greatly reduced. Such reduction in apparent gp32 binding might be attributed to increased electrostatic shielding and reduced ion-condensation-dependent mixing entropy effects as the monovalent salt concentration is increased, but the observed effects are unexpectedly large, and also interpretations of this sort offer no explanation for the second FRET peak (centered at ∼0.35) that appears in these high salt histograms in both the absence and the presence of gp32. We consider that this new peak in the high salt FRET histograms likely represents a new class of ssDNA conformations that are stabilized by high salt, and that also disfavor gp32 binding.
Further insight into the salt concentration dependence of gp32 binding to ssDNA lattice sequences can be obtained by investigating how changing salt conditions affect the association/dissociation kinetics of gp32 to the 3′-tail-pdT15-Cy3/Cy5 substrate. In Figure 8, we compare representative single-molecule trajectories taken from samples containing 0.1 μM gp32 (Figure 8A) and 1.0 μM gp32 (Figure 8B) in 10 mM NaCl. As discussed above, we found that the 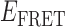 histograms for 0.1 μM gp32 exhibited a single broad feature with peak value
histograms for 0.1 μM gp32 exhibited a single broad feature with peak value 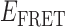 = ∼0.45, suggesting that binding interactions between gp32 and the p/t DNA substrate are slightly disrupted at reduced salt concentration in comparison to physiological salt concentrations. Individual smFRET trajectories revealed that the fluctuations occurred primarily between long-lived bound states with relatively low FRET efficiencies, and a much shorter-lived state with slightly higher efficiency. When 1 μM gp32 was used under the same low salt conditions, the low efficiency FRET state was enhanced (with peak value ∼0.35), and the frequency of low-to-high FRET transitions were reduced in comparison to those observed with the 0.1 μM gp32 sample (Figure 8B). These data indicate that under low salt conditions gp32 monomers bind more tightly to ssDNA, as expected, resulting in a reduced dissociation rate. These findings are in agreement with those of previous studies (18,37).
= ∼0.45, suggesting that binding interactions between gp32 and the p/t DNA substrate are slightly disrupted at reduced salt concentration in comparison to physiological salt concentrations. Individual smFRET trajectories revealed that the fluctuations occurred primarily between long-lived bound states with relatively low FRET efficiencies, and a much shorter-lived state with slightly higher efficiency. When 1 μM gp32 was used under the same low salt conditions, the low efficiency FRET state was enhanced (with peak value ∼0.35), and the frequency of low-to-high FRET transitions were reduced in comparison to those observed with the 0.1 μM gp32 sample (Figure 8B). These data indicate that under low salt conditions gp32 monomers bind more tightly to ssDNA, as expected, resulting in a reduced dissociation rate. These findings are in agreement with those of previous studies (18,37).
Figure 8.

smFRET trajectories (left column) and histograms (right column) for the 3′-tail-pdT15-Cy3/Cy5 substrate at low salt conditions (10 mM NaCl, with 6 mM MgCl2 and 10 mM Tris, pH 8.0). (A) 0.1 μM gp32; (B) 1 μM gp32. Cy3 fluorescence levels have been vertically displaced to clearly display the anti-correlated signal fluctuations. The red-shaded regions in the histograms show the probability distribution of background signals, which was determined from control measurements performed using biotin-Cy3 samples (see Supplementary Data).
We also performed experiments at 1 μM gp32 and high salt conditions (200 mM NaCl). In this case, the majority of smFRET trajectories (>97%) exhibited evidence of protein binding with a broad distribution of relatively high-FRET efficiencies (see Supplementary Figure S6A, S6B and Figure 7, third row, third column). Under these conditions, the equilibrium distribution of FRET efficiencies was slightly broader than that of the p/t DNA substrates in the absence of protein (see Figure 7, third row, first column).
In Figure 6B, we show the cumulative TDPs obtained from ∼50 molecular trajectories using 0.1 μM gp32 in 10 mM NaCl. The corresponding TDP constructed in the presence of 1 μM gp32 is shown in Supplementary Figure S4B. At this low-salt condition we observed, for both gp32 concentrations, a single prominent feature associated with state-to-state inter-conversion events that was significantly broadened in comparison to the individual features observed at the 100 mM NaCl condition (shown in Figure 5). The most prominent transition appears to occur between a 2-bound state and a 1-bound state, possibly reflecting the effects of reduced salt on the flexibility of the ssDNA substrate and the associated enhanced binding affinity of gp32.
We compared the average persistence times calculated from HMM analyses at 0.1 and 1.0 μM gp32 under low-salt (10 mM NaCl) and approximately physiological salt (100 mM NaCl) conditions (see Supplementary Tables S1 and S2). At both 0.1 and 1.0 μM gp32 concentrations, the average persistence times in the high efficiency FRET (0-bound) states were dramatically reduced under low-salt conditions, while the average persistence times in the low efficiency FRET (2-bound) states were increased, reflecting the shift of the equilibrium distribution to favor the cooperative gp32-ssDNA bound state under the reduced salt conditions. For the low salt 0.1 μM gp32 condition, we obtained the lifetime values 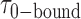 = 0.20 s and
= 0.20 s and 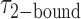 = 0.76 s, corresponding to an apparent equilibrium constant (
= 0.76 s, corresponding to an apparent equilibrium constant (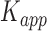 = [2-bound]/[0-bound]) of 3.8. For the 1.0-μM gp32 condition,
= [2-bound]/[0-bound]) of 3.8. For the 1.0-μM gp32 condition, 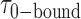 = 0.10 s and
= 0.10 s and 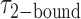 = 0.96 s corresponding, to a value of
= 0.96 s corresponding, to a value of 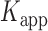 = [2-bound]/[0-bound] = 9.6, suggesting (as expected) significantly higher occupancy levels for the ssDNA tails under these latter binding conditions.
= [2-bound]/[0-bound] = 9.6, suggesting (as expected) significantly higher occupancy levels for the ssDNA tails under these latter binding conditions.
The above salt-dependent studies suggest that the conformations of the 3′-tail-p(dT)15-Cy3/Cy5 DNA substrates themselves may exist at elevated salt conditions in a number of distinct, long-lived conformations, even in the absence of protein. Although the majority of smFRET trajectories of these substrates exhibited quasi-stationary 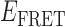 values over the course of the 120 s data set, it was possible to observe some trajectories that showed infrequent transitions between the various states represented by the histograms shown in Figure 7. An example of one such trajectory, for the DNA substrate alone at 300 mM NaCl, is shown in Supplementary Figure S6C. We see that the time scale of inter-conversion between the high- and low-FRET states is long (several seconds) in comparison to that of protein association/dissociation events (sub-second). We performed additional salt concentration-dependent smFRET measurements on a p/t DNA substrate in which the tail region was composed of a mixed base sequence [3′-tail-p(mixed-base-sequence)15-Cy3/Cy5]. The results of these measurements are shown in Supplementary Figure S7. We note that for the full range of salt concentrations investigated there is no evidence of the ∼0.36 feature that we observed for the 3′-tail-p(dT)15-Cy3/Cy5 substrates, although a number of distinct salt dependent features appear to occur over the range 0.6–0.95 of
values over the course of the 120 s data set, it was possible to observe some trajectories that showed infrequent transitions between the various states represented by the histograms shown in Figure 7. An example of one such trajectory, for the DNA substrate alone at 300 mM NaCl, is shown in Supplementary Figure S6C. We see that the time scale of inter-conversion between the high- and low-FRET states is long (several seconds) in comparison to that of protein association/dissociation events (sub-second). We performed additional salt concentration-dependent smFRET measurements on a p/t DNA substrate in which the tail region was composed of a mixed base sequence [3′-tail-p(mixed-base-sequence)15-Cy3/Cy5]. The results of these measurements are shown in Supplementary Figure S7. We note that for the full range of salt concentrations investigated there is no evidence of the ∼0.36 feature that we observed for the 3′-tail-p(dT)15-Cy3/Cy5 substrates, although a number of distinct salt dependent features appear to occur over the range 0.6–0.95 of 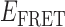 values. Future studies, perhaps conducted with sub-millisecond time resolution, should provide the needed information to determine the nature of these states. In the next section, we show that similar effects can be observed in the 5′-tail-p(dT)15-Cy3/Cy5 substrates, even under physiological salt conditions.
values. Future studies, perhaps conducted with sub-millisecond time resolution, should provide the needed information to determine the nature of these states. In the next section, we show that similar effects can be observed in the 5′-tail-p(dT)15-Cy3/Cy5 substrates, even under physiological salt conditions.
The influence of ssDNA strand polarity on the cooperative binding of gp32 protein
As previously discussed (14,15), gp32 binds to ssDNA in a polar fashion. Present models suggest that cooperative head-to-tail binding is required to permit multiple gp32 proteins to effectively coat the ssDNA regions of the genome in lagging strand DNA replication. Moreover, binding polarity is likely to be central in additional physiologically important processes in which gp32 is involved. Such processes include the displacement of gp32 by the helicase loading protein (gp59) during helicase loading onto the replication fork, in addition to myriad other interactions between gp32 and various replication, recombination and repair proteins at the p/t junction. These processes are likely regulated by kinetic features of competitive loading and displacement reactions, and understanding their dynamics could help to achieve a molecular understanding of central genome manipulation processes. To begin to investigate such issues, we examined the cooperative binding of the gp32 protein to p/t DNA constructs as a function of strand polarity (5′ versus 3′-tail), in addition to ssDNA p(dT)N strand length (N = 7, 15 and 22, see Figure 1). The DNA constructs used in this study are all shown in Table 1.
To initiate this investigation, we compared the binding behavior of the 3′-tail-pdT15-Cy3/Cy5 substrate discussed in previous sections to that of the 5′-tail-pdT15-Cy3/Cy5 substrate, again under approximately physiological solution conditions (100 mM NaCl, 6 mM MgCl2 in Tris buffer at pH 8.0). In Figure 9, we plot examples of smFRET trajectories and FRET efficiency histograms for the 5′-tail p/t DNA construct at the same protein concentrations as those shown for the 3′-tail p/t DNA construct in Figure 2. We found that the 5′-tail construct exhibited, in general, qualitatively similar smFRET behavior to that of the 3′-tail substrate. Nevertheless, we also observed some notable differences. In the absence of protein, the histogram of 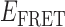 values exhibited its most prominent feature at ∼0.6. However, a secondary feature at
values exhibited its most prominent feature at ∼0.6. However, a secondary feature at 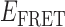 ≈ 0.33 is also evident (Figure 9A, right). Although the majority of smFRET trajectories exhibited stationary signals, it was possible to observe some trajectories that showed infrequent transitions between high- and low-FRET states. An example such a trajectory is shown in the left column of Figure 9A. Similar to the behavior we observed for the 3′ p(dT)15 substrates, the persistence times of these conformational states appear to be on the order of several seconds.
≈ 0.33 is also evident (Figure 9A, right). Although the majority of smFRET trajectories exhibited stationary signals, it was possible to observe some trajectories that showed infrequent transitions between high- and low-FRET states. An example such a trajectory is shown in the left column of Figure 9A. Similar to the behavior we observed for the 3′ p(dT)15 substrates, the persistence times of these conformational states appear to be on the order of several seconds.
Figure 9.

Representative smFRET trajectories of the 5′-tail-pdT15-Cy3/Cy5 p/t DNA substrate at various gp32 concentrations. (A) p/t DNA substrates alone; (B) in the presence of 0.1 μM gp32; (C) 1.0-μM gp32; and (D) 10 μM gp32. Histograms of the smFRET efficiencies [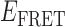 = IA/(ID + IA)], which are based on the compilation of thousands of individual smFRET trajectories, are shown in the right column. The trajectory for the DNA substrate alone [shown in Panel (A)] exhibits slow fluctuations between a high-FRET state centered at 0.6 and a low-FRET state centered at 0.35. The red-shaded regions in the histograms show the probability distribution of background signals, which was determined from control measurements performed using biotin-Cy3 samples (see Supplementary Data). The buffer solution in this figure and the next contained 100 mM NaCl, 6 mM MgCl2 and 10 mM Tris (pH 8.0).
= IA/(ID + IA)], which are based on the compilation of thousands of individual smFRET trajectories, are shown in the right column. The trajectory for the DNA substrate alone [shown in Panel (A)] exhibits slow fluctuations between a high-FRET state centered at 0.6 and a low-FRET state centered at 0.35. The red-shaded regions in the histograms show the probability distribution of background signals, which was determined from control measurements performed using biotin-Cy3 samples (see Supplementary Data). The buffer solution in this figure and the next contained 100 mM NaCl, 6 mM MgCl2 and 10 mM Tris (pH 8.0).
At 0.1 μM gp32, we observed that the majority of smFRET trajectories (>70% of the molecules within the sample imaging area) exhibited anti-correlated Cy3/Cy5 signal fluctuations, indicating the presence of protein-induced state-to-state transitions (Figure 9B). The FRET efficiency histogram at this protein concentration was broad and shifted, with a peak value of 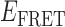 of ∼0.4, similar to the histograms we had observed for the 3′-tail p/t DNA substrate (see Figure 2B). However, a notable difference for the 5′-tail construct was that the individual smFRET trajectories exhibited a more complex pattern of state-to-state transitions, suggesting the involvement of multiple coupled intermediate states. At 1.0 μM gp32 concentration, the FRET efficiency histogram exhibited a broad feature that was further shifted to a lower peak value, with
of ∼0.4, similar to the histograms we had observed for the 3′-tail p/t DNA substrate (see Figure 2B). However, a notable difference for the 5′-tail construct was that the individual smFRET trajectories exhibited a more complex pattern of state-to-state transitions, suggesting the involvement of multiple coupled intermediate states. At 1.0 μM gp32 concentration, the FRET efficiency histogram exhibited a broad feature that was further shifted to a lower peak value, with 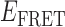 ≈ 0.32 (see Figure 9C). Individual smFRET trajectories at this protein concentration exhibited longer persistence times in the low-
≈ 0.32 (see Figure 9C). Individual smFRET trajectories at this protein concentration exhibited longer persistence times in the low-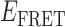 state, similar to the behavior that we observed previously for the 3′-tail p/t DNA constructs. At this higher protein concentration, the pattern of state-to-state transitions appeared to be more complex than those obtained from the 3′-tail substrates, suggesting the presence of multiple short-lived intermediate states with relatively low-
state, similar to the behavior that we observed previously for the 3′-tail p/t DNA constructs. At this higher protein concentration, the pattern of state-to-state transitions appeared to be more complex than those obtained from the 3′-tail substrates, suggesting the presence of multiple short-lived intermediate states with relatively low-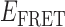 values. At protein concentrations well above saturation (10 μM gp32), individual smFRET trajectories exhibited quasi-stationary FRET signals with average peak value
values. At protein concentrations well above saturation (10 μM gp32), individual smFRET trajectories exhibited quasi-stationary FRET signals with average peak value 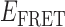 ≈ 0.29. Under such saturation conditions we observed no evidence for the occurrence of protein-induced state-to-state transitions.
≈ 0.29. Under such saturation conditions we observed no evidence for the occurrence of protein-induced state-to-state transitions.
Using our HMM analysis of smFRET trajectories for the 5′-tail-pdT15 p/t DNA substrate in the presence of protein, we calculated the average persistence times of bound and unbound states. Following our analysis of the 3′-tail-pdT15 p/t DNA substrate above, we made the simplified interpretation that the primary transitions we observe between high- and low-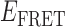 values at low protein concentration can be assigned to 0-bound and 2-bound states, respectively (see Supplementary Tables S1 and S2 for 0.1 and 1.0 μM gp32, respectively. At 0.1 μM gp32 concentrations, the average persistence times in the high- and low-efficiency FRET states are very similar to those we observed for the 3′-tail-pdT15 p/t DNA substrate, in which the unbound states were favored relative to the bound states. However, at 1.0 μM gp32 concentrations, we do not observe that the bound states become favored at these higher protein concentrations, as we do for the substrates of opposite polarity. This result suggests that cooperative gp32 cluster binding may proceed more expeditiously for the ssDNA tail with 3′ polarity. For the 0.1 μM gp32 condition, we obtained the lifetime values
values at low protein concentration can be assigned to 0-bound and 2-bound states, respectively (see Supplementary Tables S1 and S2 for 0.1 and 1.0 μM gp32, respectively. At 0.1 μM gp32 concentrations, the average persistence times in the high- and low-efficiency FRET states are very similar to those we observed for the 3′-tail-pdT15 p/t DNA substrate, in which the unbound states were favored relative to the bound states. However, at 1.0 μM gp32 concentrations, we do not observe that the bound states become favored at these higher protein concentrations, as we do for the substrates of opposite polarity. This result suggests that cooperative gp32 cluster binding may proceed more expeditiously for the ssDNA tail with 3′ polarity. For the 0.1 μM gp32 condition, we obtained the lifetime values 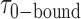 = 0.97 s and
= 0.97 s and 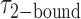 = 0.26 s, corresponding to an apparent equilibrium (occupancy) constant
= 0.26 s, corresponding to an apparent equilibrium (occupancy) constant 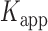 = [2-bound]/[0-bound] = 0.26. For the 1.0 μM gp32 condition,
= [2-bound]/[0-bound] = 0.26. For the 1.0 μM gp32 condition, 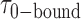 = 0.54 s and
= 0.54 s and 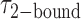 = 0.26 s, corresponding to a value of
= 0.26 s, corresponding to a value of 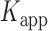 = 0.48.
= 0.48.
To identify the transition pathways associated with the cooperative binding of gp32 to the 5′-tail-pdT15-Cy3/Cy5 substrate, we constructed cumulative TDPs from ∼50 smFRET trajectories (corresponding to ∼5000 state-to-state transitions). In Figure 6C, we present the cumulative TDPs taken from smFRET trajectories recorded in the presence of 0.1 μM gp32. The corresponding TDP constructed in the presence of 1 μM gp32 is shown in Supplementary Figure S4C. We note that the patterns of state-to-state transitions indicated by the TDPs at both protein concentrations for the 5′-tail p/t DNA substrate versus the 3′-tail p/t DNA substrate are qualitatively similar, yet quantitatively different (compare with Figure 5). For the 5′-tail p/t DNA substrate at the lower protein concentration (0.1 μM gp32, Figure 6C), the transition density distribution is divided into two ‘families’ of low- and high-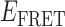 values, with a gap occurring near the value
values, with a gap occurring near the value 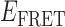 ∼0.4. Following the procedure we adopted for the 3′-tail p/t DNA substrate in previous sections, we assign the transition density features with
∼0.4. Following the procedure we adopted for the 3′-tail p/t DNA substrate in previous sections, we assign the transition density features with 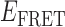 < 0.4 to the 2-bound states, and the features with
< 0.4 to the 2-bound states, and the features with 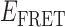 > 0.4 to the 0-bound state. For the 5′-tail p/t DNA substrate, our data indicate that two types of transitions occur between distinct 2-bound and 0-bound states (indicated by horizontal and vertical gray dashed line segments in Figure 6C).
> 0.4 to the 0-bound state. For the 5′-tail p/t DNA substrate, our data indicate that two types of transitions occur between distinct 2-bound and 0-bound states (indicated by horizontal and vertical gray dashed line segments in Figure 6C).
Of course, there is no a priori reason to expect the 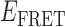 values corresponding to the various lattice conformations of the 5′-tail-pdT15 p/t DNA substrate to be the same, or even closely related, to those of the 3′-tail-pdT15 p/t DNA substrate. These values must depend on lattice polarity, and will additionally be affected by the preferred orientations of non-cooperatively and cooperatively bound proteins, and the attachment of the Cy3 and Cy5 probes at the distal ends of the p(dT)15 tails and the p/t junctions. Nevertheless, the interpretation we have adopted is a useful starting point to compare the behavior of the two different polarity substrates.
values corresponding to the various lattice conformations of the 5′-tail-pdT15 p/t DNA substrate to be the same, or even closely related, to those of the 3′-tail-pdT15 p/t DNA substrate. These values must depend on lattice polarity, and will additionally be affected by the preferred orientations of non-cooperatively and cooperatively bound proteins, and the attachment of the Cy3 and Cy5 probes at the distal ends of the p(dT)15 tails and the p/t junctions. Nevertheless, the interpretation we have adopted is a useful starting point to compare the behavior of the two different polarity substrates.
For the 5′-tail p/t DNA substrate at still higher protein concentration (1.0 μM gp32, Supplementary Figure S4C), the cumulative TDP appeared to be more heavily weighted towards smaller 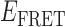 values in comparison to that of the 3′-tail p/t DNA substrate (compare to Figure 5B). We note the appearance of a pronounced feature with a peak value of
values in comparison to that of the 3′-tail p/t DNA substrate (compare to Figure 5B). We note the appearance of a pronounced feature with a peak value of 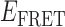 ≈ 0.4, which we assign to a 1-bound state. As before, we assign features with
≈ 0.4, which we assign to a 1-bound state. As before, we assign features with 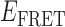 < 0.4 to 2-bound states, and those with
< 0.4 to 2-bound states, and those with 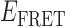 > 0.4 to 0-bound states. These data indicate the presence of a prominent transition pathway between a distinct 2-bound state and a 1-bound state. Our comparison between 3′ and 5′-tail-pdT15-Cy3/Cy5 constructs suggests that gp32 binding and the assembly of cooperatively bound clusters likely follows different detailed pathways for ssDNA templates with opposite polarities. The weak dependence of binding stability on gp32 concentration for the 5′ substrate suggests that the cooperative binding mechanism might be altered relative to that for the 3′ substrate. There appears to be only one primary assembly/disassembly pathway indicated by the TDPs for the 5′-tail-pdT15-Cy3/Cy5 substrate at low and high protein (0.1 μM gp32) concentration (Figure 6C and Supplementary Figure S4C, respectively), while there appear to be four competing pathways for the 3′-tail-pdT15-Cy3/Cy5 substrate (Figure 5A). This may be due to the higher stability for the 3′ substrate of the two cooperatively bound conformations (at positions 1–14 and 2–15) of 2-bound states, for which assembly and disassembly may be facilitated by the presence of a nascent 3′-tail. For the case of the 5′ substrate, the stability of 2-bound states may be disrupted, due in part to the absence of the nascent 3′-tail. Disruption of the cooperative binding mechanisms is consistent with shorter persistence times of 2- and 1-bound states for the 5′ substrates, as we have observed.
> 0.4 to 0-bound states. These data indicate the presence of a prominent transition pathway between a distinct 2-bound state and a 1-bound state. Our comparison between 3′ and 5′-tail-pdT15-Cy3/Cy5 constructs suggests that gp32 binding and the assembly of cooperatively bound clusters likely follows different detailed pathways for ssDNA templates with opposite polarities. The weak dependence of binding stability on gp32 concentration for the 5′ substrate suggests that the cooperative binding mechanism might be altered relative to that for the 3′ substrate. There appears to be only one primary assembly/disassembly pathway indicated by the TDPs for the 5′-tail-pdT15-Cy3/Cy5 substrate at low and high protein (0.1 μM gp32) concentration (Figure 6C and Supplementary Figure S4C, respectively), while there appear to be four competing pathways for the 3′-tail-pdT15-Cy3/Cy5 substrate (Figure 5A). This may be due to the higher stability for the 3′ substrate of the two cooperatively bound conformations (at positions 1–14 and 2–15) of 2-bound states, for which assembly and disassembly may be facilitated by the presence of a nascent 3′-tail. For the case of the 5′ substrate, the stability of 2-bound states may be disrupted, due in part to the absence of the nascent 3′-tail. Disruption of the cooperative binding mechanisms is consistent with shorter persistence times of 2- and 1-bound states for the 5′ substrates, as we have observed.
CONCLUSIONS AND BIOLOGICAL RELEVANCE
In this work we have used smFRET trajectories, gathered over a 2-min time window with 100-ms resolution, to begin to investigate the dynamics of the non-cooperative and cooperative binding of gp32 to short (N = 7–22 nts) oligo-dT lattices attached to the 3′- and 5′-ends of dsDNA ‘stems’ to form ssDNA ‘tails’ with defined polarities at p/t junctions. These constructs—in the context of gp32 binding—can effectively serve as ‘single-tailed’ models of DNA replication forks and p/t junctions. As pointed out in the Introduction, studies of such systems are relevant to biological mechanisms because gp32 molecules in bacteriophage T4 DNA replication complexes [and other ssDNA binding (ssb) proteins in the replication complexes of higher organisms] must bind to and assemble at such junctions correctly in order to permit DNA replication to proceed along the genome with appropriate overall processivity and fidelity. This requires that gp32 molecules bind at the replication fork (initially as monomers, and subsequently as cooperatively-bound clusters) in proper temporal and structural relationships with the primosome helicase (helicase-primase complex) and the lagging- (and possibly the leading-) strand DNA polymerases. These cooperatively-bound gp32 clusters must also be subsequently displaced to permit reformation of the DNA duplex, and this overall cycle of binding and displacement is likely to depend crucially on the dynamics of the interactions of these gp32 molecules with one another, with other protein regulatory factors, and with the ssDNA lattice at the p/t junction. Earlier studies (14,15,36,37,40) have provided some basic thermodynamic and structural information on the binding of gp32 to such model constructs. Our present studies, using model p/t junctions labeled with Cy3/Cy5 FRET pairs (see Figure 1A–C), now permit us to begin to interpret fluctuations of smFRET efficiencies as measures of the dynamics of non-cooperative and cooperative gp32 binding, rearrangement and dissociation reactions on the ssDNA ‘tails’ of such p/t constructs.
The details of these studies, and their tentative molecular interpretations in the context of each set of experiments, have been described in the Results and Discussion sections above. Here, we summarize the context of these experiments in terms of the overall T4 DNA replication system to provide an overview of how these results might eventually fit into a full structural, thermodynamic and kinetic picture of how the components of the elongating replication complex interact and work together.
Figure 10 provides two cartoon views of the context in which such interactions must occur. In Figure 10A we show a schematic of the main protein components of the T4 DNA replication system as they are distributed across a two-dimensional ‘snapshot’ of the DNA framework of the elongating complex. The gp32 proteins, which are centrally involved in integrating the functions of the polymerase, helicase and clamp loader subassemblies, are shown in Figure 10A bound in a polar fashion to the transient single-stranded portions of the DNA framework of the T4 DNA replication complex. Clearly, this represents a particular, rather than an average, distribution of gp32 molecules, since the numbers of gp32 molecules bound at the various positions shown will depend on the lengths of the ssDNA segments exposed, and also the stage of Okazaki fragment extension in the lagging strand DNA synthesis cycle.
Figure 10.

Schematic views of the T4 DNA replication complex with and without its protein components. (A) An ‘exploded’ two-dimensional view of the functional sub-assemblies of replication complex, specifically the polymerases, the helicase and the clamp-loader sub-assemblies, together with representative bound gp32 molecules and clusters (see text). (B) The same view, but here of the DNA framework of the replication complex with the proteins removed, in order to visualize the replication forks and p/t constructs within the overall complex to which gp32 can bind, and which are modeled, in part, by the ‘tailed’ DNA constructs used in this study and shown in Table 1.
Figure 10B shows the same DNA framework of the T4 replication complex, but now devoid of the protein components. This view is intended to reveal the positions and polarities of the various replication fork and p/t junctions that are present within the replication complex, but are partially obscured in the protein-containing view shown in Figure 10A. These junctions represent the positions to which gp32 must bind initially as monomers, and subsequently as cooperatively bound clusters. The model p/t DNA constructs used in this study and shown in Table 1 are intended to mimic these various critical junction positions, and are used in this paper to initiate a characterization of the binding dynamics of gp32 to these various potential targets of gp32 binding. It seems very likely that the binding dynamics revealed in this study, and their dependencies on gp32 binding polarity relative to the ssDNA ‘tails’, will play a role in determining the positioning and binding dynamics of the functional sub-assemblies of the replication complex.
Supplementary Material
Acknowledgments
The authors are grateful to Steve Weitzel and Miya Mary Michael for preparing the gp32 used in these studies, and to our laboratory colleagues, in particular Dr Carey Phelps, for many useful discussions of this work. In addition, we thank Morgan Marsh for help with performing some of the smFRET experiments. The authors also thank Dr Larry Scatena for his support of the laser equipment used for our single-molecule fluorescence experiments. The authors are also grateful to Dr Walter Baase for his detailed and helpful comments on several iterations of this manuscript. This work was supported by grants from the National Institutes of Health/National Institute of General Medical Sciences Grant (GM-15792 - to P.H.v.H.) and from the National Science Foundation, Chemistry of Life Processes Program (CHE-1608915 - to A.H.M.). B.I. was supported as a predoctoral trainee on NIH-NIGMS Institutional Research Service Award in Molecular Biology and Biophysics (GM-07759). P.H.v.H. is an American Cancer Society Research Professor of Chemistry.
Footnotes
Present addresses:
Wonbae Lee, Department of Physics and Astronomy, University of California, Irvine, CA 92697, USA.
John P. Gillies, Division of Biological Sciences, University of California at San Diego, La Jolla, CA 92093, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health/National Institute of General Medical Sciences [GM-15792 to P.H.v.H.]; National Science Foundation, Chemistry of Life Processes Program [CHE-1608915 to A.H.M.]; NIH-NIGMS Institutional Research Service Award in Molecular Biology and Biophysics [GM-07759 to B.I. as a predoctoral trainee]; P.H.v.H. is an American Cancer Society Research Professor of Chemistry. Funding for open access charge: National Institutes of Health/National Institute of General Medical Sciences [GM-15792 to P.H.v.H.].
Conflict of interest statement. None declared.
REFERENCES
- 1.Alberts B.M. Prokaryotic DNA replication mechanisms. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1987;317:395–420. doi: 10.1098/rstb.1987.0068. [DOI] [PubMed] [Google Scholar]
- 2.Nossal N.G. In: Molecular Biology of Bacteriphage T4. Karam JD, Kreuzer KN, Hall DH, editors. Washington, D.C.: American Society for Microbiology; 1994. pp. 43–54. [Google Scholar]
- 3.von Hippel P.H., Delagoutte E. A general model for nucleic acid helicases and their ‘coupling’ within macromolecular machines. Cell. 2001;104:177–190. doi: 10.1016/s0092-8674(01)00203-3. [DOI] [PubMed] [Google Scholar]
- 4.Mueser T.C., Hinerman J.M., Devos J.M., Boyer R.A., Williams K.J. Structural analysis of bacteriophage T4 DNA replication: A review in the Virology Journal series on bacteriophage T4 and its relatives. Virol. J. 2010;7:1–10. doi: 10.1186/1743-422X-7-359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jose D., Weitzel S.E., Jing D., von Hippel P.H. Assembly and subunit stoichiometry of the functional helicase-primase (primosome) complex of bacteriophage T4. Proc. Natl. Acad. Sci. U.S.A. 2012;109:13596–13601. doi: 10.1073/pnas.1210040109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee W., Jose D., Phelps C., Marcus A.H., Hippel P.H.v. A single-molecule view of the assembly pathway, subunit stoichiometry and unwinding activity of the bacteriophage T4 primosome (Helicase-Primase) complex. Biochemistry. 2013;52:3157–3170. doi: 10.1021/bi400231s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sinha N.K., Morris C.F., Alberts B.M. Efficient in vitro replication of double-stranded DNA templates by a purified T4 bacteriophage replication system. J. Biol. Chem. 1980;255:4290–4293. [PubMed] [Google Scholar]
- 8.Nossal N.G., Makhov A.M., Chastain P.D. 2nd, Jones C.E., Griffith J.D. Architecture of the bacteriophage T4 replication complex revealed with nanoscale biopointers. J. Biol. Chem. 2007;282:1098–1108. doi: 10.1074/jbc.M606772200. [DOI] [PubMed] [Google Scholar]
- 9.Pietroni P., von Hippel P.H. Multiple ATP binding is required to stabilize the ‘activated’ (clamp open) clamp loader of the T4 DNA replication complex. J. Biol. Chem. 2008;283:28338–28353. doi: 10.1074/jbc.M804371200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kelch B.A., Makino D.L., O'Donnell M., Kuriyan J. How a DNA polymerase clamp loader opens a sliding clamp. Science. 2011;334:1675–1680. doi: 10.1126/science.1211884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kelch B.A. The lord of the rings: Structure and mechanism of the sliding clamp loader. Biopolymers. 2016;105:532–546. doi: 10.1002/bip.22827. [DOI] [PubMed] [Google Scholar]
- 12.Gold L., O'Farrell P.Z., Russel M. Regulation of gene 32 expression during bacteriophage T4 infection of Escherichia coli. J. Biol. Chem. 1976;251:7251–7262. [PubMed] [Google Scholar]
- 13.von Hippel P.H., Kowalczykowski S.C., Lonberg N., Newport J.W., Paul L.S., Stormo G.D., Gold L. Autoregulation of gene expression: Quantitative evaluation of the expression and function of the bacteriophage T4 gene 32 (single-stranded DNA binding) protein system. J. Mol. Biol. 1982;162:6795–6818. doi: 10.1016/0022-2836(82)90548-4. [DOI] [PubMed] [Google Scholar]
- 14.Jose D., Weitzel S.E., Baase W.A., Michael M.M., von Hippel P.H. Mapping the interactions of the single-stranded DNA binding protein of bacteriophage T4 (gp32) with DNA lattices at single nucleotide resolution: polynucleotide binding and cooperativity. Nucleic Acids Res. 2015;43:9291–9305. doi: 10.1093/nar/gkv818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jose D., Weitzel S.E., Baase W.A., von Hippel P.H. Mapping the interactions of the single-stranded DNA binding protein of bacteriophage T4 (gp32) with DNA lattices at single nucleotide resolution: gp32 monomer binding. Nucleic Acids Res. 2015;43:9276–9290. doi: 10.1093/nar/gkv817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Alberts B.M., Frey L. T4 bacteriophage gene 32: A structural protein in the replication and recombination of DNA. Nature. 1970;227:1313–1318. doi: 10.1038/2271313a0. [DOI] [PubMed] [Google Scholar]
- 17.Jensen D.E., Kelly R.C., von Hippel P.H. DNA ‘melting’ proteins. II. Effects of bacteriophage T4 gene 32-protein binding on the conformation and stability of nucleic acid structures. J. Biol. Chem. 1976;251:7215–7228. [PubMed] [Google Scholar]
- 18.McGhee J.D., von Hippel P.H. Theoretical aspects of DNA-protein interactions: Cooperative and non-cooperative binding of large ligands to a one-dimensional homogeneous lattice. J. Mol. Biol. 1974;86:469–489. doi: 10.1016/0022-2836(74)90031-x. [DOI] [PubMed] [Google Scholar]
- 19.Record M.T., Jr, Lohman M.L., De Haseth P. Ion effects on ligand-nucleic acid interactions. J. Mol. Biol. 1976;107:145–158. doi: 10.1016/s0022-2836(76)80023-x. [DOI] [PubMed] [Google Scholar]
- 20.Selvin P.R. The renaissance of fluorescence resonance energy transfer. Nat. Struct. Biol. 2000;7:730–734. doi: 10.1038/78948. [DOI] [PubMed] [Google Scholar]
- 21.Murphy M.C., Rasnik I., Cheng W., Lohman T.M., Ha T. Probing single-stranded DNA conformational flexibility using fluorescence spectroscopy. Biophys. J. 2004;86:2530–2537. doi: 10.1016/S0006-3495(04)74308-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee W., von Hippel P.H., Marcus A.H. Internally Labeled Cy3 / Cy5 DNA Constructs Show Greatly Enhanced Photostability in Single-Molecule FRET Experiments. Nucleic Acids Res. 2014;42:5967–5977. doi: 10.1093/nar/gku199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Roy R., Kozlov A.G., Lohman T.M., Ha T. SSB protein diffusion on single-stranded DNA stimulates RecA filament formation. Nature. 2009;461:1092–1097. doi: 10.1038/nature08442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nguyen B., Sokoloski J., Galletto R., Elson E.L., Wold M.S., Lohman T.M. Diffusion of human replication protein A along single-stranded DNA. J. Mol. Biol. 2014;426:3246–3261. doi: 10.1016/j.jmb.2014.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Joo C., McKinney S.A., Nakamura M., Rasnik I., Myong S., Ha T. Real-time observation of RecA filament dynamics with single monomer resolution. Cell. 2006;126:515–527. doi: 10.1016/j.cell.2006.06.042. [DOI] [PubMed] [Google Scholar]
- 26.Koh H.R., Kidwell M.A., Ragunathan K., Doudna J.A., Myong S. ATP-independent diffusion of double-stranded RNA binding proteins. Proc. Natl. Acad. Sci. U.S.A. 2013;110:151–156. doi: 10.1073/pnas.1212917110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Qiu Y., Antony E., Doganay S., Koh H.R., Lohman T.M., Myong S. Srs2 prevents Rad51 filament formation by repetitive motion on DNA. Nat. Commun. 2013;4:1–10. doi: 10.1038/ncomms3281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Myong S., Bruno M.M., Pyle A.M., Ha T. Spring-loaded mechanism of DNA unwinding by Hepatitis C virus NS3 helicase. Science. 2007;317:513–516. doi: 10.1126/science.1144130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Myong S., Rasnik I., Joo C., Lohman T.M., Ha T. Repetitive shuttling of a motor protein on DNA. Nature. 2005;437:1321–1325. doi: 10.1038/nature04049. [DOI] [PubMed] [Google Scholar]
- 30.Yodh J.G., Stevens B.C., Kanagaraj R., Janscak P., Ha T. BLM helicase measures DNA unwound before switching strands and hRPA promotes unwinding reinitiation. EMBO J. 2009;28:405–416. doi: 10.1038/emboj.2008.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Park J., Myong S., Niedziela-Majka A., Lee K.S., Yu J., Lohman T.M., Ha T. PcrA helicase dismantles RecA filaments by reeling in DNA in uniform steps. Cell. 2010;142:544–555. doi: 10.1016/j.cell.2010.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Morten M.J., Peregrina J.R., Figueira-Gonzalez M., Ackermann K., Bode B.E., White M.F., Penedo J.C. Binding dynamics of a monomeric SSB protein to DNA: a single-molecule multi-process approach. Nucleic Acids Res. 2015;43:10907–10924. doi: 10.1093/nar/gkv1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Carroll R.B., Neet K., Goldthwait D.A. Studies of the self-association of bacteriophage T4 gene 32 protein by equilibrium sedimentation. J. Mol. Biol. 1975;91:275–291. doi: 10.1016/0022-2836(75)90380-0. [DOI] [PubMed] [Google Scholar]
- 34.Epstein I.R. Cooperative and non-cooperative binding of large ligands to a finite one-dimensional lattice. A model for ligand-oligonucleotide interactions. Biophys. Chem. 1978;8:327–339. doi: 10.1016/0301-4622(78)80015-5. [DOI] [PubMed] [Google Scholar]
- 35.McKinney S.A., Joo C., Ha T. Analysis of single-molecule FRET trajectories using hidden Markov modeling. Biophys. J. 2006;91:1941–1951. doi: 10.1529/biophysj.106.082487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Newport J.W., Lonberg N., Kowalczykowski S.C., von Hippel P.H. Interactions of bacteriophage T4-coded gene 32 protein with nucleic acids. II. Specificity of binding to DNA and RNA. J. Mol. Biol. 1981;145:105–121. doi: 10.1016/0022-2836(81)90336-3. [DOI] [PubMed] [Google Scholar]
- 37.Kowalczykowski S.C., Lonberg N., Newport J.W., von Hippel P.H. Interactions of bacteriophage T4-coded gene 32 protein with nucleic acids. I. Characterization of the binding interactions. J. Mol. Biol. 1981;145:75–104. doi: 10.1016/0022-2836(81)90335-1. [DOI] [PubMed] [Google Scholar]
- 38.Lohman T.M., Kowalczykowski S.C. Kinetics and mechanism of the association of the bacteriophage T4 gene 32 (helix destabilizing) protein with single-stranded nucleic acids. Evidence for protein translocation. J. Mol. Biol. 1981;152:67–109. doi: 10.1016/0022-2836(81)90096-6. [DOI] [PubMed] [Google Scholar]
- 39.Pant K., Karpel R.L., Rouzina I., Williams M.C. Salt dependent binding of T4 gene 32 protein to single and double-stranded DNA: Single molecule force spectroscopy measurements. J. Mol. Biol. 2005;349:317–330. doi: 10.1016/j.jmb.2005.03.065. [DOI] [PubMed] [Google Scholar]
- 40.Jones C.E., Mueser T.C., Nossal N.G. Bacteriophage T4 32 protein is required for helicase-dependent leading strand synthesis when the helicase is loaded by the T4 59 helicase-loading protein. J. Biol. Chem. 2004;279:12067–12075. doi: 10.1074/jbc.M313840200. [DOI] [PubMed] [Google Scholar]
- 41.Bittner M., Burke R.L., Alberts B.M. Purification of the T4 gene 32 protein free from detectable deoxyribonuclease activities. J. Biol. Chem. 1979;254:9565–9572. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.