

Abstract
Drugs of abuse cause changes in the prefrontal cortex (PFC) and associated regions that impair inhibitory control over drug-seeking. Breaking the contingencies between drug-associated cues and the delivery of the reward during extinction learning reduces rates of relapse. Here we used vagus nerve stimulation (VNS) to induce targeted synaptic plasticity to facilitate extinction of appetitive behaviors and to reduce relapse. Rats self-administered cocaine and were given VNS during extinction. Relapse to drug-seeking was assessed in a cued reinstatement session. We used immunohistochemistry to measure changes in the expression of the phosphorylated transcription factor cAMP response-element binding protein (pCREB) in the PFC and the basolateral amygdala (BLA), which regulate cue learning and extinction. In vivo recordings of evoked field potentials measured drug- and VNS-induced changes in metaplasticity in the pathway from the PFC to the BLA. VNS-treated rats showed improved rates of extinction and reduced reinstatement. Following reinstatement, pCREB levels were reduced in the IL and BLA of VNS-treated rats. Evoked responses in the BLA were greatly reduced in VNS-treated rats, and these rats were also resistant to the induction of LTD. Taken together, these results show that VNS facilitates extinction and reduces reinstatement. Changes in the pathway between the PFC and the amygdala may contribute to these beneficial effects.
Cocaine addiction is characterized by an impaired ability to develop adaptive behaviors that can compete with cocaine seeking, implying a deficit in the ability to induce plasticity in networks that regulate motivated behavior (Moussawi et al. 2009). As a result of this, recovery from drug addiction is frequently hampered by craving and relapse. Learning to inhibit, or extinguish, drug-seeking in response to drug-associated cues can reduce relapse. Extinction can reverse neuroadaptations caused by drug self-administration (Self et al. 2004; Millan et al. 2011). However, extinction training alone is often insufficient to prevent drug relapse (Weiss et al. 2001; Conklin and Tiffany 2002), potentially because the corticolimbic networks that are important for cue and reward processing, which include the nucleus accumbens, the prefrontal cortex (PFC), and the amygdala (Jentsch and Taylor 1999), become dysregulated by chronic drug use themselves (Fowler et al. 2007; Sinha and Li 2007; Liu et al. 2009; Nic Dhonnchadha and Kantak 2011). Modulating extinction processes to better consolidate the new-formed memories thus has clinical potential to reshape maladaptive behavior and to prevent relapse (Taylor et al. 2009). Vagus nerve stimulation (VNS) is a minimally invasive neuroprosthetic treatment which can induce targeted plasticity in active networks (Hays et al. 2013). VNS is FDA-approved for the treatment of epilepsy and depression, and it can improve sensory and motor function in models of tinnitus and stroke (Engineer et al. 2011; Porter et al. 2012; Hays et al. 2014a,b). VNS has also already been shown to facilitate extinction of conditioned fear (Peña et al. 2013, 2014), but an important open question is whether these effects also apply to the extinction of appetitive behaviors.
In addition, little is known about how VNS modulates synaptic plasticity that underlies extinction learning. Cue-learning and extinction of both aversive and rewarding stimuli involves activity in the pathway between the medial PFC and the basolateral nucleus of the amygdala (BLA) (Quirk and Mueller 2008; Sierra-Mercado et al. 2011). We have previously shown that pairing VNS with extinction of conditioned fear leads to metaplasticity (changes in the ability to induce LTP or LTD) in the pathway from the infralimbic cortex (IL) to the BLA (Peña et al. 2014). Here we tested the effect of VNS on extinction from cocaine self-administration and examined changes in synaptic plasticity in a part of the extinction network that controls cue-induced reinstatement. Our results show that VNS reduces cue-induced reinstatement without affecting ongoing appetitive behaviors. To examine VNS-induced changes in the network important for cue-induced reinstatement of drug-seeking, we used immunohistochemistry for the phosphorylated form of the transcription factor cAMP response-element binding protein (pCREB), as well as in vivo electrophysiological recordings of synaptic plasticity in the IL → BLA pathway following cued reinstatement. VNS-treated rats had reduced pCREB expression in the IL and BLA compared with sham-stimulated rats. Recordings of evoked local field potentials in the IL → BLA pathway furthermore showed that pairing VNS with extinction from cocaine seeking (but not extinction by itself) reduced the ability to induce LTD in this pathway. Taken together, our results suggest that VNS can reduce cocaine seeking by regulating activity in the mPFC–BLA network.
Results
VNS enhances extinction from drug-seeking and reduces reinstatement
To study the effect of VNS on extinction from drug-seeking, rats were first trained to self-administer cocaine, which was then followed by extinction training paired with sham-stimulation, ncVNS, cVNS, or a period of abstinence. After 12 d of extinction or abstinence, reinstatement to drug-seeking was measured in a cued reinstatement session by presenting the conditioned drug cues (Fig. 1). Over 10 d of drug self-administration rats in all groups (sham, n = 20; ncVNS, n = 20; cVNS, n = 8; abstinent, n = 9; drug only, n = 10) showed similar rates of responses on the active lever (F(1,61) = 0.76, P = 0.555), and responses on the active lever exceeded responses on the inactive lever in all groups (sham, F(1,38) = 73. 233, P < 0.001; ncVNS, F(1,38) = 68.296, P < 0.001; cVNS, F(1,14) = 23.371, P = <0.001; abstinent, F(1,16) = 13.091, P = 0.002; drug only, F(1,18) = 124.808, P < 0.001). During extinction, responses to the previously active lever were measured and used as an indicator of extinction learning. Sham-stimulated rats showed higher levels of responding during the 12 d extinction period than ncVNS rats (F(1,38) = 8.203, P = 0.007, Fig. 1A). No difference was found between sham-stimulated and cVNS rats during extinction (F(1,25) = 2.71, P = 0.112, Fig. 1B). Twenty-four hours after the last extinction session, drug-seeking was reinstated in a cued reinstatement session. Abstinent rats showed higher response rates during reinstatement than sham-stimulated rats (t(27) = 3.264, P = 0.003, Fig. 1C), however, pairing extinction training with VNS significantly facilitated extinction learning and further reduced relapse during cue-primed reinstatement. Both ncVNS rats (t(38) = 2.769, P = 0.009, Fig. 1A) and cVNS rats (t(25) = 2.254, P = 0.033, Fig. 1B) showed fewer responses at the active lever during cued reinstatement than sham-stimulated rats.
Figure 1.
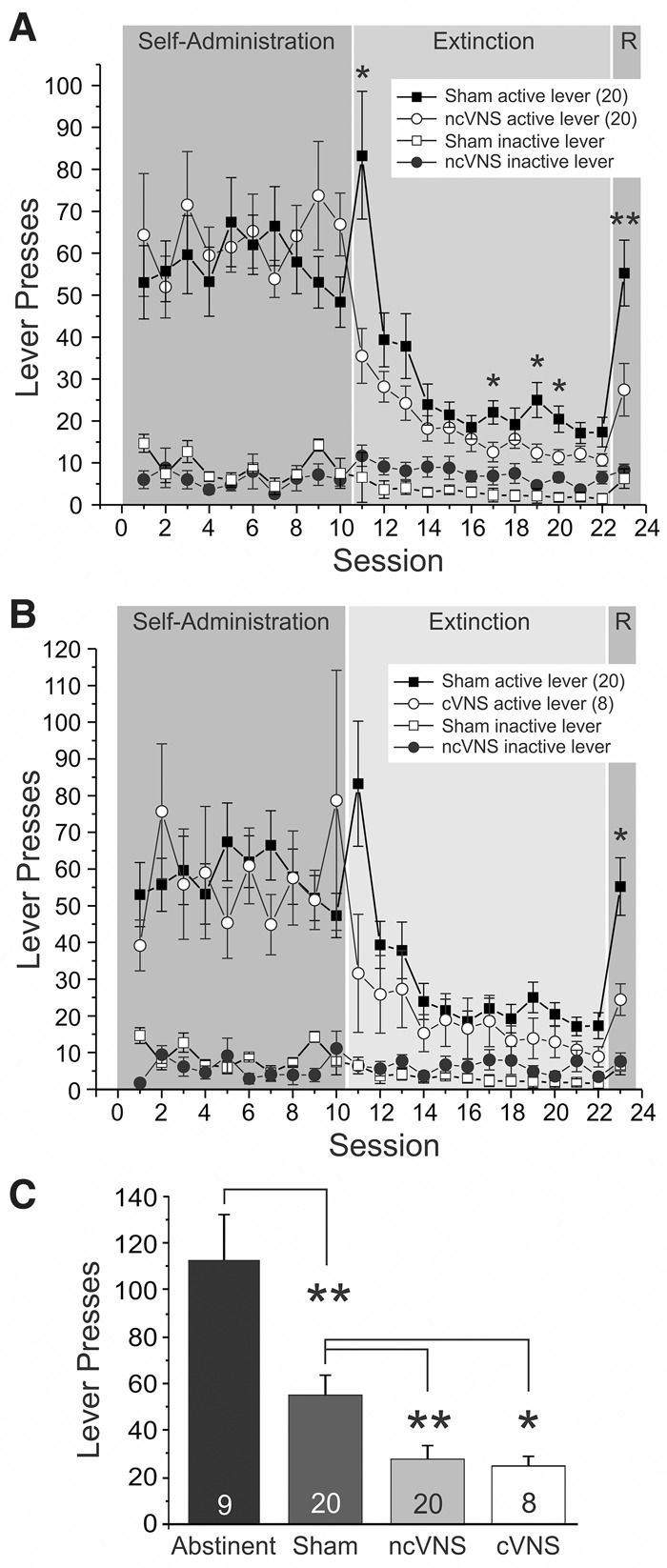
Vagus nerve stimulation (VNS) facilitates extinction from cocaine seeking and reduces cue-induced reinstatement. Rats were trained to self-administer cocaine and then underwent extinction training for 12 d while either receiving VNS (circles) or sham-stimulation (squares) before drug-seeking was reinstated by presentation of the previously drug-paired cues during a reinstatement session (R). (A) Comparison of sham-stimulated rats and rats receiving noncontingent VNS (ncVNS; see Materials and Methods for details of the stimulation parameters). (B) Comparison of sham-stimulated rats and rats receiving contingent VNS delivered with each lever press during extinction (cVNS; see Materials and Methods for details of the stimulation parameters). Rats receiving either ncVNS or cVNS showed reduced active lever presses during extinction and reinstatement. Inactive lever presses are shown as open circles. (C) Number of active lever presses during the reinstatement session for abstinent, sham-stimulated, and VNS treated rats. P values are (*) <0.05 and (**) <0.001.
VNS does not impact ongoing appetitive behavior
To exclude the possibility that the immediate and precipitous reduction in drug-seeking behavior on the first day of extinction resulted from unspecific motor or aversive effects of VNS, we applied VNS during appetitive responding to either food- or drug rewards. We trained rats to self-administer food (n = 6), or cocaine (n = 6), and applied VNS at different points during self-administration (Fig. 2). Because rats showed very high response rates during food self-administration, we used this opportunity to test the effects of VNS delivered contingently with each lever press (cVNS). Cocaine self-administering rats received ncVNS throughout their training sessions. We compared differences in response rates over a baseline period of self-administration without VNS (5 d food/10 d drug) to a 5 d period of self-administration with VNS. Response rates at the active lever were unaltered by VNS during both cocaine (F(1,10) < 0.001, P = 0.985) and food self-administration (F(1,10) = 0.630, P = 0.446). These results suggest that application of VNS is neither rewarding nor punishing, and does not affect ongoing appetitive behavior.
Figure 2.
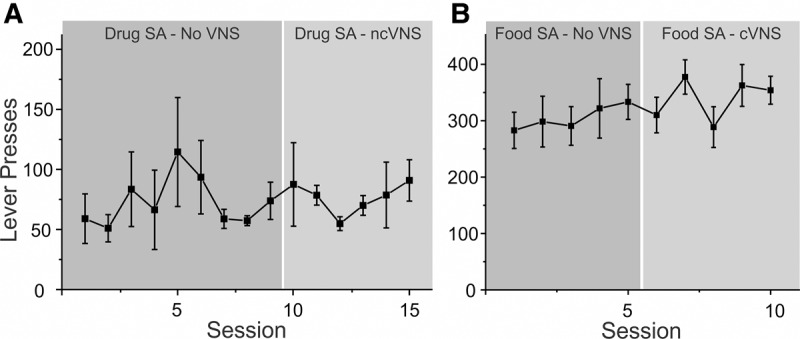
Vagus nerve stimulation does not alter ongoing appetitive behavior. Rats were trained to self-administer cocaine (A) or food pellets (B). After a stable rate of response was reached, rats were given VNS during self-administration sessions to see whether this changes response patterns. Rats that self-administered cocaine received noncontingent VNS, while rats that self-administered food received contingent VNS with each lever press. Responses at the active lever during self-administration were not altered by VNS.
VNS enhances extinction from food-seeking
To test whether the effects of VNS on extinction learning are specific to drug-seeking, we also delivered VNS to rats that extinguished from operant food self-administration (Fig. 3). Rats were allowed to self-administer food pellets for 5 d. No difference was found in rates of food self-administration between sham-stimulated (n = 10) and VNS (n = 10) rats (F(1,18) < 0.001, P = 0.990). As in extinction from drug self-administration, VNS delivered during extinction from food self-administration facilitated extinction learning of the operant response. During 5 d of extinction, sham-stimulated rats had more responses on the previously active lever than ncVNS rats (F(1,18) = 6.365, P = 0.021). Similarly, sham-stimulated rats showed higher absolute response rates during the cue-induced reinstatement session compared with ncVNS rats (t(18) = 2.511, P = 0.022). However, overall rats in both groups showed relatively little reinstatement, so that response rates at the active lever during the reinstatement session were not significantly different from those on the last day of extinction. These results imply that VNS may facilitate extinction across a wide range of reinforcers, including heroin (Liu et al. 2011), and food (present data), as well as aversive stimuli (Peña et al. 2013, 2014).
Figure 3.
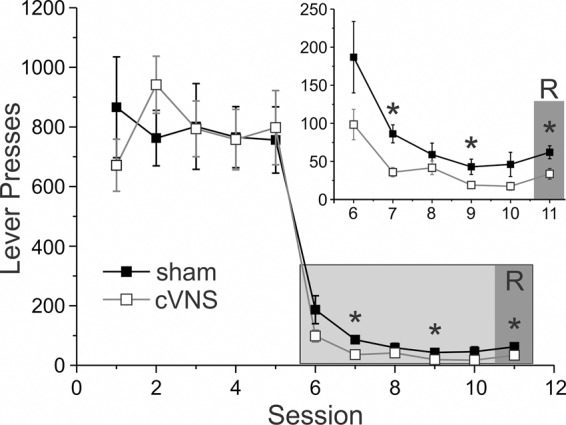
Vagus nerve stimulation facilitates extinction from food seeking. Rats were trained to self-administer food pellets for 5 d and then underwent extinction training for 5 d while they received either VNS (open squares) or sham-stimulation (black squares). Food seeking was reinstated by presentation of the previously food-paired cues during a reinstatement session (R). Compared with sham-stimulated rats VNS rats made fewer responses at the active lever during extinction and during the cue-induced reinstatement session. However, rats in both groups showed only small, nonsignificant increases in responses during cue-induced reinstatement when compared with the last day of extinction . P values are (*) <0.05. Inset shows a detail of the extinction and reinstatement days.
VNS reduces pCREB expression in IL and BLA
Changes in the expression of pCREB occur after drug-administration, extinction, and reinstatement (Zhou and Zhu 2006). To determine how VNS affects network activity in regions important for cue extinction, we measured levels of pCREB in the PL, IL, and BLA immediately after cue-induced reinstatement (Fig. 4). A repeated-measures ANOVA revealed a main effect of treatment in the PL (F(1,23) = 6.174, P = 0.003). Drug-naïve rats (n = 6) expressed less pCREB than drug-only and sham-stimulated groups (BonferroniDrugOnly P = 0.029, n = 5; BonferroniSham P = 0.029, n = 7). There was no difference between drug-naive and VNS rats in the PL (BonferroniVNS P = 0.162, n = 9). No difference was found in any other comparisons in the PL (Bonferroni P > 0.05). In the IL, treatment also showed a main effect (F(1,23) = 8.956, P = 0.0004), with drug-only and sham-stimulated groups showing higher expression levels of pCREB than drug-naïve rats (BonferroniDrugOnly P = 0.009; BonferroniSham P = 0.0004). No difference was found between drug-naïve rats and VNS rats (BonferroniVNS P = 0.198). VNS rats expressed less pCREB than sham-stimulated rats (Bonferroni P = 0.039), but were not different from drug-only subjects (Bonferroni P = 0.518). In the BLA, treatment also showed a main effect (F(1,23) = 7.988, P = 0.001). Post hoc analysis showed that drug-naive rats expressed less pCREB in the BLA than drug-only and sham-stimulated rats (BonferroniDrugOnly P = 0.031, BonferroniSham P = 0.015). VNS rats were not different from drug-naïve rats (Bonferronidrug-naïve P = 1), but expressed significantly less pCREB in the BLA than drug-only and sham-stimulated rats (BonferroniDrugOnly P = 0.015; BonferroniShamP = 0.006). Across all three regions (PL, IL, BLA) drug-only and sham-stimulated rats showed similar trends (Bonferroni = 1). Importantly, following VNS, pCREB expression in the BLA was different from both drug-only and sham-stimulated rats, and was reduced to levels similar to those seen in drug-naïve rats. In the somatosensory cortex (S1) no differences were found between any treatment groups (F(1,20) = 0.836, P = 0.490), suggesting that treatment-dependent differences seen in the other brain regions are specific to the extinction circuit.
Figure 4.

Vagus nerve stimulation reduces pCREB expression in infralimbic cortex and basolateral amygdala following reinstatement of drug-seeking. Photomicrographs (20× magnification) of phosphorylated CREB expression (red) in the prelimbic (PL, top row) and infralimbic (IL, second row) prefrontal cortex, the basolateral amygdala (BLA, third row), as well as the primary somatosensory cortex (S1, bottom row) in four treatment groups (drug-naïve, drug-only, sham-stimulated, and noncontingent VNS). Bargraphs on the right summarize the data for each row (PL, IL, BLA, S1). pCREB expression is shown as the percentage of cell bodies that showed pCREB immunoreactivity among all DAPI-stained cells. Rats trained to self-administer cocaine (drug-only, light gray bars) and sham-stimulated rats (dark gray bars) showed increased pCREB expression in the PL, IL, and BLA compared with drug-naïve rats (white bars). VNS treatment (black bars) given during extinction reduced pCREB expression in the IL and BLA compared with sham-stimulated rats to similar levels as seen in drug-naïve rats. P values are (*) <0.05 and (**) <0.001.
VNS alters plasticity in the extinction circuit
We have previously shown that VNS facilitates extinction of conditioned fear and alters the synaptic plasticity in the pathway between the IL and the BLA (12). The projection between the IL and the BLA is also important for the extinction of cue-induced drug-seeking behavior (26). To test whether VNS induces changes in synaptic plasticity in this pathway when given during extinction from drug-seeking, we stimulated the IL and recorded eLFPs in the BLA following reinstatement (Fig. 5). A two-way ANOVA of the relationship between stimulation intensity (0.4 mA–2.0 mA) and amplitude of the baseline eLFP revealed a main effect of treatment on these I–O curves (F(1,21) = 3.460, P = 0.025), with VNS rats having significantly smaller baseline responses than drug-naïve and abstinent rats (LSDdrug-naive P = 0.006; LSDAbstinent P = 0.006, LSDSham P = 0.390, LSDDrugOnly P = 0.068). Next, we applied low-frequency stimulation (LFS) designed to induce LTD. In drug-naïve (n = 6) and sham-stimulated (n = 8) rats, LFS reliably induced LTD (drug-naïve, F(1,10) = 8.128, P = 0.017; sham-stimulated, F(1,14) = 5.535, P = 0.034). In contrast, rats that received ncVNS during extinction training (n = 9) were resistant to the induction of LTD, showing little change in eLFP amplitude from baseline (F(1,16) = 2.605, P = 0.170). Abstinent rats (n = 5) also showed no difference between baseline and post-LFS amplitude (F(1,8) = 2.063, P = 0.189). A fifth group (drug-only) was recorded 24 h after the last drug self-administration session, without receiving any extinction training (n = 7). This group also showed no differences between the baseline and post-LFS response (F(1,12) = 0.503, P = 0.492, Fig. 5D). Taken together, these data show that combining extinction training with VNS alters the responsiveness of the BLA to IL inputs and changes the parameters for induction of LTD in the IL → BLA pathway. However, drug taking alone also alters synaptic plasticity in this pathway.
Figure 5.

VNS treatment alters the responsiveness of the basolateral amygdala to stimulation of the infralimbic cortex. (A) Diagram of the stimulation and recording sites in the infralimbic (IL) PFC and the BLA. (B) Single-pulse stimulation targeted to the IL elicited negative field potentials in the BLA that peaked after 20–25 msec. Representative traces (average of six consecutive sweeps) of an input–output curve from a drug-naive rat. (C) Input–output curves from rats in five treatment groups (drug-naïve, black squares; drug-only, triangles; abstinent, gray circles; sham-stimulated, white squares; non-contingent VNS, black circle). VNS rats showed reduced amplitudes of the evoked local field potential compared with drug-naïve and abstinent rats. (D) Low-frequency (1 Hz) stimulation applied at baseline stimulation intensity (40% of the stimulation intensity that evoked a maximum field response) induced LTD in drug-naïve and sham-stimulated rats. All other groups show no significant change from baseline following low-frequency stimulation, despite differences in the range of field potential amplitudes that could be evoked under baseline conditions (C).
Discussion
One strategy for reducing relapse is to promote self-regulation by extinguishing responses to drug-associated environmental stimuli. However, this type of exposure therapy for drug addiction is not consistently effective (Dutra et al. 2008), potentially because the brain areas required for extinction learning may become impaired by drug use (Nic Dhonnchadha et al. 2008; Liu et al. 2009; Kantak and Nic Dhonnchadha, 2011). Extinction alone can reverse some of the maladaptive changes caused by drug use (Self et al. 2004); thus, enhancing extinction learning may improve clinical outcomes (Taylor et al. 2009). Here, we administered VNS during extinction from cocaine- and food-seeking and found enhanced extinction and reduced reinstatement compared with sham-stimulated rats. Importantly, ongoing appetitive behavior was not altered by VNS, supporting previous reports that VNS is tolerated well (Handforth et al. 1998).
We previously found that VNS enhances extinction from conditioned fear (Peña et al. 2014). Here we show that VNS can also facilitate extinction from drug-seeking and from food self-administration. A recent study also indicated that VNS can reduce relapse to heroin seeking (Liu et al. 2011). Taken together, these data suggest that VNS can be used to modulate extinction from a wide range of reinforcers. This is important for both our understanding of the mechanisms of VNS, as well as the prevention of relapse to drug seeking, because the mechanisms and networks that underlie extinction from psychostimulant and opiate self-administration are thought to be at least partially different, with the ventromedial prefrontal cortex acting as neural OFF switch for cocaine seeking, but an ON switch for heroin seeking (Ettenberg et al. 1982; Peters et al. 2013).
In addition, VNS administered either contingent with nonreinforced lever presses or noncontingently throughout the extinction sessions reduced reinstatement to equal degrees, suggesting that VNS may function both by enhancing learning about the operant response, and/or by enhancing contextual learning, as previously suggested (Peña et al. 2014). The fact that VNS can facilitate extinction from various reinforcers and across a range of stimulation parameters is consistent with the idea that VNS promotes plasticity by altering neurotransmission selectively in those brain regions actively engaged in a task (Kilgard 2012; Childs et al. 2015). Temporally precise modulation of active networks could explain how the effects of VNS can generalize to motor functions, as well as a range of conditioned and unconditioned stimuli (Engineer et al. 2011; Porter et al. 2012; Hays et al. 2014a,b).
Activation of the mesocorticolimbic system, including dopaminergic and glutamatergic pathways that connect the ventral tegmental area, nucleus accumbens, amygdala, and PFC, constitutes a common pathway through which various drugs of abuse mediate their reinforcing effects (Nestler 2009). Long-term neuroadaptations in this circuitry underlie the transition to drug dependence and cycles of relapse. As an initial test of the mechanisms that underlie VNS-enhanced extinction, we measured levels of pCREB in the mPFC and BLA to infer activity levels during reinstatement. Decreases in pCREB expression in the PFC were previously found following drug-induced reinstatement of morphine-conditioned place preference (Zhou and Zhu 2006). Deactivation studies using local application of drugs or focal lesions (Weissenborn et al. 1997; McFarland and Kalivas 2001; McLaughlin and See 2003; Peters et al. 2008) suggest that in the mPFC of rodents a dorsal-ventral distinction exists, wherein the PL drives the expression of both fear and drug-seeking, whereas the IL suppresses these behaviors after extinction (Peters et al. 2008, 2009). The distinct effects of IL and PL on extinction and reinstatement of drug-seeking are mediated, at least in part, through neuroadaptations in anatomically distinct projections to the nucleus accumbens core (from PL) and shell (from IL), respectively (Self et al. 2004; Peters et al. 2008, 2009; Moussawi et al. 2009; Knackstedt et al. 2010). In addition, recent electrophysiological analyses show that neuronal networks within PL and IL can contribute to both response execution and inhibition, suggesting that neurons in both regions encode context-appropriate responses, firing more strongly for a “Go” response during reward seeking and for a “No-Go” response during extinction (Moorman and Aston-Jones 2014; Moorman et al. 2015). Our immunohistochemical analysis of pCREB expression is seemingly at odds with a simple functional dichotomy of the mPFC subregions: While our main finding was that pairing VNS with extinction reduced pCREB expression in the extinction network compared with sham-stimulated subjects, the pattern of these changes did not differ between IL and PL. However, it is important to note that we studied pCREB expression after the cue-induced reinstatement session, which shows characteristics of both drug-seeking (initiated by presentation of the cues) and intra-session extinction (caused by the absence of drug reinforcement). Thus while pCREB expression can serve as a general marker for VNS-induced changes in activity in the extinction circuit, it may not be sufficient to distinguish the different roles of multiple overlapping networks in the IL and PL. When pairing VNS with extinction we observed the largest reduction of pCREB expression in the BLA. To further examine how VNS changes plasticity in the extinction circuit, we performed in vivo recordings in the IL → BLA pathway, which is important for the formation of discrete stimulus–reward associations (See 2005) and the expression of extinction behavior (Peters et al. 2008; Rogers et al. 2008; Kruzich and See 2001). In VNS-treated rats, which showed reduced reinstatement of drug seeking, we observed diminished responsiveness of the BLA to IL stimulation. Furthermore, pairing extinction with VNS caused a shift in metaplasticity, making the IL → BLA pathway resistant to LTD induction. This parallels VNS-induced changes in the extinction circuit for conditioned fear (Peña et al. 2014). The reduction in the baseline response may result in a “floor effect” that prevents further depotentiation during LTD induction. In rats that only underwent self-administration (drug-only) and abstinent subjects that had no extinction training, the IL → BLA pathway was also resistant to LTD induction, despite larger baseline responses seen in these groups. This suggests differences in the ability to induce plasticity in the BLA during each of the phases of the self-administration/extinction paradigm, that are caused by either the drug itself, learned cue-reward associations, or extinction learning. This is again similar to what is seen in the IL → BLA pathway during fear conditioning and extinction from conditioned fear (Vouimba and Maroun 2011). There, the response in the BLA increases after fear conditioning and remains elevated unless subjects undergo extinction, at which point the response becomes smaller than the baseline response.
Bidirectional changes in synaptic strength between nodes of the extinction circuit have also been reported before in rats that underwent drug self-administration and extinction (Moussawi et al. 2011). In the pathway from the mPFC to the nucleus accumbens core the strength of the synaptic connection reflected whether the animal was drug-taking, extinguished, or withdrawn. These changes in the baseline response also resulted in altered metaplasticity (Knackstedt et al. 2010; Moussawi et al. 2011). Finally, our observations of reduced pCREB expression in the BLA and reduced baseline responses in the IL → BLA pathway are also consistent with the idea of reduced excitability of neurons within the BLA. Such reduced excitability could result in reduced drive in the reciprocal connection from the BLA to the PL, which contributes to drug-seeking via the projection from the PL to the nucleus accumbens (Peters et al. 2008).
VNS has been shown to enhance cortical and subcortical plasticity (Zuo et al. 2007; Engineer et al. 2011; Porter et al. 2012; Peña et al. 2014). Our electrophysiological data add further evidence for that VNS can be used to promote targeted plasticity. The mechanisms that underlie these effects are less clear. Stimulation of ascending fibers of the cervical vagus nerve leads to activation of the nucleus of the solitary tract, indirectly causing the release of several neuromodulators and widespread cortical and subcortical activation (Chase et al. 1966, Krahl and Clark 2012). In drug-naïve rats, acute VNS increases levels of norepinephrine in the BLA and hippocampus (Hassert et al. 2004). Similarly, long-term VNS upregulates serotonin levels in the mPFC and BLA (Manta et al. 2009). VNS also induces fos expression in the amygdala, as well as the habenula, locus coeruleus, and various cortical areas (Naritoku et al. 1995). Changes in neurotransmission and gene expression indicate that VNS may be acting as a primer, readying the system for plasticity in response to specific inputs that occur during sensory stimulation (Engineer et al. 2011), motor learning (Porter et al. 2012), or extinction learning (Peña et al. 2013, 2014). This tight temporal control and network-specificity that electrical stimulation of the vagus nerve allows, reflects a major advantage of VNS over pharmacological treatments. This idea is further supported by our finding that even brief VNS delivered contingent with nonreinforced lever presses during extinction reduced drug-seeking during reinstatement. On the other hand, even repeated long lasting VNS given outside of a behaviorally relevant context (e.g., in the home cage), does not induce changes in behavior or synaptic plasticity (Peña et al. 2014). The targeted nature of VNS may reduce the threshold for efficacy in current behavioral therapies where pharmacological approaches have yielded mixed results. Recent advances in transcutaneous VNS (Aihua et al. 2014) could further increase the versatility of VNS in clinical settings.
Materials and Methods
Subjects
We used male Sprague-Dawley rats (Charles River, Wilmington, MA) that were at least 60 d old (250–300 g) at the time of surgery. Rats were individually housed and kept on a 12 h reverse light/dark cycle, with free access to food and water until surgery, when food was restricted to 25 g/d standard rat chow. All protocols were approved by the IACUC of The University of Texas at Dallas and were conducted in compliance with the NIH Guide for the Care and Use of Laboratory Animals.
Jugular vein catheterization and vagus nerve electrode implant
Rats were anesthetized with ketamine (85 mg/kg) and xylazine (5 mg/kg) and implanted with a catheter in the right external jugular vein for drug administration. Atropine (1 mg/kg) was administered to reduce secretions, and all incision sites were pretreated with Marcaine. Catheters consisted of silicon tubing (Silastic; 0.51 mm I.D., 0.94 mm O.D.) connected to a back-mounted cannula (Plastics One). During the same surgery, a custom-made cuff electrode was placed around the left vagus nerve for the delivery of VNS (Childs et al. 2015). Catheters were flushed daily with heparinized saline (0.2 mL of 100 IU) and gentamycin (0.2 mL of 0.1 g/mL) to maintain catheter patency, which was assessed weekly by infusing 1.0 mg/0.1 mL of the fast-acting anesthetic methohexital sodium and noting the presence or absence of a brief period of sedation.
Drug self-administration and extinction training
Five to 7 d following surgery, rats were trained in a single overnight session to self-administer food pellets (45 mg, Bio Serv) in an operant conditioning chamber (Med Associates). Drug self-administration training took place in the same chamber, which was equipped with two levers, a house light, a cue light, and a tone. Each active lever press produced a 0.05 mL infusion of 0.5 mg/kg of cocaine (Sigma) in saline, and the presentation of drug-paired cues (illumination of the light over the active lever and the presentation of a 2900 Hz tone), followed by a 20 sec timeout. Self-administration sessions ended after 2 h or at a maximum of 200 infusions. Both right and left levers were available for the duration of the session and drug-seeking behavior was quantified as active lever presses. Rats self-administered cocaine over at least 10 d, during which they had to achieve at least 10 infusions per session. Rats that did not reach this criterion were excluded from further analysis. Following the last self-administration session rats in the drug-only group were either sacrificed immediately for immunohistochemical experiments or were used for in vivo recordings on the following day. Subjects in the extinction groups underwent 12 d of extinction training in which lever presses on the previously active lever no longer produced cocaine or presentation of drug-paired cues. During extinction training rats received either sham-stimulation or VNS under one of two conditions: in rats that received contingent VNS (cVNS), each press of the previously drug-paired lever triggered VNS (0.8 mA 15 biphasic pulses,100 µsec, at 30 Hz, 500 msec) (Rush et al. 2000; Elliott et al. 2011). In rats that received noncontingent VNS (ncVNS), stimulation occurred every 5 min for 30 sec at 0.4 mA for the duration of the training session. These stimulation parameters mimic those used for depression (Sackheim et al. 2001), and are also similar to those used to enhance retention performance in rats (Clark et al. 1995). An additional group of rats underwent forced abstinence in their home cages. After 12 d of extinction training or abstinence, drug-seeking behavior was reinstated by presentation of the drug-associated cues in the operant conditioning chambers. During the reinstatement session presses on the “active” lever led to presentation of the previously drug-associated tone and light, but did not result in drug delivery or VNS. Following the reinstatement session, subjects were then sacrificed immediately for immunohistochemistry experiments, or were used the following day for in vivo LFP recordings.
Food self-administration and extinction training
To assess the specificity of VNS’s effects on extinction of drug-seeking, we trained additional groups to self-administer food. Rats were allowed to self-administer food pellets for 5 d in 2 h sessions and were then extinguished with either ncVNS stimulation or sham-stimulation for an additional 5 d. After extinction, operant responding was reinstated by presentation of the previously food-associated cues, and the number of responses at the active lever were taken as a measure of reinstatement of food-seeking behavior.
Appetitive behavior
To ensure that the effects of VNS on extinction learning were not due to discomfort (punishment), and to more generally assess the impact of VNS on appetitive behavior, we also tested the effects of VNS administered during food or drug self-administration. Rats were allowed to self-administer cocaine for 10 d as described above. On days 11–15, ncVNS was administered throughout the 2 h self-administration sessions. Rats working for food rewards received a pellet in response to every active lever press without any timeouts. To avoid a possible “cost benefit” determination masking any punishment effects, in this group VNS (500 msec, 0.8 mA) was administered contingently with each lever press. Rats were allowed to self-administer food pellets with sham-stimulation for 5 d, followed by cVNS for an additional 5 d.
Immunohistochemistry
A subset of sham-stimulated, ncVNS, Drug-only, and drug-naive rats were sacrificed for immunohistochemical analysis of pCREB in slices of the IL, PL, BLA, and the primary sensory cortex (S1). S1 was included in this analysis as a negative control, because we expected that our experimental treatments would only minimally alter pCREB expression in this area. After the last treatment session (either reinstatement or drug self-administration), rats were anesthetized with 30% urethane (2 g/kg bodyweight) and transcardially perfused with 0.9% saline followed by cold 4% paraformaldehyde in 0.12 M phosphate buffered saline (PBS). Brains were post-fixed for 1 h in 4% paraformaldehyde and 30% sucrose and then transferred to 30% sucrose in PBS for 24 h at 4°C. Coronal slices (40 µm) were cut on a freezing microtome and free floating sections were incubated in primary rabbit anti-pCREB (working dilution 1:2000; Millipore) for 24 h in PBS and 0.3% Triton X-100 (Sigma). Slices were washed three times in PBS and incubated in secondary anti-rabbit 594 (working dilution 1:1000; Jackson Laboratories) for 2 h at room temperature. Slices were washed, mounted, and coverslipped with ProLong Gold antifade reagent with DAPI (Life Technologies). Fluorescence images were taken at 20× magnification on a laser scanning confocal microscope (Fluoview 1000, Olympus Corporation). Cells expressing DAPI and pCREB were counted by experimenters that were blind to the experimental condition. pCREB expression is presented as a ratio of pCREB-labeled cells over DAPI-labeled cells.
In vivo LFP recording
Twenty-four hours after the last treatment, a bipolar matrix stimulation electrode (FHC) was placed in the IL (D/V: 4.6, A/P: 3.0, M/L: 0.6 from bregma) and evoked local field potentials (eLFPs) were recorded in the BLA (D/V: 7.3, A/P:−2.76, M/L: −4.9 from bregma) of urethane-anesthetized (1.5 g/kg IP) rats, using glass microelectrodes (2 M KCl; 1–2 MOhms resistance), following previously published protocols (Peña et al. 2014; Childs et al. 2015). Baseline data were collected for a minimum of 10 min before long-term depression (LTD) was induced using low-frequency stimulation (1 Hz for 15 min) at the same stimulation intensity used for baseline recordings (23). The amplitude of the EFP was measured as the difference between the mean of a 5 msec window before the stimulation artifact and the mean of a 5 msec window around 20–25 msec after the stimulation artifact, corresponding to the negative peak of the field potential. Data were normalized to a 10 min baseline, and the period 40–50 min after plasticity induction was used to analyze long-term changes.
Data analysis
Self-administration and extinction data were analyzed using two-way repeated-measures ANOVA and single trial reinstatement data were analyzed using a one way ANOVA. Analyses of VNS’ effects on ongoing behavior used repeated-measures ANOVA. Immunohistochemical analyses used repeated measures ANOVA and LSD post hoc testing. Changes in evoked field potential amplitudes following induction of synaptic plasticity were compared using a partially repeated-measures ANOVA with a treatment group × time interaction. Input–output curves of evoked field potentials were analyzed using a two-way repeated measures ANOVA and LSD post hoc testing. P values <0.05 were considered significant.
Competing interst statement
The authors declare no competing financial interests.
Acknowledgments
S.K. wishes to acknowledge institutional support from The University of Texas at Dallas.
Footnotes
Article is online at http://www.learnmem.org/cgi/doi/10.1101/lm.043539.116.
References
- Aihua L, Lu S, Liping L, Xiuru W, Hua L, Yuping W. 2014. A controlled trial of transcutaneous vagus nerve stimulation for the treatment of pharmacoresistant epilepsy. Epilepsy Behav 39: 105–110. [DOI] [PubMed] [Google Scholar]
- Chase HM, Sterman MB, Clemente CD. 1966. Cortical and subcortical patters of response to afferent vagal stimulation. Exp Neurol 16: 36–49. [DOI] [PubMed] [Google Scholar]
- Childs JE, Alvaraz-Dieppa A, McIntyre C, Kroener S. 2015. Vagus nerve stimulation as a tool to induce plasticity in pathways relevant for extinction learning. J Vis Exp 102: e53032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark KB, Krahl SE, Smith DC, Jensen RA. 1995. Post-training unilateral vagal stimulation enhances retention performance in the rat. Neurobiol Learn Mem 63: 213–216. [DOI] [PubMed] [Google Scholar]
- Conklin CA, Tiffany ST. 2002. Applying extinction research and theory to cue-exposure addiction treatments. Addiction 97: 155–167. [DOI] [PubMed] [Google Scholar]
- Dutra L, Stathopoulou G, Badsen SL, Leyro TM, Powers MB, Otto MW. 2008. A meta-analytic review of psychosocial interventions for substance use disorders. Am J Psychiatry 165: 179–187. [DOI] [PubMed] [Google Scholar]
- Elliott RE, Morsi A, Kalhorn SP, Marcus J, Sellin J, Kang M, Silverberg A, Rivera E, Geller E, Carlson C, et al. 2011. Vagus nerve stimulation in 436 consecutive patients with treatment resistant epilepsy: long-term outcomes and predictors of response. Epilepsy Behav 20: 57–63. [DOI] [PubMed] [Google Scholar]
- Engineer ND, Riley JR, Seale JD, Vrana WA, Shetake JA, Sudanagunta SP, Borland MS, Kilgard MP. 2011. Reversing pathological neural activity using targeted plasticity. Nature 470: 101–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ettenberg A, Pettit HO, Bloom FE, Koob GF. 1982. Heroin and cocaine intravenous self-administration in rats: mediation by separate neural systems. Psychopharmacology 78: 204–209. [DOI] [PubMed] [Google Scholar]
- Fowler JS, Volkow ND, Kassed CA, Chang L. 2007. Imaging the addicted human brain. Sci Pract Perspect 3: 4–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handforth A, DeGiorgio CM, Schachter SC, Uthman BM, Naritoku DK, Tecoma ES, Henry TR, Collins SD, Vaughn BV, Gilmartin RC, et al. 1998. Vagus nerve stimulation therapy for partial onset seizures: a randomized active-control trial. Neurology 51: 48–55. [DOI] [PubMed] [Google Scholar]
- Hassert DL, Miyashita T, Williams CL. 2004. The effects of peripheral vagal nerve stimulation at a memory-modulating intensity of norepinephrine output in the basolateral amygdala. Behav Neurosci 118: 79–88. [DOI] [PubMed] [Google Scholar]
- Hays SA, Rennaker RL, Kilgard MP. 2013. Targeting plasticity with vagus nerve stimulation to treat neurological disease. Prog Brain Res 207: 275–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hays SA, Khodaparast N, Hulsey DR, Ruiz A, Sloan AM, Rennaker RL, Kilgard MP. 2014a. Vagus nerve stimulation during rehabilitative training improves functional recovery after intracerebral hemorrhage. Stroke 45: 3097–3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hays SA, Khodaparast N, Ruiz A, Sloan AM, Hulsey DR, Rennaker RL, Kilgard MP. 2014b. The timing and amount of vagus nerve stimulation during rehabilitative training affect poststroke recovery of forelimb strength. Neuroreport 25: 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jentsch JD, Taylor JR. 1999. Impulsivity resulting from frontostriatal dysfunction in drug abuse: implications for the control of behavior by reward-related stimuli. Psychopharmacology 146: 373–390. [DOI] [PubMed] [Google Scholar]
- Kantak KM, Nic Dhonnchadha BÁ. 2011. Pharmacological enhancement of drug cue extinction learning: translational challenges. Ann N Y Acad Sci 1216: 122–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilgard MP. 2012. Harnessing plasticity to understand learning and treat disease. Trends Neurosci 35: 715–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knackstedt LA, Moussawi K, Lalumiere R, Schwendt M, Klugmann M, Kalivas PW. 2010. Extinction training after cocaine self-administration induces glutamatergic plasticity to inhibit cocaine seeking. J Neurosci 30: 7984–7992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krahl SE, Clark KB. 2012. Vagus nerve stimulation for epilepsy: a review of central mechanisms. Surg Neurol Int 3: 255–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruzich PJ, See RE. 2001. Differential contributions of the basolateral and central amygdala in the acquisition and expression of conditioned relapse to cocaine-seeking behavior. J Neurosci 21: RC155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Liang J, Qin W, Tian J, Yuan K, Bai L, Zhang Y, Wang W, Wang Y, Li Q, et al. 2009. Dysfunctional connectivity patterns in chronic heroin users: an fMRI study. Neurosci Lett 460: 72–77. [DOI] [PubMed] [Google Scholar]
- Liu H, Liu Y, Yu J, Lai M, Zhu H, Sun A, Chen W, Zhou W. 2011. Vagus nerve stimulation inhibits heroin-seeking behavior induced by heroin priming or heroin-associated cues in rats. Neurosci Lett 494: 70–74. [DOI] [PubMed] [Google Scholar]
- Manta S, Dong J, Debonnel G, Blier P. 2009. Enhancement of the function of rat serotonin and norepinephrine neurons by sustained vagus nerve stimulation. J Psychiatry Neurosci 34: 372–280. [PMC free article] [PubMed] [Google Scholar]
- McFarland K, Kalivas PW. 2001. The circuitry mediating cocaine-induced reinstatement of drug-seeking behavior. J Neurosci 21: 8655–8663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin J, See RE. 2003. Selective inactivation of the dorsomedial prefrontal cortex and the basolateral amygdala attenuates conditioned-cued reinstatement of extinguished cocaine-seeking behavior in rats. Psychopharmacology (Berl) 168: 57–65. [DOI] [PubMed] [Google Scholar]
- Millan ZE, Marchant NJ, McNally GP. 2011. Extinction of drug seeking. Behav Brain Res 271: 454–462. [DOI] [PubMed] [Google Scholar]
- Moorman DE, Aston-Jones G. 2014. Orbitofrontal cortical neurons encode expectation-driven initiation of reward-seeking. J Neurosci 34: 10234–10246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moorman DE, Jame MH, McGlinchey EM, Aston-Jones G. 2015. Differential roles of medial prefrontal subregions in the regulation of drug seeking. Brain Res 1628: 130–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moussawi K, Pacchioni A, Moran M, Olive MF, Grass JT, Lavin A, Kalivas PW. 2009. N-Acetylcysteine reverses cocaine-induced metaplasticity. Nat Neurosci 12: 182–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moussawi K, Zhou W, Shen H, Reichel CM, See RE, Carr DB, Kalivas PW. 2011. Reversing cocaine-induced synaptic potentiation provides enduring protection from relapse. Proc Natl Acad Sci 108: 385–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naritoku DK, Terry WJ, Helfer RH. 1995. Regional induction of fos immunoreactivity in the brain by anticonvulsant stimulation of the vagus nerve. Epilepsy Res 22: 53–62. [DOI] [PubMed] [Google Scholar]
- Nestler EJ. 2009. Cellular and molecular mechanisms of drug addiction. In Neurobiology of Mental Illness (ed. Charney DS, Nestler EJ) pp. 775–785. Oxford University Press, New York. [Google Scholar]
- Nic Dhonnchadha BÁ, Cunningham KA. 2008. Serotonergic mechanisms in addiction-related memories. Behav Brain Res 195: 39–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nic Dhonnchadha BÁ, Kantak KM. 2011. Cognitive enhancers for facilitating drug cue extinction: insights from animal models. Pharmacol Biochem Behav 99: 229–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peña DF, Engineer ND, McIntyre CK. 2013. Rapid remission of conditioned fear expression with extinction training paired with vagus nerve stimulation. Biol Psychiatry 73: 1071–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peña DF, Childs JE, Willet SM, Vital A, McIntyre CK, Kroener S. 2014. Vagus nerve stimulation enhances extinction of conditioned fear and modulates plasticity in the pathway from the ventromedial prefrontal cortex to the amygdala. Front Behav Neurosci 8: 327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters J, LaLumiere RT, Kalivas PW. 2008. Infralimbic prefrontal cortex is responsible for inhibiting cocaine seeking in extinguished rats. J Neurosci 28: 6046–6053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters J, Kalivas PW, Quirk GJ. 2009. Extinction circuits for fear and addiction overlap in prefrontal cortex. Learn Mem 16: 279–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters J, Pattij T, De Vries TJ. 2013. Targeting cocaine versus heroin memories: divergent roles within ventromedial prefrontal cortex. Trends Pharmacol Sci 34: 689–695. [DOI] [PubMed] [Google Scholar]
- Porter BA, Khodaparast N, Fayyaz T, Cheung RJ, Ahmed SS, Vrana WA, Rennaker RL II, Kilgard MP. 2012. Repeatedly pairing vagus nerve stimulation with a movement reorganizes primary motor cortex. Cereb Cortex 22: 2365–2374. [DOI] [PubMed] [Google Scholar]
- Quirk GJ, Mueller D. 2008. Neural mechanisms of extinction learning and retrieval. Neuropsychopharmacology 33: 56–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers JL, Ghee S, See RE. 2008. The neural circuitry underlying reinstatement of heroin-seeking behavior in an animal model of relapse. Neuroscience 151: 579–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rush AJ, George MS, Sackeim HA, Marangell LB, Husain MM, Giller C. 2000. Vagus nerve stimulation (VNS) for treatment-resistant depression: a multicenter study. Biol Psychiatry 47: 276–286. [DOI] [PubMed] [Google Scholar]
- Sackheim HA, Rush AJ, George MS, Marangell LB, Husain MM, Nahas Z. 2001. Vagus nerve stimulation (VNS) for treatment-resistant depression: efficacy, side effects, and predictors of outcome. Neuropsychopharmacology 25: 713–728. [DOI] [PubMed] [Google Scholar]
- See RE. 2005. Neural substrates of cocaine-cue associations that trigger relapse. Eur J Pharmacol 526: 140–146. [DOI] [PubMed] [Google Scholar]
- Self DW, Choi K, Simmons D, Walker JR, Smagula CS. 2004. Extinction training regulates neuroadaptive responses to withdrawal from chronic cocaine self-administration. Learn Mem 11: 648–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sierra-Mercado D, Padilla-Coreano N, Quirk GJ. 2011. Dissociable roles of prelimbic and infralimbic cortices, ventral hippocampus and basolateral amygdala in the expression and extinction of conditioned fear. Neuropsychopharmacology 36: 529–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha R, Li CS. 2007. Imaging stress- and cue-induced drug and alcohol craving: association with relapse and clinical implications. Drug Alcohol Rev 26: 25–31. [DOI] [PubMed] [Google Scholar]
- Taylor JR, Olausson P, Quinn JJ, Torregrossa MM. 2009. Targeting extinction and reconsolidation mechanisms to combat the impact of drug cues on addiction. Neuropharmacology 56: 186–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vouimba RM, Maroun M. 2011. Learning-induced changes in mPFC-BLA connections after fear conditioning, extinction and reinstatement of fear. Neuropsychopharmacology 36: 2276–2285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss F, Martin-Fardon R, Ciccocioppo R, Kerr TM, Smith DL, Ben-Shahar O. 2001. Enduring resistance to extinction of cocaine-seeking behavior induced by drug-related cues. Neuropsychopharmacology 25: 361–372. [DOI] [PubMed] [Google Scholar]
- Weissenborn R, Robbins TW, Everitt BJ. 1997. Effects of medial prefrontal or anterior cingulate cortex lesions on responding for cocaine under fixed-ratio and second-order schedules of reinforcement in rats. Psychopharmacology (Berl) 134: 242–257. [DOI] [PubMed] [Google Scholar]
- Zhou LF, Zhu YP. 2006. Changes of CREB in rat hippocampus, prefrontal cortex and nucleus accumbens during three phases of morphine induced conditioned place preference in rats. J Zhejiang Univ Sci B 7: 107–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo Y, Smith DC, Jensen RA. 2007. Vagus nerve stimulation potentiates hippocampal LTP in freely moving rats. Physiol Behav 90: 583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]