

Abstract
Atypical PKM, a persistently active form of atypical PKC, is proposed to be a molecular memory trace, but there have been few examinations of the role of PKMs generated from other PKCs. We demonstrate that inhibitors used to inhibit PKMs generated from atypical PKCs are also effective inhibitors of other PKMs. In contrast, we demonstrate that dominant-negative PKMs show isoform-specificity. A dominant-negative PKM from the classical PKC Apl I blocks activity-dependent intermediate-term facilitation (a-ITF) when expressed in the sensory neuron, while a dominant-negative PKM from the atypical PKC Apl III does not. Consistent with a specific role for PKM Apl I in activity-dependent facilitation, live imaging FRET-based cleavage assays reveal that activity leads to cleavage of the classical PKC Apl I, but not the atypical PKC Apl III in the sensory neuron varicosities of Aplysia. In contrast, massed intermediate facilitation (m-ITF) induced by 10 min of 5HT is sufficient for cleavage of the atypical PKC Apl III in the motor neuron. Interestingly, both cleavage of PKC Apl I in the sensory neuron during a-ITF and cleavage of PKC Apl III in the motor neuron during m-ITF are inhibited by a dominant-negative form of a penta-EF hand containing classical calpain cloned from Aplysia. Consistent with a role for PKMs in plasticity, this dominant-negative calpain also blocks both a-ITF when expressed in the sensory neuron and m-ITF when expressed in the motor neuron. This study broadens the role of PKMs in synaptic plasticity in two significant ways: (i) PKMs generated from multiple isoforms of PKC, including classical isoforms, maintain memory traces; (ii) PKMs play roles in the presynaptic neuron.
The identification of pharmacological agents capable of disrupting long-established memories suggests that an ongoing, active process is required to maintain memories over prolonged periods (Sossin 2008; Sacktor 2011). A leading candidate for this active process is PKMζ, a persistently active atypical protein kinase composed of the isolated catalytic domain of PKCζ (Sacktor 2012). Indeed, the first identified amnesic agent capable of disrupting memory maintenance, the zeta inhibitor peptide (ZIP), was designed to inhibit PKMζ (Ling et al. 2002). In vertebrates, PKMζ is encoded by an alternative mRNA transcript, which is transported to dendrites and then translated during plasticity to generate the persistently active kinase (Hernandez et al. 2003; Muslimov et al. 2004). The specific role of PKMζ in memory has been challenged due to reports of intact late-long term potentiation (L-LTP) and memory in mice lacking PKC/PKMζ (Lee et al. 2013; Volk et al. 2013). However, other lines of evidence, including dominant negatives (Shema et al. 2011) and acute knock-out approaches using siRNA (Dong et al. 2015; Tsokas et al. 2016) support a role for PKMζ in memory maintenance. Interestingly, compensation from the other atypical PKC in vertebrates, PKC iota (PKCι), in PKC/PKMζ knock-out animals has been demonstrated (Tsokas et al. 2016). This raises the question of whether PKMs generated from other isoforms of PKC normally play roles in memory maintenance. Indeed, very few previous experiments have considered the isoform specificity of either pharmacological inhibitors or dominant negatives.
We have been examining the role of PKMs in the synaptic changes underlying the maintenance of memory in Aplysia californica. In this model, specific neurons involved in simple, modifiable behaviors have been identified and characterized, and the relevant changes in synaptic strength can be reproduced in cultured neurons. This allows for the molecular, optical, and electrophysiological analysis of synaptic connections involved in learning and memory in a reduced preparation (Kandel 2001). Three PKCs are expressed in the Aplysia nervous system, PKC Apl I, PKC Apl II, and PKC Apl III, belonging to the classical, novel and atypical PKC families, respectively (Sossin 2007). No alternative transcript producing a PKM has been identified in this system consistent with the alternative transcript for PKMζ first appearing in early chordates (Bougie et al. 2009). Nevertheless, the same inhibitors used in the vertebrate system, ZIP and chelerythrine (Serrano et al. 2005, 2008), block the maintenance of a number of forms of synaptic plasticity and memory in Aplysia, including activity-dependent intermediate-term facilitation (a-ITF) (Sutton and Carew 2000), activity-dependent long-term facilitation (Hu and Schacher 2015), site-specific sensitization (Sutton et al. 2004), massed intermediate-term facilitation (m-ITF) (Villareal et al. 2009), massed operant conditioning (Michel et al. 2012), long-term facilitation and long-term sensitization (Cai et al. 2011). Moreover, a dominant-negative form of the atypical PKM Apl III has been shown to block m-ITF (Bougie et al. 2012). The evidence that transcription-independent forms of plasticity, such as a-ITF and m-ITF can be reversed during their maintenance phase by inhibitors of PKMs (Sutton and Carew 2000; Villareal et al. 2009) allows for experimental analysis of the targets of these inhibitors in simpler forms of plasticity.
In Aplysia, PKMs are proposed to be formed by calpain-mediated cleavage of PKC (Sutton et al. 2004; Bougie et al. 2009) and consistent with this concept, calpain inhibitors block induction of several of the putatively PKM-dependent forms of plasticity described above (Sutton et al. 2004; Villareal et al. 2009). This raises the question of isoform specificity since calpain can cleave all the PKC isoforms into PKMs in vitro (Sutton et al. 2004; Bougie et al. 2009).
While the mammalian calpains CAPN1 (µ-calpain) and CAPN2 (m-calpain) are the best-studied isoforms in regulating synaptic plasticity (Baudry and Bi 2016), numerous diverse calpains are encoded in Metazoan genomes, most of which fall into four highly conserved subfamilies. These are the classical subfamily, which includes CAPN1, CAPN2 and is defined by the presence of EF hands at the carboxy terminal, the SOL calpain family (name derived from the Drosophila mutant phenotype Small Optic Lobes), the PalB family (named after the screen for acid-sensitive phosphatase mutants in the fungi, Asperilligus) and the Tra family (named after the sex-determining transformer protein 3 from Caenorhabditis elegans) subfamilies. While the human genome encodes 15 calpains and includes members of all four subfamilies (including nine classical calpains), the calpain isoforms present in Aplysia have not previously been determined.
In this study, using isoform-specific dominant negatives and cleavage assays, we find that distinct PKCs in presynaptic and postsynaptic neurons are cleaved into PKMs by an Aplysia classical calpain orthologous to mammalian classical calpains in response to different physiologically relevant stimuli. Cleavage of the classical PKC Apl I into PKM Apl I by this classical calpain plays a role in the presynaptic neuron during a-ITF whereas cleavage of the atypical PKC Apl III into PKM Apl III by the same classical calpain plays a role in the postsynaptic neuron during m-ITF. Thus, PKMs derived from classical calpain cleavage of not only atypical, but also classical PKCs play a role in memory maintenance and PKMs are formed in the presynaptic as well as the postsynaptic cell.
Results
Neither chelerythrine, nor pseudosubstrate-based inhibitors distinguish between isoforms of PKM
Two inhibitors of PKMζ, ZIP (based on the pseudosubstrate of atypical PKCs) and chelerythrine, erase memories in both vertebrates and Aplysia (Serrano et al. 2008; Cai et al. 2011), but no studies have examined the specificity of these inhibitors for distinct PKMs. To examine this question, we performed kinase assays using purified PKM Apl I or PKM Apl III. The PKMs were purified from baculovirus-infected SF9 cells and the amount of kinase normalized using a common amino-terminal His-tag antibody (Fig. 1A). Both myristoylated ZIP and chelerythrine inhibited PKM Apl III activity at slightly lower concentrations than required for inhibition of PKM Apl I activity, but this difference is unlikely to provide specificity required for use in cells (Fig. 1B,C). A myristoylated version of the pseudosubstrate from classical PKCs required higher concentrations for inhibition than myristoylated ZIP, but similar to ZIP, inhibited the atypical PKM Apl III at slightly lower concentrations than required to inhibit the classical PKM Apl I (Fig. 1D). Interestingly, the myristoylation of ZIP is important for its affinity as an inhibitor since the concentration required for inhibition was much larger for ZIP without the myristoylation (Fig. 1E) even though these assays were conducted in a system without membranes or lipids. An inhibitor based on the ATP binding site, Bis-I, inhibited PKM Apl I at a much lower concentration than it inhibited PKM Apl III, consistent with earlier results using PKCs (Fig. 1F; Villareal et al. 2009). Thus, the inhibitors used to target PKMζ also target PKMs made from other types of PKC and inhibitors targeted to the ATP binding site of classical PKCs only weakly inhibit atypical PKMs.
Figure 1.
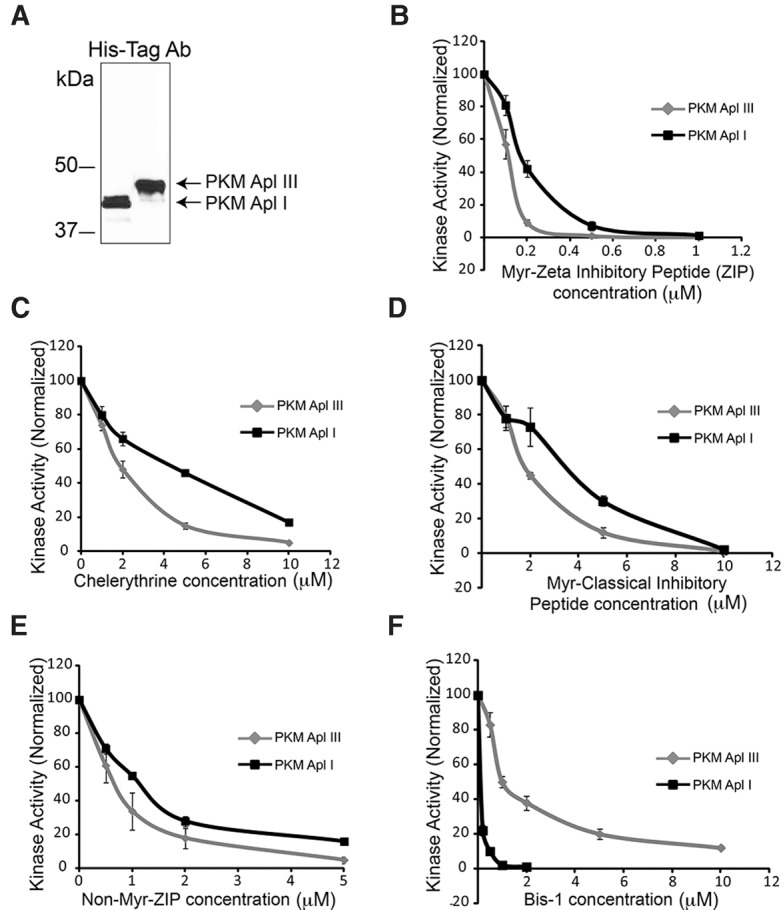
Kinase assays. (A) Sf9 cells were infected with recombinant baculovirus containing either His-Tagged PKM Apl I or His-Tagged PKM Apl III. At 72 h post-infection, cells were lysed by sonication and His-tagged proteins purified (see Materials and Methods). An aliquot of the purified proteins were separated on SDS-PAGE gels, and then transferred to a nitrocellulose membrane that was immunostained using an anti-His-Tag antibody. The membrane was used to quantify the relative amounts of PKM Apl I and PKM Apl III in order to assess PKM inhibitors using the same molar concentration of PKMs. The immunoblot displayed had an intervening lane that was removed (black line). (B–F) Protein Kinase C activity assays were performed as described in methods using 10 μM of PKC epsilon substrate for both enzymes and differing amounts of (B) myristoylated zeta inhibitory peptide (Myr-ZIP), (C) chelerythrine, (D) myristolated classical inhibitory peptide, (E) non-myristoylated zeta inhibitory peptide (Non-Myr-ZIP), and (F) bisindolylmaleimide 1 (Bis-1). N = 3–4 separate kinase assays from two independent preparations of the enzymes for B–F. Values represent the mean ± SEM.
A dominant-negative PKM Apl I blocks a-ITF
Site-specific sensitization is the increased defensive response observed when the animal is tested at the site of the shock as opposed to testing the animal in nonshocked locations (Walters 1987). A-ITF is the cellular basis for site-specific sensitization and is induced by the coincidence of sensory neuron action potential activity (the touch) and serotonin (5-hydroxytryptamine, 5HT) release (shock). The maintenance of site-specific sensitization is blocked by the ATP-based inhibitor Bis-1 (Sutton et al. 2004) suggesting that memory in this system is not retained by the atypical PKM Apl III which is relatively insensitive to this inhibitor (Fig. 1F). However, the maintenance is likely to be mediated by a PKM, since both maintenance of site-specific sensitization and its cellular analog, a-ITF is reversed by chelerythrine, which inhibits PKMs better than PKCs (Sutton and Carew 2000; Villareal et al. 2009). The isoform of PKC required for the induction of a-ITF in the sensory neuron is the classical PKC Apl I as a dominant-negative form of PKC Apl I, but not a dominant-negative form of the novel PKC Apl II, blocks a-ITF when expressed in the sensory neuron (Zhao et al. 2006). To determine if PKM Apl I plays an important role in a-ITF, the cellular analog of site-specific sensitization, we used a dominant-negative strategy. The dominant-negative form of PKM Apl III with an aspartic acid (D) in the ATP binding pocket converted to alanine (A) has previously been shown to block m-ITF when expressed in the motor neuron (Bougie et al. 2012). We generated a construct with the same mutation in PKM Apl I. We expressed either mRFP-DN PKM Apl I or mRFP-DN PKM Apl III in sensory neurons paired with motor neurons, sensory neurons were stimulated to produce action potentials in the presence of 5HT as previously described (see Materials and Methods), and then tested for a-ITF at 60 min after stimulation. Consistent with a role for PKM Apl I in a-ITF, there was no significant a-ITF at 60 min when mRFP-DN PKM Apl I was expressed, while significant a-ITF was observed after expressing either mRFP or mRFP-DN PKM Apl III (Fig. 2). The inhibition of a-ITF with mRFP-DN PKM Apl I only occurred with high expression levels (see Materials and Methods). At the synaptic connections used in Figure 2 with high dominant-negative PKM expression in the sensory neuron, levels of mRFP-DN PKM Apl I and mRFP-DN PKM Apl III were similar, as judged by mRFP fluorescence intensity (Fig. 2A, inset).
Figure 2.

PKM Apl I is required for activity-dependent ITF. (A) Images of sensory (SN) to motor neuron (MN) pairs in phase contrast (PC) and with mRFP fluorescence filters (Red) showing mRFP tagged PKM expression in the sensory neuron (scale bar is 40 µm). Inset, bar graph showing the average mRFP intensity in mRFP-DN PKM Apl I and mRFP-DN PKM Apl III groups used in the experiment. mRFP intensity measured in the SN soma and expressed as a fold increase above the average autofluorescence intensity observed in SN somata. There is no significant difference between the mRFP intensity in the two groups (P > 0.05 Student's unpaired t-test). (B) PSP amplitude 60 min after evoking 20 action potentials at 10 Hz in the sensory neuron at 1, 2, 3, and 4 min of a 5 min 5HT application. Representative traces of PSPs from each group before 5HT application (PSP1), the first PSP produced from the stimulation during 5HT (PSP2) and the PSP 60 min after 5HT application (PSP 60 min). Scale bars are 5–5–2.5 mV/15 msec. PSP amplitude was significantly increased at 60 min with mRFP alone or mRFP-DN PKM Apl III, but not with mRFP-DN PKM Apl I expression in the sensory neuron (comparing before to 60 min following 5HT and sensory neuron activity with a paired Student's t-test, n = 13, 12, and 11 synaptic connections in each group), (*) P < 0.05 after Bonferroni correction for multiple t-tests. An ANOVA of the normalized PSP amplitudes gave a value of (F(33,2) = 2.35, P = 0.112. Initial PSP amplitudes were 21.3 ± 3.9, 27.8 ± 5.5, 23.8 ± 6.3 mV; initial MN input resistances were 64.1 ± 5.3, 72.4 ± 6.4, 67.8 ± 3.7 MΩ; and the changes in input resistance at 60 min as 98.8%, 90.7%, 88.5% of initial in mRFP, mRFP-DN PKM Apl I, and mRFP-DN PKM Apl III, respectively. There were no significant differences between the groups for any of these parameters (P > 0.05 ANOVA).
PKC Apl I is cleaved by calpain in response to activity in the presynaptic sensory neuron
Since there is no alternative start site identified in PKC Apl I to generate a PKM Apl I encoding mRNA, we examined whether PKC Apl I could be cleaved in the sensory neuron during a-ITF, similar to previously described cleavage of PKC Apl III by 5HT in the motor neuron (Bougie et al. 2012). PKCs are tagged with CFP on the carboxy terminus and YFP on the amino terminus. Due to the proximity of the CFP and YFP on one molecule of PKC, these constructs show FRET and cleavage is measured as a loss of intramolecular FRET. We first tested the integrity of the protein by confirming that CFP-PKC Apl I-YFP could be activated by measuring translocation to the plasma membrane following the pairing of membrane depolarization with 5HT application as previously described for eGFP-PKC Apl I (Shobe et al. 2009). The membrane was depolarized with 100 mM KCl to mimic activity in the sensory neuron in this paradigm as has previously been used for PKC Apl I translocation (Shobe et al. 2009). We found that CFP-PKC Apl I-YFP translocates to the plasma membrane in isolated sensory neurons in response to KCl (100 mM) and 5HT (10 µM) with translocation ratios similar to the ones previously reported for eGFP-PKC Apl I (Shobe et al. 2009) (Fig. 3A) (translocation ratio (post/pre) of 1.58 ± 0.12 for CFP and 1.42 ± 0.02 for YFP, n = 3 cells from two independent experiments).
Figure 3.
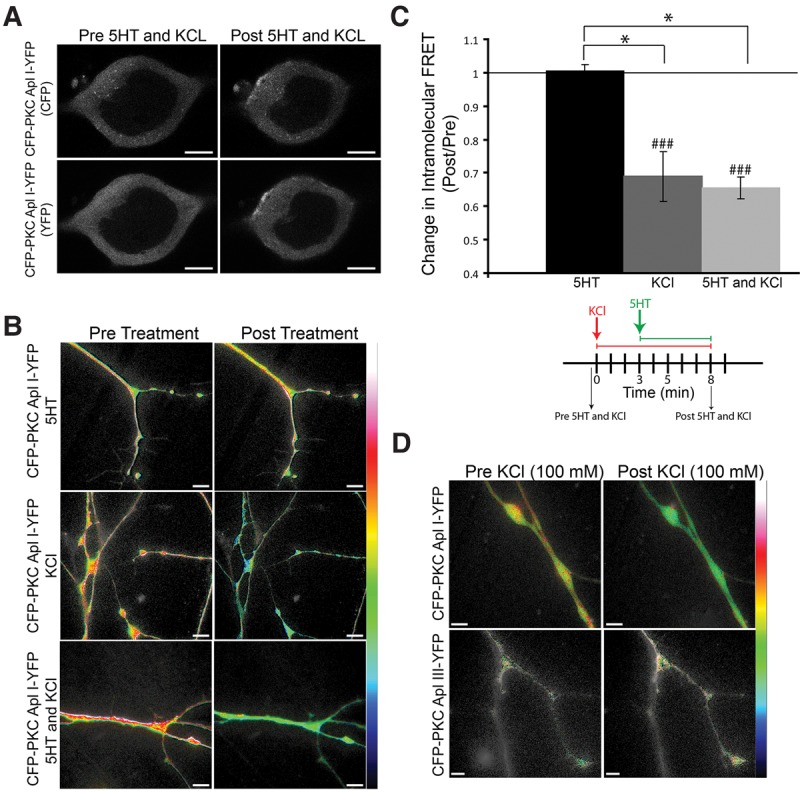
KCl treatment alone, not 5HT, causes a loss of intramolecular FRET for CFP-PKC Apl I-YFP, but not for CFP-PKC Apl III-YFP, in sensory neuron varicosities. (A) Representative confocal fluorescence images of Aplysia sensory neurons expressing CFP-PKC Apl I-YFP before 5HT (10 µM) and KCl (100 mM) treatment or 5 min following 5HT (10 µM) and KCl (100 mM) treatment. CFP-PKC-Apl I-YFP translocates to the plasma membrane in response to 5HT and KCl treatment. (B) Representative images with color-coded FRET maps of Aplysia sensory neuron varicosities expressing CFP-PKC Apl I-YFP pre- and post-treatment with 5HT alone (10 µM) for 5 min, KCl alone (100 mM) for 8 min or 5HT (10 µM) and KCl (100 mM) [KCl was added first for 3 min followed by 5HT in the presence of KCl for an additional 5 min as previously described (Shobe et al. 2009)]. The FRET maps display color-coded images of the measured FRET signal, in which warm colors represent higher levels of FRET and cooler colors represent lower levels of FRET. Scale is from 0 to 0.4 for the 5HT alone and the KCl alone groups and scale is from 0 to 0.5 for the 5HT and KCl group. Loss of intramolecular FRET is seen in the KCl- and the (5HT and KCl)-treated sensory neurons expressing CFP-PKC Apl I-YFP post-treatment, not in the 5HT-treated sensory neurons. (C) Change in intramolecular FRET (Post/Pre) was calculated for the 5HT alone group post 5 min treatment (n = 8 varicosities, 4 cells, 2 independent experiments), the KCl alone group post 8 min treatment (n = 32 varicosities, 16 cells, 2 independent experiments) and the 5HT and KCl group post 3 min KCl and an additional 5 min KCl and 5HT as described above (n = 18 varicosities, 9 cells, 2 independent experiments). For the cells treated with 5HT alone, similar results were obtained with shorter (3 min; 1.09 + 0.05, n = 8 varicosities, 4 cells, 3 independent experiments) and longer applications of 5HT (8 min; 1.08 + 0.04, n = 7 varicosities, 4 cells, 15 min; 0.91 + 0.09, n = 7 varicosities, 4 cells and 30 min; 1.04 + 0.06, n = 9 varicosities, 5 cells, 3 independent experiments). A paired Student's t-test was performed to compare raw FRET signal post-treatment to pretreatment for each condition, (###) P < 0.005 with Bonferroni corrections for multiple t-tests. One-Way ANOVA with Student–Newman–Keuls multiple comparisons post-test was performed on normalized FRET ratio (Post/Pre) to compare the KCl alone group, the KCl and 5HT group and the 5HT alone group and groups that were significantly different are shown (*) P < 0.05. (D) Representative images with color-coded FRET maps of Aplysia sensory neuron varicosities expressing CFP-PKC Apl I-YFP or CFP-PKC Apl III-YFP pre- and post-treatment with KCl (100 mM) for 3 min. Scale is from 0 to 0.4. Cleavage is indicated as a loss of intramolecular FRET as seen in the KCl-treated sensory neurons expressing CFP-PKC Apl I-YFP post-treatment. Error bars indicate SEM and scale bar is 10 μm for all parts of the figure. The image shown in D for CFP-PKC Apl I- YFP is from the same set of neurons quantified for KCl treatments in C. Only the subset of KCl treated neurons where CFP-PKC Apl I-YFP and CFP-PKC Apl III-YFP were examined in the same experiment, are used in the quantification shown in the text.
We then determined whether this stimulus led to a loss of FRET in varicosities of isolated sensory neurons. There was a significant loss of FRET measured immediately after washout of 5HT and KCl, (Fig. 3B). 5HT alone did not cause a loss of FRET for CFP-PKC Apl I-YFP, while KCl (100 mM) treatment alone did cause a significant loss of FRET that was not different than that seen by the combination of KCl (100 mM) with 5HT (Fig. 3B,C). Since KCl is not sufficient to induce translocation of PKC Apl I (Shobe et al. 2009), and the loss of FRET lasts for at least 30 min after the washout of KCl (see below), it is unlikely that the loss of FRET is due to transient conformational changes in the full-length PKC. Moreover, KCl (100 mM) treatment did not cause a loss of FRET for CFP-PKC Apl III-YFP in sensory neurons, suggesting specific cleavage of PKC Apl I, but not PKC Apl III with sensory neuron depolarization (Fig. 3D). The change in intramolecular FRET (Post/Pre) for the CFP-PKC Apl I-YFP group was 0.74 ± 0.05 (n = 25 varicosities, 13 cells, 2 independent experiments) and 0.96 ± 0.11 for the CFP-PKC Apl III-YFP group (n = 15 varicosities, 8 cells, 2 independent experiments) 3 min post KCl (100 mM) treatment (*) P < 0.05 by an unpaired Student's t-test performed on the normalized FRET ratios. A paired Student's t-test was performed to compare raw FRET signal post-treatment to pretreatment for each construct, (###) P < 0.001 for the CFP-PKC Apl I-YFP group and non-significant for the CFP-PKC Apl III-YFP group.
Cleavage of CFP-PKC Apl I-YFP also occurred following evoked action potential activity in the sensory neuron (similar to the protocol used to induce a-ITF, see Materials and Methods) although the reduction in FRET was delayed compared with KCl treatment (Fig. 4A). Similar to KCl, evoking action potential activity in the sensory neurons in the presence of 5HT did not enhance the reduction of FRET. The change in intramolecular FRET (Post/Pre) was 0.62 ± 0.02 for the sensory neurons with activity (four trains of action potentials, 10 Hz 2 sec, 1 min ITI; n = 8 varicosities, 4 cells, 2 independent experiments) and 0.58 ± 0.03 for the 5HT and activity group (n = 13 varicosities, 7 cells, 2 independent experiments) 30 min post-treatment (nonsignificant by an unpaired Student's t-test performed on the normalized FRET ratios). A paired Student's t-test was performed to compare FRET signal post-treatment to pretreatment for each construct, (###) P < 0.001 for both groups.
Figure 4.
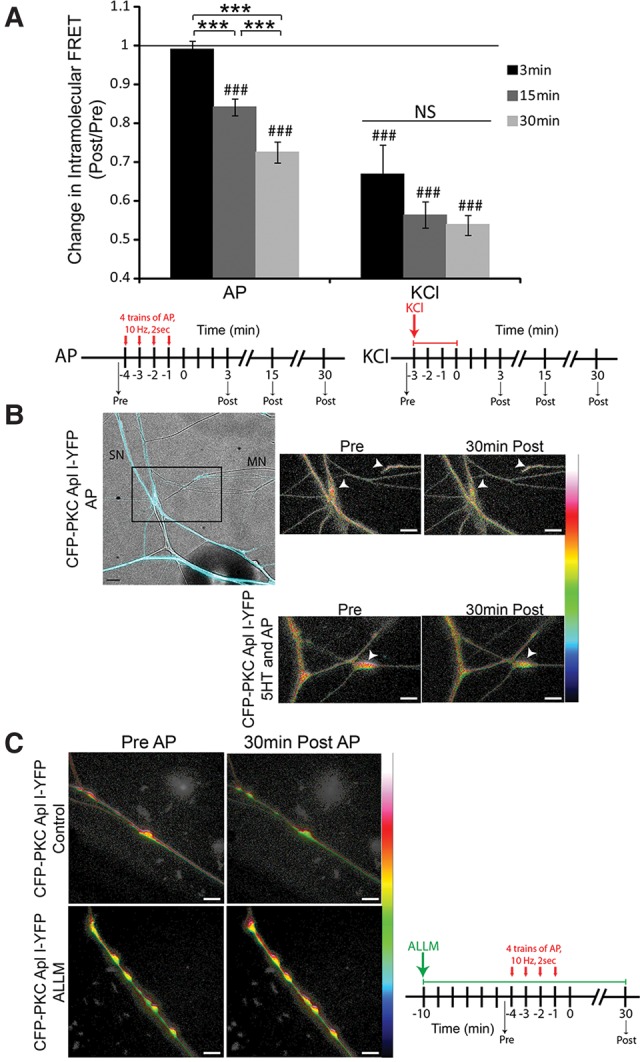
Evoking action potentials in the sensory neuron also induces cleavage of CFP-PKC Apl I-YFP in sensory neuron varicosities and the calpain inhibitor ALLM blocks this cleavage. (A) Quantification of sensory neuron varicosities expressing CFP-PKC Apl I-YFP. Sensory neurons were either depolarized to evoke action potentials (four trains of action potentials, 10 Hz 2 sec, 1 min ITI) or treated with KCl (100 mM) and pictures were taken pre- and 3 min, 15 min and 30 min post-treatment. Change in intramolecular FRET was calculated for the firing alone group (n > 7 varicosities, 8–14 cells per time point, 2–7 independent experiments depending on the time point) and the KCl alone group (n > 12 varicosities, 7–19 cells per time point, 1–3 independent experiments depending on the time point). A paired Student's t-test was performed to compare raw FRET signal post-3 min, 15 min, and 30 min to prefiring or pre-KCl, (###) P < 0.005 with Bonferroni correction for multiple t-tests. One-Way ANOVA with Student–Newman–Keuls multiple comparisons post-test was performed to compare the normalized FRET ratio (Post/Pre) at the 3 min, 15, and 30 min time points post-treatment in the Activity group and the KCl group and groups that were different are shown, (***) P < 0.001. Error bars indicate SEM; NS, nonsignificant. (B) Representative images with color-coded FRET maps of Aplysia sensory neurons expressing CFP–PKC Apl I–YFP cocultured with motor neurons. The top left panel is a merge of the CFP channel (to visualize the sensory neuron) and bright-field images (to visualize the motor neuron). Sensory neurons were depolarized to induce action potentials (four trains of action potentials, 10 Hz 2 sec, 1 min ITI) in the absence or presence of 5HT (10 μM) and images were taken before and 30 min post-treatment. White arrowheads point to presynaptic varicosities. For activity, scale is from 0 to 0.8. For 5HT and activity, scale is from 0 to 0.6. (C) Representative images with color-coded FRET maps of Aplysia sensory neuron varicosities expressing CFP–PKC Apl I–YFP treated with vehicle (control) or the calpain inhibitor ALLM pre-, immediately post-action potentials, and 30-min post-action potentials the sensory neuron. ALLM (50 μM) was added to the dish 10 min prior to evoked action potentials and was present throughout the experiment. Scale is from 0 to 1. Cleavage, as observed by a loss of intramolecular FRET, is seen in the control sensory neurons expressing CFP-PKC Apl I-YFP post-firing but not in the ALLM treated neurons; AP, action potential. Scale bar is 10 μm for all parts of the figure.
The previous results were all done in isolated sensory neurons, but synapse formation may change some actions of serotonin (Farah et al. 2015). The loss of intramolecular FRET was also observed with action potential activity at presynaptic varicosities of sensory neurons paired with motor neurons and similar to isolated sensory neurons, serotonin did not increase the loss of FRET at synapses (Fig. 4B). The change in intramolecular FRET (Post/Pre) was 0.80 ± 0.04 for the activity alone group (n = 6 varicosities, 3 cells, 3 independent experiments) and 0.91 ± 0.02 for the 5HT and activity group (n = 6 varicosities, 3 cells, 3 independent experiments) 30 min post-treatment. A paired Student's t-test was performed to compare raw FRET signal post-treatment to pretreatment for each condition, (###) P < 0.001 for the activity alone group and (##) P < 0.01 for the 5HT and activity group. An unpaired Student's t-test performed on the normalized FRET ratios showed a significant difference between the two groups (*) P < 0.05 indicating that at synapses, 5HT slightly inhibited the decrease in intramolecular FRET.
While the delayed loss of FRET and the dissociation from PKC translocation (activity alone vs. activity + 5HT) argue against loss of FRET due to conformational changes, they cannot rule this out. To obtain additional evidence that the loss of FRET was due to cleavage, we examined the effects of Calpain inhibitor II (N-acetyl-Leu-Leu-Methional; ALLM), which blocked site-specific sensitization (Sutton et al. 2004). Incubation with ALLM significantly inhibited the loss of intramolecular FRET induced by activity (Fig. 4C). The change in intramolecular FRET (Post/Pre) was 0.74 ± 0.04 for the control group (n = 10 varicosities, 5 cells, 2 independent experiments) and 0.82 ± 0.02 for the ALLM group (n = 8 varicosities, 4 cells, 2 independent experiments) 30 min post-activity (*P < 0.05 by an unpaired Student's t-test performed on the normalized FRET ratios). A paired Student's t-test was performed to compare raw FRET signal post-treatment to pretreatment for each condition, (###) P < 0.001 for the control group and the ALLM group. While the significant inhibition by ALLM strongly indicates that there is loss of FRET due to cleavage, it is not clear if the loss of FRET still observed in the presence of ALLM is due to conformational changes in the PKC or incomplete inhibition of cleavage by ALLM (see Discussion).
The classical calpain is important for cleavage of PKC Apl I into PKM Apl I during a-ITF and cleavage of PKC Apl III into PKM Apl III during m-ITF
There are four main families of calpains present in vertebrates and conserved over Metazoan evolution: classical calpains with penta-EF hand domains at their carboxy-terminus, PalB calpains, Tra calpains, and Sol calpains. Bioinformatics search of NCBI and Aplysia transcriptome (http://aplysiagenetools.org/) databases revealed that Aplysia contains orthologues of all the major classes of calpains as well as an atypical calpain with two dIII domains. While Calpain 10 in vertebrates also has this structure, there is lower homology in the protease domains of these two calpains than the other orthologous calpains (Table 1) and it is not clear if these are orthologues or the results of convergent evolution. The Aplysia genome encodes multiple classical calpains with EF hands, but using phylogenetic analysis one of them is significantly closer to the mammalian classical calpains than the others (Hastings et al. in prep.) and this is the only one examined in this study. The mammalian genome contains nine classical calpain genes (Capn 1, 2, 3, 8, 9, 11–14) some of which dimerize with the small subunit (Capn 4). All of these calpains, including the small subunit, appear to have originated from duplication of one or two classical calpain genes present before jawed fish (Macqueen and Wilcox 2014; Hastings et al. in prep.) and thus all are equally similar to the Aplysia classical calpain and Drosophila Calpain A and B which are also members of this family (Hastings et al. in prep.).
Table 1.
Structure of the mammalian and the Aplysia calpain isoforms and % amino acid homology in the protease domain. (Apl) Aplysia, (Capn) Calpain, (CCal) Classical Calpain

Representatives of all four calpain families and the atypical calpain are expressed in the nervous system and two of them, the classical and Sol are enriched in sensory neurons and motor neurons (Fig. 5A). To determine which isoform of calpain is important for cleavage of CFP-PKC Apl I-YFP with action potential activity, we coexpressed protease-inactive calpains with the catalytic cysteine (amino acid 152 in the classical calpain and amino acid 446 in the Sol calpain) converted to a serine (C152-S; C446-S) with the intention that these should act as dominant-negative calpains (Huang and Forsberg 1998), replacing the endogenous calpain in signaling complexes important for cleavage. Indeed, there was no significant loss of intramolecular FRET (Post/Pre) of CFP-PKC Apl I-YFP by evoked action potentials after expression of the Aplysia dominant-negative classical calpain (DN-CCal) in the presynaptic sensory neuron, while significant loss of intramolecular FRET was observed both for control conditions and after expression of the Aplysia dominant-negative SOL calpain (DN-SOL) (Fig. 5B–D). The effect of the DN-CCal was significantly different from both the control and the DN-SOL groups (Fig. 5D). We confirmed expression of the dominant-negative calpains with specific antibodies raised to the Aplysia calpains (Fig. 6).
Figure 5.

The dominant-negative Classical calpain, but not the dominant-negative SOL calpain, blocks activity-dependent cleavage of CFP-PKC Apl I-YFP in sensory neuron varicosities. (A) RT-PCR of nervous system cDNA or motor neuron or sensory neuron cDNA using primers designed to amplify fragments of the Aplysia calpain family members. (B,C) Representative images with color-coded FRET maps of Aplysia sensory neuron varicosities expressing CFP–PKC Apl I–YFP and (B) dominant-negative Classical calpain (DN-CCal) or (C) dominant-negative SOL calpain (DN-SOL). Images were taken in the sensory neuron before and 30 min after evoked action potentials (four trains of action potentials, 10 Hz 2 sec, 1 min ITI). Scale is from 0 to 1. (D) Change in intramolecular FRET was calculated for the control group (n = 10 varicosities, 10 cells, 2–3 independent experiments), the DN-SOL group (n = 5 varicosities, 4 cells, 2 independent experiments) and the DN-CCal group (n = 7 varicosities, 7 cells, 3 independent experiments). A paired Student's t-test was performed to compare raw FRET signal post- to pretreatment for each condition, (###) P < 0.001, (#) P < 0.05. One-way ANOVA with Student–Newman–Keuls multiple comparisons post-test was performed to compare the normalized FRET ratio (Post/Pre) in the DN-SOL group and the DN-CCal group to the control group, (*) P < 0.05. AP, action potential. Scale bar is 10 μm for all parts of the figure.
Figure 6.
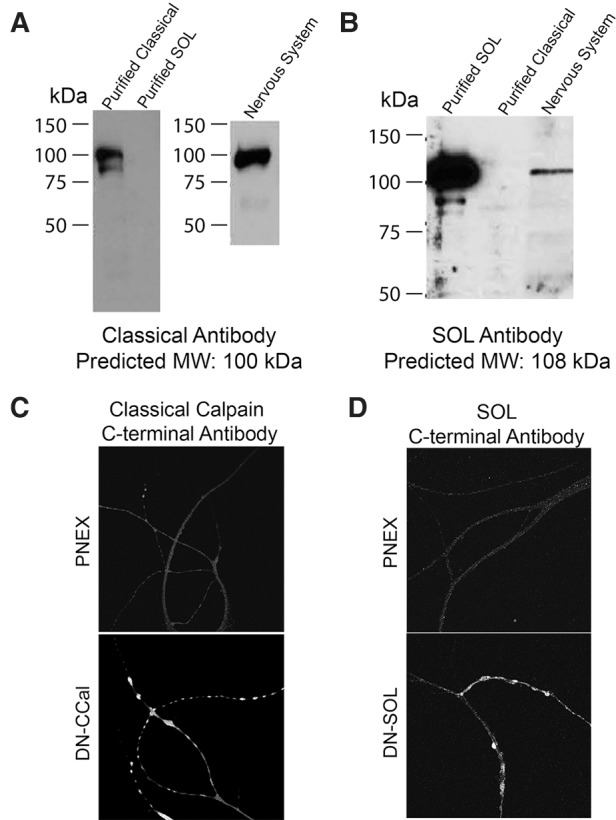
Characterization of antibodies raised against the carboxy terminal of Aplysia Classical Calpain (A) and Aplysia SOL calpain (B). Classical calpain and SOL calpain were purified from SF9 cells that were transduced with baculovirus encoding the His-tagged Classical calpain or the His-tagged SOL calpain. Nervous system homogenate was prepared as described in Materials and Methods and 10 µg of total protein were loaded per lane. Predicted molecular weights based on the cloned calpains are indicated. (C,D) Overexpression of (C) the dominant-negative Classical calpain (cells from Fig. 5B) and (D) the dominant-negative SOL calpain (cells from Fig. 5C) was confirmed using immunocytochemistry with the antibodies raised against the Classical calpain and the SOL calpain, respectively.
We have previously shown serotonin-induced cleavage of CFP-PKC Apl III-YFP in a model of m-ITF in isolated motor neurons (Bougie et al. 2012). To determine whether the same calpain could cleave both PKC Apl I and PKC Apl III and to confirm that cleavage of CFP-PKC Apl III-YFP also occurs after synapse formation, we re-examined this cleavage in sensorimotor neuron pairs in the presence or absence of the dominant-negative calpains. Similar to isolated motor neurons, we observe a robust decrease in intramolecular FRET (Post/Pre) for CFP-PKC Apl III-YFP in response to 5HT at sites of sensorimotor neuron contact (Fig. 7A–C), and this was also observed in the presence of the DN-SOL calpain, but no significant loss of intramoleclar FRET was observed in the presence of DN-CCal consistent with a role for the classical calpain in the motor neuron. The effect of the DN-CCal was significantly different from both the control and the DN-SOL groups (Fig. 7C).
Figure 7.
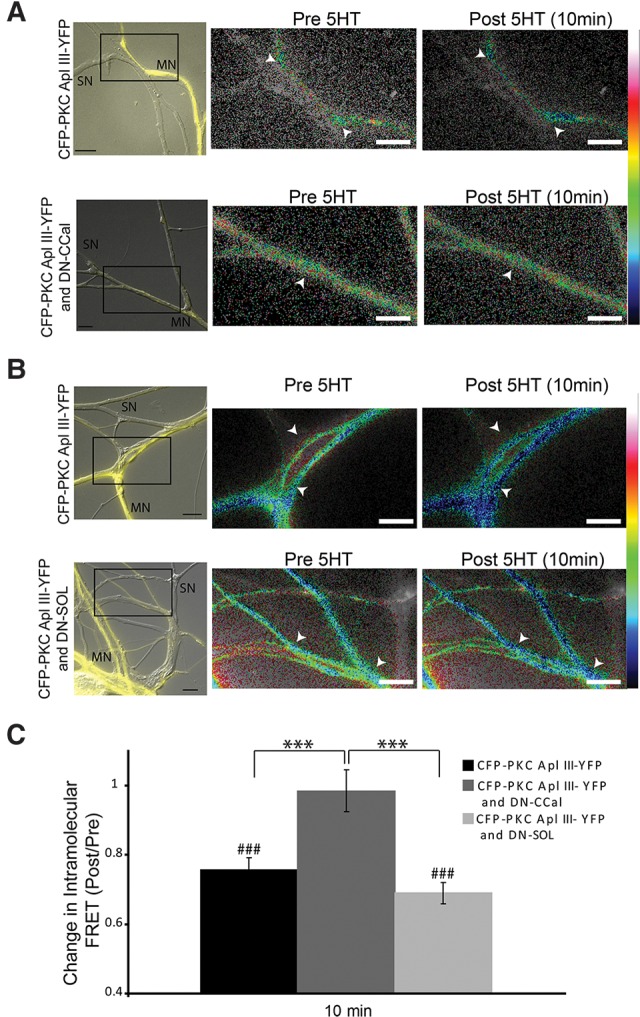
Dominant-negative Classical calpain, but not dominant-negative SOL calpain, blocks cleavage of CFP-PKC Apl III-YFP in the motor neuron. (A,B) Representative images with color-coded FRET maps of Aplysia sensorimotor neuron cocultures in which the motor neuron is expressing CFP–PKC Apl III–YFP and (A) dominant-negative Classical calpain (DN-CCal) or (B) dominant-negative SOL calpain (DN-SOL). Images were taken before and 10 min post-treatment with 5HT (10 µM). White arrowheads indicate points of contact used for quantification. Scale is from 0 to 1 for eGFP, 0 to 2 for the DN-CCal group and 0–0.8 for the DN-SOL group. The left panels show a merge of the YFP channel (to visualize the motor neuron) and bright-field images (to visualize the sensory neuron). (C) Change in intramolecular FRET was calculated for the control group (n = 72 varicosities, 13 cells, 3 independent experiments), the DN-SOL group (n = 47 varicosities, 10 cells, 3 independent experiments) and the DN-CCal group (n = 34 varicosities, 8 cells, 3 independent experiments). A paired Student's t-test was performed to compare raw FRET signal post- to pretreatment for each condition, (###) P < 0.001. One-way ANOVA with Student–Newman–Keuls multiple comparisons post-test was performed on normalized FRET ratio (Post/Pre) to compare the DN-SOL group and the DN-CCal group to the control group, (***) P < 0.001. Scale bar is 10 μm for all parts of the figure.
Classical calpain is required for a-ITF and m-ITF
If the classical calpain is required for PKM formation, it should also be important for the plasticity retained by the PKM. To directly test this, synaptic strength was measured at sensory to motor neuron synaptic pairs before and 60 min after a 10 min treatment of 5HT. m-ITF was not observed when the motor neuron was expressing DN-CCal, but was observed when the motor neuron was expressing DN-SOL or eGFP (Fig. 8A). The normalized EPSP at 60 min was significantly different when DN-CCal was expressed compared with DN_SOL or eGFP (Fig. 8A). Initial PSP amplitude, short-term facilitation, and postsynaptic input resistance were not affected by expression of either DN-CCal, DN-SOL, or eGFP alone in the motor neuron (Fig. 8A). Overall, the data suggest that classical calpain in the postsynaptic motor neuron is involved in PKC Apl III cleavage and the expression of m-ITF at sensory to motor neuron synapses following a 10 min stimulation with 5HT.
Figure 8.

The dominant-negative Classical calpain, but not the dominant-negative SOL calpain, blocks both m-ITF and a-ITF. (A) PSP amplitude 60 min after the start of a 10 min application of 5HT (10 µM), as a percentage of the initial amplitude prior to 5HT. Representative traces of PSPs before 5HT, at 5 min during 5HT application, and after 60 min (scale bars are 5-10-5 mV/20 msec). The postsynaptic motor neuron was injected with PNEX vectors for expression of either eGFP alone (n = 12), DN-CCal and eGFP (n = 14) or DN-SOL and eGFP (n = 15) 18–24 h prior to recording. The eGFP and DN-SOL group both showed significant ITF (paired Student's t-test with Bonferroni correction for multiple t-test, (*) P < 0.05). One-Way ANOVA with Student–Newman–Keuls multiple comparisons post-test was performed to compare the different groups (F(2,37) = 3.969; P = 0.027). Facilitation at 60 min in motor neurons expressing DN-CCal and eGFP, but not DN-SOL and eGFP, was significantly less than facilitation at 60 min in neurons expressing eGFP alone, P < 0.05. Initial PSP amplitudes were 22.0 ± 4.3, 24.0 ± 4.8, 29.0 ± 5.0 mV; initial MN input resistances were 102.9 ± 11.4, 112.8 ± 11.3, 105.7 ± 7.8 MΩ; and the changes in input resistance at 60 min as 84.1%, 81.4%, 80.1% of initial in eGFP, DN-Ccal, and DN-SOL, respectively. None of these measures were significantly different from each other (ANOVA, P > 0.1). (B) PSP amplitude 60 min after the start of a 5 min application of 5HT (10 µM) combined with firing 20 action potentials at 10 Hz in the presynaptic sensory neuron at 1, 2, 3, and 4 min into the 5HT application (as a percentage of PSP#1, before 5HT). Representative traces of PSPs before 5HT and after 60 min are superimposed above each condition (the larger traces in each group are from 60 min, scale bars are 10 mV/40 msec). The presynaptic sensory neuron was injected with PNEX vectors for expression of either eGFP alone (n = 7), DN-CCal and eGFP (n = 8), or DN-SOL and eGFP (n = 7) 18–24 h prior to recording. The eGFP and DN-SOL group both showed significant ITF (paired Student's t-test with Bonferroni correction for multiple t-test ((*) P < 0.05). One-way ANOVA with Student–Newman–Keuls multiple comparisons post-test was performed to compare the different groups (F(2,19) = 5.433; P = 0.014). Facilitation at 60 min in sensory neurons expressing DN-CCal and eGFP was significantly less than facilitation at 60 min in neurons expressing DN- SOL and eGFP or eGFP alone, P < 0.05. Initial PSP amplitudes were 8.4 ± 2.9, 3.8 ± 1.5, 13.0 ± 3.9; initial MN input resistances were 45.3 ± 4.0, 60.2 ± 6.8, 50.2 ± 7.1 MΩ; and the changes in input resistance at 60 min as 98.8%, 90.7%, 88.5% of initial in eGFP, DN-CCal, and DN-SOL, respectively. None of these groups were significantly different from each other (ANOVA, P > 0.1).
Similarly, the increase in synaptic strength 60 min following the pairing of sensory neuron action potential activity and 5HT application was not observed with expression of the dominant-negative classical calpain in the presynaptic sensory neuron (DN-CCal; Fig. 8B) despite normal short-term facilitation (Fig. 8B). This inhibition of a-ITF was not observed with the expression of eGFP or the dominant-negative SOL calpain (DN-SOL; Fig. 8B). The normalized EPSP at 60 min was significantly different when DN-CCal was expressed compared with DN-SOL or eGFP (Fig. 8B). While there was a trend for DN-CCal to reduce initial synaptic strength when expressed in the sensory neuron, the reduction was not significant (Fig. 8B), and there was no correlation between initial synaptic strength and the extent of a-ITF (for the n = 22 synaptic pairs examined in Fig. 8B Pearson r = −0.2501, P = 0.26).
Discussion
A PKM formed from a classical PKC plays a role in the maintenance of synaptic strength
While the existence of a unique transcript encoding PKMζ in vertebrates and the effectiveness of ZIP suggest an important role for this PKM isoform, there are limited data testing the isoform specificity of PKMζ for synaptic plasticity or memory. As we demonstrate, neither ZIP, nor chelerythrine, are selective inhibitors of atypical PKMs. Thus, we have used a number of nonpharmacological strategies to examine isoform specific roles of PKMs in synaptic plasticity. In the presynaptic sensory neuron, a dominant-negative PKM Apl I, but not a dominant-negative PKM Apl III blocked a-ITF, suggesting an isoform specific role of PKM Apl I in the sensory neuron. Consistent with this, CFP-PKC Apl I-YFP, but not CFP-PKC Apl III-YFP was cleaved in the sensory neuron by stimuli that induced a-ITF. Thus, both the dominant-negative PKMs and the measurement of cleavage by FRET show isoform-specificity. These results show relatively conclusively that for some forms of plasticity, PKMs other than the atypical PKM maintain plasticity, and thus greatly widen the scope of the role that PKMs play in maintaining memory.
The isoform specificity of dominant negatives depends to a great extent on the mechanism of action of the dominant negative. We have used a strategy to generate catalytically inactive PKMs that are properly primed, and thus should not sequester common upstream regulators of PKCs (Cameron et al. 2009). PKCs do not form dimers. Thus, the action of the dominant negative in this case is not to suppress the endogenous protein through direct binding. Since most kinases require correct subcellular localization through protein scaffolds to function (Sossin 2007), the dominant negatives probably work by competing for scaffolds with endogenous kinases and thus, may lead to a lack of isoform-specificity if PKMs share a common scaffold. In the sensory neuron, the dominant-negative PKM Apl I blocked a-ITF, but the dominant-negative PKM Apl III did not, suggesting specificity for the role of PKM Apl I in a-ITF. Dominant negatives can also be misleading if they sequester binding sites used by other signaling molecules and thus, provide only suggestive evidence for function. This is particularly important in this case, since high levels of the dominant-negative PKM Apl I were required to block a-ITF (see Materials and Methods). However, in this case there is also a great deal of additional evidence for a role of PKM Apl I in maintaining a-ITF including pharmacological data (Sutton et al. 2004), the specificity of cleavage for the FRET construct of PKC Apl I over PKC Apl III (Fig. 3) and the requirement of the classical calpain for both cleavage of the FRET construct and generation of a-ITF (Figs. 5, 8). Taken together, our data make a strong case for concluding that PKM Apl I is important for a-ITF. There are many models of presynaptic-mediated long-term synaptic plasticity in vertebrates (Yang and Calakos 2013; Padamsey and Emptage 2014), but there is no consensus on how these forms of plasticity are maintained. While some targets, such as Rim, Rab3A, and Munc18 have been shown to be involved in presynaptically driven synaptic plasticity (Yang and Calakos 2013), it will be interesting to determine whether calpain-mediated cleavage of classical PKCs is upstream of these targets.
It was surprising that activity alone caused cleavage of CFP-PKC Apl I-YFP and did not require 5HT, since induction of a-ITF requires the coincidence of activity and 5HT. This suggests that PKM Apl I is not by itself sufficient for increases in activity when expressed in the sensory neuron. Indeed, expression of PKM Apl I in the sensory neuron is not sufficient to increase synaptic strength (S. Schacher, personal communication). It is also surprising that the loss of FRET was delayed. This delay could be due to delayed activation of calpain by evoked action potentials, as opposed to depolarization by KCl that caused a faster loss of FRET. It may also be that the delayed loss of FRET is due to a smaller but prolonged activation of calpain by evoked action potentials. This question cannot be resolved without a better understanding of how calpains are activated in this system.
Changes in FRET in similar PKC constructs have also been used to indicate changes in conformation of the kinase without cleavage (Braun et al. 2005; Antal et al. 2014). While we have shown that, in our case, the loss of FRET was inhibited by both pharmacological and molecular manipulations of calpain, we cannot rule out some contribution from conformational changes. Indeed, the Calpain inhibitor, ALLM, only partially blocked cleavage by activity. Since calpain cleavage, unlike for example phosphorylation, is an irreversible reaction, one might expect pharmacological inhibitors to have limited effectiveness. Moreover, we do not know how effective this inhibitor is on the Aplysia calpain isoform. Nevertheless, this data is also consistent with a contribution from conformational changes. The blockade of cleavage by the dominant-negative classical calpain was more complete as no significant loss of FRET was seen in the presence of the dominant negative. Together, the partial effect by ALLM and the complete block by the dominant-negative calpain lead us to conclude that the loss of FRET we measure after activity is mainly caused by the cleavage of PKC Apl I.
Why does the same calpain cleave distinct substrates in different forms of plasticity?
The substrates for the classical calpain are distinct in the presynaptic terminal and the postsynaptic terminal as PKC Apl I is cleaved by classical calpain in the presynaptic terminal during a-ITF, while PKC Apl III is cleaved by classical calpain in the postsynaptic terminal during m-ITF. This suggests that the calpain complexes in the presynaptic and postsynaptic cell are distinct and are differentially activated by different stimuli. Similar to the dominant-negative PKCs, the isoform specificity of the dominant negatives depends on their mechanism of action. At this point, it is unclear whether classical calpains or Sol calpains in Aplysia form dimers, or whether the dominant negatives act by sequestering upstream activators or scaffolding sites. We also do not have information about how the different stimuli activate calpain in Aplysia. One possibility is that PKCs may need to be activated to allow for calpain cleavage and PKC Apl I is known to be specifically activated in the sensory neuron during a-ITF. However, if activation was required for cleavage of PKC Apl I, it is surprising that cleavage was seen only with activity, since translocation of PKC, which is associated with the conformational changes required to free the pseudosubstrate from the regulatory domain, requires both serotonin and activity (Zhao et al. 2006; Shobe et al. 2009).
While the evidence that cleavage occurs with activity in the absence of 5HT, there have been previous suggestions that activity alone is sufficient for some PKC action. Bursts of action potentials are sufficient to prevent homosynaptic depression and this is mediated by PKC Apl I (Wan et al. 2012). Thus, while activity is not sufficient to translocate fluorescently tagged PKC, it may activate a pretargeted pool of PKC Apl I and this could also be the pool cleaved by calpain. Activity alone (in this case 20 Hz for 2 sec) was sufficient for an increase in sensorin levels and the effects of activity were blocked by chelerythrine (Hu et al. 2007), suggesting that PKM Apl I may be sufficient to activate translation of sensorin, although this would not be important for a-ITF which is not blocked by antibodies against sensorin (Hu et al. 2007).
Conclusion
The idea that persistent protein kinases can act as molecular memory traces has been a topic of interest for thirty years (Lisman 1985; Greenberg et al. 1987). Recently, a particular PKM, PKMζ, has received considerable interest. In mice, PKC/PKMζ knock-out mice retain memory and sensitivity to the ZIP peptide (Lee et al. 2013), but recent evidence suggests PKCι may be compensating for this loss (Tsokas et al. 2016). Overall, our observations implicating multiple PKMs in memory, combined with the promiscuity of ZIP and chelerythrine, extend the pool of candidate compensatory kinases to include members of the classical PKC family, suggesting that the ability to form a PKM and maintain synaptic enhancement may be a more general role of PKCs than previously thought. It is also conceivable that forms of memory previously thought to rely on PKMζ based on pharmacological sensitivity are actually mediated by other PKM isoforms formed by calpain-dependent cleavage. Indeed, classical calpains have been shown to be required for the induction of some forms of LTP in vertebrates (Amini et al. 2013).
Materials and Methods
Constructs
The mRFP-PKM Apl III and mRFP-DN PKM Apl III (D392A) construct were previously described (Bougie et al. 2012). mRFP-PKM Apl I was generated using PCR and PKM starts at amino acid 300 from PKC Apl I (YPEP). mRFP-DN PKM Apl I (D444A) [this mutation removes >95% of kinase activity, but allows for stabilizing phosphorylation sites (Cameron et al. 2009; Bougie et al. 2012)] was generated with overlap PCR at the conserved residue in PKM Apl I (D444). The CFP-PKC Apl III-YFP was previously described (Bougie et al. 2012). To generate CFP-PKC Apl I-YFP, enhanced CFP (eCFP) was amplified by PCR using primers containing NcoI and BsrGI sites. The product of this amplification was then cut with NcoI and BsrGI and used to replace the eGFP from eGFP-PKC Apl I (Zhao et al. 2006). Enhanced YFP (eYFP) was then amplified by PCR using primers containing Nde and Blp1 sites with the nucleotides encoding a putative PDZ binding domain (FVVTV) at the end of PKC Apl I added on at the 3′ end of eYFP. The product of this amplification was then cut with Nde1 and BlpI and ligated to the CFP–PKC Apl I vector cut with the same enzymes.
Aplysia small optic lobes (SOL) calpain and classical calpain were amplified by PCR from an Aplysia nervous system cDNA library. Classical calpain was amplified with nested primers: outer CCTTCACCTTCTTCACCAATC (5′) and CAGCCAGAATATGCAGGCAT (3′); Inner: CTTCTTCACCAATCATGGA (5′) and GCCAGAATATGCAGGCATCT (3′). SOL calpain was amplified in two fragments, then assembled using overlap PCR. Primer for 5′ fragment: GTCACGATGGGTGACTGGAT (5′) and TCTTCGGGTAATGGGGTACGA; 3′ fragment: ACCGGGTACCCATTTGTGGA (5′) and TCCTAGACGTAGGGTCGTG (3′). PCR products were cloned into the pJET1.2 vector using the Thermo-Scientific CloneJET PCR cloning kit. Baculovirus was generated using the Bac-to-Bac cloning system (Life Technologies Inc.). Classical calpain was excised from pJET with Not1 and XbaI and inserted into pFastBacHT-A cut with the same enzymes. SOL was amplified from pJET with extended primers containing restriction sites for BamHI 5′ primer: AATTGGATCCGTCACGATGGGTGACTGGAT; and EcoRI 3′ primer: AATTGAATTCTCCTAGACGTAGGGTCGTGG), digested with BamHI and EcoRI and ligated into FastBacHT-B cut with the same enzymes. Baculovirus was raised in Sf9 cells according to the manufacturer's instructions. To make neuronal overexpression constructs, classical calpain was excised from pJET1.2 with SspI and XhoI and ligated into pNEX3 vector cut with SmaI and SalI. SOL was amplified from pJET using extended primers containing restriction sites for SalI 5' primer: AATTGTCGACGTCACGATGGGTGACTGGAT and BamHI 3' primer: AATTGGATCCTCCTAGACGTAGGGTCGTG), and ligated into pNEX3 cut with the same enzymes. C to S mutations were introduced by PCR amplifying two overlapping fragments from the calpain-pNEX3 constructs, with the C to S mutation encoded in primers corresponding to the overlap. The classical calpain overlap PCR product was digested with SbfI and StuI and ligated into the classical calpain-pNEX3 construct cut with the same enzymes or for Sol calpain the product was digested with NaeI and BtgZI and ligated into the SOL calpain-pNEX3 construct cut with the same enzymes.
Kinase assays
Constructs were generated to express His-tagged PKM Apl III (beginning at amino acid 248, MTAK) (Villareal et al. 2009) and His tagged PKM Apl I (beginning at amino acid 300, YPEP) using PCR and the vector BBACHis2. These constructs were then used to make baculovirus as previously described (Lim and Sossin 2006). SF9 cells were infected with these baculoviruses and PKCs were purified on nickel columns (Lim and Sossin 2006). The relative amount of enzyme was quantified by immunoblotting a serial dilution of enzymes from the same preparation of purified enzyme used in the assay with the His antibody to allow for equal molar amounts of PKM Apl I and PKM Apl III to be used in the kinase assays. Dilutions from each kinase preparation were made to ensure the kinase assay was in the linear range. The specific activities (counts per minute of 32P-ATP incorporated into substrate/µg PKM) of the PKMs in this assay were comparable (PKM Apl III had 1.5 ± 0.9 more specific activity of PKM Apl I, n = 3 preparations). Enzymatic activity of the kinases was assayed in an in vitro reaction, with all parameters previously described (Sossin et al. 1996). Briefly, PKMs were incubated with 10 µM substrate A-ε peptide (Sossin et al. 1996), radioactive 32P-ATP, 50 µM ATP, 50 mM Tris pH 7.5, 10 mM MgCl2, and various concentrations of PKC inhibitors or water. Reactions were performed at 20°C for 30 min. Each reaction was blotted onto P81 Whatman filter paper, stopped in a 2% w/v cold ATP solution, washed 4× with 0.425% v/v phosphoric acid, and then counted in a scintillation counter to determine levels of radioactivity incorporated into the substrate. All determinations were done in duplicate. All values were normalized to the amount of activity in the absence of the inhibitor.
PCR screen for calpain expression
Motor neuron and total nervous system cDNA preparations were screened with two sequential rounds of PCR using either nested (Tra, Atypical, Sol, Classical) or identical (PalB) primers: Aplysia Tra outer: 5p: AGACATAGTGTGGAAGAGGC, 3p: CTCTTCGGATTTGAACTCGG, inner 5p: GGTAACTGCTGGTTTGTGGC, 3p: GTGAGAGCTTCGTAACAGCC. PalB 5p: ATAGCTGTGCAGCATGACAG, 3p: AGGTTGATTGCCTCGTCTAG (identical primers used for second PCR). Atypical outer 5p: TGAAAATGAGGAGACTCTGG, 3p: CGTTGTAGTGGTCATACAAG, inner 5p: CTCTGGAAATTAACGGCAGA, 3p: ACAAGTAGTTGATGTCCAGG. SOL outer 5p: GCAAAACTACAGGTGTGTCC, 3p: GACAGGGTTGGCAGTCTAGA, inner 5p: GTGTCCAATGGTGCTTCCA, 3p: ATCTCCGAGGAGACTCACAGA. Classical outer 5p: TCTCGGATTTCAAAGACACG, 3p: GGTCCTCGAAGAGAATATCC, inner 5p: CACGTTCGACAAATACAAGG, 3p: ATCCTCGTCCTGGCACTTCT. Sensory neuron cDNA was screened with a single round of PCR with the outer primers only.
Expression and purification of recombinant proteins
SF9 cells in suspension were infected with baculovirus. Three days after infection, His-tagged calpain was purified using Pro-bond His-Affinity resin (Life Technologies Inc.), in modified purification buffer (20 mM HEPES pH 7.5, 1 mM EDTA, 1 mM DTT, 100 mM KCl, 10% glycerol). Proteins were eluted in elution buffer (Purification buffer with 0.25 M Imidazole). DTT was added to a final concentration of 11 mM, and the sample was concentrated and stored at −80°C.
Antibody production
Polyclonal antibodies against the carboxy termini of classical and SOL calpain were raised in rabbit using the following epitopes: classical: GFAPYDIDSFIQATMYS, SOL: ISPQMESIHQPRPYV. To affinity purify the antibodies, the pMAL Protein Fusion and Purification System (New England Biolabs) was used to express a fusion protein consisting of the epitope peptide fused to maltose-binding protein in Escherichia coli. The fusion protein was then purified by incubation with amylose beads, subjected to SDS-PAGE and transferred to a PVDF membrane, which was then incubated with the antibody-containing serum overnight at 4°C. After three washes with 50 mM Tris pH 7.4 with 500 mM NaCl and one wash with 50 mM Tris pH 7.4 with 100 mM NaCl, bound antibody was eluted from the membrane with Glycine 50 mM pH 2.5. After adjusting to pH 7 or 8 with Tris 2 M pH 8.0, antibodies were concentrated, glycerol added to a final concentration of 50% and aliquots frozen at −80°C.
Nervous system homogenization for antibody testing
Pleuropedal ganglia were dissected from anesthetized animals, treated with Dispase and desheathed as described above. Ganglia were then homogenized with a disposable tissue grinder in ice-cold homogenization buffer (20 mM HEPES pH 7.5, 0.5 mM EDTA, 0.5 mM EGTA, 2.6 mM 2-mercaptoethanol, 50 mM NaF, 5 mM Sodium Pyrophosphate, 10% glycerol, Roche Complete EDTA-free protease inhibitor cocktail). After a 30 sec centrifugation at 13000 rpm, the supernatant was collected and concentration was determined by Bradford assay. Proteins were denatured by addition of Laemmli sample buffer and boiling before being subjected to SDS-PAGE and immunoblotting. Ten to 20 µg of total protein were loaded.
Aplysia cell culture
Aplysia californica were obtained from the University of Miami Aplysia Resource Facility (RSMAS) and sensory neurons, motor neurons and sensorimotor neuron cocultures were prepared as previously described (Zhao et al. 2006; Farah et al. 2008, 2015). Animals were anesthetized by an injection of 50–100 mL of 400 mM (isotonic) MgCl2. Abdominal and/or pleuropedal ganglia were removed and digested in L15 medium containing 10 mg/mL of Dispase II (Roche Diagnostics) for either 2 h at 37°C or 18 h at 19°C. L15 media (Sigma-Aldrich) was supplemented with 0.2 M NaCl, 26 mM MgSO4, 35 mM dextrose, 27 mM MgCl2, 4.7 mM KCl, 2 mM NaHCO3, 9.7 mM CaCl2, and 15 mM HEPES, pH of 7.4. Sensory neurons and LFS motor neurons were isolated from pleural and abdominal ganglia, respectively, that were dissected from adult Aplysia californica (60–100 g which are hermaphrodite). The sensory neurons were manually paired with motor neurons and maintained in coculture at 19°C in a high-humidity chamber for 5 d (Hu et al. 2010) in L15 medium containing 50% Aplysia hemolymph on MatTek glass-bottom culture dishes (MatTek Corporation) with a glass surface of 14 mm and a coverslip thickness of 0.085–0.13 mm. In experiments involving kinase-dependent cleavage, cells were plated in L15 medium containing 25% hemolymph. Dishes were pretreated with poly-L-lysine (molecular weight >300 000; Sigma-Aldrich).
Plasmid microinjection
Forty-eight to 72 h after isolation, solutions of plasmids in distilled water containing 0.2% fast green were microinjected into the nuclei of sensory neurons or LFS motor neuronsusing back-filled glass micropipettes. The tip of the micropipette was inserted into the cell nucleus. A short pressure pulse was delivered until the nucleus of the sensory neuron or LFS became uniformly green (Farah and Sossin 2011) Injected construct concentrations were as follows (in ng/μL): mRFP 200 and 300, mRFP-DN PKM Apl I 450, mRFP-DN PKM Apl III 200, eGFP 100, eGFP+DN-CCal 100 and 250, eGFP+DN-SOL 100 and 250, eCFP 100, eYFP 100, CFP-PKC Apl I-YFP 100, 200 and 400, CFP-PKC Apl III-YFP 50 and 100. Total DNA concentration was kept constant between groups using pNEX3 vector. The cells were incubated for 4–5 h at room temperature and then kept in a 19°C incubator until use. For experiments involving PKC Apl I translocation, cells were kept at 4°C ON and used on the following day. The fluorescent images of construct expression were captured using a Zeiss Axioobserver Z1 fluorescent microscope attached to a QuantEM 512SC EMCCD camera or a Nikon Diaphot microscope attached to a silicon-intensified target (SIT) (Dage 68; Dage-MTI) video camera.
Sample sizes
Sample size was not computed during study design as the variability of the measures was not known before beginning the experiments. For each experiment, sample size was estimated based on the variability of the measure. If, based on this estimate, trends were visible, but results were not statistically significant, additional experiments were performed.
FRET and image quantification
Right before imaging, the culture media was replaced with artificial sea water (ASW). Cells were imaged using a Zeiss Axioobserver Z1 fluorescent microscope using Axiovision Microscopy software for image acquisition as previously described (Bougie et al. 2012). Cells expressing CFP alone or YFP alone were used to calculate the background FRET signal from each channel. Each cell was imaged in the CFP, YFP, and FRET channels using CFP (Zeiss 47 BP 436/20, FT 455, BP 480/40), YFP (Zeiss 46-HE BP 500/25 DMR 25, FT 515 HE, BP 535/30 DMR 25) and CFP-YFP FRET (Zeiss 48 BP 436/20, FT 455, BP 535/30) filter sets and exposure times were kept constant for all groups within each experiment. Zeiss AxioVision software was used to quantify the images and FRET Xia formula with background subtraction used to calculate the FRET signal (Xia and Liu 2001). FRET calculation yields a value that is illustrated as a color-coded FRET value map in which lower values are assigned cooler colors and higher values are assigned warmer colors. Images were taken pretreatment and at multiple time points post-treatment. The change in the intramolecular FRET value was then calculated (post/pre). In isolated sensory neurons, one to three varicosities were chosen per cell. In cocultures of sensory and motor neurons, one to three sensory neuron varicosities adjoining the motor neuron were chosen. Similarly, one to three regions of interest in the motor neuron that were adjoining presynaptic varicosities were chosen. Statistical analysis was performed using Student's t-test when comparing two groups and One-way ANOVA followed by a Student–Newman–Keuls post hoc test when comparing three groups. In each experiment, a paired Student's t-test was performed to compare the raw FRET ratio post- and pretreatment for each condition and P values were corrected for multiple t-tests with the Bonferroni correction. ANOVAs were then performed on the change in intramolecular FRET (post/pre) values. Cells showing an obvious change of focus in the post image compared with pre were not included.
Electrophysiology
Presynaptic sensory neurons and postsynaptic motor neurons (LFS) were paired as previously described (Zhao et al. 2006; Farah et al. 2008, 2015). Pairs of sensory and motor neurons in individual culture dishes were incubated in culture media and placed in a 19°C incubator until use. After 3 d in culture, nuclear injections of plasmids occurred, allowing for at least 18 h of expression before electrophysiological recordings. Cell pairs were removed from the incubator and all solutions brought to room temperature at least 1 h prior to recording. Before electrophysiological recordings, the culture media was replaced with a recording saline consisting of in (mM): NaCl (460), MgCl2 (55), CaCl2 (10), KCl (10), D-Glucose (10), HEPES (10), pH 7.6.
Membrane potentials were recorded and controlled in current clamp mode with sharp, intracellular electrodes attached to an Axoclamp 900A and Digidata 1440A (Molecular Devices). Electrodes (∼15 MΩ) were backfilled with 2 M potassium acetate and bridge-balanced before and after membrane penetration. The postsynaptic cell was impaled first, so that if entry into the presynaptic cell resulted in generation of an action potential, the resultant postsynaptic potential (PSP) would be recorded. Injection of hyperpolarizing current during electrode entry aided in preventing this activity and was used to keep both neurons (pre and postsynaptic) at −80 mV during the experiment. Postsynaptic input resistance was measured with 500 msec, −0.5 nA pulses. Action potentials in the presynaptic neuron were generated by 50 msec depolarizing pulses of increasing current until a single action potential is fired. PSP amplitude was measured directly for PSPs <20 mV, for stronger synapses initial PSP rise-rate was measured as described (Dunn and Sossin 2013). Measuring initial rise rate avoids the nonsynaptic influence that voltage-dependent currents have on the PSP waveform at strong synapses.
Measurement of mITF and aITF
In a-ITF experiments, 20 action potentials at 10 Hz were generated at 1, 2, 3, and 4 min in the presence of 5HT (10 µM) with 12 msec pulses through intracellular electrodes at current amplitudes set to generate single action potentials per pulse in the sensory neuron (Sutton and Carew 2000). For m-ITF 5HT (10 µM) was added for 10 min, before wash out. PSP amplitude was measured as described (Dunn and Sossin 2013). All PSPs are expressed as a percentage of the measurement of the first PSP (%PSP1), prior to 5HT addition.
Since the magnitude of ITF can vary between synapses in cultures made from different animals, controls (pNEX alone or a pNEX plasmid expressing a fluorescent protein only) were always included for each recording day. Periods occur where animals do not show ITF, so to prevent this data from reducing the overall ITF measurements, when all trials show no facilitation at 60 min (<100% of PSP1) on a particular recording day, the data from this day were not included. All sensory neurons used on one recording day were isolated from the same animal, while the postsynaptic motor neurons were isolated from between 1–4 animals for each recording day. In each group, to determine whether ITF had occurred, a paired Student's t-test was performed to compare the EPSP post- and pretreatment for each condition and P values were corrected for multiple t-tests with the Bonferroni corrections. For experiments with three groups, an ANOVA was performed on the normalized PSP amplitude after facilitation followed by a Student–Newman–Keuls post hoc test when comparing three groups. For the expression of dominant-negative PKMs, only cells with >100-fold the average intensity of the somatic autofluorescence were used. In the trials where mRFP-DN PKM Apl I intensity was <100-fold greater than autofluorescence, a-ITF was observed (PSP at 60 min 157.7 ± 15.1% PSP#1, a significant increase as measured with a paired t-test, P < 0.01, n = 10) similar to mRFP-DN PKM Apl III and mRFP expression alone. All data presented are mean values ± the standard error of the mean (SEM).
Acknowledgments
This work was supported by CIHR grant MOP 12046 and 340328 to W.S.S. W.S.S. is a James McGill Professor. M.H.H. is supported by a FRSQ studentship. We thank Joanna Bougie for initial work on cloning of the classical calpain and Xiaotang Fan for technical support.
Footnotes
Article is online at http://www.learnmem.org/cgi/doi/10.1101/lm.043745.116.
References
- Amini M, Ma CL, Farazifard R, Zhu G, Zhang Y, Vanderluit J, Zoltewicz JS, Hage F, Savitt JM, Lagace DC, et al. 2013. Conditional disruption of calpain in the CNS alters dendrite morphology, impairs LTP, and promotes neuronal survival following injury. J Neurosci 33: 5773–5784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antal CE, Violin JD, Kunkel MT, Skovso S, Newton AC. 2014. Intramolecular conformational changes optimize protein kinase C signaling. Chem Biol 21: 459–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baudry M, Bi X. 2016. Calpain 1 and Calpain 2: the Yin and Yang of synaptic plastictiy. Trends Neurosci 39: 234–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bougie JK, Lim T, Farah CA, Manjunath V, Nagakura I, Ferraro GB, Sossin WS. 2009. The atypical protein kinase C in Aplysia can form a protein kinase M by cleavage. J Neurochem 109: 1129–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bougie JK, Cai D, Hastings M, Farah CA, Chen S, Fan X, McCamphill PK, Glanzman DL, Sossin WS. 2012. Serotonin-induced cleavage of the atypical protein kinase C Apl III in Aplysia. J Neurosci 32: 14630–14640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun DC, Garfield SH, Blumberg PM. 2005. Analysis by fluorescence resonance energy transfer of the interaction between ligands and protein kinase Cdelta in the intact cell. J Biol Chem 280: 8164–8171. [DOI] [PubMed] [Google Scholar]
- Cai D, Pearce K, Chen S, Glanzman DL. 2011. Protein kinase M maintains long-term sensitization and long-term facilitation in Aplysia. J Neurosci 31: 6421–6431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron AJ, Escribano C, Saurin AT, Kostelecky B, Parker PJ. 2009. PKC maturation is promoted by nucleotide pocket occupation independently of intrinsic kinase activity. Nat Struct Mol Biol 16: 624–630. [DOI] [PubMed] [Google Scholar]
- Dong Z, Han H, Li H, Bai Y, Wang W, Tu M, Peng Y, Zhou L, He W, Wu X, et al. 2015. Long-term potentiation decay and memory loss are mediated by AMPAR endocytosis. J Clin Invest 125: 234–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn TW, Sossin WS. 2013. Inhibition of the Aplysia sensory neuron calcium current with dopamine and serotonin. J Neurophys 110: 2071–2081. [DOI] [PubMed] [Google Scholar]
- Farah CA, Sossin WS. 2011. Live-imaging of PKC translocation in Sf9 cells and in Aplysia sensory neurons. J Vis Exp 10.3791/2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farah CA, Nagakura I, Weatherill D, Fan X, Sossin WS. 2008. Physiological role for phosphatidic acid in the translocation of the novel protein kinase C Apl II in Aplysia neurons. Mol Cell Biol 28: 4719–4733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farah CA, Naqib F, Weatherill DB, Pack CC, Sossin WS. 2015. Synapse formation changes the rules for desensitization of PKC translocation in Aplysia. Eur J Neurosci 41: 328–340. [DOI] [PubMed] [Google Scholar]
- Greenberg SM, Castellucci VF, Bayley H, Schwartz JH. 1987. A molecular mechanism for long-term sensitization in Aplysia. Nature 329: 62–65. [DOI] [PubMed] [Google Scholar]
- Hernandez AI, Blace N, Crary JF, Serrano PA, Leitges M, Libien JM, Weinstein G, Tcherapanov A, Sacktor TC. 2003. Protein kinase M ζ synthesis from a brain mRNA encoding an independent protein kinase C ζ catalytic domain. Implications for the molecular mechanism of memory. J Biol Chem 278: 40305–40316. [DOI] [PubMed] [Google Scholar]
- Hu J, Schacher S. 2015. Persistent associative plasticity at an identified synapse underlying classical conditioning becomes labile with short-term homosynaptic activation. J Neurosci 35: 16159–16170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu JY, Chen Y, Schacher S. 2007. Protein kinase C regulates local synthesis and secretion of a neuropeptide required for activity-dependent long-term synaptic plasticity. J Neurosci 27: 8927–8939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu JY, Chen Y, Bougie JK, Sossin WS, Schacher S. 2010. Aplysia cell adhesion molecule and a novel protein kinase C activity in the postsynaptic neuron are required for presynaptic growth and initial formation of specific synapses. J Neurosci 30: 8353–8366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Forsberg NE. 1998. Role of calpain in skeletal-muscle protein degradation. Proc Natl Acad Sci 95: 12100–12105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandel ER. 2001. The molecular biology of memory storage: a dialogue between genes and synapses. Science 294: 1030–1038. [DOI] [PubMed] [Google Scholar]
- Lee AM, Kanter BR, Wang D, Lim JP, Zou ME, Qiu C, McMahon T, Dadgar J, Fischbach-Weiss SC, Messing RO. 2013. Prkcz null mice show normal learning and memory. Nature 493: 416–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim T, Sossin WS. 2006. Phosphorylation at the hydrophobic site of protein kinase C Apl II is increased during intermediate term facilitation. Neuroscience 141: 277–285. [DOI] [PubMed] [Google Scholar]
- Ling DS, Benardo LS, Serrano PA, Blace N, Kelly MT, Crary JF, Sacktor TC. 2002. Protein kinase Mζ is necessary and sufficient for LTP maintenance. Nat Neurosci 5: 295–296. [DOI] [PubMed] [Google Scholar]
- Lisman JE. 1985. A mechanism for memory storage insensitive to molecular turnover: a bistable autophosphorylating kinase. Proc Natl Acad Sci 82: 3055–3057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macqueen DJ, Wilcox AH. 2014. Characterization of the definitive classical calpain family of vertebrates using phylogenetic, evolutionary and expression analyses. Open Biol 4: 130219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel M, Green CL, Gardner JS, Organ CL, Lyons LC. 2012. Massed training-induced intermediate-term operant memory in Aplysia requires protein synthesis and multiple persistent kinase cascades. J Neurosci 32: 4581–4591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muslimov IA, Nimmrich V, Hernandez AI, Tcherepanov A, Sacktor TC, Tiedge H. 2004. Dendritic transport and localization of protein kinase Mζ mRNA: implications for molecular memory consolidation. J Biol Chem 279: 52613–52622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padamsey Z, Emptage N. 2014. Two sides to long-term potentiation: a view towards reconciliation. Philos Trans R Soc London B Biol Sci 369: 20130154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacktor TC. 2011. How does PKMζ maintain long-term memory? Nat Rev Neurosci 12: 9–15. [DOI] [PubMed] [Google Scholar]
- Sacktor TC. 2012. Memory maintenance by PKMζ--an evolutionary perspective. Mol Brain 5: 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrano P, Yao Y, Sacktor TC. 2005. Persistent phosphorylation by protein kinase Mζ maintains late-phase long-term potentiation. J Neurosci 25: 1979–1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrano P, Friedman EL, Kenney J, Taubenfeld SM, Zimmerman JM, Hanna J, Alberini C, Kelley AE, Maren S, Rudy JW, et al. 2008. PKMζ maintains spatial, instrumental, and classically conditioned long-term memories. PLoS Biol 6: 2698–2706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shema R, Haramati S, Ron S, Hazvi S, Chen A, Sacktor TC, Dudai Y. 2011. Enhancement of consolidated long-term memory by overexpression of protein kinase Mζ in the neocortex. Science 331: 1207–1210. [DOI] [PubMed] [Google Scholar]
- Shobe JL, Zhao Y, Stough S, Ye X, Hsuan V, Martin KC, Carew TJ. 2009. Temporal phases of activity-dependent plasticity and memory are mediated by compartmentalized routing of MAPK signaling in Aplysia sensory neurons. Neuron 61: 113–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sossin WS. 2007. Isoform specificity of protein kinase Cs in synaptic plasticity. Learn Mem 14: 236–246. [DOI] [PubMed] [Google Scholar]
- Sossin WS. 2008. Molecular memory traces. Prog Brain Res 169: 3–25. [DOI] [PubMed] [Google Scholar]
- Sossin WS, Fan X, Saberi F. 1996. Expression and characterization of Aplysia protein kinase C: a negative regulatory role for the E region. J Neurosci 16: 10–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton MA, Carew TJ. 2000. Parallel molecular pathways mediate expression of distinct forms of intermediate-term facilitation at tail sensory-motor synapses in Aplysia. Neuron 26: 219–231. [DOI] [PubMed] [Google Scholar]
- Sutton MA, Bagnall MW, Sharma SK, Shobe J, Carew TJ. 2004. Intermediate-term memory for site-specific sensitization in Aplysia is maintained by persistent activation of protein kinase C. J Neurosci 24: 3600–3609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsokas P, Hsieh C, Yao Y, Lesburgue Y, Wallace EJC, Tcherepanov A, Jothianandan D, Hartley BR, Pan L, Rivard B, et al. 2016. Compensation for PKMζ in long-term potentiation and spatial long-term memory in mutant mice. Elife 5 10.7554/eLife.14846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villareal G, Li Q, Cai D, Fink AE, Lim T, Bougie JK, Sossin WS, Glanzman DL. 2009. Role of protein kinase C in the induction and maintenance of serotonin-dependent enhancement of the glutamate response in isolated siphon motor neurons of Aplysia californica. J Neurosci 29: 5100–5107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volk LJ, Bachman JL, Johnson R, Yu Y, Huganir RL. 2013. PKM-ζ is not required for hippocampal synaptic plasticity, learning and memory. Nature 493: 420–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walters ET. 1987. Site-specific sensitization of defensive reflexes in Aplysia: a simple model of long-term hyperalgesia. J Neurosci 7: 400–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan Q, Jiang XY, Negroiu AM, Lu SG, McKay KS, Abrams TW. 2012. Protein kinase C acts as a molecular detector of firing patterns to mediate sensory gating in Aplysia. Nat Neurosci 15: 1144–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Z, Liu Y. 2001. Reliable and global measurement of fluorescence resonance energy transfer using fluorescence microscopes. Biophys J 81: 2395–2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Calakos N. 2013. Presynaptic long-term plasticity. Front Synaptic Neurosci 5: 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Leal K, Abi-Farah C, Martin KC, Sossin WS, Klein M. 2006. Isoform specificity of PKC translocation in living Aplysia sensory neurons and a role for Ca2+-dependent PKC APL I in the induction of intermediate-term facilitation. J Neurosci 26: 8847–8856. [DOI] [PMC free article] [PubMed] [Google Scholar]