

Abstract
Nucleotide-free kinesin motor domains from several kinesin families convert reversibly to a refractory conformation that cannot rapidly rebind ADP. In the absence of glycerol, the refractory conformation of Drosophila kinesin motor domains is favored by 50-fold with conversion of the active to the refractory species at ~0.052 s−1 and reactivating in the presence of ADP at ~0.001 s−1. This reactivation by ADP is due to conformational selection rather than induced fit because ADP is not bound to the refractory species at concentrations of ADP that are sufficient to saturate the rate of reactivation. Glycerol stabilizes the active conformation by reducing the rate of inactivation, while having little effect on the reactivation rate. Circular dichroism indicates a large conformational change occurs on formation of the refractory species. The refractory conformation binds ANS (8-anilino-1-napthalenesulfonic acid) with a large increase in fluorescence, indicating that it has molten globule character. High ANS binding is also observed with the refractory forms of Eg5 (a kinesin-5) and Ncd (a kinesin-14), indicating that a refractory conformation with molten globule characteristics may be a common feature of nucleotide-free kinesin motor domains.
Graphical abstract
Introduction
Kinesins are a large superfamily of proteins that share a common motor domain that catalyzes microtubule-activated ATP hydrolysis [1,2]. Most kinesins use the energy of ATP hydrolysis to produce movement along microtubules (MTs), although some also function to depolymerize MTs. The kinesin-1 family was the first to be discovered and is the most extensively studied. In the absence of MTs, kinesin-1 (hereafter just called kinesin) hydrolyzes ATP and releases Pi rapidly, but can only slowly release ADP. Interaction with MTs induces a conformational change that accelerates ADP release [3,4]. Fast binding of ATP or ADP coupled with slow release as ADP results in very tight net binding of ADP. In fact when native heterotetrameric kinesin was isolated from bovine brain, it was found to have retained an ADP at its active site in spite of ATP not having been included in the solutions used in purification [3]. ADP itself, in the absence of Mg2+, binds only weakly to kinesin and has a fast dissociation rate of ~18 s−1 compared to ~0.03 s−1 for release of Mg2+ from the Mg2+ complex of E•MgADP and ~0.005 s−1 for concerted release of MgADP [5,6]. In the presence of millimolar Mg2+ concentrations, the initial release of Mg2+ at 0.03 s−1 is usually followed by rapid Mg2+ rebinding to E•ADP before release of Mg2+-free ADP can occur. In the presence of excess EDTA, the released Mg2+ is trapped by binding to EDTA and cannot rebind to E•ADP to suppress ADP release. Consequently the net binding of ADP is weak in the presence of excess EDTA. Gel filtration or other methods are now sufficient in the presence of excess EDTA to strip kinesin of its bound ADP. With full length bovine kinesin, this nucleotide-free form was able to rapidly rebind ATP with burst kinetics [4].
A striking early observation was that isolated nucleotide-free motor domains of Drosophila kinesin prepared in this way were, however, unable to rapidly rebind ADP, although they did slowly regain ADP binding during prolonged incubation with ATP [7]. This result is in contrast to the relatively stable apo-form of full length bovine kinesin discussed above. Ma and Taylor were able to stabilize isolated human motor domains in an active nucleotide-free conformation by use of 20% glycerol and high salt or by rapid removal of released ADP by apyrase [8,9]. Although human nucleotide-free motor domains also undergo reversible conversion to a refractory form, the rate of inactivation with human motor domains is slower than for Drosophila motor domains, which allowed for the study of the kinetics of nucleotide binding to active apo-motor domains. Members of the kinesin-5 and kinesin-14 families have also been shown to produce an inactive apo-form that can slowly reactivate [10–13].
This report demonstrates that even the more labile Drosophila kinesin motor domains can be stabilized by high concentrations of glycerol and further presents a detailed characterization of the reversible transition between active and refractory conformations of nucleotide-free motor domains. The refractory conformation has properties consistent with that of a molten globule and this is also true of the refractory conformation of Eg5 and Ncd as examples of kinesin-5 and kinesin-14 families. These properties help to explain why crystallization of nucleotide-free kinesin motor domains has been difficult.
Materials and Methods
Protein preparations
General methods for expression of motor domains in E. coli and purification were as previously described [14]. All buffers used in protein purification contained ≥ 0.1 mM MgATP. K346 is a monomeric Drosophila motor domain of amino acids 2–346 with an N-terminal extension of GS and a C-terminal extension of GGTS that was obtained by cleavage with Tev protease of a fusion with thioredoxin (modified pET32 plasmid with a linker containing a 6xHis-tag and a Tev protease cleavage site and with the two cysteine residues of thioredoxin converted to serines). The fusion protein was expressed in E. coli with induction by isopropyl-1-β-D-thiogalactose and lysed essentially as described [14]. Kinesin was first batch purified by adsorption to phosphocellulose (P11, Markson LabSales) at pH 6.3; resuspended in 20 mM MES/NaOH pH 6.3 with 100 mM NaCl, 2 mM MgCl2 and 2 mM mercaptoethanol; refiltered; and then kinesin was eluted with T20 buffer (20 mM Tris/Cl pH 8, 2 mM MgCl2) with 800 mM NaCl and 2 mM mercaptoethanol. The high salt phosphocellulose filtrate was adsorbed to an Ni-NTA column (Sigma) and eluted with a 0–200 mM imidazole gradient. His-tagged Tev protease [15] was added to the NTA eluent before dialysis against T20 buffer with 1000 mM NaCl and 2 mM MgCl2 to remove imidazole so that the cleavage mixture could be reapplied to an Ni-NTA column. In the absence of imidazole, cleaved motor domains bind weakly to Ni-NTA and were eluted with a 0–20 mM imidazole gradient (the His-tagged thioredoxin and Tev protease require much higher concentrations of imidazole for elution). K357 is the monomeric motor domain of amino acids 2–357 with an N-terminal extension of GS and a C-terminal extension of GGTS. It was cloned as a fusion protein with maltose binding protein (derived from pMal5c with a linker containing a 6xHis-tag and a Tev protease cleavage site) and was purified and cleaved with Tev protease as described for K346. All the data presented here for Drosophila kinesin-1 was obtained with K346 except for the circular dichroism experiments with K357 that undergoes reversible inactivation at similar rates (not shown). K346 and K357 are expected to have similar properties to those of the corresponding non-fusion constructs DKH346 and DKH357 studied previously [16]. H349 is the untagged human Kif5B motor domain consisting of amino acids 1–349 and was purified as previously described [5]. His-tagged human Eg5 motor domain is residues 2–386 [17]. Ncd motor domain was cloned as amino acids 330–700 with N-and C-terminal extensions of MGSSHHHHHHGT and TS respectively. Eg5 and Ncd were purified on phosphocellulose and Ni-NTA essentially as described for K346 except that Ncd was initially adsorbed to P11 at pH 8 and not 6.3.
Because ATP was present throughout the purifications at ≥ 0.1 mM, these procedures yield the E•ADP complex. Motor domains were stored in small aliquots at −80 °C following dialysis versus A25 buffer (25 mM ACES/KOH (pH 6.9), 2 mM magnesium acetate, 2 mM potassium EGTA, 0.1 mM potassium EDTA) that was supplemented with 50% glycerol, 100–300 mM KCl, 0.1 mM MgATP and 2 mM DTT. Kinesin concentrations were determined by absorption at 280 nm in 6 M guanidine hydrochloride [18]. An extinction coefficient of 2310 M−1cm−1 at 280 nm was used for bound ADP in preparations with ADP. Microtubules (MTs) were prepared from porcine tubulin and stabilized with paclitaxel, as previously described [19]. All reactions with MTs contained 10 μM paclitaxel.
Nucleotide-free kinesins were prepared from E•ADP by chromatography in the presence of glycerol and excess EDTA at 4 °C. E•ADP was diluted with 20 mM Mops pH 7.2, 2 mM EDTA, 2 mM DTT and 50% glycerol (v/v); adsorbed to a MacroPrep High S cation exchange column (Bio-Rad); washed extensively with the same 50% glycerol buffer to remove ADP and then eluted with stabilization buffer of 800 mM KCl, 50% glycerol, 20 mM Mops pH 7.2 and 2 mM DTT. All superfamily members prepared by this procedure had negligible MT-ATPase in the absence of added ATP (see Fig. 1B), consistent with essentially complete removal of bound ADP.
Figure 1. Evaluation of residual ADP in apo-K346.
A. Spectra of K346•ADP and apo-K346 at 15 μM in 6 M guanidine hydrochloride. K346•ADP at >600 μM was dialyzed against buffer with 100 μM free ADP to ensure saturation with ADP and to allow correction for the contribution of free ADP by subtracting the spectrum of an equivalent dilution of the solution outside the dialysis bag. B. MT-ATPase of apo-K346. The reaction was initiated by addition of 0.87 μM apo-K346 to 1.5 μM MTs in an ATPase reaction mixture with 2 mM PEP, 0.2 mM NADH, pyruvate kinase and lactic dehydrogenase, but without ATP. A low ionic strength buffer was used to increase the affinity of kinesin for MTs (20 mM Mops pH7.0, 2 mM MgCl2 and 0.1 mM EDTA). Addition of ATP to 0.1 and 0.2 μM produced a linear increase in rate. Based on this slope, the rate in the absence of added ATP corresponds to that expected for 0.019 μM ATP or 2% of the 0.87 μM K346 sites. This represents an upper limit because the MTs likely contain some free GDP/GTP that would be a substrate for both pyruvate kinase and K346. Note that apo-K346 is not inactivated over time in this assay because it is stabilized by binding to the MTs.
Kinetics analysis
All experiments were conducted at 25 °C in A25 buffer with 50 mM KCl and variable glycerol and nucleotide as indicated. Binding curves were fit to the full quadratic equation for tight binding (mutual depletion)
where P and X are the total concentrations and Y(bar) is the fraction of P with X bound. FRET from kinesin to mdADP (2′-deoxy-3′-mant-ADP) was measured using excitation at 280 nm and a 418 nm long pass filter for emission. mdADP was prepared by the method of Hiratsuka [20]. Stopped flow experiments were performed on a SX-20 instrument (Applied Photophysics). Fluorescence measurements in a stirred cuvet format were conducted in a custom designed thermostated chamber with fiber optics linking it to the SX-20 instrument that was used as the excitation light source and for data acquisition. ATPase rates were determined from the rate of decrease in absorbance of NADH at 340 nm using a coupled assay of pyruvate kinase and lactic dehydrogenase [21]. Nucleotides were added as the 1:1 complex with magnesium.
Rates of nucleotide binding and release
The kinetics of nucleotide binding and release from kinesin were determined as previously described [5]. This method uses the FRET signal from enzyme tyrosines to the mant group of bound mdADP that is lost when mdADP dissociates. For release of mdADP from E•mdADP, a concentrated stock of E•mdADP was prepared by equilibration of E•ADP with excess mdADP and then diluted to 0.84 μM E•mdADP in A25 buffer containing 50 mM KCl, 400 μM MgATP and glycerol as indicated. The falling FRET transient as bound mdADP is replaced with ATP during the chase was fit to a single exponential. The rate of release of ADP from E•ADP was determined similarly with E•ADP being diluted to 0.63 μM in buffer containing a chase of 13.2 μM mdADP and the rate of the rising FRET transient determined. An observed pseudo-first-order rate (kobs) for binding of mdADP to K346 was determined from the rising FRET transient on mixing apo-K346 in the stopped flow with a ≥10-fold excess of mdADP. Because inactivation is fast in 5% glycerol, the mdADP binding rate in this case only was determined by mixing apo-K346 in 10% glycerol with mdADP in the absence of glycerol for a final concentration of 5% glycerol. The bimolecular association rate was calculated as kobs/[mdADP]. Rates are reported as ±SD for ≥3 transients.
Circular Dichroism (CD)
The CD experiments were performed on a Jasco 715 spectropolarimeter using a 1 mm path-length cell at 20° C. The spectra were scanned at 100 nm/min with a 4 second response time and 2 nm bandwidth. Spectra were obtained following dilution of apo-K357 in stabilization buffer to 4 μM in 20 mM Mops/NaOH pH 7.2 with 2 mM MgCl2 and with KCl, glycerol and ADP as indicated. Spectra with protein were corrected for blank spectra with ADP as indicated, but in the absence of protein.
ANS Fluorescence
For ANS (8-anilino-1-napthalenesulfonic acid) binding studies, apoenzymes in stabilization buffer were first diluted to 2 μM without glycerol in either the presence or absence of 1 mM ADP. ANS was then added to 20 μM after a delay of 10 minutes to allow trapping of EA by ADP when ADP was present or maximum conversion to ER in the absence of added ADP. Eg5 and Ncd have significant amounts of the ER conformation even in stabilization buffer and a 5 minute delay after adding mdADP also provides sufficient time for conversion of ER to EA and trapping by mdADP. Samples with Ncd were supplemented with extra KCl to 500 mM total. Spectra were recorded on a PTI QM-400 fluorimeter. Excitation was at 365 nm and emission was monitored over 400–600 nm using band widths of 1 and 4 nm for excitation and emission respectively. A blank spectrum in the absence of ANS was subtracted.
Results
Generation of nucleotide-free apo-K346
Nucleotide-free Drosophila motor domain K346 was prepared by chromatography with excess EDTA. Briefly, kinesin was adsorbed at low ionic strength to a cation exchange column with EDTA to weaken ADP binding and 50% glycerol to stabilize the active conformation; the column was washed extensively with 50% glycerol/EDTA until the eluent had minimal absorbance at 260 nm; and then apo-K346 was eluted with 800 mM KCl in 50% glycerol (stabilization buffer). The absorbance spectrum of this preparation (Fig. 1A) indicates the loss of absorbance at 260 nm due to the removal of the starting stoichiometric amount of bound ADP. The maximum is at 277 nm as expected for a protein with tyrosines but no tryptophan. An estimate of ≤ 2% residual ADP was obtained from the ATPase rate in the absence of added nucleotide (Fig. 1B). Nucleotide-free H349, Eg5and Ncd motor domains prepared by this method also had negligible levels of MT-stimulated ATPase in the absence of added ATP.
Rebinding of ADP to apo-K346
This analysis employs the fluorescent ADP analog 2′-deoxy-3′-mant-ADP (mdADP) [20]. Mant-labeled nucleotides have been widely employed to monitor nucleotide binding to kinesins because they generate a fluorescence signal on binding and they can have binding and hydrolysis kinetics that are similar to those of unmodified nucleotides (see [5] for characterization of the interaction of mant nucleotides specifically with Drosophila kinesin). Use of the 2′-deoxy-3′-mant version (mdADP) avoids complications produced by differences in binding affinity and fluorescence intensity between the 2′ versus 3′ isomers of mant-(ribo)ADP. Binding of mdADP to kinesin does not produce a large net fluorescence change of the mant group itself on binding to kinesin [8], but does produce a FRET signal on binding due to energy transfer from protein tyrosines to bound mdADP that is similar to that observed with mant-(ribo)ADP [5]. When increasing amounts of apo-K346 in stabilization buffer are added to a fixed amount of mdADP in 20% glycerol to stabilize the active species, the fluorescence due to FRET increases linearly to an equivalence point at 1.07 kinesins per mdADP (Fig. 2). Thus apo-K346 added from stabilization buffer can still rebind an approximately stoichiometric amount of mdADP and the binding is very tight in 20% glycerol (mutual depletion), consistent with a Kd for mdADP of 0.4 nM calculated from the ratio of the dissociation and association rates of mdADP in 20% glycerol of 2.4 μM−1s−1 and 0.001 s−1 respectively (see below). The absence of a slow rise in fluorescence after the immediate increase on addition of mdADP indicates that little ER or EA•ADP is present in the preparation of apo-K346 because these species would slowly convert to EA•mdADP with increase in fluorescence.
Fig. 2. Titration of apo-K346 and mdADP.
The increase in fluorescence due to FRET from enzyme tyrosines to bound mdADP was determined for addition of varying amounts of apo-K346 from stabilization buffer into a stirred cell containing a fixed concentration of 0.67 μM mdADP in 20% glycerol. Traces begin with buffer only, followed by addition of mdADP at 10 seconds and then apo-K346 at 20 seconds to 0.22, 0.44, 0.53 and 1.31 μM for bottom to top trace respectively. Insert is dependence of the fluorescence increase on the apo-K346 concentration. Small corrections were applied for the fluorescence of apo-K346 alone. The vertical line is at the concentration of mdADP. The theoretical plot is for an unrestrained fit to the full quadratic equation for mutual depletion that gives an equivalence point at 0.72 μM. Insert contains data for addition concentrations of apo-K346 that were not included in the main figure for clarity.
ATPase activity of apo-K346 in the absence of glycerol
When diluted from stabilization buffer into an ATPase reaction mixture without glycerol, but containing ATP and MTs (Fig. 3), the activity of the apo-K346 (curve a) is essentially identical to that of E•ADP (curve b) from which the bound ADP has never been removed (ratio of the rate for apo-K346 to that for E•ADP is 1.049 ±0.045). Thus this preparation of apo-K346 is fully capable of rapidly rebinding nucleotide with full ATPase activity in the absence of glycerol. However, if apo-K346 is diluted from stabilization buffer into buffer alone and incubated for 5 minutes before addition of MTs and ATP (curve c), the initial rate is close to zero but activity is slowly recovered. This slow recovery of MT-ATPase is consistent with the slow recovery of nucleotide binding by inactivated apo-DKH340 determined using [32P]ATP [7]. Dilution of apo-K346 into buffer containing only 50 μM ATP (curve d) or 1 μM MTs (curve e), followed by incubation for 8 minutes before addition of a MTs or 1 mM ATP respectively is sufficient to stabilize apo-K346 against inactivation. Thus apo-K346 in stabilization buffer is predominately in a fully active conformation EA and this active form can either convert to the inactive ER form or be trapped and stabilized by binding to nucleotide or MTs (see Scheme for partitioning of EA between conversion to ER and ADP dependent trapping as EA•ADP).
Figure. 3. Inactivation of MT-ATPase of active apo-K346 by dilution without glycerol.
Stocks of E•ADP and apo-K346 in 50% glycerol were prepared at the same concentration and then diluted 100-fold into a complete reaction mixture (25 mM KCl) with taxol and the ATPase rate monitored by loss of absorbance of NADH at 340 nm as previously described [21] using the coupled enzyme system of PEP, pyruvate kinase and lactic dehydrogenase. Curves a and b were initiated by addition of 0.36 μM E•ADP or apo-K346 respectively to a complete reaction mix containing 1 mM MgATP and 1.0 μM MTs. Curve c was initiated by dilution of 0.36 μM apo-K346 into 0.9 volumes of buffer only for 5 minutes, followed by addition of a 10-fold concentrated stock containing ATP, MTs and the other reaction components. Curve d is for dilution of apo-K346 into buffer with 50 μM ATP only for 8 minutes followed by addition of MTs and ATP to 1 mM. Curve e is for dilution of apo-K346 into buffer with MTs but without ATP for 8.5 minutes before addition of ATP to 1 mM. Small offsets were applied so all curves start at an absorbance of 1.0
Scheme.
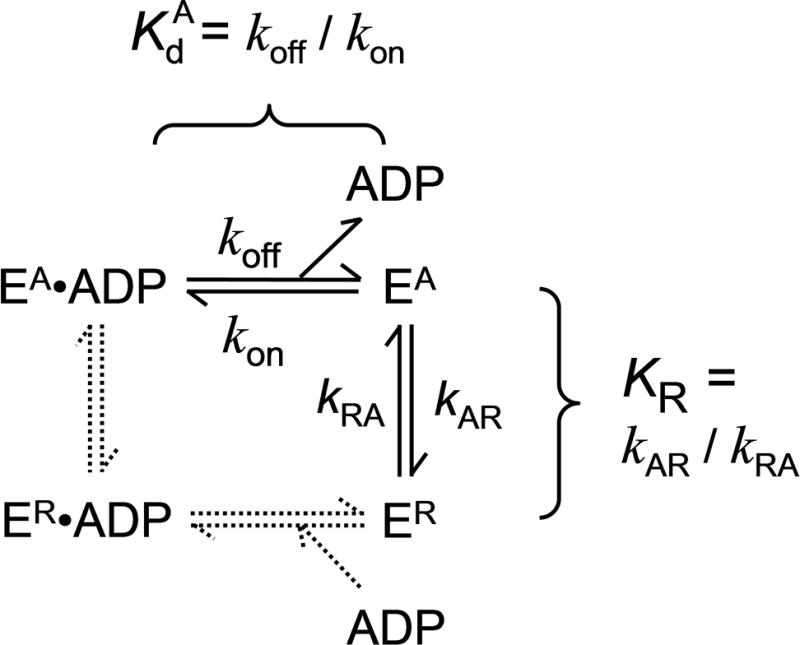
Scheme for binding of ADP to EA and ER
Kinetics of inactivation in absence of glycerol
The kinetics of inactivation can be measured by the loss of the ability of EA to bind mdADP following dilution of apo-K346 out of stabilization buffer into buffer without glycerol as indicated in Fig. 4. Curve a gives the fluorescence versus time starting with buffer alone followed by sequential addition of mdADP to 1.8 μM at 10 seconds and apo-K346 to 0.875 μM at 20 seconds. In this case with excess mdADP present, EA will partition predominately to EA•mdADP rather than to ER (Scheme) because kAR is slow at 0.052 s−1 as shown below whereas binding of mdADP is fast with a pseudo-first order rate for binding with increased FRET of ≥ 2.7 s−1 (the bimolecular rate for binding of mdADP is 3 μM−1s−1 as determined below and the free mdADP concentration is always ≥ 0.9 μM). This results in an immediate increase in FRET as EA•mdADP is formed during the mixing time. Curve b is for the reverse order of addition with apo-K346 added at 10 seconds and mdADP added at 20 seconds (a 10 second delay). In this case, the initial EA has partially converted to ER during the 10 second delay and the fluorescence increase on adding mdADP is smaller because less EA is present when mdADP is added after a delay. Longer delays of 20, 100, 300 and 600 seconds for curves c-e respectively give progressively lower amounts of initial fluorescence increase on adding mdADP. The horizontal line is the amount of fluorescence expected for combination of the fluorescence of kinesin and mdADP separately in the absence of binding and FRET. The limiting value for the initial fluorescence at long delays approaches this value, indicating little increased FRET in the burst phase and thus almost complete conversion of EA to ER during the longer delays in the absence of glycerol. The loss in initial FRET signal obeys first order kinetics and represents a rate for conversion of EA to ER of 0.055 s−1 for Fig. 4C and 0.052 ±0.005 SD including a second independent determination.
Fig. 4. Time course of inactivation and reactivation in absence of glycerol.
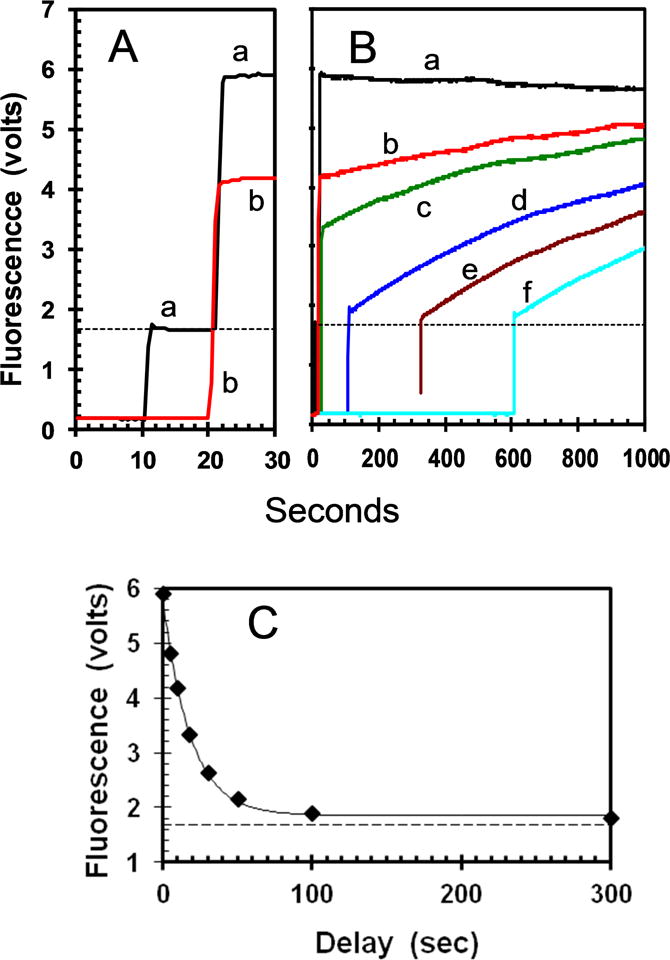
A. Curve a is for sequential addition of mdADP to 1.8 μM and EA to 0.875 μM at 10 and 20 seconds respectively. The order of addition was reversed for curve b. The horizontal line indicates the level expected in the absence of FRET calculated as sum of the values for mdADP and EA separately. B. Full time course with additional delays between addition of EA and mdADP of 10, 100, 310 and 600 seconds for the curves c-f respectively. C. Plot of the initial FRET amplitude versus the delay between addition of EA and mdADP with a first order rate constant of 0.055 s−1. Panel C contains data for additional delays not included in panel B for clarity.
The regain in fluorescence following addition of mdADP after a delay is due to slow conversion of ER back to EA. The EA is then trapped by binding to the excess mdADP before significant conversion back to ER can occur. The kinetics are too slow to follow to completion, but modeling (Fig. S1) indicates a value of ~0.0009 s−1 for reactivation is most consistent with the data. This data predicts an equilibrium constant KR for ER/EA of 0.052 s−1/0.0009 s−1 = 58 and ~2% as EA at equilibrium. The similar values for the rates of reactivation with long delays in Fig. 4 indicate that ER is stable over this time range.
Kinetics of mdADP binding and reversible inactivation by stopped flow FRET in 5–20% glycerol
The experimental design is analogous to that used previously to show stabilization of apo-DKH405 by tail domains [22]. The inactivation reaction was initiated by dilution of active apo-K346 from stabilization buffer into buffer with 5–20% glycerol and then rapidly loaded into one syringe of the stopped flow. The other syringe contained excess mdADP in the same concentration of glycerol. Mixing produces a fast transient as mdADP binds to apo-EA with increase in FRET (Fig. 5A). With increasing delay between dilution of apo-K346 and mixing with mdADP, the amplitudes of the transients decrease as EA converts to ER with reduced extent of rapid binding of mdADP. However, the rate constants do not change (Fig. 5C) because only EA can bind mdADP in the burst phase. The sample mixed at 1800 seconds in 20% glycerol was observed over a longer period (Fig. 5B) and the fluorescence recovers slowly as ER converts to EA and is trapped by mdADP at a rate of ~0.0006 s−1. With 20% glycerol, the amplitudes reach a plateau value of 19% of the extrapolated value immediately after dilution (Fig. 5C). The plateau value of 19% as EA at equilibrium corresponds to an equilibrium constant KR of 4.3 for conversion of EA to ER. The observed rate constant kobs for the approach to equilibrium is ~0.0031 s−1. For a reversible reaction KR = kAR/kRA and kobs = kAR + kRA and thus kAR = 0.0025 s−1 and kRA = 0.00059 s−1. A kRA value of this magnitude is consistent with the value of ~0.0006 s−1 observed above (Fig. 5B). Thus glycerol reduces the rate of inactivation much more strongly (0.052 s−1 versus 0.0025 s−1 for 0 versus 20% glycerol respectively or over 20-fold) than it reduces the rate of reactivation (0.009 s−1 versus 0.006 s−1 or 1.5-fold). Glycerol concentrations of 10 and 5% have progressively faster rates of inactivation and less EA at equilibrium (Fig. 5C).
Fig. 5. Time course of inactivation and reactivation in 5–20 % glycerol.
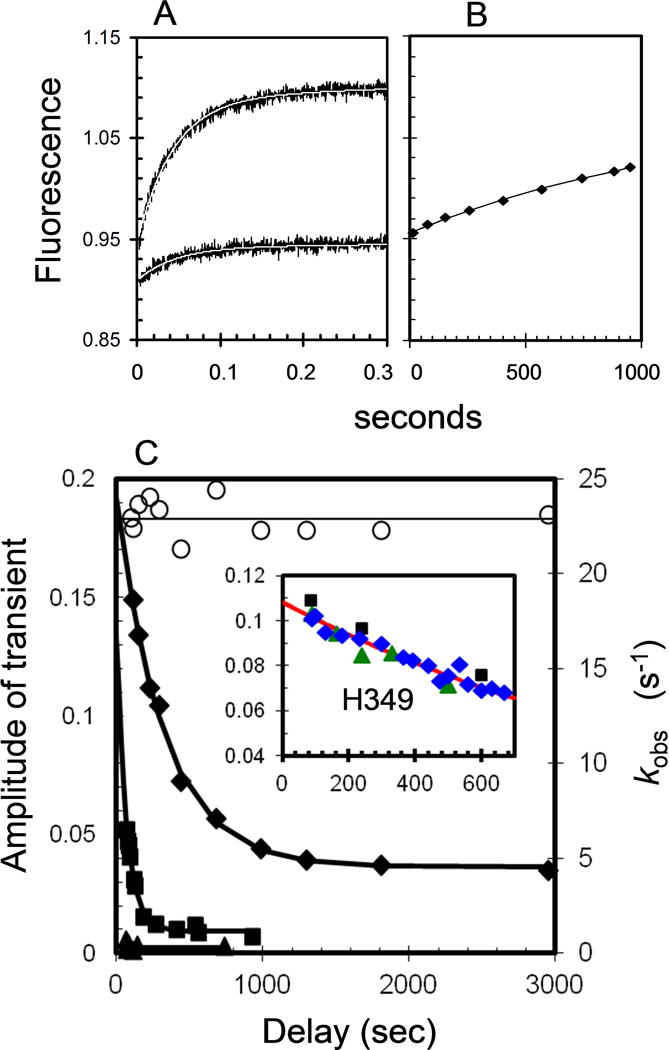
Apo-K346 in stabilization buffer was diluted with 5, 10 or 20 % glycerol and immediately loaded into the stopped flow syringe and mixed with mdADP in the same concentration of glycerol. Final concentrations after mixing were 0.5 μM apo-K346 and 10 μM mdADP. (A) Representative traces in 20% glycerol after a delay of 95 (upper) and 2950 (lower) seconds before mixing apo-K346 with mdADP. Thin white lines are fits to a single exponential function with rates (kobs) and amplitudes included in Fig. 5C. (B) Increasing fluorescence due to recovery of mdADP binding when mixed with mdADP after a delay of 1800 seconds. The line is a fit to a single exponential function with a rate constant of 0.0006 s−1 for recovery of the starting amplitude. (C) Left axis, filled symbols: Decay of amplitude of the transients on adding mdADP (after a delay) in 5% (triangles); 10% (squares); or 20% glycerol (diamonds). Right axis, open circles: the rates for the transients (kobs) in 20% glycerol. The fitted line for 20 % glycerol is described in the text. Insert is a plot of the amplitude of the transients versus time for an experiment performed similarly with apo-H349 in the absence of glycerol (final concentrations of 0.24 and 2.5 μM for H349 and mdADP respectively). Plot includes results of 3 independent experiments indicated by the different symbols. The line is the fit to a single exponential function (At = Ao e−kt with At and Ao the amplitudes of the transients at time t and zero) with rate constant of 0.00072 s−1 for initial conversion of EA to ER.
Influence of glycerol on kinetics of nucleotide binding and release
During the inactivation of apo-K346 in 20% glycerol the bimolecular rate of mdADP binding (kon = kobs/[mdADP]) remains constant at 2.39 ±0.17 μM−1s−1 (Fig. 5C for kobs at 10 μM mdADP). The rate of mdADP binding is also independent of the delay time in 10 and 5 % glycerol with values of 2.57 ±0.24 and 2.75 ±0.07 μM−1s−1 respectively (not shown). Inactivation is too fast to allow measurement of mdADP binding in the absence of glycerol, but a value of ~3 μM−1s−1 can be estimated by extrapolation. The rate of release of ADP from E•ADP can be determined from the exponential rise in FRET on addition of excess mdADP to E•ADP to chase off the ADP. Conversely, the rate of release of mdADP can be determined from the decrease in FRET on addition of excess ADP to E•mdADP to chase off the mdADP. With K346 the rate of dissociation of ADP from E•ADP and of mdADP from E•mdADP in the absence of glycerol are similar at 0.0043 ±0.0005 s−1 and 0.0060 ±0.0004 s−1 respectively (Fig. S2), indicating that ADP and mdADP have similar kinetics. In contrast to its weak influence on the rate of mdADP binding, glycerol strongly reduces the rate of nucleotide release with the rate of release of mdADP from E•mdADP decreasing from 0.0060 ±0.0004 s−1 in the absence of glycerol to 0.00104 ±0.00003 s−1 in 20% glycerol (Fig. S2).
ADP reactivates ER by conformational selection and not induced fit
The two limiting models for net binding of mdADP to ER are induced fit, in which mdADP initially binds reversibly to ER and then ER •mdADP slowly converts to EA•mdADP (dashed lines in Scheme), or conformational selection in which ER first converts to EA which can then bind mdADP (solid lines in Scheme). At concentrations of mdADP that saturate the rate of net conversion of ER to EA•mdADP, the predominant species following addition of mdADP would be ER for a conformational selection model because there would not yet be any EA to bind mdADP and ER does not bind mdADP. In this case the rate limiting step would be conversion of ER to EA which is then trapped by mdADP binding. Conversely, the major species for an induced fit model at saturating mdADP would be ER•mdADP with the rate limiting step being conversion of ER•mdADP to EA•mdADP. As indicated in Fig. 6, the rate of reactivation of ER is independent of the concentration of mdADP at ≥1 μM and consistent with the rates estimated in Fig. 4. Although mdADP may be held more tightly in the EA•ADP complex than in ER•ADP, there is not likely to be large changes in the distances between the mant acceptor and the tyrosine donors and both EA•mdADP and ER•mdADP should be high FRET states. Thus at concentrations of mdADP that saturate the rate of reactivation of ER to EA, induced fit predicts a large initial increase in fluorescence following addition of mdADP due to accumulation of ER•mdADP, whereas conformational selection predicts no initial increase in fluorescence but rather just a slow rise as ER slowly converts to EA and only then is able to bind mdADP. The observed initial increase in fluorescence on adding 0.9 μM mdADP (thick versus thin lines of curve a) is ~3% of the total fluorescence increase expected for complete binding of mdADP. A significant part of even this small increase is due to rapid mdADP binding to the ~2% EA present at equilibrium and to the fluorescence of kinesin itself. There are additional small increases in initial fluorescence at higher mdADP in Fig. 6, but this could be due to non-specific binding or to a weak specific interaction with ER that does not accelerate the rate of reactivation. The lack of a significant increase in FRET on initial mixing of ER with saturating mdADP indicates that ER•mdADP cannot be the predominant species. Thus ER without bound mdADP is the predominant intermediate and reactivation occurs by conformational selection. Results with Ncd are also consistent with activation by conformational selection [12]. The need for conversion of ER to EA by conformational selection before mdADP can bind, however, does not preclude an induced fit mechanism for binding of mdADP to EA once it is formed.
Fig. 6. Dependence of reactivation rate on concentration of mdADP.
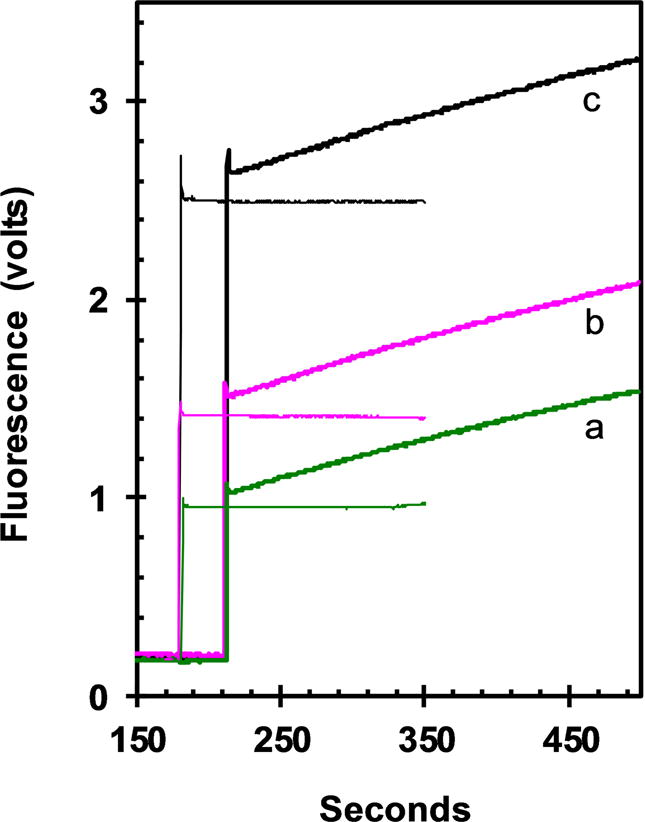
Performed similarly to Fig. 4 with 0.5 μM apo-K346 added at 10 seconds to buffer alone, followed by a delay of 200 seconds for conversion of EA to ER and finally addition of mdADP at 210 seconds to 0.9, 1.35 and 2.7 μM respectively for curves a-c (thick lines). The thin lines are for addition of the corresponding amount of mdADP, but without addition of apo-K346 as a control for the fluorescence due to mdADP alone in the absence of binding to apo-K346.
Affinity of ER for MTs
Active kinesin motor domains bind tightly to MTs in the absence of nucleotide whereas E•ADP binds very weakly. Quantitative analysis of the binding of ER however is complicated by the conversion free ER to EA and trapping of EA by MTs at a rate comparable to the time needed for binding assays such as ultracentrifugation to remove MTs and bound heads. To avoid this complication, the turbidity increase due to head binding to MTs was determined immediately after addition of ER to MTs (before significant conversion to EA could occur). As indicated in Fig. 7, EA binds tightly and stoichiometrically, whereas E•ADP binds very weakly as expected. Binding of ER is intermediate with a Kd of 1.8 μM.
Fig. 7. Binding of K346 to MTs.
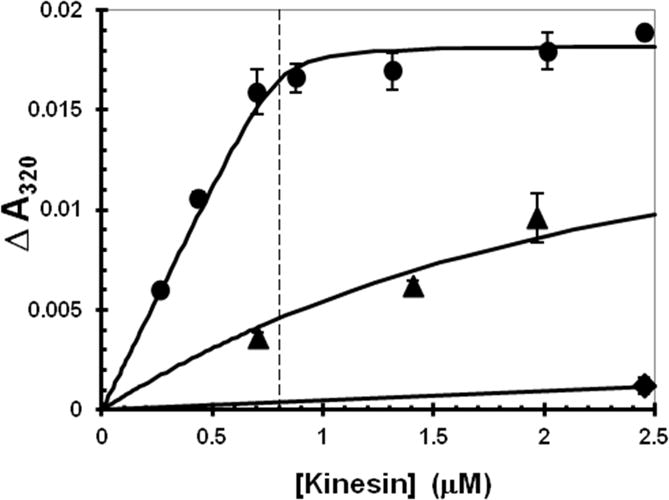
K346 was added to a gently stirred suspension of MTs and the turbidity increase was determined by the difference in absorbance at 320 nm between before and immediately after addition of K346. A small correction was applied for the turbidity of K346 in the absence of MTs. The MT concentration was 0.8 μM (indicated by the vertical line) after correction for depolymerized tubulin that remained in the supernatant after ultracentrifugation to remove MTs. Apo-K346 as EA in stabilization buffer into buffer with 1 mM ADP (diamonds) or without ADP (circles), apo-K346 as ER without glycerol into buffer without ADP (triangles). The fitted curves were calculated using a mutual depletion scheme and assuming that the MT complex of ER has the same maximum turbidity value as observed for EA.
Human kinesin has a slower rate of inactivation
When the experimental protocol of Fig. 5 was performed with human apo-H349 instead of Drosophila apo-K346, the initial rate for loss of EA in the absence of glycerol was ~ 0.0007 s−1 (Fig. 5C insert). Thus inactivation of apo-H349 is over 70-fold slower than for apo-K346.
Circular Dichroism (CD)
Far-UV CD can provide information about the folding of kinesin and how it changes between EA and ER. Dilution of apo-K357 from stabilization buffer into buffer without glycerol, but with 20 μM ADP, results in rapid trapping of apo-K357 as EA•ADP. The CD spectrum under this condition shows a broad minimum between 210–222 nm (Fig. 8A). When ADP is omitted from the buffer, rapid conversion to apo-ER occurs before the spectrum is obtained and a decrease in negative amplitude at 222 nm and an increase at 208 nm is observed. However, when apo-K357 is diluted into buffer with 50% glycerol and 800 mM KCl, the spectra with and without ADP (Fig. 8B) are more similar to each other and also similar to the spectrum for EA•ADP in the absence of glycerol. The small observed shift between apo-K357 and K357•ADP in Fig. 8B is consistent with a low level of ER in equilibrium with EA in the absence of ADP even in 50% glycerol.
Fig. 8. Circular dichroism of K357.
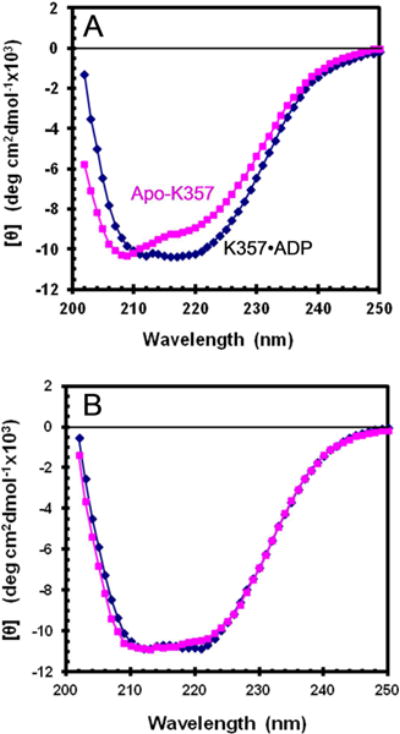
Spectra with 20 μM ADP (diamonds) or without ADP (squares) in 20 mM Mops/NaOH pH 7.2 with 2 mM MgCl2. (A) apo-K357 in the absence of glycerol with 25 mM KCl and (B) K357 in 50% glycerol and 800 mM KCl
ANS binding
The fluorescent dye ANS is often used to detect the presence of molten globules [23]. ANS has low fluorescence in water and in the presence of compact folded proteins that do not allow the dye access to their hydrophobic interior, but becomes highly fluorescent when bound to molten globules that largely retain secondary structure but have a loose ternary conformation that allow access of the dye to the hydrophobic interior[24,25]. Conversion of a molten globule to a fully unfolded protein, however, reduces the fluorescence of ANS, likely because hydrophobic clusters are no longer present. When the EA•ADP complex is formed by dilution of EA from stabilization buffer into buffer with excess ADP, the fluorescence of ANS is not greatly increased over that in the absence of K346 (Fig. 9A). However6, when ADP is omitted and the starting EA is allowed to convert to ER before addition of ANS, the fluorescence is greatly enhanced. This indicates that EA•ADP is tightly folded and cannot bind ANS extensively, whereas ER has molten globule character.
Fig. 9. ANS fluorescence.
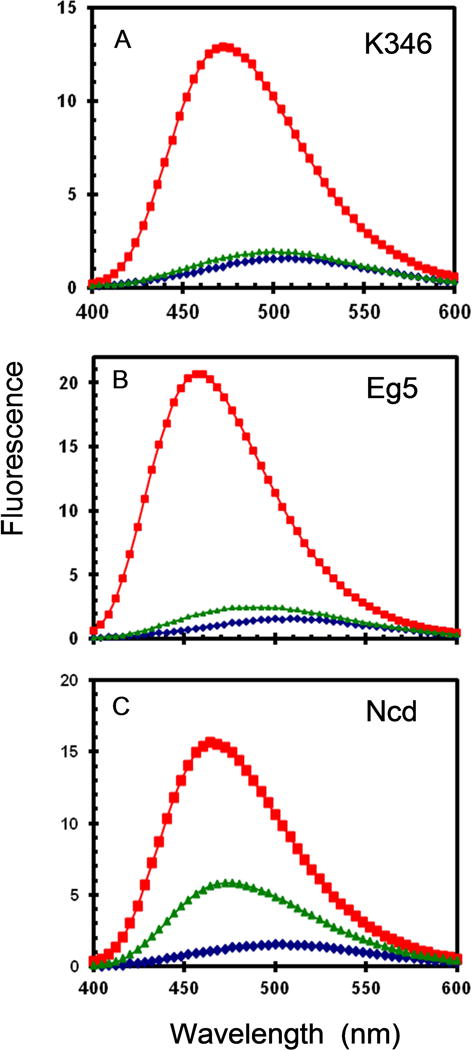
Fluorescence emission spectra for 20 μM ANS and 2 μM apoprotein in buffer without glycerol. (A) K346; (B) Eg5; (C) Ncd. ANS alone (diamonds); ANS with protein and 1 mM ADP (trianges); ANS with protein in absence of ADP (squares) are bottom to top lines respectively.
Eg5 and Ncd are examples of kinesin-5 and kinesin-14 families respectively that also form nucleotide-free motor domains conformations that are refractory to binding of ADP, but can regain activity slowly after addition of nucleotide [10–13]. When tested for ANS binding, nucleotide-free forms of both Eg5 and Ncd exhibit high fluorescence in the absence of ADP (Fig. 9B,C), consistent with a molten globule ER conformation. Ncd motor domains are known to be highly unstable and recovery of EA•ADP is only partial in Fig. 9C on addition of ADP to ER.
Discussion
The work presented here establishes that the refractory ER form of kinesin-1 is a stable species in equilibrium with the active EA form. The equilibrium between the active and refractory states is strongly dependent on the concentration of stabilizing agents such as glycerol. For Drosophila apo-K346 in the absence of glycerol, the equilibrium favors the inactive ER form with an equilibrium constant KR for conversion of EA to ER of ~58. Just 20% glycerol is sufficient to shift this equilibrium 13-fold to ~4.4. In 50% glycerol KR further decreases to < 0.1 with EA the predominant component as indicated by i) the close to stoichiometric rebinding of ADP by apo-K346 (Fig. 2); ii) the similar MT-ATPase rates of apo-K346 diluted from stabilization buffer and control E•ADP (Fig. 3); and iii) the similar CD spectra in stabilization buffer with and without ADP (Fig. 8B).
The large shift in CD spectra between EA and ER indicates that there is a major change in conformation on reversible conversion of EA to ER. The large enhancement of ANS fluorescence observed here for ER versus EA also indicates a large change in conformation from the ordered EA to a significantly disordered ER with characteristics of a molten globule. Molten globular character in the absence of bound nucleotide may be a general feature of the kinesin superfamily as members of two divergent families (kinesin-5 and kinesin-14) also bind ANS (Fig. 9). This is consistent with previous results that apo-Eg5 binds Sypro Orange [26]. Further work will be needed to determine whether this is the result of a global shift to a molten globule conformation or if ER retains a core of intact ternary structure. It is also likely that both the ER and EA states are composed of multiple subconformations whose properties and kinetics of interconversion remain to be determined.
The CD spectrum for Drosophila K357 reported here is in good agreement with that reported by Shimizu and Morii [27] for the E•ADP complex of the equivalent human homolog. However they also reported the apparently contradictory results that the CD spectrum of apo-H349, even in the absence of glycerol, was that expected for the EA conformation and that the CD spectra for Ncd was that expected for ER both with and without ADP. The result with H349 is likely a consequence of the increased stability of the human EA conformation with limited conversion to ER, especially as the spectrum of apo-H349 was obtained in high salt that could further stabilize the EA conformation. The spectrum of Ncd without ADP is similar to that for ER and indicates a molten globule conformation in agreement with the high ANS binding. The Ncd spectrum with ADP may be biased towards the ER conformation by the high instability of Ncd as indicated by significant ANS fluorescence even after addition of ADP (Fig. 9C) and by a low ADP concentration of 20 μM.
Glycerol decreases the rate of conversion of EA to ER while having little effect on the rate of ER to EA (Table I). Thus the interaction of glycerol with both ER and the transition state (TS) are similar (ΔΔG = 1.1 kJ/mole) whereas the difference in interaction of glycerol between EA and TS is large (ΔΔG = 7.5 kJ/mole) as illustrated in Fig. 10. A reference ΔG=0 for either EA or ER in cases (A) and (B) respectively is arbitrary because the absolute ΔG for interaction of glycerol EA or ER is not known. Fixing the absolute free energy at some level other than zero for EA or ER however will not change the ΔΔG values (shifting the red or green curve vertically relative to the other due to an interaction with glycerol that is the same for EA, TS and ER will not change the ΔΔG values). The similar interaction energy of glycerol with the TS and ER, but not with EA suggests that the TS may lie closer structurally to the molten globule like ER than to the EA conformation. A TS with significant disorder is also supported by the lack of ADP binding by ER during conversion of ER → EA•ADP by conformational selection at saturating [ADP] (Fig. 7). This is consistent with a tight nucleotide binding site not being formed until after the transition state, although other models such as steric blocking of ADP binding in ER are also possible.
Table I.
Kinetic Summaryc
| Glycerol | kAR | kRA | KR |
koff (ADP) |
koff (mdADP) |
kon (mdADP) |
KdA (mdADP) |
|---|---|---|---|---|---|---|---|
| (% v/v) | (s−1) | (s−1) | (s−1) | (s−1) | (μM−1s−1) | (μM) | |
| Drosophila | |||||||
| 0 | 0.052±0.005 | ~0.0009 | ~58 a | 0.0043±0.0005 | 0.0060±0.0004 | ~3 | 0.002 b |
| 5 | 2.75 ±0.07 | ||||||
| 10 | 2.57 ±0.24 | ||||||
| 20 | 0.0025 | ~0.0006 | ~4.3 a | 0.00104±0.00003 | 2.39 ±0.17 | 0.0004 b | |
| 50 | <0.1 | ||||||
| Human | |||||||
| 0 | ~0.0007 |
Calculated using KR = kAR/kAR
Calculated using KdA = koff/kon for mdADP
Missing values were not determined
Fig. 10. Diagram for free energy versus reaction coordinate for EA, TS and ER in 0 and 20% glycerol.
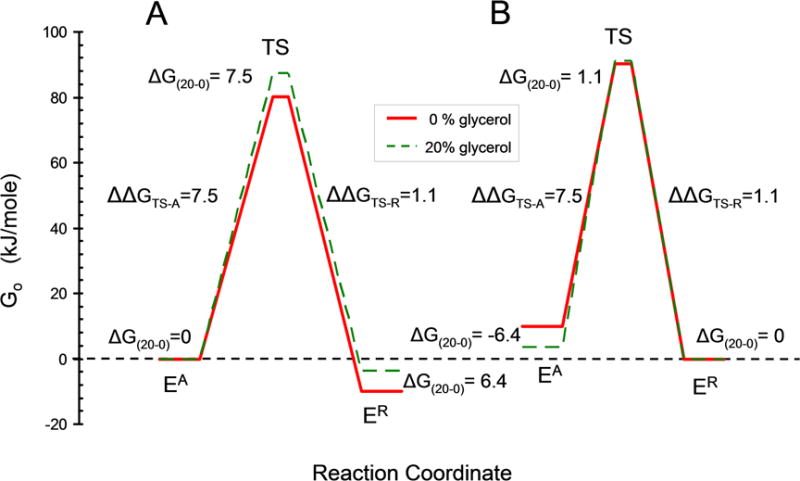
A. Preferential destabilization of ER by glycerol. The free energies for EA in 20% and 0% glycerol (dashed and solid lines respectively) were first fixed at the same zero value with ΔG(20–0) = 0 for the difference in free energy between EA in 20 and 0 % glycerol. Preferential destabilization of ER by glycerol was then modeled by lowering the free energy of ER by the amount required by the KR values in Table I using ΔGAR(0%) = −RT ln(KR(0%)) = −10.0 kJ/mole and ΔGAR(20%) = −RT ln(KR(20%)) = −3.6 kJ/mole. The free energy of the transition states were calculated from the velocity values in Table I using velocity = (kBT/h)e−ΔG╪/RT where kB is the Boltzmann constant and h is the Planck constant. B. Preferential stabilization of EA by glycerol. Calculated similarly to (A) but starting with the free energy of ER in 0 and 20 % glycerol fixed at zero and glycerol preferentially stabilizing EA.
A widely accepted model for the ability of osmolytes such as glycerol to stabilize proteins is that they selectively decrease the solvation of the exposed peptide backbone in the unfolded conformation compared to the folded conformation in which much of the backbone is not exposed to solvent [28]. Penetration of water and osmolyte into the interior of a molten globule could also result in selective destabilization of the molten globule backbone and explain the smaller ΔΔG value for ER and TS between 0 and 20 % glycerol if they both had similar significant disorder, whereas the ΔΔG between a compact EA and disordered TS would be larger.
Inactivation of EA by conversion to ER is not likely to be a factor in vivo. After ADP release, nucleotide-free apo-EA will rapidly bind ATP in vivo because the ATP binding rate is fast and the ATP concentration in cells is high. However, in vitro experiments with purified proteins are often performed at much lower concentrations of ATP or ADP where kinesin can convert into the inactive refractory conformation, particularly with superfamily members such as Eg5 and Ncd that much more readily convert to the refractory form. Characterization of this refractory transition will allow for the design of experiments that minimize its accumulation and allow analysis of the influence of the refractory state on interpretation of previous work. One example is that the molten globule refractory ER conformation is likely a factor in the difficulty in crystallization of nucleotide-free kinesin. Under many crystallization conditions, nucleotide-free Drosophila kinesin would be predominately in the molten globule ER conformation. Even the more stable human kinesin could accumulate significant amounts of the ER conformation over the time scale required for crystal growth. Screening of crystallization conditions for formation of a dominant EA species at equilibrium could facilitate crystallization in the absence of nucleotide or microtubules.
The slower rate of inactivation of human H349 compared to Drosophila K346 also helps to explain an apparent contradiction in previous work. Nucleotide-free kinesin was first prepared by chromatography in excess EDTA of native full length kinesin that had been isolated from bovine brain. Full length bovine kinesin remained in a state competent for rapid ATP binding for an extended period even in the absence of glycerol, as indicated by the approximately stoichiometric burst of Pi produced by hydrolysis after addition of ATP [4]. This contrasted with motor domains of Drosophila kinesin that rapidly inactivated without ADP and did not show a burst of ADP binding [7]. Bovine kinesin motor domains are much more homologous to the human than Drosophila motor domains and are likely to be similarly stabilized against inactivation. An additional factor in stabilization of even Drosophila full length kinesin is the binding of C-terminal tail domains to the motor domains to produce the autoinhibited conformation [29]. Binding of tail domains to apo-kinesin motor domains does in fact strongly inhibit the refractory transition from EA to ER [6]. The key feature of the tail-motor domain complex is that it forces the neck linker [30] to remain docked against the motor domain [31] and this indicates that neck linker docking, even in the absence of bound ADP, can also significantly stabilize EA.
Conclusions and Perspectives
Kinesin motor domains in the absence of nucleotide convert reversibly between an active state and an inactive state that has characteristics of a molten globule. This molten globule state is an excellent candidate for more extensive structural studies because it is i) stable; ii) the predominant species under physiological conditions and iii) readily formed without the use of chaotropic agents or extremes of pH. Furthermore, the kinetics and mechanism of the interconversion between native and molten globule states are experimentally accessible because the equilibrium between the native and molten globule states can be shifted by nucleotides and osmolytes such as glycerol.
Supplementary Material
Fig. S1. Simulation of time course for reactivation of K346 by mdADP. Plot is the data of Fig. 4 with the results of a simulation using the model in the Scheme overlaid as thin lines. Simulation was calculated by numerical integration in Excel assuming that the starting apo-K346 was 95% EA and that the fluorescence increase when mdADP was added first was due to all of the 95% EA binding mdADP. The starting amounts of EA and ER after a delay were calculated for a decay rate of 0.055 s−1. The kAR value during reactivation was fixed at 0.055 s−1 and the kRA values were 0.0014, 0.0009 and 0.0006 s−1 for panels A-C respectively.
Fig. S2. ADP binding and release kinetics. (A) ADP chase out in absence of glycerol. Reaction initiated by adding K345•ADP to buffer with excess mdADP. Final concentrations were 0.63 and 13 μM for K346 and mdADP respectively. (B) mdADP chase out in absence of glycerol. Reaction initiated by adding K345•mdADP to buffer with excess ATP. Final were concentrations 0.84 and 400 μM for K345•mdADP and ATP respectively. (C) mdADP chase out in 20% glycerol. As in B but in 20% glycerol and shutter was only opened intermittently. Thin magenta line is fit to a single exponential with rate constants of 0.00432, 0.00591 and 0.00102 s−1 for A-C respectively. A and B are average of three traces. The similar values reported in Table I are averages of traces fit individually.
Highlights.
-
*
Apo-kinesin reversibly converts to an inactive conformation.
-
*
Inactive conformation has characteristics of a molten globule.
-
*
Glycerol stabilizes active form by selectively reducing the rate of inactivation.
-
*
Reactivation in the presence of ADP occurs by conformational selection.
Acknowledgments
Supported by NIH grant NS058848. The technical assistance of Calvin Lin, James Hopkins and Benjamin Bryant in cloning, expression and purification of proteins is gratefully acknowledged.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Endow SA, Kull FJ, Liu H. Kinesins at a glance. J Cell Sci. 2010;123:3420–3424. doi: 10.1242/jcs.064113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hirokawa N, Noda Y, Tanaka Y, Niwa S. Kinesin superfamily motor proteins and intracellular transport. Nat Rev Mol Cell Biol. 2009;10:682–696. doi: 10.1038/nrm2774. [DOI] [PubMed] [Google Scholar]
- 3.Hackney DD. Kinesin ATPase: rate-limiting ADP release. Proc Natl Acad Sci U S A. 1988;85:6314–6318. doi: 10.1073/pnas.85.17.6314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hackney DD, Malik AS, Wright KW. Nucleotide-free kinesin hydrolyzes ATP with burst kinetics. J Biol Chem. 1989;264:15943–15948. [PubMed] [Google Scholar]
- 5.Cheng JQ, Jiang W, Hackney DD. Interaction of mant-adenosine nucleotides and magnesium with kinesin. Biochemistry. 1998;37:5288–5295. doi: 10.1021/bi972742j. [DOI] [PubMed] [Google Scholar]
- 6.Hackney DD, Stock MF. Kinesin tail domains and Mg2+ directly inhibit release of ADP from head domains in the absence of microtubules. Biochemistry. 2008;47:7770–7778. doi: 10.1021/bi8006687. [DOI] [PubMed] [Google Scholar]
- 7.Huang TG, Hackney DD. Drosophila kinesin minimal motor domain expressed in Escherichia coli. Purification and kinetic characterization. J Biol Chem. 1994;269:16493–16501. [PubMed] [Google Scholar]
- 8.Ma YZ, Taylor EW. Kinetic mechanism of kinesin motor domain. Biochemistry. 1995;34:13233–13241. doi: 10.1021/bi00040a039. [DOI] [PubMed] [Google Scholar]
- 9.Ma YZ, Taylor EW. Kinetic Mechanism of a Monomeric Kinesin Construct. J Biol Chem. 1997;272:717–723. doi: 10.1074/jbc.272.2.717. [DOI] [PubMed] [Google Scholar]
- 10.Crevel IMTC, Lockhart A, Cross RA. Weak and strong states of kinesin and ncd. J Mol Biol. 1996;257:66–76. doi: 10.1006/jmbi.1996.0147. [DOI] [PubMed] [Google Scholar]
- 11.Lockhart A, Cross RA. Kinetics and motility of the Eg5 microtubule motor. Biochemistry. 1996;35:2365–2373. doi: 10.1021/bi952318n. [DOI] [PubMed] [Google Scholar]
- 12.Shimizu T, Morii H. Comparison of ncd and kinesin motor domains by circular dichroism spectroscopy. J Biochem (Tokyo) 1996;120:1176–1181. doi: 10.1093/oxfordjournals.jbchem.a021538. [DOI] [PubMed] [Google Scholar]
- 13.Shimizu T, Sablin E, Vale RD, Fletterick R, Pechatnikova E, Taylor EW. Expression, purification, ATPase properties, and microtubule-binding properties of the ncd motor domain. Biochemistry. 1995;34:13259–13266. doi: 10.1021/bi00040a042. [DOI] [PubMed] [Google Scholar]
- 14.Stock MF, Hackney DD. Methods Mol Biol – Kinesin Protoc. Humana Press; Totowa: 2001. Expression of kinesin in E. coli; pp. 43–48. [DOI] [PubMed] [Google Scholar]
- 15.Tropea JE, Cherry S, Waugh DS. Expression and purification of soluble His(6)-tagged TEV protease. Methods Mol Biol Clifton NJ. 2009;498:297–307. doi: 10.1007/978-1-59745-196-3_19. [DOI] [PubMed] [Google Scholar]
- 16.Jiang W, Stock MF, Li X, Hackney DD. Influence of the kinesin neck domain on dimerization and ATPase kinetics. J Biol Chem. 1997;272:7626–7632. doi: 10.1074/jbc.272.12.7626. [DOI] [PubMed] [Google Scholar]
- 17.DeBonis S, Simorre JP, Crevel I, Lebeau L, Skoufias DA, Blangy A, Ebel C, Gans P, Cross R, Hackney DD, Wade RH, Kozielski F. Interaction of the mitotic inhibitor monastrol with human kinesin Eg5. Biochemistry. 2003;42:338–349. doi: 10.1021/bi026716j. [DOI] [PubMed] [Google Scholar]
- 18.Gill SC, von Hippel PH. Calculation of protein extinction coefficients from amino acid sequence data. Anal Biochem. 1989;182:319–326. doi: 10.1016/0003-2697(89)90602-7. [DOI] [PubMed] [Google Scholar]
- 19.Hackney DD. Pathway of ADP-stimulated ADP release and dissociation of tethered kinesin from microtubules. Implications for the extent of processivity. Biochemistry. 2002;41:4437–4446. doi: 10.1021/bi0159229. [DOI] [PubMed] [Google Scholar]
- 20.Hiratsuka T. New ribose-modified fluorescent analogs of adenine and guanine nucleotides available as substrates for various enzymes. Biochim Biophys Acta. 1983;742:496–508. doi: 10.1016/0167-4838(83)90267-4. [DOI] [PubMed] [Google Scholar]
- 21.Hackney DD, Jiang W. Assays for kinesin microtubule-stimulated ATPase activity. Methods Mol Biol Clifton NJ. 2001;164:65–71. [PubMed] [Google Scholar]
- 22.Hackney DD, Stock MF. Kinesin tail domains and Mg2+ directly inhibit release of ADP from head domains in the absence of microtubules. Biochemistry. 2008;47:7770–7778. doi: 10.1021/bi8006687. [DOI] [PubMed] [Google Scholar]
- 23.Semisotnov GV, Rodionova NA, Razgulyaev OI, Uversky VN, Gripas’ AF, Gilmanshin RI. Study of the “molten globule” intermediate state in protein folding by a hydrophobic fluorescent probe. Biopolymers. 1991;31:119–128. doi: 10.1002/bip.360310111. [DOI] [PubMed] [Google Scholar]
- 24.Baldwin RL, Rose GD. Molten globules, entropy-driven conformational change and protein folding. Curr Opin Struct Biol. 2013;23:4–10. doi: 10.1016/j.sbi.2012.11.004. [DOI] [PubMed] [Google Scholar]
- 25.Naiyer A, Hassan MI, Islam A, Sundd M, Ahmad F. Structural characterization of MG and pre-MG states of proteins by MD simulations, NMR, and other techniques. J Biomol Struct Dyn. 2015;33:2267–2284. doi: 10.1080/07391102.2014.999354. [DOI] [PubMed] [Google Scholar]
- 26.Yokoyama H, Sawada J, Katoh S, Matsuno K, Ogo N, Ishikawa Y, Hashimoto H, Fujii S, Asai A. Structural basis of new allosteric inhibition in Kinesin spindle protein Eg5. ACS Chem Biol. 2015;10:1128–1136. doi: 10.1021/cb500939x. [DOI] [PubMed] [Google Scholar]
- 27.Shimizu T, Morii H. Comparison of ncd and kinesin motor domains by circular dichroism spectroscopy. J Biochem (Tokyo) 1996;120:1176–1181. doi: 10.1093/oxfordjournals.jbchem.a021538. [DOI] [PubMed] [Google Scholar]
- 28.Bolen DW, Rose GD. Structure and energetics of the hydrogen-bonded backbone in protein folding. Annu Rev Biochem. 2008;77:339–362. doi: 10.1146/annurev.biochem.77.061306.131357. [DOI] [PubMed] [Google Scholar]
- 29.Stock MF, Guerrero J, Cobb B, Eggers CT, Huang TG, Li X, Hackney DD. Formation of the compact confomer of kinesin requires a COOH-terminal heavy chain domain and inhibits microtubule-stimulated ATPase activity. J Biol Chem. 1999;274:14617–14623. doi: 10.1074/jbc.274.21.14617. [DOI] [PubMed] [Google Scholar]
- 30.Rice S, Lin AW, Safer D, Hart CL, Naber N, Carragher BO, Cain SM, Pechatnikova E, Wilson-Kubalek EM, Whittaker M, Pate E, Cooke R, Taylor EW, Milligan RA, Vale RD. A structural change in the kinesin motor protein that drives motility. Nature. 1999;402:778–784. doi: 10.1038/45483. [DOI] [PubMed] [Google Scholar]
- 31.Kaan HYK, Hackney DD, Kozielski F. The structure of the kinesin-1 motor-tail complex reveals the mechanism of autoinhibition. Science. 2011;333:883–885. doi: 10.1126/science.1204824. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Simulation of time course for reactivation of K346 by mdADP. Plot is the data of Fig. 4 with the results of a simulation using the model in the Scheme overlaid as thin lines. Simulation was calculated by numerical integration in Excel assuming that the starting apo-K346 was 95% EA and that the fluorescence increase when mdADP was added first was due to all of the 95% EA binding mdADP. The starting amounts of EA and ER after a delay were calculated for a decay rate of 0.055 s−1. The kAR value during reactivation was fixed at 0.055 s−1 and the kRA values were 0.0014, 0.0009 and 0.0006 s−1 for panels A-C respectively.
Fig. S2. ADP binding and release kinetics. (A) ADP chase out in absence of glycerol. Reaction initiated by adding K345•ADP to buffer with excess mdADP. Final concentrations were 0.63 and 13 μM for K346 and mdADP respectively. (B) mdADP chase out in absence of glycerol. Reaction initiated by adding K345•mdADP to buffer with excess ATP. Final were concentrations 0.84 and 400 μM for K345•mdADP and ATP respectively. (C) mdADP chase out in 20% glycerol. As in B but in 20% glycerol and shutter was only opened intermittently. Thin magenta line is fit to a single exponential with rate constants of 0.00432, 0.00591 and 0.00102 s−1 for A-C respectively. A and B are average of three traces. The similar values reported in Table I are averages of traces fit individually.