

Abstract
Despite the fact that actinomycetes harbor the genetic potential to produce terpenes, terpenoid natural products tend to be a rare occurrence in fermentation broths. Here we report two new halimane-type diterpenoids, micromonohalimanes A (1) and B (2), that were isolated from a Micromonospora sp. cultivated from the marine ascidian Symplegma brakenhielmi. This is the first report of the halimane-type diterpenoids from Micromonospora. The structures were determined using spectroscopic methods including X-ray crystallography to establish the absolute configuration. Micromonohalimane B demonstrated moderate antibacterial activity against methicillin-resistant Staphylococcus aureus.
TOC Image
Traditional methods used for bacterial natural products drug discovery have become time- and labor-intensive, in part, due to high rediscovery rates. In order to overcome this historical weakness that has led to high rates of rediscovery, our group has used LCMS-based metabolomics.1–4
We and others have demonstrated that LCMS-based metabolomics provides a high-throughput platform to prioritize bacterial strains and drive discovery of novel natural products. Using LCMS–principal component analysis (PCA), we have identified strains with unique metabolite profiles, and in our experience those metabolic outliers tend to yield novel natural products. One of the strengths of PCA versus other statistical methods is that common background ions have little effect on the analysis. In other words, background subtraction, which can be challenging, is not necessary. Therefore, PCA provides an analysis option that is robust to small variations within the data sets and does not rely on fragmentation. The strain identified in this report had a unique metabolic profile as determined by PCA, but the molecules presented here were not the drivers of the statistical variance within the analyses. Overall, this is not surprising since many metabolites detected by mass spectrometry are below isolation thresholds. In our experience, unique molecular signatures often identify a bacterium as having potential for production of novel natural products.
Strain WMMC-218 was identified as having unique metabolite markers by PCA in an analysis of 29 actinomycetes (Supporting Information, Figure S1). Our current discovery workflow involves identification of unique bacteria by LCMS–PCA followed by extract production from three distinct media and subsequent screening for biological activity. WMMC-218 was prioritized based on broad-spectrum antibiotic activity. Although the most potent compound had antibacterial activity against methicillin-resistant Staphylococcus aureus (MRSA), we also discovered two new halimane-type diterpenoids. Unfortunately, the compound responsible for the potent antibacterial activity was not stable and decomposed.
WMMC-218 was cultivated from the marine ascidian Symplegma brakenhielmi (Shirley Parker-Nance, 2013). Micromonospora spp. are a well-known group of Gram-positive, spore-forming microbes, and many classes of compounds are known to be produced by Micromonospora.5 Several Micromonospora spp. have previously been cultivated from ascidians.6–8 However, this is the first report of halimane-type diterpenoids from Micromonospora. The most closely related diterpenoid was from a marine invertebrate gorgonian, Echinomuricea sp.9
Micromonospora sp. WMMC-218 was prioritized due to unique m/z signatures identified via LCMS-based metabolomics in conjunction with bioactivity (PCA in Supporting Information, Figure S1). Following fractionation and screening, some of the active fractions appeared to contain terpenoid compounds. Because terpenes tend to be rather rare in actinomycete extracts and activity was observed in the terpene-containing fractions, the terpenes were targeted for isolation and structure determination.
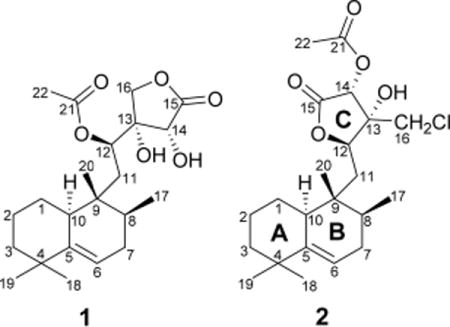
Micromonohalimane A (1) was obtained as colorless crystals and gave a molecular formula of C22H34O6 with six degrees of unsaturation, as determined by HR-ESIMS at m/z 417.2247 [M + Na]+. The IR spectrum showed absorptions typical of hydroxy (3395 cm−1) and ester carbonyl (1787 and 1708 cm−1) groups.
The 1H NMR spectrum of micromonohalimane A (1) (Table 1) revealed five methyls [δH 0.66 (3H, s), 0.83 (3H, d, J = 6.7 Hz), 0.95 (3H, s), 1.05 (3H, s), 2.13 (3H, s)], one olefinic proton [δH 5.40 (1H, m)], and three (of five) resolved methine protons [(δH 4.46 (1H, s), 5.31 (1H, d, J = 10.3 Hz), 2.04 (1H, m)], along with a resolved diastereotopic methylene [(δH 4.27 (1H, d, J = 10.1 Hz), 4.13 (1H, d, J = 10.1 Hz)]. The 13C NMR and HSQC spectra of compound 1 (Table 1) revealed the presence of two ester carbonyls (δC 175.3 and 170.9), two olefinic carbons (δC 145.5 and 115.8), three sp3 quaternary carbons (δC 36.0, 37.2, and 78.5), four sp3 methines (δC 33.4, 40.4, 71.8, and 71.8), six sp3 methylenes (δC 22.3, 27.5, 31.6, 34.6, 40.8, and 71.8), and five methyls (δC 15.5, 16.4, 21.2, 28.8, and 29.6) in the structure. The NMR spectroscopic features (Table 1) were characteristic of an acetoxy ester containing a halimane-type diterpene containing an acetoxy ester.10 COSY and HSQC analyses revealed three isolated spin systems: (a) C(10)H–C(1)H2–C(2)H2–C(3)H2, (b) C(6)H–C(7)H2–C(8)H–C(17)H3, and (c) C(11)H2–C(12)H. The HMBC correlations from H3-18/H3-19 to C-3/C-4/C-5, H3-18 to C-19, H-6 to C-4/C-5/C-10/C-7/C-8, H3-20 to C-8/C-9/C-10/C-11, and H3-17 to C-7/C-8/C-9, together with the spin systems (a) and (b) deduced above, led to the assignment of rings A and B.
Table 1.
1H NMR (600 MHz) and 13C NMR (125 MHz) Spectral Data of Compounds 1 and 2 in CDCl3
| 1
|
2
|
|||
|---|---|---|---|---|
| position | δC, type | δH (J in Hz) | δC, type | δH (J in Hz) |
| 1 | 27.5, CH2 | 1.85, m 1.03, m |
27.5, CH2 | 1.74, m 0.99, m |
| 2 | 22.3, CH2 | 1.62, m 1.45, m |
21.9, CH2 | 1.56, m 1.18, m |
| 3 | 40.8, CH2 | 1.40, m 1.21, m |
40.7, CH2 | 1.41, d (12.0) 1.19, m |
| 4 | 36.0, C | 36.1, C | ||
| 5 | 145.5, C | 146.2, C | ||
| 6 | 115.8, CH | 5.40, m | 115.5, CH | 5.45, d (2.2) |
| 7 | 31.6, CH2 | 1.93, m 1.77, m |
31.5, CH2 | 1.91, m 1.81, m |
| 8 | 33.4, CH | 1.58, m | 33.4, CH | 1.64, m |
| 9 | 37.2, C | 37.2, C | ||
| 10 | 40.4, CH | 2.04, m | 40.8, CH | 2.35, d (13.0) |
| 11 | 34.6, CH2 | 2.08, m 1.28, m |
35.1, CH2 | 2.06, dd (16.1, 1.1) 1.72, dd (16.1, 10.3) |
| 12 | 71.8, CH | 5.31, d (10.3) | 79.3, CH | 4.63, dd (10.3, 1.1) |
| 13 | 78.5, C | 77.6, C | ||
| 14 | 71.8, CH | 4.46, s | 73.6, CH | 5.60, s |
| 15 | 175.3, C | 168.6, C | ||
| 16 | 71.8, CH2 | 4.27, d (10.1) 4.13, d (10.1) |
46.6, CH2 | 3.75, d (11.9) 3.67, d (11.9) |
| 17 | 15.5, CH3 | 0.83, d (6.7) | 15.2, CH3 | 0.83, d (6.7) |
| 18 | 28.8, CH3 | 0.95, s | 29.1, CH3 | 1.03, s |
| 19 | 29.6, CH3 | 1.05, s | 29.7, CH3 | 1.07, s |
| 20 | 16.4, CH3 | 0.66, s | 16.3, CH3 | 0.70, s |
| 21 | 170.9, C | 170.0, C | ||
| 22 | 21.2, CH3 | 2.13, s | 20.5, CH3 | 2.24, s |
| –OH | 3.20, s | |||
The HMBC correlations from H-12 to C-9/C-11/C-14/C-16, from H-16 to C-14/C-15, and from H-11 to C-13, as well as the spin system (c) C(11)H2–C(12)H, allowed for the assignment of ring C. Additionally, the HMBC correlations from H-12 to C-21 suggested that the ester carbonyl group was connected to C-12.
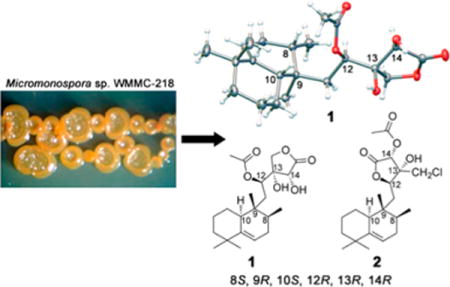
The relative and absolute configurations of compound 1 were determined by X-ray crystallographic analysis (Figure 1). The X-ray analysis allowed for assignment of the absolute configurations of the stereogenic carbons to be 8S, 9R, 10S, 12R, 13R, and 14R. Therefore, compound 1 was named (8S,9R,10S,12R,13R,14R)-micromonohalimane A.
Figure 1.
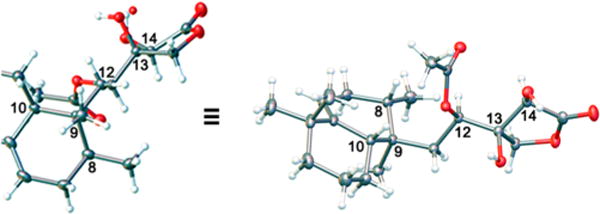
Molecular drawing of the X-ray crystal structure of 1.
Compound 2 had the molecular formula C22H33ClO5, as established by HR-ESIMS, with six degrees of unsaturation. A comparison of NMR (Table 1) and HR-ESIMS data with compound 1 suggested that the structures were similar in rings A and B, but differed in ring C. The HMBC correlations from H-12 to C-9/C-16/C-14/C-15/C-13, from H-16 to C-14/C-12/C-13, from H-11 to C-13, from H-14 to C-15, and from –OH to C-12/C13/C-14/C-16, as well as the spin system (c) C(11)H2–C(12)H, allowed for the assignment of ring C, which connected with ring B through C-11. Additionally, the HMBC correlations from H-14 to C-21 suggested that the ester carbonyl group was connected to C-14 of ring C. The placement of the Cl was determined largely based on the downfield shift of the diastereotopic methylene protons (H-16, δ 3.67, 3.75), in conjunction with carbon resonance for C-16 at δC 46.6.
The relative configuration of compound 2 was investigated by ROESY, 1D-DPFGSE-NOE, and DFT studies. Correlations of H-10 with H3-18/H-8/H-11a, the absence of ROESY correlation between H-10 and H3-20, and the same chemical shifts of rings A and B as compound 1 confirmed the relative configurations of rings A and B were the same as for compound 1. Although a ROESY correlation was observed between H-12 and H-16, molecular modeling indicated that these protons would be sufficiently close for NOE regardless of configuration, in part due to the flat nature of the lactone. From a biosynthesis perspective, the configuration of compound 2 around the lactone ring should be identical to that found in 1 since compound 2 appears to simply be a regioisomer in terms of the lactone. To help confirm the biosynthetic hypothesis, we used molecular modeling and DFT shielding tensor calculations to compare carbon chemical shifts around the lactone ring for all eight of the possible diastereomers. Spartan’1411 was used to find an equilibrium conformer using molecular mechanics, and Gaussian 0912 was used for further geometry optimization and NMR calculations (B3LYP/6-31G(d,p)).13 NMR shifts were referenced to tetramethylsilane (TMS) and benzene using the multistandard approach.14 After referencing, the calculated chemical shifts were compared to those experimentally observed for all atoms in the lactone ring. The structure shown fit best with the experimental data. Furthermore, the best fit from the DFT studies possessed stereochemical configurations identical to those found in compound 1, as would be predicted. We also evaluated the conformational flexibility with 2 to ensure that only one conformation would be observed for the lactone ring. To that end, we examined 10 000 conformers via molecular mechanics and selected the 16 that were below 40 kJ/mol followed by subsequent local geometry optimization and DFT energy calculations to determine a Boltzmann distribution. Although molecular mechanics provides easy access to conformers, molecular mechanics does not provide accurate energy calculations relative to DFT. Hence, our final Boltzmann distribution was based on energies determined by DFT calculations. Only two relevant conformations were found. Both closely resembled the equilibrium conformer used for the DFT NMR calculations (Supporting Information, Figure S15). Therefore, compound 2 was named (8S*,9R*,10S*,12R*,13R*,14R*)-micromonohalimane B.
Micromonohalimanes A (1) and B (2) were tested for antibacterial activity against methicillin-resistant Staphylococcus aureus, and the minimum inhibitory concentration (MIC) was determined for each compound. Micromonohalimane A (1) was determined to have an MIC of greater than 200 μg/mL against MRSA. Micromonohalimane B (2) was determined to have an MIC of 40 μg/mL against MRSA. In order to identify if micromonohalimane B (2) was bactericidal or bacteriostatic, a sterile swab was dipped into each well that showed inhibition of bacterial growth and was inoculated on an LB plate. A bactericidal agent would show no growth on the LB plate, while a bacteriostatic agent would show bacterial growth.15 Bacterial growth of each sample on the LB plate was seen, which suggests that micromonohalimane B (2) is a bacteriostatic agent.
In conclusion, two diterpenes were isolated from a Micromonospora sp. Although the isolation of terpenes from actinomyctes has been rare, genomics suggests that most actinomycetes have the potential to biosynthesize terpenes. Nonetheless, this is the first report of a halimine-type diterpene from an actinomycete.
EXPERIMENTAL SECTION
General Experimental Procedures
Melting points were measured on a MEL-TEMP micromelting point apparatus and were uncorrected. Optical rotations were measured on a PerkinElmer 241 polarimeter. UV spectra were recorded using an Aminco/OLIS UV–vis spectrophotometer. IR spectra were measured with a Bruker Equinox 55/S FT-IR spectrophotometer. NMR spectra were obtained in CDCl3 with a Bruker Avance 600 MHz spectrometer equipped with a 1H{13C/15N} cryoprobe and a Bruker Avance 500 MHz spectrometer equipped with a 13C/15N{1H} cryoprobe. HRMS data were acquired with a Bruker MaXis 4G QTOF mass spectrometer. Normal-phase flash chromatography was performed using a Combi Flash Rf 200 HPLC system and a 4 g gold silica gel column (Teledyne Isco).
Biological Material
Strain WMMC-218 was isolated from the ascidian Symplegma brakenhielmi collected on August 7, 2013, in Florida at Stanblum State Park (27.479326, −80.311687). A voucher specimen for the ascidian was identified by one of the authors (S.P.-N.).
WMMC-218 displayed over 99% identity to Micromonospora sp. HK160111. The 16S sequence for WMMC-218 was deposited in GenBank (accession number KU183007).
Fermentation, Extraction, and Isolation
Strain WMMC-218 was fermented in 25 × 150 mm culture tubes (4 × 10 mL) in medium ASW-A (20 g soluble starch, 10 g glucose, 5 g peptone, 5 g yeast extract, 5 g CaCO3 per liter of artificial seawater) for 1 week at 28 °C. Baffled flasks (250 mL, 12 × 50 mL) containing ASW-A were inoculated with 2 mL from the culture tube and shaken at 200 rpm at 28 °C for 7 days. Flasks (4 L, 8 × 500 mL) containing medium ASW-A with Diaion HP20 (4% by weight) were inoculated with 25 mL and shaken at 200 rpm at 28 °C for 7 days. Filtered HP20 was washed with H2O and extracted with acetone. The acetone extract (11 g) was subjected to a liquid–liquid partitioning using 10% aqueous MeOH and hexane (1:1), which then increased to 30% aqueous methanol, and partitioned using CHCl3 (1:1). The CHCl3-soluble partition (3.1 g) was subjected to ENV+ column chromatography (ISOLUTE, 3 × 500 mg) with H2O and MeOH (10%, 50%, 75%, 100%). Fraction 100% (0.88 g) was chromatographed through a Sephadex LH-20 column with MeOH/H2O (7:3) and was further purified by 4 g HP silica column chromatography (Teledyne Isco) with a linear gradient solvent system consisting of solvent A (hexane) and solvent B (2-propanol) at room temperature as follows: 0–5 min, 0% B; 5–25 min, 0% to 30% B; 25–30 min, 30% B, yielding 2 (3.0 mg, tR 9.1 min) and 1 (1.6 mg, tR 10.5 min), respectively. We cultured strain WMMC-218 in three media (ASW-A, RAM2, GOT), analyzed the extract of each media, and then selected the media ASW-A.
(8S,9S,10S,12R,13R,14R)-Micromonohalimane A (1)
Colorless needles [hexane/2-propanol/MeOH, (2:1:4)]; mp 163–164 °C; [α]20D +18 (c 0.03, MeOH); UV (MeOH) λmax (log ε) 210 (3.59) nm; IR (ATR) νmax 3395, 3314, 2927, 2854, 1787, 1708, 1462, 1373 cm−1; 1H NMR (600 MHz, CDCl3) and 13C NMR (125 MHz, CDCl3) data, Table 1; HR-ESIMS m/z 417.2246 [M + Na]+ (calcd for C22H34NaO6, 417.2248).
X-ray Crystallographic Analysis of Compound 1.16
Crystallization from hexane/2-propanol/MeOH (2:1:4) using the vapor diffusion method yielded colorless crystals of compound 1. A colorless crystal with approximate dimensions 0.40 × 0.15 × 0.15 mm3 was selected under oil under ambient conditions and attached to the tip of a MiTeGen MicroMount. The crystal was mounted in a stream of cold nitrogen at 100(1) K and centered in the X-ray beam by using a video camera. The crystal evaluation and data collection were performed on a Bruker SMART APEXII diffractometer with Cu Kα (λ = 1.541 78 Å) radiation and a diffractometer to crystal distance of 4.03 cm. Crystal data for C22H36O7 (M = 412.51 g/mol): monoclinic, space group P21 (no. 4), a = 9.2381(5) Å, b = 6.9763(6) Å, c = 16.8910(8) Å, β = 90.987(3)°, V = 1088.43(13) Å3, Z = 2, T = 100.01 K, μ(Cu Kα) = 0.759 mm−1, Dcalc = 1.259 g/cm3, 23 887 reflections measured (5.232° ≤ 2θ ≤ 146.796°), 4036 unique (Rint = 0.0195, Rsigma = 0.0121), which were used in all calculations. The final R1 was 0.0297 (I > 2σ(I)) and wR2 was 0.0790 (all data).
(8S,9S,10S,12S,13R,14S)-Micromonohalimane B (2)
Colorless oil; [α]20D +110 (c 0.01, MeOH); UV (MeOH) λmax (log ε) 210 (4.09) nm; IR (ATR) νmax 3390, 3312, 2927, 2854, 1780, 1700, 1460, 1372 cm−1; 1H NMR (600 MHz, CDCl3) and 13C NMR (125 MHz, CDCl3) data, Table 1; HR-ESIMS m/z 435.1906 [M + Na]+ (calcd for C22H33ClNaO5, 435.1909).
Molecular Modeling Calculations
Molecular modeling calculations were performed on a Dell Precision T5500 Linux workstation with a Xeon processor (3.3 GHz, 6-core). Low-energy conformers were obtained using Spartan’14 software (MMFF, 10000 conformers examined).11 The low-energy conformer for each compound was analyzed using Gaussian 09 for geometry optimization and NMR calculations (B3LYP/6-31G(d,p)). NMR shifts were referenced to TMS and benzene using the multistandard approach.17 Molecules were modeled in the gas phase.
Antibacterial Assay
Micromonohalimanes A (1) and B (2) were tested for antibacterial activity against MRSA (ATCC #33591), and MICs were determined using a dilution antimicrobial susceptibility test for aerobic bacteria.18 Micromonohalimanes A (1) and B (2) were dissolved in DMSO, serially diluted to 10 concentrations (0.125–64 μg/mL), and tested in a 96-well plate. Vancomycin was used as a control and exhibited an MIC of 1 μg/mL against MRSA. Micromonohalimanes A (1) and B (2) and vancomycin were tested in triplicate. Six untreated media controls were included on each plate. The plates were incubated at 33 °C for 18 h. The MIC was determined as the lowest concentration that inhibited visible growth of bacteria.
Supplementary Material
Acknowledgments
This work was funded by the National Institutes of Health, U19-AI109673. We thank A. I. Vinokur for assistance with the crystallography work. We thank the Analytical Instrumentation Center at the University of Wisconsin–Madison School of Pharmacy for the facilities to acquire spectroscopic data, especially NMR and MS. This study made use of the National Magnetic Resonance Facility at Madison (NMRFAM), which is supported by NIH grant P41GM103399 (NIGMS). Equipment was purchased with funds from the University of Wisconsin–Madison, the NIH (P41GM103399, S10RR02781, S10RR08438, S10RR023438, S10RR025062, S10RR029220), the NSF (DMB-8415048, OIA-9977486, BIR-9214394), and the USDA.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jnatprod.6b00555.
1D and 2D NMR spectra for compounds 1 and 2 and 13C calculation data, as well as PCA data representation, and a depiction of the two possible conformers of structure 2 (PDF)
Crystallographic data (CIF)
Notes
The authors declare no competing financial interest.
References
- 1.Hou YP, Braun DR, Michel CR, Klassen JL, Adnani N, Wyche TP, Bugni TS. Anal Chem. 2012;84:4277–4283. doi: 10.1021/ac202623g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Adnani N, Michel CR, Bugni TS. J Nat Prod. 2012;75:802–806. doi: 10.1021/np300034c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wyche TP, Piotrowski JS, Hou YP, Braun DR, Deshpande R, McIlwain S, Ong IM, Myers CL, Guzei IA, Westler WM, Andes DR, Bugni TS. Angew Chem, Int Ed. 2014;53:11583–11586. doi: 10.1002/anie.201405990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hou YP, Tianero MaDB, Kwan JC, Wyche TP, Michel CR, Ellis GA, Vazquez-Rivera E, Braun DR, Rose WE, Schmidt EW, Bugni TS. Org Lett. 2012;14:5050–5053. doi: 10.1021/ol3022758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jimenez PC, Ferreira EG, Araújo LA, Guimarães LA, Sousa TS, Pessoa ODL, Lotufo TMC, Costa-Lotufo LV. Lat Am J Aquat Res. 2013;41:335–343. [Google Scholar]
- 6.He H, Ding WD, Bernan VS, Richardson AD, Ireland CM, Greenstein M, Ellestad GA, Carter GT. J Am Chem Soc. 2001;123:5362–5363. doi: 10.1021/ja010129o. [DOI] [PubMed] [Google Scholar]
- 7.Sousa T da S, Jimenez PC, Ferreira EG, Silveira ER, Braz-Filho R, Pessoa ODL, Costa-Lotufo LV. J Nat Prod. 2012;75:489–493. doi: 10.1021/np200795p. [DOI] [PubMed] [Google Scholar]
- 8.Khan ST, Izumikawa M, Motohashi K, Mukai A, Takagi M, Shin-Ya K. FEMS Microbiol Lett. 2010;304:89–96. doi: 10.1111/j.1574-6968.2009.01886.x. [DOI] [PubMed] [Google Scholar]
- 9.Chung HM, Hu LC, Yen WH, Su JH, Lu MC, Hwang TL, Wang WH, Sung PJ. Mar Drugs. 2012;10:2246–2253. doi: 10.3390/md10102246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nagashima F, Tanaka H, Kan Y, Huneck S, Asakawa Y. Phytochemistry. 1995;40:209–212. [Google Scholar]
- 11.Spartan’14, Wavefunction, Inc. Irvine, CA. Except for molecular mechanics and semi-empirical models, the calculation methods used in Spartan have been documented in:; Shao Y, Molnar LF, Jung Y, Kussmann J, Ochsenfeld C, Brown ST, Gilbert ATB, Slipchenko LV, Levchenko SV, O’Neill DP, DiStasio RA, Jr, Lochan RC, Wang T, Beran GJO, Besley NA, Herbert JM, Lin CY, VanVoorhis T, Chien SH, Sodt A, Steele RP, Rassolov VA, Maslen PE, Korambath PP, Adamson RD, Austin B, Baker J, Byrd EFC, Dachsel H, Doerksen RJ, Dreuw A, Dunietz BD, Dutoi AD, Furlani TR, Gwaltney SR, Heyden A, Hirata S, Hsu C-P, Kedziora G, Khalliulin RZ, Klunzinger P, Lee AM, Lee MS, Liang WZ, Lotan I, Nair N, Peters B, Proynov EI, Pieniazek PA, Rhee YM, Ritchie J, Rosta E, Sherrill CD, Simmonett AC, Subotnik JE, Woodcock HL, III, Zhang W, Bell AT, Chakraborty AK, Chipman DM, Keil FJ, Warshel A, Hehre WJ, Schaefer HF, Kong J, Krylov AI, Gill PMW, Head-Gordon M. Phys Chem Chem Phys. 2006;8:3172–3191. doi: 10.1039/b517914a. [DOI] [PubMed] [Google Scholar]
- 12.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA, Jr, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam NJ, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas Ö, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ. Gaussian 09. Gaussian, Inc; Wallingford, CT: 2009. Revision A.1. [Google Scholar]
- 13.Stappen I, Buchbauer G, Robien W, Wolschann P. Magn Reson Chem. 2009;47:720–726. doi: 10.1002/mrc.2452. [DOI] [PubMed] [Google Scholar]
- 14.Sarotti AM, Pellegrinet SC. J Org Chem. 2009;74:7254–7260. doi: 10.1021/jo901234h. [DOI] [PubMed] [Google Scholar]
- 15.Pankey GA, Sabath LD. Clin Infect Dis. 2004;38:864–870. doi: 10.1086/381972. [DOI] [PubMed] [Google Scholar]
- 16.Crystallographic data for compound 1 have been deposited with the Cambridge Crystallographic Data Centre (deposition number CCDC 1443011).
- 17.Sarow AM, Pellegrinet SC. J Org Chem. 2009;74:7254–7260. doi: 10.1021/jo901234h. [DOI] [PubMed] [Google Scholar]
- 18.National Committee for Clinical Laboratory Standards. Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria that Grow Aerobically. 7th. NCCLS; Villanova, PA: 2006. Approved standard M7-A7. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.