


Abstract
During an analysis of DNA enzymes by gel electrophoresis, we found that some DNA enzymes can adopt more than one conformation. The DNA enzyme Dz31 that formed more than one conformer contained a stretch of G residues. Further investigations, involving kinetic analysis and measurements of circular dichroism, indicated that this DNA enzyme and its derivatives formed G-quadruplexes. Moreover, we found that some derivative oligomers were capable of forming dimeric G-quadruplexes. We also compared the catalytic activities of Dz31 and its mutant derivatives. The present findings suggest that DNA enzymes with five or more continuous G residues are less favorable than those without G5 in the association step in the enzymatic reaction and, thus, the choice of targets that contain a continuous stretch of C residues downstream of the cleavage site should be avoided. In addition, we found that negative charge–charge repulsion disrupted the dimerization of G-quadruplexes when a phosphate group was added directly to the 5′-terminal G of oligomers with continuous guanosine residues. In the case of 5′-phosphorylated G5CTA, direct attachment of a phosphate group to the continuous G5 sequence inhibited dimerization of G-quadruplexes, at least during electrophoresis on a denaturing gel.
INTRODUCTION
Unlike double-stranded DNA, single-stranded DNA can fold into well-defined, sequence-dependent tertiary structures; it can bind specifically to a variety of target molecules and it can exhibit catalytic activity similar to that of ribozymes or protein enzymes (1–3) even though no natural catalytic DNAs have been identified. DNA enzymes were discovered by in vitro selection procedures and were shown to catalyze chemical reactions, such as phosphoester transfer, phosphoester formation, porphyrin metalation, DNA capping and the cleavage of RNA or DNA, in the presence of divalent cations (4–10). Joyce's group and Breaker's group have developed DNA enzymes that can cleave RNAs in a sequence-specific manner and, thus, DNA enzymes may prove useful as inactivators of mRNAs of interest (11,12). The catalytic domain of a DNA enzyme is flanked by two substrate-recognition domains of seven or eight deoxyribonucleotides each, and the RNA substrate binds to the DNA enzyme through Watson–Crick base pairing (Figure 1). DNA enzymes can be complementary to ribozymes, such as hammerhead and hairpin ribozymes, and they have broad sequence-specificity and higher stability than RNA enzymes in cells. Therefore, it should be possible to utilize DNA to build new enzymes for applications both in vitro (13) and in vivo (14,15).
Figure 1.
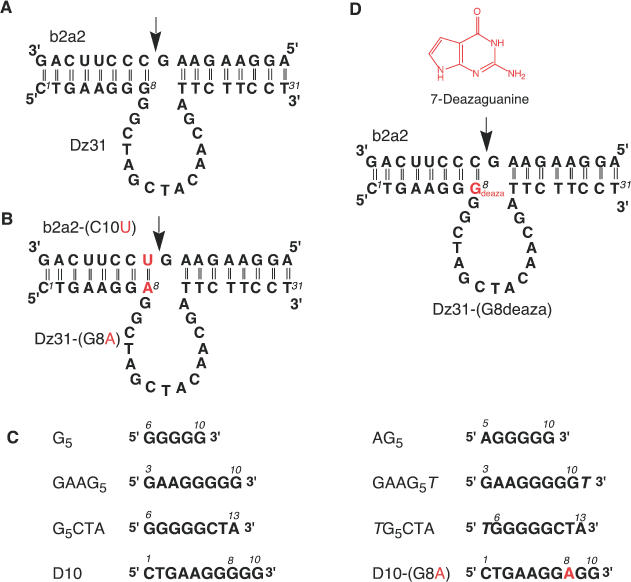
(A) Schematic representation of the DNA enzyme (Dz31) and its substrate (b2a2) [this enzyme was designated Dz3; (15)]. (B) The mutated DNA enzyme Dz31-(G8A) and its substrate b2a2-(C10U). (C) Segments of DNA enzymes that were examined by gel electrophoresis. (D) 7-Deazaguanine, the modified DNA enzyme Dz31-(G8deaza) and its substrate (b2a2), which were used in comparisons with wild-type Dz31.
DNA enzymes have been engineered to act in trans against other RNA molecules and to catalyze the cleavage of phosphodiester bonds at specific sites to generate products with a 2′,3′-cyclic phosphate and a 5′-hydroxyl group (16). In previous studies, we showed that a DNA enzyme, designated as DNAzyme(31mer) or Dz3 (this DNA enzyme is referred to as Dz31 in the present paper; Figure 1) and directed against an mRNA(17mer) motif (b2a2), successfully cleaved its substrate at a specific position in vivo (14,15). The 17mer substrate designated b2a2 is a part of the b2a2 mRNA that is transcribed from the Philadelphia chromosome, which results from reciprocal chromosomal translocations and produces cytogenetically abnormal cells in patients with chronic myelogenous leukemia (17). For our subsequent studies of structure–activity relationships in the active core of DNA enzymes, we have focused on Dz31 and, during the present study, we found an unexpected complex, with lower mobility than expected, when we were checking the purity of Dz31 by gel electrophoresis.
In this study, we used gel electrophoresis and circular dichroism (CD) to identify the unexpected complex. We also investigated the activity of this complex in cleavage reactions. To compare the catalytic activities of Dz31, a mutated DNA enzyme(31mer) [Dz31-(G8A)] in which the central G within the continuous stretch of guanine nucleotides was replaced by A, and a modified DNA enzyme(31mer) [Dz31-(G8deaza)] in which hydrogen bonding within the stretch of G residues was disrupted, we designed substrates with an mRNA(17mer) motif (b2a2) and a modified mRNA(17mer) motif (b2a2-C10U) (Figure 1). We compared the activities of Dz31 and Dz31-(G8deaza) against the substrate with the mRNA(17mer) motif (b2a2) and examined the activity of Dz31-(G8A) against an mRNA(17mer) motif (b2a2-C10U). Dz31-(G8A), which differed from Dz31 only at the position of the eighth nucleotide (from the 5′ end) that is very important for the enzymatic reaction, was 50 times more active than Dz31 andDz31-(G8deaza). The present findings suggest that DNA enzymes with a continuous stretch of five or more G residues participate inefficiently in the association step in the enzymatic reaction and that an adenosine at position 8 at the margin of the catalytic core of the DNA enzyme appears more suitable for effective activity than a guanine nucleotide at this position.
Guanine-rich sequences are found in biologically significant regions of the human genome, such as telomeres (18), immunoglobulin switch regions (19), gene promoter regions (20,21) and sequences associated with human diseases (22). A guanine quartet (G-quartet) is a cyclic array of four hydrogen-bonded guanine bases, in which each base is both the donor and acceptor of two hydrogen bonds with its neighbors through Hoogsteen-type base pairs. The core structure of the G-quadruplexes can be formed in many different ways, and many different structures have been observed. G-quadruplexes are known to be stable under certain conditions. In addition, G-quadruplexes are preferentially stabilized by metal ions. Monovalent ions typically interact predominantly with the negatively charged phosphate groups on DNA. However, in G-quartets, monovalent ions interact with eight carbonyl oxygen atoms. It is likely that the ion-binding properties of G-quadruplexes might be the most important contributor to their unique behavior (23).
The DNA enzyme(31mer) Dz31 and segments of this enzyme, namely, DNA(10mer) (D10), DNA(8mer) (GAAG5) and DNA(6mer) (AG5), which had a stretch of five continuous guanine nucleotides, each formed a band of lower mobility (upper band) during gel electrophoresis, as well as the expected band, whereas a modified DNA enzyme(31mer) [Dz31-(G8deaza)], a mutated DNA enzyme(31mer) [Dz31-(G8A)], and part of this enzyme DNA(10mer) [D10-(G8A)], which had no similar continuous stretch of guanine nucleotides, did not form an upper band. The measurements of CD revealed spectra typical of a G-quadruplex only in the case of oligomers with a continuous stretch of five guanine nucleotides. The CD data supported the results of gel electrophoresis and strongly suggested that the upper band on gels was due to the formation of a G-quadruplex. Importantly, some upper bands of derivative oligomers were found to be dimeric G-quadruplexes and their dimerization could be inhibited by 5′ phosphorylation.
MATERIALS AND METHODS
Synthesis and purification of substrates RNA(17mer) b2a2, RNA(17mer) b2a2-(C10U) and DNA oligomers
RNA oligonucleotides were synthesized on a DNA/RNA synthesizer (model 394; PE Applied Biosystems, Foster City, CA). The reagents for RNA synthesis were purchased from Glen Research (Sterling, VA). The crude deprotected oligonucleotides were purified on an open column and then purified on a 20% polyacrylamide gel that contained 7 M urea. Each band of interest was excised under ultraviolet irradiation, and RNA was eluted with milli-Q water (Millipore, Billerica, MA) followed by ethanol precipitation and desalted by passage through a NAP10 column (Amersham Pharmacia Biotech AB, Uppsala, Sweden) that had been equilibrated with milli-Q water. Dz31-(G8deaza) was obtained from Japan Bio Service, Inc. (Saitama, Japan). Other DNAs were obtained from Hokkaido System Science Co., Ltd (Sapporo, Hokkaido, Japan) and Espec Oligo Service Corp. (Ibaraki, Japan). The DNAs were purified on a 20% polyacrylamide gel and preparations were desalted on a NAP10 column.
Gel electrophoresis
Gel electrophoresis was performed with 32P-labeled or non-labeled oligonucleotides. For the preparations of labeled samples, oligonucleotides were 5′ end-labeled with [γ-32P]ATP using T4 polynucleotide kinase (Takara Bio, Inc., Shiga, Japan) and then mixed with unlabeled oligonucleotides to give appropriate final concentrations. Samples were analyzed on a 20% denaturing polyacrylamide gel that contained 100 mM KCl and have been equilibrated at 4°C for 3 h. Electrophoresis was performed in 100 mM Tris-borate buffer that contained 100 mM KCl overnight at room temperature. In the experiments with non-labeled oligomers, DNAs in 100 mM KCl were loaded onto 20% polyacrylamide gels without further processing, and then stained with Mupid-Blue (ADVANCE-BIO, Tokyo, Japan).
CD measurements
Samples were dissolved in 10 mM sodium cacodylate, 100 mM KCl or 1 M urea, as necessary. The pH of each solution was adjusted to 6.7. CD spectra were recorded on a spectropolarimeter (J-820; Jasco, Tokyo, Japan) equipped with a temperature controller (PTC-423L; Jasco). For each sample, scans were performed over the range of wavelengths from 220 to 350 nm and a temperature range of 5–95°C in a cell with a 1 cm path length. The scan of each respective buffer was subtracted from the scan of each sample. The spectra were plotted in units of millidegrees versus wavelength and normalized to the total concentration of each species. The cell-holding chamber was flushed with a constant stream of dry nitrogen gas to avoid condensation of water on the exterior of the cell. All the experiments were performed at least twice to confirm the reproducibility of results.
Measurements of kinetic parameters
In general, reactions were initiated by the addition of metal ions and were stopped by the removal, at appropriate intervals, of aliquots of the reaction mixture, which were mixed with an equivalent volume of a solution that contained 100 mM EDTA, 9 M urea, 0.1% xylene cyanol and 0.1% bromophenol blue. The substrates were labeled with [γ-32P]ATP by T4 polynucleotide kinase. Substrates and 5′-cleaved products were separated by electrophoresis on a 20% polyacrylamide/7 M urea denaturing gel and were detected by autoradiography. The extent of cleavage was determined by quantitation of radioactivity in the bands of substrate and product with a Bio-Image Analyzer (STORM; Molecular Dynamics, Sunnyvale, CA).
Assays of activities of Dz31 and Dz31-(G8deaza) were performed in 25 mM MgCl2 and 50 mM Tris–HCl (pH 8.0) at 37°C, under single-turnover conditions, with incubations for 20, 40, 60, 120, 240 and 420 min. In the case of Dz31-(G8A), 50 mM Bis-Tris–HCl (pH 6.0) buffer was used and reaction mixtures were incubated for 5, 10, 20, 30, 60 and 120 min. To obtain kinetic parameters, we performed reactions under multiple-turnover conditions using 0.05–10 μM DNA enzyme, 1–200 μM RNA, 25 mM MgCl2 and 50 mM Tris–HCl buffer (pH 8.0) at 37°C. Values of Km and kcat were calculated from Eadie–Hofstee plots.
RESULTS AND DISCUSSION
Formation and identification of a low-mobility form of DNA enzyme
We examined the formation of low-mobility forms (upper bands) of DNA enzymes and of segments of these enzymes by non-denaturing and denaturing PAGE (Figure 2). Figure 1 shows the sequences of DNA enzymes and the segments of these enzymes that we used in these experiments and their corresponding mobilities on a denaturing polyacrylamide gel are shown in Figure 2A and B. The mutant DNA enzyme Dz31-(G8A) (Figure 1B) and the modified DNA enzyme Dz31-(G8deaza) (Figure 1D) differed from the parental Dz31 (Figure 1A) in that the continuous stretch of five G residues was disrupted. In the experiments for which results are shown in Figure 2A and B, the oligonucleotides were denatured completely by heating at 96°C for 3 min, then they were allowed to equilibrate at the desired temperature for 4 h in the presence of K+ ions before being loaded onto 20% non-denaturing and denaturing polyacrylamide gels that were maintained at a specific temperature during electrophoresis. The samples were initially radiolabeled at their 5′ termini with [γ-32P]ATP and purified by gel electrophoresis. Then, unlabeled oligonucleotides were mixed with labeled oligonucleotides to give the appropriate concentrations.
Figure 2.
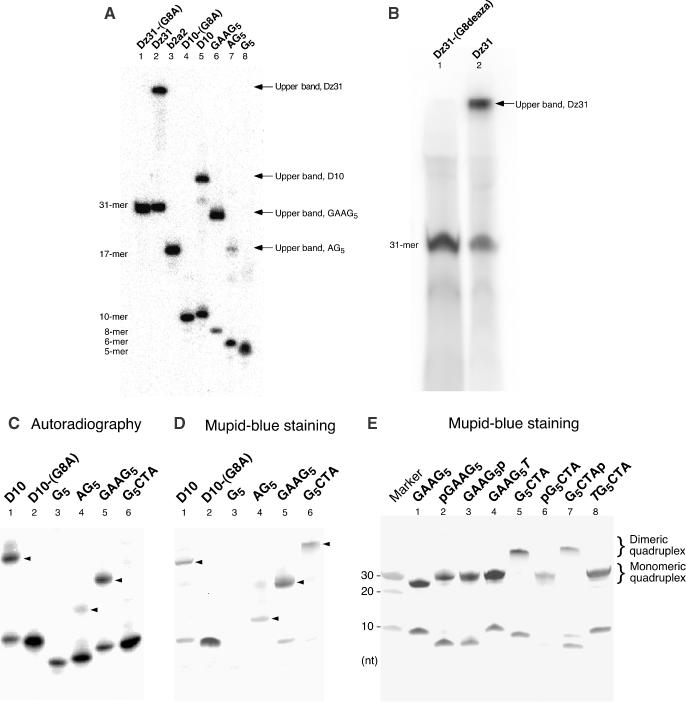
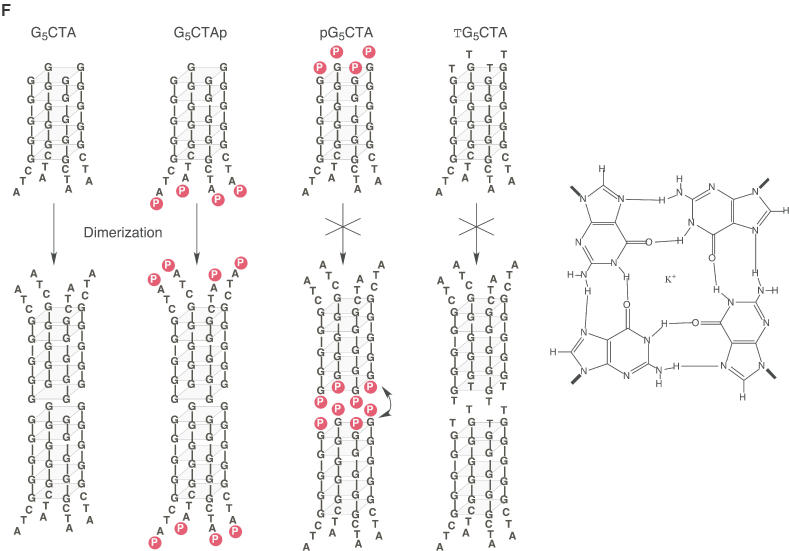
Gel electrophoretic analysis of the formation of G-quadruplexes. (A–C) 5′-32P-labeled nucleotides were incubated at 4°C for 12 h in the presence of 100 mM KCl. Samples were subjected to electrophoresis on a denaturing 20% polyacrylamide gel in TBE buffer and detected by autoradiography. (D) The same gel used in (C) was stained with Mupid-Blue. Upper bands are indicated by arrowheads. (E) Non-labeled DNAs were subjected to electrophoresis and the gel was stained with Mupid-Blue. (F) Schematic representations of the effects of phosphorylation on the dimerization of a G-quadruplex. Phosphates at 5′-G terminus interfere the dimerization of G-quadruplexes.
Dz31 and its derivatives, D10, GAAG5 and AG5, each yielded two bands during electrophoresis on a 20% non-denaturing gel in the absence of urea (data not shown). They also yielded two bands on a denaturing polyacrylamide gel in the presence of urea (Figure 2A). In contrast, Dz31-(G8A) and its derivative D10-(G8A) each yielded a single band (Figure 2A, lanes 1 and 4), as did Dz31-(G8deaza), on a denaturing gel (Figure 2B, lane 1). The mobilities of the lower bands indicated that the free oligonucleotides were of the expected size relative to that of the marker. The mobility of the upper band suggested that it was composed of material of 4 times the molecular weight of the material in the lower band. In other words, the mobility of a reference oligonucleotide with 4 times the molecular weight of Dz31 was almost the same as that of the material in the upper band (data not shown). According to the literature (23–25), our data suggested that the material in the upper band might have been a tetramer formed by intermolecular hydrogen bonding (Figure 2F).
To examine this hypothesis, we mutated to yield Dz31-(G8A) and its derivative D10-(G8A) at G8, the middle guanine nucleotide in the continuous stretch of five guanosine, changing G to A. In Dz31-(G8deaza) (Figure 1D), this G residue was modified by 7-deazaguanine, which breaks the hydrogen bond between the imidazole nitrogen (N7) and the exocyclic-amine group of another guanine nucleotide. This mutation prevented the appearance of an upper band (Figure 2B). Therefore, it appeared that the upper bands were due to the five adjacent guanine nucleotides in the sequences, which should easily form a G-quadruplex as a result of intermolecular aggregation (23–28). Thawing and melting increased the intensity of the upper band of both Dz31 and its derivatives (data not shown), in agreement with previously reported results (29).
Interference with dimerization of G-quadruplexes by terminal phosphate groups
To enhance our understanding of the formation of G-quadruplexes, we also examined several oligonucleotides whose sequences were derived from Dz31. For the preparation of the phosphorylated oligomers shown in Figure 2C, we used T4 polynucleotide kinase to label oligomers with [γ-32P]ATP. As shown in Figure 2C, GAAG5 formed an upper band, but G5CTA and G5 did not. It is probable that negative charge–charge repulsion disrupted the formation of a G-quadruplex when a phosphate group was added directly to the 5′-terminal G of a stretch of four or more guanosine residues. In fact, Figure 2C shows that no upper bands were formed by G5CTA and G5, which might have a phosphate group at the 5′-terminal end of the G5 region.
In the experiments for which results are shown in Figure 2D, we used the new reagent Mupid-Blue for the high-sensitivity detection of non-labeled oligomers. In contrast to the apparent absence of a G-quadruplex in Figure 2C, staining with Mupid-Blue indicated that GAAG5 and G5CTA yielded upper bands in each case, probably a result of the formation of a parallel G-quadruplex (Figure 2D, lanes 5 and 6). Therefore, the absence of upper bands that corresponded to G5CTA and G5 in Figure 2C was due to the fact that the T4 polynucleotide kinase was unable to attach phosphate groups to the termini of the G-quadruplexes and it was not due to disruption of the formation of a G-quadruplex by negative charge–charge repulsion.
To analyze the effect of phosphate groups, we prepared several oligomers with and without a phosphate group. The phosphorylated oligomers used in experiments for which results are shown in Figure 2E were synthesized by chemical addition of a phosphate group at the 5′ or 3′ end. As shown in Figure 2E, upper bands were detected in cases of GAAG5 (lanes 1–3) and G5CTA (lanes 5–7) with or without a terminal phosphate.
Although the formation of these G-quadruplexes was very reasonable with ‘G-continuous’ DNAs, we did note an unexpectedly large difference in terms of electrophoretic mobility between G5CTA and pG5CTA (Figure 2E, lanes 5 and 6). We postulated that the G-quadruplexes formed by four molecules of G5CTA might form a dimeric G-quadruplex (two molecules of G-quadruplexes), as shown in Figure 2F. Since this kind of head-to-head dimerization of two sets of G-quadruplexes is known to be inhibited by the addition of one thymine nucleotide at the 5′ terminus (30–32) and in an attempt to explain the difference in mobilities, we prepared a one-thymine-nucleotide-supplemented oligomer (TG5CTA) and subjected it to gel electrophoretic analysis. We found that the mobility of TG5CTA was almost identical to that of pG5CTA, a monomeric G-quadruplex (Figure 2E). It appears that both TG5CTA and pG5CTA formed a simple parallel G-quadruplex.
While the reasons for these interesting phenomena are now somewhat clearer, several questions remain. The mobility of GAAG5, which is potentially capable of forming a tail-to-tail dimeric G-quadruplex, was nearly the same as that of the monomeric G-quadruplex of GAAG5T (Figure 2E, lanes 1 and 4). It appears that GAAG5 formed a simple parallel G-quadruplex in the same way as did GAAG5T, despite the fact that most of the corresponding oligonucleotides examined in earlier similar studies formed a dimeric G-quadruplex with a 3′-G terminus (30–32). We are at present trying to examine this issue by analyzing many related derivatives.
Finally, although the newly discovered dimerization of the G-quadruplexes formed by G5CTA is not unexpected, considering the G–G face interactions, it is noteworthy that the addition of a phosphate group at the ‘G-terminal’ end (pG5CTA) interfered with the dimerization of G-quadruplexes, just as the addition of a thymidine disrupted the formation of dimers (Figure 2E and F). To our knowledge, this is the first demonstration of interference in the G–G face interactions by terminal phosphate groups.
CD spectra of various oligomers
As noted above, the unexpected upper bands seen after gel electrophoresis seemed to be due to the formation of monomeric and dimeric G-quadruplexes. A simple way to confirm such a possibility is to record the CD spectra of such oligomers since a four-stranded quadruplex has a very specific spectrum that is due to the formation of a G-quadruplex. For example, parallel four-stranded quadruplexes are known to have a characteristic strong positive CD band at ∼262 nm and a negative band at 240 nm, whereas antiparallel folded quadruplexes have a positive band at 295 nm and a negative band at 260 nm (25). We applied this technique to the oligomers used in this study and confirmed that G5 yielded spectral pattern of a parallel four-stranded quadruplex, as a positive control. As shown in Figure 3A(a), the spectra that we recorded coincided with those in previous reports by other groups, with a characteristic strong positive CD peak at 260–263 nm and a negative peak at 240 nm in the presence of 100 mM of KCl, at neutral pH, over a wide range of temperatures. In the presence of 1 M urea and ∼10 mM NaCl (a component of the buffered solution) instead of 100 mM KCl, these strong peaks disappeared at higher temperatures, as shown in Figure 3A(b). This phenomenon was not unexpected since urea breaks hydrogen bonds between base pairs by interacting with functional groups of nucleobases. We defined the spectra and the changes in spectra with changes in temperature in the presence of 100 mM KCl or 1 M urea without K+ ions as the criteria by which the presence or absence of a four-stranded quadruplex is assessed.
Figure 3.
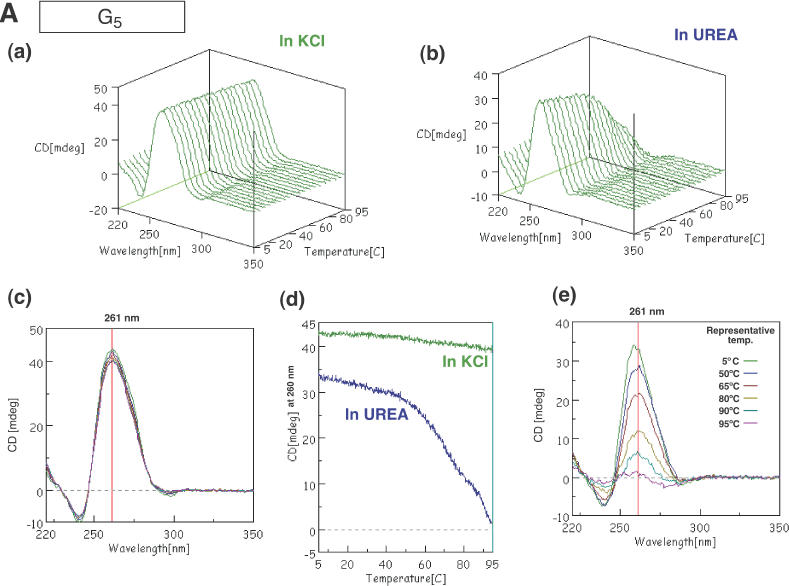
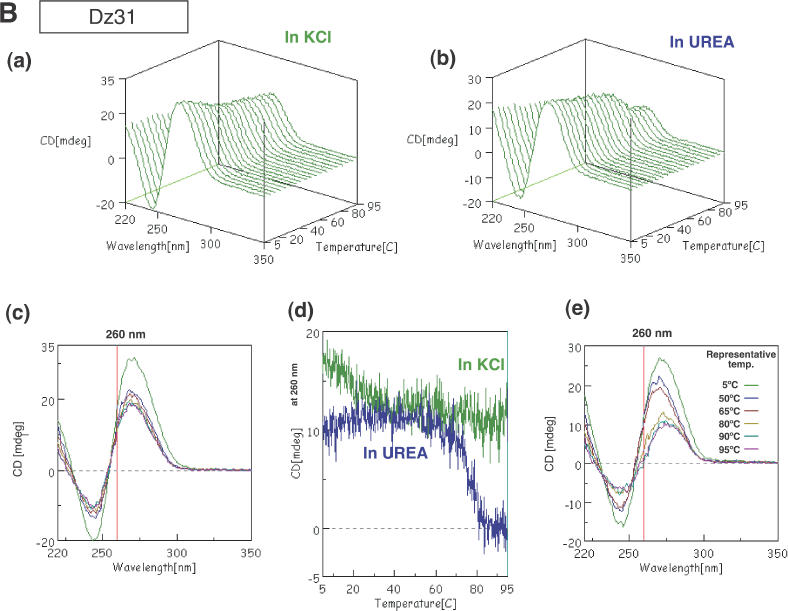
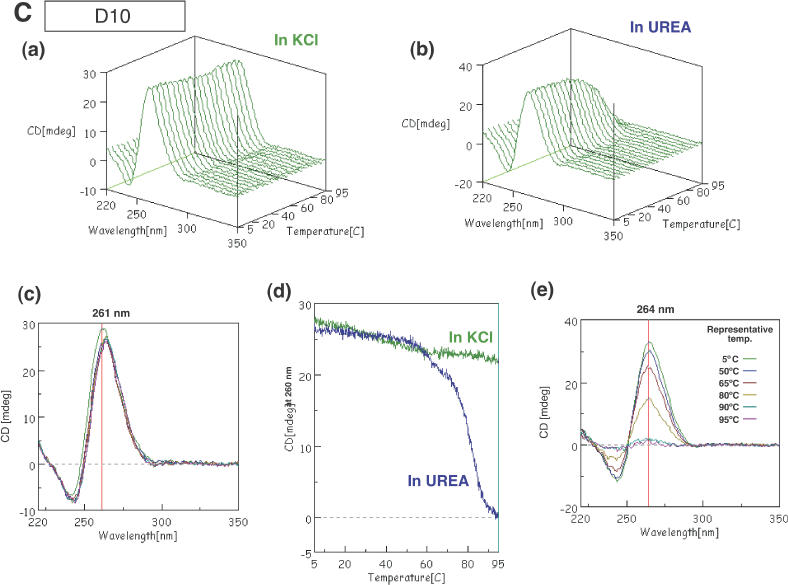
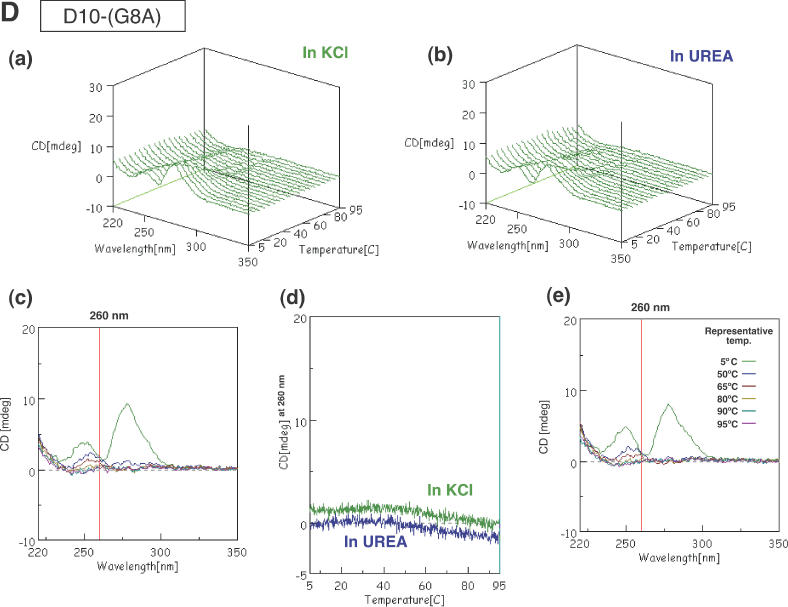
CD spectra of G5 (A), Dz31 (B), D10 (C) and D10-(G8A) (D). Changes in spectra in KCl (a) and in urea (b) upon changes in temperature are shown. (c) The spectra in KCl are shown in two-dimensional graphs of CD (mdeg) versus wavelength (nm) at representative temperatures of 5, 50, 65, 80, 90 and 95°C. The vertical line exhibits the indicated wavelength, 260 or 261 nm. (d) Comparison of peak heights (mdeg) at 260 nm upon changes in temperature in KCl and in urea. (e) The spectra in urea as two-dimensional graphs of peak height (mdeg) versus wavelength (nm) at representative temperatures of 5, 50, 65, 80, 90 and 95°C. The vertical line exhibits the indicated wavelength, 260 or 261 nm.
We monitored the CD spectra of Dz31 with changes in temperature in the presence of 100 mM KCl and in 1 M urea without K+ ions. As shown in Figure 3B, strong positive and negative peaks were observed above 270 nm and above 240 nm, respectively, over the entire range of temperatures examined. These peaks did not disappear, even at 95°C and in the presence of 1 M urea. However, the value at 260 nm decreased dramatically, falling to zero in the case of urea, with a smaller decrease in the case of KCl, as shown in Figure 3B(c–e). Although the spectral patterns of Dz31 upon changes in temperature were somewhat ambiguous, the difference between the changes in intensities at 260 nm suggests the existence of a four-stranded quadruplex in the presence of KCl. The ambiguity might have been due to the existence of several other spectral forms, e.g. the catalytic core.
To facilitate our analysis, we decided to monitor the spectra of D10 at various temperatures. The sequence of the D10 motif is the same as that of the first 10 nt from the 5′ end of Dz31. The spectrum had strong positive and negative peaks at 261 and 240 nm respectively, as seen also in the spectrum of G5 (Figure 3C). Thus, the region adjacent to the continuous stretch of five guanosine nucleotides in D10 did not affect the formation of parallel four-stranded quadruplexes in KCl. In the presence of 1 M urea without KCl, the heights of peaks at 264 and 240 nm fell to zero at higher temperatures, as seen also in the case of G5 itself [Figure 3C(c–e)].
Comparison of the results of CD spectroscopy and gel electrophoresis
As noted in our discussion of the results of gel electrophoresis, several oligomers containing five adjacent guanosine nucleotides, such as Dz31, D10, GAAG5 and AG5, yielded unexpected bands with reduced mobility, as well as the expected bands, during electrophoresis, namely, ‘upper bands’ (Figure 2A). These oligomers yielded similar CD spectral patterns and changes in patterns in KCl and urea, even though the spectra of Dz31 were rather complicated. Since the patterns of CD spectra were due, apparently, to parallel four-stranded quadruplexes that were associated with the formation of G-quadruplexes, it seems likely that the upper bands were due to the complexes that consisted of parallel four-stranded quadruplexes.
In contrast to the above-described results, D10-(G8A) did not form an upper band on the gel (Figure 2A) and did not yield a CD spectrum typical of a parallel four-stranded quadruplex (Figure 3D). This result was to be expected since D10-(G8A) did not include five adjacent guanosine nucleotides: the middle guanosine was replaced by adenosine and the formation of a G-quadruplex was inhibited. Comparing the CD spectra and results of electrophoresis for Dz31 and D10 with those for Dz31-(G8A) and D10-(G8A), we can clearly see that four or more adjacent guanosine nucleotides are responsible for the formation of a G-quadruplex by the DNA enzyme.
Interference in reactions by the G-quadruplex
The sequences of the DNA enzymes and the substrates that bind through Watson–Crick base pairing are shown in Figure 1. We found earlier that Dz31, which is 31 nt long, was the most effective DNA enzyme; it cleaved the substrate between the indicated guanosine and cytidine residues of b2a2 mRNA (14,15). To investigate the influence of the G-quadruplex on catalysis by the DNA enzyme, we prepared two analogs, Dz31-(G8A) and Dz31-(G8deaza). Dz31-(G8A) included adenosine instead of guanosine at position 8 in Dz31. The replacement of G by A in the middle of the five adjacent guanosine residues hinders the formation of a parallel four-stranded quadruplex. Dz31-(G8deaza) had an N7-deaza-guanosine moiety at position 8 and it too failed to yield an upper band on gels (Figure 2B). D10-(G8A) also did not produce CD patterns characteristic of the formation of a G-quadruplex (Figure 3D). We adjusted the substrate of Dz31-(G8A), by changing C to U, to allow the formation of a base pair between Dz31-(G8A) and its substrate [b2a2-(C10U)] (Figure 1).
We calculated the kinetic parameters of the DNA enzymes by performing RNA-cleavage reactions under single- and multiple-turnover conditions. The cleavage products were analyzed by electrophoresis on a 20% polyacrylamide/7 M urea denaturing gel, as shown in Figure 4. The reactions with Dz31 and Dz31-(G8deaza) under single-turnover conditions were performed in 50 mM Tris–HCl (pH 8.0) plus 25 mM Mg2+ at 37°C. We used a lower pH, namely pH 6.0, for Dz31-(G8A) in order to slow down the rates of reactions so that we could make accurate measurements of kinetic parameters under single-turnover conditions in 50 mM Bis-Tris–HCl (pH 6.0). To obtain the kinetic parameters Km and kcat under single-turnover conditions, we used the equation kobs = kcleav − (kobs × Kd)/[E] (Figure 5A–C) and the results are summarized in Table 1.
Figure 4.
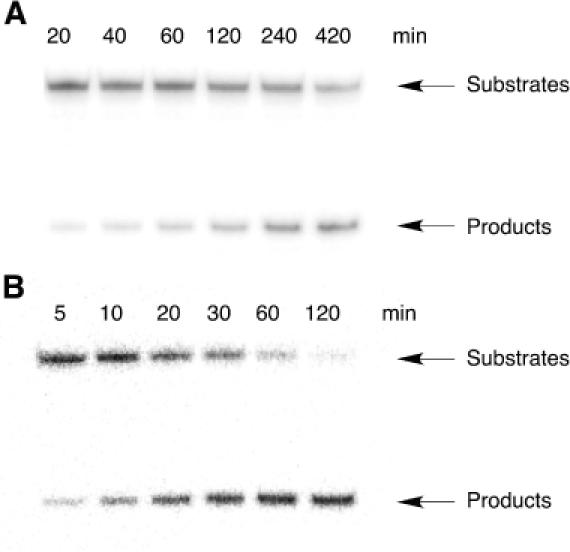
Autoradiograms showing the products of reactions in the presence of 25 mM Mg2+ at 37°C: (A) 50 mM Tris (pH 8.0) with catalysis by Dz31; (B) 50 mM Bis-Tris (pH 6.0) with catalysis by Dz31-(G8A).
Figure 5.
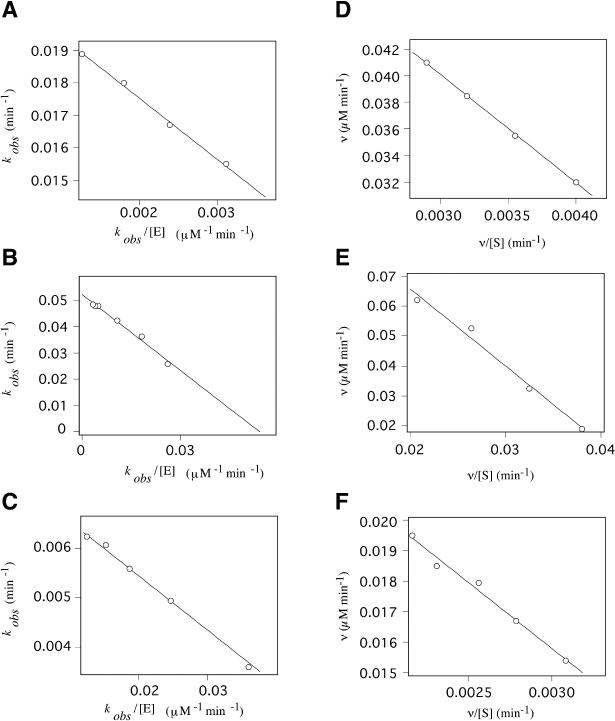
Determination of kinetic parameters from the equation kobs = kcleav − (kobs × Kd)/[E] (A–C) and Eadie–Hofstee plots (D–F). Reactions were performed in the presence of 25 mM Mg2+ at 37°C, under single-turnover conditions (A–C) and in the presence of 25 mM Mg2+ in 50 mM Tris (pH 8.0), at 37°C under multiple-turnover conditions (D–F). (A) Kinetic data for Dz31 were obtained from reactions in 50 mM of Tris (pH 8.0) and yielded a Km of 1.89 μM and a kcat of 0.02 min−1. (B) Values of Km of 0.96 μM and of kcat of 0.05 min−1 were obtained for Dz31-(G8A) from reactions in 50 mM of Bis-Tris (pH 6.0). (C) Kinetic data for Dz31-(G8deaza) were obtained from reactions in 50 mM Tris (pH 8.0) and yielded a Km of 0.11 μM and a kcat of 0.016 min−1. (D) Data for Dz31 yielded a Km of 8.2 μM and a kcat of 0.05 min−1. (E) Data for Dz31-(G8A) yielded a Km of 2.93 μM and a kcat of 2.35 min−1. (F) Data for Dz31-(G8deaza) yielded a Km of 4.3 μM and a kcat of 0.10 min−1.
Table 1. Kinetic parameters of cleavage of an RNA (17mer) substrate.
| Enzyme | Substrate | kcat (min−1) | Km (μM) |
|---|---|---|---|
| Dz31 (31mer) | b2a2 (17mer) | 0.020 ± 0.003 | 1.89 ± 0.35 |
| Dz31-(G8A) (31mer)a | b2a2-(C10U) (17mer) | 0.050 ± 0.003 | 0.96 ± 0.12 |
| Dz31-(G8deaza) (31mer) | b2a2 (17mer) | 0.016 ± 0.001 | 0.11 ± 0.02 |
Determined in 50 mM of Tris–HCl (pH 8.0) and 25 mM of MgCl2 under enzyme-saturating (single-turnover) conditions at 37°C.
apH was 6.0.
The mixtures for multiple-turnover reactions contained 0.5–10 μM DNA enzyme [Dz31 or Dz31-(G8deaza)] plus 1–50 μM b2a2 mRNA and 0.05–1 μM Dz31-(G8A) plus 0.5–15 μM b2a2-(C10U) mRNA. All these reactions were performed in 50 mM Tris–HCl (pH 8.0) plus 25 mM Mg2+ ions at 37°C. Cleavage rates were obtained from the initial slopes of the curves of the time courses of reactions, and Km and kcat were calculated from Eadie–Hofstee plots (Figure 5D–F). The results are summarized in Table 2.
Table 2. Kinetic parameters of cleavage of an RNA (17mer) substrate.
| Enzyme | Substrate | kcat (min−1) | Km (μM) |
|---|---|---|---|
| Dz31 (31mer) | b2a2 (17mer) | 0.050 ± 0.013 | 8.20 ± 0.30 |
| Dz31-(G8A) (31mer) | b2a2-(C10U) (17mer) | 2.34 ± 0.02 | 2.93 ± 0.35 |
| Dz31-(G8deaza) (31mer) | b2a2 (17mer) | 0.10 ± 0.01 | 4.30 ± 0.51 |
Determined in 50 mM of Tris–HCl (pH 8.0) and 25 mM of MgCl2 under multiple-turnover conditions at 37°C.
The Km values of Dz31-(G8deaza) and Dz31-(G8A) were smaller than that of Dz31, as shown in Tables 1 and 2. Since Dz31-(G8deaza) and Dz31-(G8A) do not form quadruplexes, they can bind easily to their substrates. In contrast, Dz31 can form a quadruplex and, thus, the association of the DNA enzyme with its substrate is impaired. Since the preformed G-quadruplex seemed to be extremely stable (inert), it is difficult to unfold Dz31-quadruplex to accommodate its substrate RNA (33). As a result, the observed Km of Dz31 was larger than those of Dz31-(G8deaza) and Dz31-(G8A). The kcat value of Dz31-(G8deaza) was similar to that of Dz31, as shown in Tables 1 and 2. This result is reasonable since kcat reflects the cleavage rate under saturating conditions, implying that the formation of a G-quadruplex does not affect the chemical step during cleavage.
The kcat value of Dz31-(G8A) was much higher than that of Dz31 and Dz31-(G8deaza), as shown in Figure 4 and summarized in Tables 1 and 2. The cleavage reaction catalyzed by Dz31-(G8A) was too fast for measurements of kinetic parameters at pH 7 and 8, and the reaction seemed to be pH dependent, as is the case for reactions catalyzed by hammerhead ribozymes (3,34). The difference between the reactions catalyzed by Dz31-(G8deaza) and Dz31-(G8A) involves the difference in sequence at position 8 in the DNA enzyme and the complementary sequence in the substrate. In the former case, there is an N7-deazaG–C base pair and in the latter case there is an A–U base pair. In neither case does a quadruplex form. The different base pairs produce extremely different kcat values that reflect the chemical step during cleavage. It is possible that the combination of the A–U base pair moves the conformation in the vicinity of the catalytic site closer to the transition state with lower energy and, thus, the A–U base pair is more effective than the G–C base pair at this position (35).
It seems probable that the difference in kinetic parameters between Dz31 and Dz31-(G8A), as shown in Tables 1 and 2, was due to at least two factors: (i) the formation of a G-quadruplex, which affects Km and (ii) the conformation in the transition state, which affects kcat.
CONCLUSION
We have analyzed in detail the formation of quadruplexes by DNA oligomers. Adjacent G residues, even if they are within the catalytic site of the DNA enzyme, form a G-quadruplex, as indicated by gel mobility, CD measurements and kinetic parameters. Addition of negative charges (phosphorylation) to the ends of G-quadruplex-forming oligomers inhibited dimerization of G-quadruplexes. To our knowledge, this is the first example of inhibition of the dimerization of G-quadruplexes by direct phosphorylation of a continuous terminal stretch of G residues. Furthermore, we compared the catalytic activities of Dz31 and its mutant derivatives, such as Dz31-(G8A) and Dz31-(G8deaza), in which hydrogen bonding within the stretch of G residues was disrupted. The values of kcat for Dz31 and Dz31-(G8deaza) were almost the same under single-turnover conditions, but Dz31-(G8deaza) was a slightly more efficient catalyst under multiple-turnover conditions, reflecting the presence of a higher concentration of a more reactive species in the latter case. Dz31-(G8A), which did not form a G-quadruplex and possessed the favorable A–U base pair, was 50 times more reactive than Dz31 and Dz31-(G8deaza). Reflecting the formation of a G-quadruplex, Dz31 had a significantly higher Km than Dz31-(G8A) and Dz31-(G8deaza). As expected, G-quadruplexes within DNA enzymes inhibited catalysis and, thus, selection of corresponding target sequences should be avoided in an effort to maintain high rates of cleavage by DNA enzymes.
Acknowledgments
ACKNOWLEDGEMENTS
The authors thank Ms Clair Price for critical reading of the manuscript. M.K.U. was supported by the Japan Society for the Promotion of Science (JSPS).
REFERENCES
- 1.Bock L.C., Griffin,L.C., Latham,J.A., Vermaas,E.H. and Toole,J.J. (1992) Selection of single-stranded DNA molecules that bind and inhibit human thrombin. Nature, 355, 564–566. [DOI] [PubMed] [Google Scholar]
- 2.Ellington A.D. and Szostak,J.W. (1992) Selection in vitro of single-stranded DNA molecules that fold into specific ligand-binding structures. Nature, 355, 850–852. [DOI] [PubMed] [Google Scholar]
- 3.He Q.-C., Zhou,J.-M., Zhou,D.-M., Nakamatsu,Y., Baba,T. and Taira,K. (2002) Comparison of metal-ion-dependent cleavages of RNA by a DNA enzyme and a hammerhead ribozyme. Biomacromolecules, 3, 69–83. [DOI] [PubMed] [Google Scholar]
- 4.Cuenoud B. and Szostak,J.W. (1995) A DNA metalloenzyme with DNA ligase activity. Nature, 375, 611–614. [DOI] [PubMed] [Google Scholar]
- 5.Carmi N., Shultz,L.A. and Breaker,R.R. (1996) In vitro selection of self-cleaving DNAs. Chem. Biol., 3, 1039–1046. [DOI] [PubMed] [Google Scholar]
- 6.Li Y. and Sen,D. (1997) Toward an efficient DNA enzyme. Biochemistry, 36, 5589–5599. [DOI] [PubMed] [Google Scholar]
- 7.Li Y. and Breaker,R.R. (1999) Deoxyribozymes: new players in the ancient game of biocatalysis. Curr. Opin. Struct. Biol., 9, 315–323. [DOI] [PubMed] [Google Scholar]
- 8.Li Y., Liu,Y. and Breaker,R.R. (2000) Capping DNA with DNA. Biochemistry, 39, 3106–3114. [DOI] [PubMed] [Google Scholar]
- 9.Feldman A.R. and Sen,D. (2001) A new and efficient DNA enzyme for the sequence-specific cleavage of RNA. J. Mol. Biol., 313, 283–294. [DOI] [PubMed] [Google Scholar]
- 10.Wang D.Y., Lai,B.H.Y., Feldman,A.R. and Sen,D. (2002) A general approach for the use of oligonucleotide effectors to regulate the catalysis of RNA-cleaving ribozymes and DNA enzymes. Nucleic Acids Res., 30, 1735–1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Breaker R.R. and Joyce,G.F. (1994) A DNA enzyme that cleaves RNA. Chem. Biol., 1, 223–229. [DOI] [PubMed] [Google Scholar]
- 12.Santoro S.W. and Joyce,G.F. (1997) A general purpose RNA-cleaving DNA enzyme. Proc. Natl Acad. Sci. USA, 94, 4262–4266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cox J.C., Cohen,D.S. and Ellington,A.D. (1999) The complexities of DNA computation. Trends Biotechnol., 17, 151–154. [DOI] [PubMed] [Google Scholar]
- 14.Kuwabara T., Warashina,M., Tanabe,T., Tani,K., Asano,S. and Taira,K. (1997) Comparison of the specificities and catalytic activities of hammerhead ribozymes and DNA enzymes with respect to the cleavage of BCR-ABL chimeric L6 (b2a2) mRNA. Nucleic Acids Res., 25, 3074–3081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Warashina M., Kuwabara,T., Nakamatsu,Y. and Taira,K. (1999) Extremely high and specific activity of DNA enzymes in cells with Philadelphia chromosome. Chem. Biol., 6, 237–250. [DOI] [PubMed] [Google Scholar]
- 16.Santoro S.W. and Joyce,G.F. (1998) Mechanism and utility of an RNA-cleaving DNA enzyme. Biochemistry, 37, 13330–13342. [DOI] [PubMed] [Google Scholar]
- 17.Nowell P.C. and Hungerford,D.A. (1960) A minute chromosome in human chronic granulocytic leukemia. Science, 132, 1497–1499. [DOI] [PubMed] [Google Scholar]
- 18.Blackburn E.H. (1994) Telomeres: no end in sight. Cell, 77, 621–623. [DOI] [PubMed] [Google Scholar]
- 19.Sen D. and Gilbert,W. (1988) Formation of parallel four-stranded complexes by guanine-rich motifs in DNA and its implications for meiosis. Nature, 334, 364–366. [DOI] [PubMed] [Google Scholar]
- 20.Evans T., Schon,E., Gora-Maslak,G., Patterson,J. and Efstratiadis,A. (1984) S1-hypersensitive sites in eukaryotic promoter regions. Nucleic Acids Res., 12, 8043–8058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kilpatrick M.W., Torri,A., Kang,D.S., Engler,J.A. and Wells,R.D. (1986) Unusual DNA structures in the adenovirus genome. J. Biol. Chem., 261, 11350–11354. [PubMed] [Google Scholar]
- 22.Fry M. and Loeb,L.A. (1994) The fragile X syndrome d(CGG)n nucleotide repeats form a stable tetrahelical structure. Proc. Natl Acad. Sci. USA, 91, 4950–4954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Williamson J.R. (1993) Guanine quartets. Curr. Opin. Struct. Biol., 3, 357–362. [Google Scholar]
- 24.Williamson J.R. (1994) G-quartet structures in telomeric DNA. Annu. Rev. Biophys. Biomol. Struct., 23, 703–730. [DOI] [PubMed] [Google Scholar]
- 25.Keniry M.A. (2001) Quadruplex structures in nucleic acids. Biopolymers, 56, 123–146. [DOI] [PubMed] [Google Scholar]
- 26.Henderson E., Hardin,C.C., Walk,S.K., Tinoco,J.I. and Blackburn,E.H. (1987) Telomeric DNA oligonucleotides form novel intramolecular structures containing guanine–guanine base pairs. Cell, 51, 899–908. [DOI] [PubMed] [Google Scholar]
- 27.Gueschlbauer W., Chantot,J.-F. and Thiele,D. (1990) Four-stranded nucleic acid structures 25 years later: from guanosine gels to telomer DNA. J. Biomol. Struct. Dyn., 8, 491–511. [DOI] [PubMed] [Google Scholar]
- 28.Balagurumoorthy P. and Brahmachari,S.K. (1994) Structure and stability of human telomeric sequence. J. Biol. Chem., 269, 21858–21869. [PubMed] [Google Scholar]
- 29.Penazova H. and Vorlickova,M. (1997) Guanine tetraplex formation by short DNA fragments containing runs of guanine and cytosine. Biophys. J., 73, 2054–2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Protozanova E. and Macgregor,R.,Jr (1996) Frayed wires: a thermally stable form of DNA with two distinct structural domains. Biochemistry, 35, 16638–16645. [DOI] [PubMed] [Google Scholar]
- 31.Sen D. and Gilbert,W. (1992) Novel DNA superstructures formed by telomere-like oligomers. Biochemistry, 31, 65–70. [DOI] [PubMed] [Google Scholar]
- 32.Wang Y. and Patel,D.J. (1992) Guanine residues in d(T2AG3) and d(T2G4) form parallel-stranded potassium cation stabilized G-quadruplexes with anti glycosidic torsion angles in solution. Biochemistry, 31, 8112–8119. [DOI] [PubMed] [Google Scholar]
- 33.Li W., Miyoshi,D., Nakano,S. and Sugimoto,N. (2003) Structural competition involving G-quadruplex DNA and its complement. Biochemistry, 42, 11736–11744. [DOI] [PubMed] [Google Scholar]
- 34.Takagi Y., Warashina,M., Stec,W.J., Yoshinari,K. and Taira,K. (2001) Recent advances in the elucidation of the mechanisms of action of ribozymes. Nucleic Acids Res., 29, 1815–1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cairns M.J., King,A. and Sun,L.-Q. (2003) Optimization of the 10–23 DNAzyme-substrate pairing interactions enhanced RNA cleavage activity at purine–cytosine target sites. Nucleic Acids Res., 31, 2883–2889. [DOI] [PMC free article] [PubMed] [Google Scholar]