


Abstract
We have used DNase I footprinting, fluorescence and ultraviolet (UV) melting experiments and circular dichroism to demonstrate that, in the parallel triplex binding motif, 2′-aminoethoxy-5-(3-aminoprop-1-ynyl)uridine (bis-amino-U, BAU) has very high affinity for AT relative to all other Watson–Crick base pairs in DNA. Complexes containing two or more substitutions with this nucleotide analogue are stable at pH 7.0, even though they contain several C.GC base triplets. These modified triplex-forming oligonucleotides retain exquisite sequence specificity, with enhanced discrimination against YR base pairs (especially CG). These properties make BAU a useful base analogue for the sequence-specific creation of stable triple helices at pH 7.0.
INTRODUCTION
Triplex-forming oligodeoxyribonucleotides (TFOs) have great potential as sequence-specific DNA recognition agents (1–6). These oligonucleotides bind in the major groove of their target sequences where they make specific hydrogen bond contacts to substituents on the exposed faces of purine residues (7–10). The third strand can be arranged either parallel or antiparallel to the purine strand of the duplex; the parallel motif has been most widely studied and is characterized by the formation of T.AT and C+.GC triplets. Although these complexes have good sequence-recognition properties, their utility is limited by the relatively weak binding of the third strand to the underlying DNA duplex. This is partly due to charge repulsion between three adjacent polyanionic DNA strands. In addition, the T.AT triplet is less stable than C+.GC, possibly because it lacks the positive charge, which partially overcomes the charge repulsion (11–16). Triplexes can be stabilized by introducing basic groups, in particular amines (17–19) or guanidines (20) into the third strand and it is likely that the increased stability stems from ionic interactions between the protonated bases and the phosphodiester backbone of DNA. It has recently been shown that introduction of the 5-propargylamino (21) group or the 2′-aminoethoxy (22,23) group onto the thymidine nucleosides of TFOs significantly increases the stability of parallel triplexes and that the bis-amino compound, 2′-aminoethoxy-5-(3-aminoprop-1-ynyl)uridine (BAU) (24,25) dramatically increases triplex stability. As well as recognizing AT base pairs, T is also the best base for binding to CG inversions, forming the T.CG triplet (10). It is therefore essential to know how BAU interacts with other base pairs, to discover whether it has enhanced or diminished selectivity relative to T. We now show by footprinting, ultraviolet (UV) and fluorescence melting studies that the BAU nucleotide in parallel TFOs (Figure 1B) binds to AT base pairs with high affinity and specificity, permitting stable and selective triplex formation at pH 7.0. This analogue does not produce enhanced binding to CG inversions. We also describe the full synthesis of the BAU phosphoramidite monomer (Figure 1A).
Figure 1.
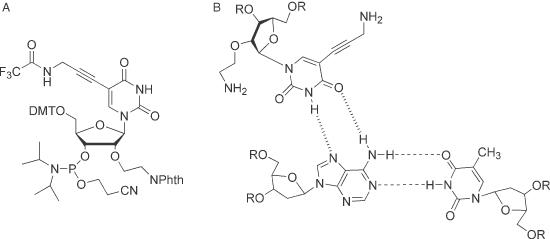
Chemical structures of (A) bis-amino uridine phosphoramidite (BAU), (B) BAU.AT triplet.
MATERIALS AND METHODS
Chemical synthesis of 2′-aminoethoxy-5-(3-aminoprop-1-ynyl)uridine phosphoramidite
The bis-aminouridine phosphoramidite monomer (Figure 1A) was synthesized from D-ribose and 5-iodouracil in 11 steps as shown in Scheme 1. The syntheses and product characterizations are described in full in the Supplementary Material. d-Ribose was first converted into methyl glycoside with 1% methanolic hydrochloric acid in methanol. The 3′ and 5′ hydroxyl groups were then protected with TIPDS using Markiewicz reagent (26) in pyridine to afford compound 3 as an anomeric mixture (α:β ∼2:1) in 72% yield. Introduction of the protected 2′-aminoethoxy substituent was achieved in three steps. First, alkylation of the 2′ position with an excess of methyl bromoacetate and sodium hydride in DMF at −5°C gave 4 (61%). Second, the ester moiety of 4 was reduced with LiBH4 in THF to give alcohol 5 (86%). Third, the hydroxyl group of 5 was substituted by phthalimide under Mitsunobu conditions (27) (91%). The phthalimide-protected 2′-aminoethoxy moiety was stable to the remaining steps of the synthesis and is compatible with oligonucleotide synthesis and deprotection (28). Treatment of compound 6 with acetic anhydride, acetic acid and concentrated sulfuric acid gave the tri-O-acetyl compound 7. This was converted to the protected nucleoside 8 (α and β-anomers 1:3 ratio) by reaction with 5-iodouracil under Vorbrüggen conditions (29). The β-anomer of 8 was deacetylated with sodium methoxide in methanol to give nucleoside 9, then protected selectively at the 5′-position by reaction with 4,4′-dimethoxytrityl chloride in pyridine to afford compound 10 in 76% yield. Addition of the 3-trifluoroacetamidoprop-1-yne (30) moiety was achieved by Sonogashira coupling (31,32) to give compound 11 which was converted to the target monomer 1 by treatment with 2-cyanoethoxy-(N,N-diisopropylamino)chlorophosphine in an argon atmosphere (70%). The above procedure was employed in the synthesis of multi-gram quantities of the phosphoramidite 1 and this monomer was used to synthesize a series of triplex-forming oligonucleotides using standard solid-phase oligonucleotide synthesis conditions. Oligonucleotides were cleaved from the solid support and deprotected using 10% methylamine in water containing 2.5 mg/ml phenol. The phenol scavenger prevents cyanoethylation of the N(3)-position of BAU with acrylonitrile, liberated by deprotection of the phosphodiesters.
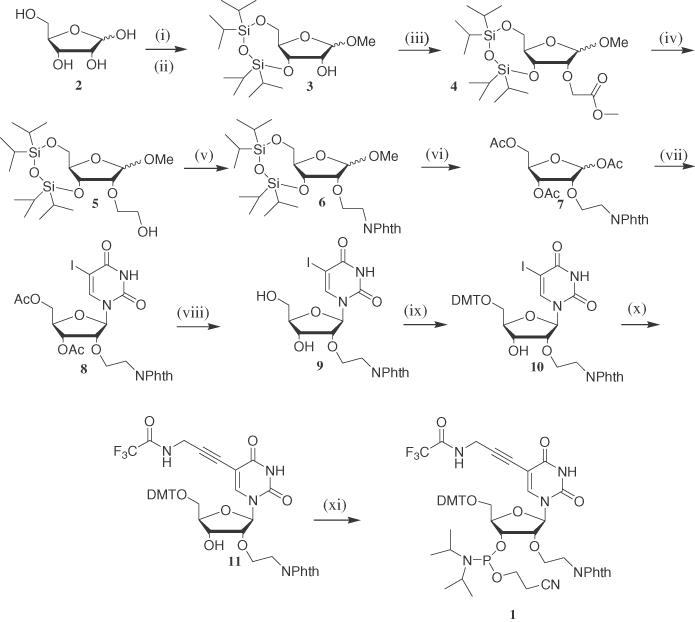
Scheme 1. Synthesis of 2′-aminoethoxy-5-(3-aminoprop-1-ynyl)uridine phosphoramidite 1 (BAU monomer). Reagents and conditions: (i) acetyl chloride, MeOH, rt, 3 h, quant.; (ii) TIPDSCl (1.2 eq), py, rt, 16 h, 72%; (iii) methyl bromoacetate (5 eq), NaH (4.4 eq), DMF, −5°C to rt, 6 h, 61%; (iv) LiBH4 (2 eq), THF, rt, 30 min, 86%; (v) Phthalimide (1.4 eq), PPh3 (1.4 eq), DEAD (1.4 eq), THF, rt, 2 h, 91%; (vi) AcOH:Ac2O (1:1), H2SO4, rt, 2 h, 79%; (vii) (a) HMDS, 5-iodouracil, TMSCl (1.0 eq), 120°C, 16 h; (b) 7 (0.3 eq), TMSOTf (3.2 eq), DCE, 0°C to rt, 2 h, 52%; (viii) NaOMe (3 eq), MeOH, rt, 2 h, quant.; (ix) DMTrCl (1.3 eq), py, rt, 3 h, 76%; (x) 3-trifluoroacetylamidoprop-1-yne (1.1 eq), CuI (0.3 eq), TEA (3 eq), Pd(PH3)4 (0.1 eq), DMF, rt, dark, 5 h, 90%; (xi) 2-cyanoethoxy-(N,N-diisopropylamino)chlorophosphine (1.2 eq), DIPEA (2.6 eq), THF, rt, 5 h, 70%.
Preparation of synthetic oligodeoxyribonucleotides
All oligonucleotides were synthesized on an Applied Biosystems 394 automated DNA/RNA synthesizer using the standard 0.2 μmol phosphoramidite cycle of acid-catalysed detritylation has, coupling, capping and iodine oxidation. Full details of the synthesis are presented in the Supplementary Material. After high-performance liquid chromatography, purification oligonucleotides were desalted using disposable NAP 10 Sephadex columns (Pharmacia), aliquoted into Eppendorf tubes and stored at −20°C. Purified oligonucleotides were analysed by matrix-assisted laser desorption ionization time-of-flight (MALDI-TOF) MS using a ThermoBioAnalysis Dynamo MALDI-TOF mass spectrometer in positive ion mode (Supplementary Material, Table S1) (33). Oligonucleotides for fluorescence melting experiments were labelled at the 5′ end of the TFO with methyl red serinol (Q) and at the 5′ end of the purine strand of the duplex target with 6-amidohexylfluorescein (F) using a commercially available monomer. The synthesis of the methyl red monomer will be described elsewhere. The sequences of the oligonucleotides used in this work are presented in Figure 2.
Figure 2.

Sequence of oligonucleotides used in this work. In each case the third strand is shown in bold and the duplex target is boxed. (A) Oligonucleotides used for fluorescence melting experiments, Q, methyl red serinol; F, 6-amidohexylfluorescein; N, BAU T, A, G or C; y.z = each base pair in turn TA, AT, CG or GC. (B) Oligonucleotides used for UV melting studies x, BAU; y.z, each base pair in turn TA, AT, CG, or GC. TFO-1 (containing one BAU residue) was identical to one of the oligonucleotides used for the fluorescence melting studies. (C) Sequence of the footprinting substrates derived from tyrT(43–59); y.z = each base pair in turn in different fragments. The DNA was labelled at the 3′ end of the EcoRI site (lower strand). The triplex target site is underlined and in bold and was targeted with TFO-4 (x = BAU).
Fluorescence melting experiments
Fluorescence melting experiments on the intermolecular triplexes were carried out using a Roche LightCycler as described previously (34). The oligonucleotides were dissolved in 50 mM sodium acetate buffer at the appropriate pH (5.0, 5.5 or 6.0) containing 200 mM NaCl. The sequences of the oligonucleotides are shown in Figure 2A. In these oligonucleotides, the purine-containing strand of the duplex was labelled with fluorescein at the 5′ end, while the third strand was labelled with methyl red at the 5′ end. These are positioned so that they are in close proximity on triplex formation, and the fluorescence is quenched. When the complex is heated, the third strand dissociates and there is a large increase in fluorescence. In this way, the dissociation of the third strand can be observed directly, without interference from dissociation of the duplex. In each case, the quencher was placed on the third strand, so that it could be added in excess, without increasing the fluorescence signal. Melting experiments were performed in a total volume of 20 μl and contained 0.25 μM duplex and 3 μM third strand. The complexes were first denatured by heating to 95°C at a rate of 0.1°C/s and maintained at this temperature for 5 min before cooling to 30°C at 0.1°C/s (or cooled to 22°C for complexes at pH 6.0). Since hysteresis was observed between the melting and annealing curves for some of the oligonucleotides, the complexes were melted and annealed again with a slower temperature gradient. In these cases, the samples were held at this lower temperature for a further 5 min before heating at a rate of 0.2°C/min to a temperature sufficiently high to ensure triplex melting (80°C). The samples were then cooled to 30°C at 0.2°C/min. Although the slowest rate of continuous temperature change in the LightCycler is 0.1°C/s, melting experiments were performed at slower rates of heating and cooling (0.2 or 0.067°C/min) by increasing the temperature in 1°C steps, leaving the samples to equilibrate for 5 or 15 min between each reading. Recordings were taken during both the annealing and denaturing steps. The LightCycler has one excitation source (488 nm) and the changes in fluorescence emission were measured at 520 nm. The Tm values were determined from the first derivatives of the melting profiles using the Roche LightCycler software. Each reaction was performed in triplicate and the Tm values usually differed by <0.5°C.
Ultraviolet melting experiments
UV melting experiments were performed on a Varian Cary 400 Scan UV-Visible spectrophotometer, in Hellma® SUPRASIL synthetic quartz, 10 mm pathlength cuvettes, monitoring at 260 nm, at a 1 μM concentration of all three oligonucleotide strands, in a volume of 1.5 ml. The sample chamber was flushed with air pumped through a dessicant to prevent condensation on the cells. Samples were prepared as follows. The third strand and the duplex were mixed in a 1:1 ratio in 2 ml Eppendorf tubes then lyophilized before resuspending in 1.5 ml of the appropriate buffer solution (10 mM sodium phosphate, pH 6.5 or 7.0 containing 1 mM EDTA and 200 mM NaCl). The samples were then filtered into the cuvettes with Kinesis regenerated cellulose 13 mm, 0.45 μM syringe filters. Following an initial heat and cool cycle (20 to 80 to 5°C at 10°C/min) to ensure uniform annealing of the strands, the UV melting curves were recorded for three consecutive heat and cool cycles (10 to 80 to 10°C at 0.5°C/min). Melting curves were then repeated over the triplex melting range at a slower rate of heating and cooling (0.06 or 0.12°C/min) to eliminate hysteresis.
Footprinting
DNA fragments
The tyrT(43–59) fragment contains a 17-base oligopurine tract between positions 43–59 (35). This was modified by site-directed mutagenesis to produce four different fragments, each containing a different base pair at position y.z (see Figure 2C). Radiolabelled fragments were produced by digesting each plasmid with EcoRI and AvaI and labelling at the 3′ end of the EcoR1 site using reverse transcriptase and [α-32P]dATP. Each fragment was separated from the remainder of the plasmid DNA on an 8% (w/v) non-denaturing polyacrylamide gel. After elution the fragment was dissolved in 10 mM Tris–HCl pH 7.5 containing 0.1 mM EDTA to give about 10 c.p.s./μl as determined on a hand-held Geiger counter (<10 nM).
DNase I footprinting
Radiolabelled DNA (1.5 μl) was mixed with TFOs (3 μl) at concentrations between 5 μM and 0.3 μM dissolved in 10 mM Tris–HCl pH 7.0 containing 50 mM sodium chloride. The complexes were left to equilibrate at 20°C for 30 min. Incubating for longer periods of time led to precipitation of the target DNA, as did the addition of 10 mM magnesium chloride. This is presumably due to the presence of multiple positive charges within the TFO. DNase I digestion was carried out by adding 2 μl of DNase I (typically 0.01 U/ml) dissolved in 200 mM NaCl containing 2 mM MgCl2 and 2 mM MnCl2. The reaction was stopped after 1 min by the addition of 4 μl of 80% formamide containing 10 mM EDTA, 10 mM NaOH and 0.1% (w/v) bromophenol blue. The products of digestion were separated on 9% polyacrylamide gels containing 8 M Urea. Samples were heated to 100°C for 3 min, before rapidly cooling on ice and loading onto the gel. Polyacrylamide gels (40 cm long, 0.3 mm thick) were run at 1500 V for about 2 h and then fixed in 10% (v/v) acetic acid. These were transferred to Whatman 3 mm paper and dried under vacuum at 86°C for 1 h. The dried gels were subjected to phosphorimaging using a Molecular Dynamics Storm phosphorimager.
The intensity of bands within each footprint was estimated using ImageQuant software. These intensities were then normalized relative to a band in the digest which is not part of the triplex target site, and which was not affected by addition of the oligonucleotides. Footprinting plots (36) were constructed from these data using Sigmaplot for windows and fitted using simple binding curves. C50 values, indicating the TFO concentration which reduces the band intensity by 50%, were then calculated from these.
Circular dichroism
Samples for circular dichroism (37) were prepared from 50 or 25 μM stock solutions of third strand and duplex in 10 mM sodium phosphate buffer pH 6.0, containing 200 mM NaCl and 1 mM EDTA. The samples were then heated at 80°C for 5 min, slowly cooled to room temperature and left at 4°C for 16 h. CD spectra were recorded on a JASCO J-710 spectropolarimeter at 25°C in a 1 mm pathlength cuvette. Spectra were collected between 320–200 nm, at 100 nm min−1, 4 s response time, 1 nm bandwidth. Each spectrum was accumulated five times, smoothed and the spectrum of the buffer was subtracted.
RESULTS AND DISCUSSION
We have previously shown that BAU forms stable triplets with AT base pairs and that the BAU.AT triplet is much more stable than T.AT (24). In the following studies, we have assessed the sequence specificity and pH dependency of triplex-forming oligonucleotides that contain this nucleotide analogue.
Fluorescence melting
We assessed the sequence selectivity of BAU in triple helix formation by determining the melting temperature (Tm) of the fluorescent-labelled intermolecular triplexes shown in Figure 2A. Tms were determined by fluorescence melting experiments and were carried out on a Roche LightCycler as previously described in (34). In the intact triplex, the methyl red and fluorescein moieties are in close proximity and collisional quenching prevents fluorescence emission. Heating the triplex causes the dissociation of the third strand, and a large increase in fluorescence is observed as the fluorophore and quencher are no longer in close proximity. In this way, the dissociation of the third strand can be observed directly, unlike UV melting, where interference from duplex dissociation can complicate the interpretation. Placing the quencher in the third strand allows us to use an excess of the third strand, without affecting the total fluorescence signal, thereby increasing the proportion of triplex that is formed.
In these experiments, a single BAU was incorporated at the centre of the 18mer TFO (indicated by N in Figure 2A), and this was targeted against four duplexes in which y.z was each base pair in turn. The stability of these complexes was compared with those formed by TFOs that contain each of the natural bases at this central position.
Representative melting profiles for the triplexes formed with the oligonucleotides that contain T or BAU in the central position are shown in Figure 3 and the results for all the triplexes at pHs between 5 and 6 are shown in Table 1. All these triplexes melted below 30°C at pH 7.0 as expected, since they contain a large number of C.GC triplets which require low pHs necessary for protonation of the third strand cytosines.
Figure 3.
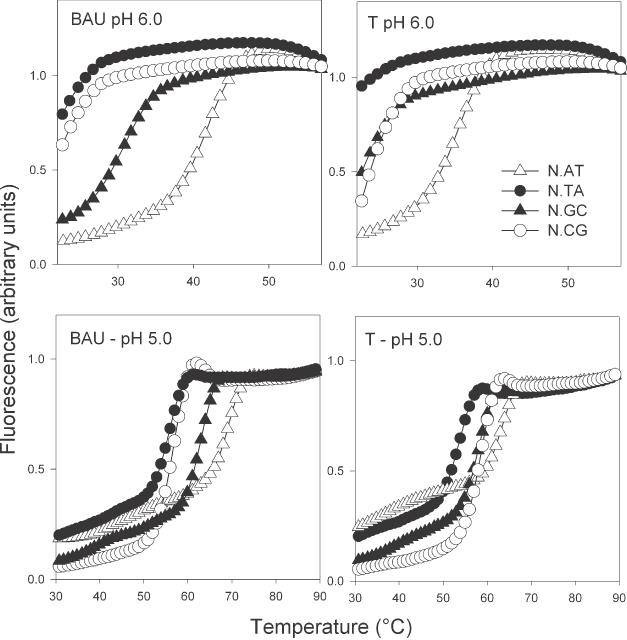
Representative fluorescence melting curves for the triplexes shown in Figure 2A. The data for pH 6.0 (upper panels) were obtained by heating at a rate of rate of 0.2°C/min, while the data at pH 5.0 (lower panels) were obtained by heating at a rate of 0.067°C/min. In these curves, the third strands contained BAU as the central base (N) targeted against duplexes containing each base pair (y.z) in turn (AT, open triangles; TA, closed circles; GC, closed triangles; CG, open circles). These experiments were performed in 50 mM sodium acetate (pH 5.0 or 6.0) containing 200 mM NaCl.
Table 1. Tm values (°C) determined for the melting of the triplexes shown in Figure 2A.
| pH | N = | A | G | C | T | BAU |
|---|---|---|---|---|---|---|
| 5.0 | N.AT | 57.3 | 54.7 | 55.3 | 63.8 | 70.8a |
| N.TA | 54.7 | 58.8 | 53.4 | 53.8 (10.0) | 55.9 (14.9) | |
| N.GC | 61.8 | 58.1 | 67.4 | 57.8 (6.0) | 62.8 (8.0) | |
| N.CG | 55.7 | 56.8 | 57.6 | 58.1 (5.7) | 56.7 (14.1) | |
| 5.5 | N.AT | 44.4 | 39.0 | 39.8 | 54.5 | 59.5 |
| N.TA | 40.7 | 47.3 | 38.7 | 39.2 (15.3) | 41.8 (17.7) | |
| N.GC | 48.4 | 44.1 | 56.6 | 43.7 (10.8) | 51.8 (7.7) | |
| N.CG | 40.7 | 41.9 | 43.7 | 44.5 (10.0) | 42.0 (17.5) | |
| 6.0 | N.AT | — | — | — | 34.8 | 41.2 |
| N.TA | — | — | — | — | — | |
| N.GC | — | — | 34.8 | — | 30.0 (11.2) | |
| N.CG | — | — | — | — | — |
The Tms were determined by the fluorescence melting technique at pH 5.0, 5.5 and 6.0 as indicated. The duplex concentration was 0.25 μM, while the third strand was 3 μM. Each value is the average of three separate determinations. The Tms shown obtained from melting profiles with a temperature change of 0.2°C/min indicates that the Tm was too low to measure (<25°C). For T and BAU, the figures in parentheses show the difference in Tm between the correct triplet (i.e. T.AT or BAU.AT) and the mismatched triplet. Tm values are reproducible to within less than 0.5°C.
aThis complex showed hysteresis between the melting and annealing profiles and the Tm for annealing was 64.4°C. The melting and annealing Tms for the BAU.AT complex were 69.0 and 66.0°C when the rate of temperature change was reduced to 0.067°C/min.
These Tm values show the expected sequence selectivity of the natural DNA bases, and the most stable complexes at the central duplex base pairs AT, GC and TA are formed with third strands containing T, C and G respectively (generating T.AT, C.GC and G.TA triplets). It can be seen that the addition of only one BAU residue greatly enhances triplex formation. However, there is significant hysteresis between the melting and annealing curves at the faster rate of temperature change (0.1°C/s). This is evident for the complexes with the central T.AT, C.GC, G.TA, BAU.AT and BAU.GC triplets (separate Tm values for melting and annealing are shown in Supplementary Material, Table S2). This is most pronounced for the BAU-AT triplet, with a 19°C difference between the melting and annealing curves, compared with an 11°C difference for T.AT. This hysteresis arises because the melting curves are not in thermodynamic equilibrium and indicates the presence of slow steps in the association and/or dissociation reactions (see below). On reducing the rate of heating to 0.2°C/min, the hysteresis is no longer evident and the Tms are approximately mid-way between the melting and annealing values obtained at the faster rate of temperature change. The only exception is for the complex that contains a single BAU.AT triplet, which still shows a 6°C difference in Tm. On reducing the heating rate further (0.067°C/min) the hysteresis was reduced, with melting and annealing Tms of 69.0 and 66.0°C.
Examination of these melting curves reveals that the most stable triplex is the one containing a single central BAU.AT triplet (Tm = 69.0°C at pH 5.0), and this is 5°C higher than the corresponding triplex with a T.AT in the same position (Tm = 63.8°C). This BAU.AT triplex is now more stable than C.GC at all pHs, in contrast to T.AT, which produces less stable complexes than C.GC. As well as showing increased triplex stability at AT, BAU produces more stable complexes than T at the GC base pairs. However, the ability of BAU to discriminate between AT and GC is similar to that of T, and the difference in Tm between BAU.GC and T.GC is very similar to that between BAU.AT and T.AT at pH 5.0 (5°C), and becomes greater at higher pHs (8°C at pH 5.5). In contrast, the difference between BAU.TA and T.TA is only 2°C, while BAU.CG is actually less stable than T.CG. The selectivity of BAU relative to T can also be seen by comparing the Tm of the correct triplets (BAU.AT and T.AT) with each of the mismatched triplets, as shown by the figures in parentheses in Table 1. In all cases (except BAU.GC at pH 5.5) the difference between the Tm values of the matched and mismatched triplets is greater for BAU than T and this effect is most pronounced for recognition of TA and CG (especially CG). These results demonstrate that, as well as producing very stable complexes at AT, BAU has enhanced discrimination against TA and CG. The enhanced discrimination against CG is especially interesting since T.CG is known to be the most stable triplet for recognition of CG interruptions (10). It therefore appears that the two amino groups in BAU are not in the correct location to enhance the interaction with CG. This emphasizes that the enhanced interaction with AT is not simply due to non-specific ionic interactions.
We also compared the relative stability of the different triplets formed with BAU and T by estimating ΔΔG values relative to the perfect matched BAU.AT and T.AT triplets from these melting profiles. These were derived from the width of the melting transitions, as described in (37,38). ΔGT can be estimated as C*(1 − T/Tm)/(1/Tm−1/T3/4) + T*X, where T3/4 is the temperature of the upper half maximum of the first derivative of the melting profile. C is 7.0 cal/mol/K and X = ln(total oligonucleotide concentration). From this, we calculated the difference in free energy (ΔΔG20°C) for the formation of triplets between BAU and each base pair relative to BAU.AT, and compared these with T interacting with each base pair relative to T.AT. These gave ΔΔG20°C values for BAU of 13, 3 and 11 kcal/mol at TA, GC and CG, respectively, relative to the BAU.AT triplet and 9, 6 and 3 kcal/mol for T relative to T.AT. These values again emphasize the increased discrimination of BAU against CG and TA.
We further examined the slow melting process by performing simple kinetic experiments on these triplexes as described previously (16). In these experiments, the complexes were first incubated at a temperature below their Tm. The temperature was then rapidly increased by 10°C (in a manner similar to that of temperature-jump relaxation kinetics) causing the equilibrium to shift towards the dissociated species, and producing a time-dependent increase in fluorescence. Under identical conditions to those used in the melting experiments (i.e. 0.25 μM duplex, 3 μM third strand, 50 mM sodium acetate containing 200 mM NaCl, pH 5.0) the complex with a central T.AT triplex dissociated with a half-life of 3.7 min at its Tm (63°C). This compared with a half-life of 10.6 min for the complex with a central BAU.AT triplet at 71°C. Clearly, the complex containing a single BAU.AT triplet dissociates much more slowly even at this elevated temperature, again demonstrating the dramatic effects produced by this single nucleotide substitution.
UV melting studies
In order to confirm that the fluorescence melting experiments were not influenced by the addition of the fluorophores, triplex melting experiments were also carried out using conventional UV spectroscopy using the oligonucleotides shown in Figure 2B. These duplexes have the same sequences as those used in the fluorescence melting studies, but lack the fluorescent groups. TFO-1 is the same oligonucleotide as used for the fluorescence melting studies and contains one BAU residue in addition to a 5′ methyl red, while TFO-2 and TFO-3 contain three and five BAU residues and lack the methyl red.
At pH 6.5, TFO-1, which contains a single central BAU residue, only shows triplex formation in the presence of the duplex with a central AT base pair (Tm = 19.5°C, Table 2). The corresponding triplexes with duplexes containing GC, CG and TA were not stable at this pH, as also were the triplexes formed with TFO-con1, which contains a T at this central position. At this pH TFO-2 and TFO-3 produced triplexes with Tms of 28.2°C and 43.6°C, respectively, at the duplex containing a central AT, while the Tms of the equivalent BAU.GC triplexes were 15.6 and 29.5°C, respectively. Representative melting profiles for these triplexes are shown in Figure 4. TFO-2 did not form stable triplexes with the duplexes containing central TA or CG base pairs, while TFO-3 produced complexes that melted at 17.0 and 15.2°C, respectively. It can be seen that at this pH the most stable alternative triplet (BAU.GC) lowers the Tm of the triplex by >13°C. At pH 7.0, only the BAU.AT triplex could be observed for both TFOs. It is possible to determine the increase in Tm per additional BAU.AT triplet by comparing the triplexes formed with TFO-2 and TFO-3. At pH 6.5, the increase is 7.7°C, and at pH 7.0 it is 5.2°C. This clearly illustrates that BAU is highly stabilizing relative to thymidine and very selective for AT base pairs. The lower melting temperatures obtained by UV melting reflect the fact that the TFO was present in a 1:1 ratio with the duplex, whereas in the fluorescence melting experiments the ratio was 12:1.
Table 2. Tm values for the triplexes shown in Figure 2B, determined by UV melting (x = BAU).
| pH 6.5 | pH 7.0 | |||||||
|---|---|---|---|---|---|---|---|---|
| x.GC | x.CG | x.AT | x.TA | x.GC | x.CG | x.AT | x.TA | |
| TFO-1 | — | — | 19.5 | — | — | — | — | — |
| TFO-2 | 15.6 | — | 28.2 | — | — | — | 21.0 | — |
| TFO-3 | 29.5 | 15.2 | 43.6 | 17.0 | — | — | 31.3 | − |
| TFO-Con1 | — | — | — | — | — | — | — | — |
The values are averages of the values determined from the melting and annealing curves. Dashes indicate Tm below 15°C. The experiments were performed in 10 mM sodium phosphate (pH 6.5 or 7.0), containing 1 mM EDTA and 200 mM NaCl. Tm values are accurate to within 0.5°C.
Figure 4.
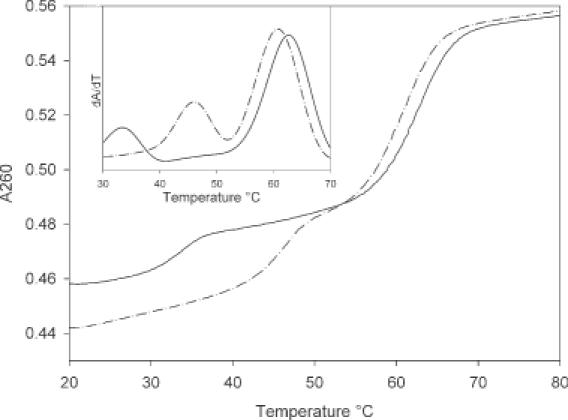
UV-melting curves for the triplex formed between TFO-3 and the duplex containing a central AT base pair (dashed line) and a central GC base pair (solid line) determined in 10 mM sodium phosphate, pH 6.5 containing 1 mM EDTA and 200 mM NaCl. The inset shows the first derivative of the melting profiles.
DNase I footprinting
We confirmed the selectivity and affinity of BAU by performing footprinting experiments with similar oligonucleotides. For these experiments, we used the footprinting substrates shown in Figure 2C and examined their interaction with TFO-4 at pH 7.0. In these experiments, we are targeting the central 12 bp of the 18 base oligopurine tract. The use of shorter oligonucleotides reduces the triplex affinity, making it easier to detect differences in affinity, as in our previous studies (13,21,24). Five BAU residues were incorporated into this oligonucleotide so as to be able to stabilize triplex formation at pH 7.0. The results are shown in Figure 5, in which a clear footprint is evident with the target containing a central AT (left hand panel). Quantitative analysis of the bands within this footprint produced a C50 value of 0.4 ± 0.1 μM. A much weaker footprint is evident with the target producing a BAU.GC triplet (at concentration of 3 μM and above), while no interaction is seen with TA or CG. The unmodified 12mer oligonucleotide d(TCTCTTTTTTCT) did not produce footprints at any of these target sequences at concentrations as high as 30 μM (data not shown) as expected, since this requires the formation of three C+.GC triplets.
Figure 5.
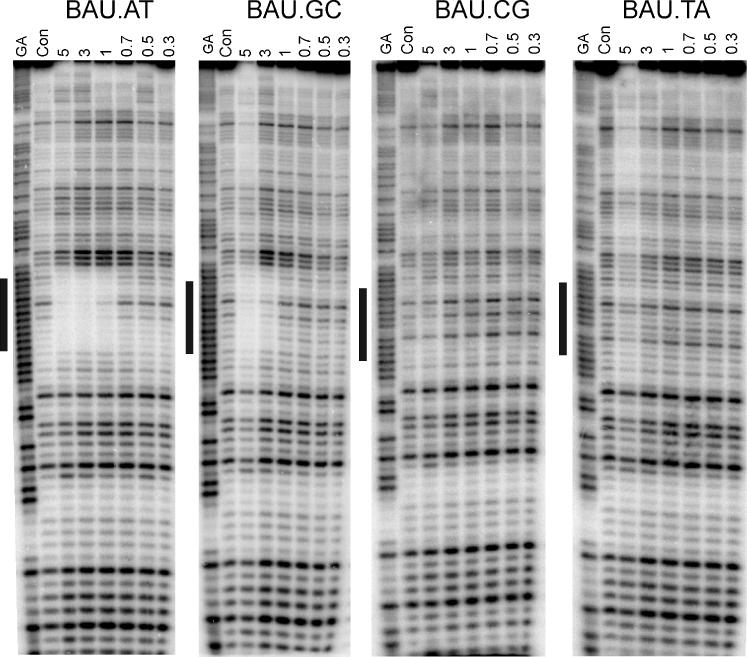
DNase I footprints showing the interaction of TFO-4 [d(xCxCxTxTxTCT-3′), x = BAU] with DNA fragments that contain each base pair in turn in the centre of the oligopurine tract, opposite the base shown in boldface. The TFO concentration (μM) is shown at the top of each lane. The experiments were performed in 10 mM Tris–HCl, pH 7.0, containing 50 mM sodium chloride and the complexes were left to equilibrate for 30 min before digestion with DNase I. The lanes labelled ‘GA’ and ‘con’ represent Maxim–Gilbert markers specific for purines and DNase I cleavage of duplex DNA in the absence of TFO, respectively.
We also examined whether BAU could stabilize triplexes that are formed in the GT motif, since the structures are known to be less stable than parallel (CT-containing) triplexes. These experiments used DNase I footprinting to examine the interaction of the 12mer oligonucleotides d(TGXGTXTXTXGT) (X = T or BAU), which are designed to form antiparallel G.GC, T.AT and BAU.AT triplets, with the target site that contains the central AT base pair. We observed no footprints at oligonucleotide concentrations as high as 10 μM for both the modified and unmodified TFOs, at pH 7.0 in the presence of 10 mM MgCl2. A similar negative results was observed using oligonucleotides with the reverse polarity, which could in theory form a parallel GT triplex. Although this result was disappointing, it demonstrates that the stabilization of parallel triplexes by BAU results from specific interactions with the amino substituents, and not from non-specific charge interactions.
Circular dichroism
The CD spectra of triplex DNA typically show an increased negative band below 220 nm, a characteristic that is associated with A-like DNA conformations (39,40). The CD spectra of the triplexes formed between the duplex target site and the unmodified oligonucleotide TFO-con2 or TFO-3 (containing five BAU residues), at pH 6.0 are shown in Figure 6. These spectra are qualitatively similar, with changes originating from the differences in UV absorption of the BAU and T bases (BAU has weaker absorption around 270 nm but absorbs more strongly above 290 nm). The CD spectra of cytosine-rich oligonucleotides are known to show evidence of helicity at low pH, and both TFOs showed this in the absence of the duplex. In both cases, the region ∼215 nm appeared to change significantly on addition of the duplex to the TFO, showing an increased negative band. The CD data suggest that the two triplexes are similar in their global structure. The observation that the CD-spectrum of TFO-3 (containing five BAU residues) is not enhanced relative to TFO-con2 suggests that the BAU residues have not significantly affected the structure of the single-stranded species and that these modifications have not conferred a special structure on the third strand.
Figure 6.
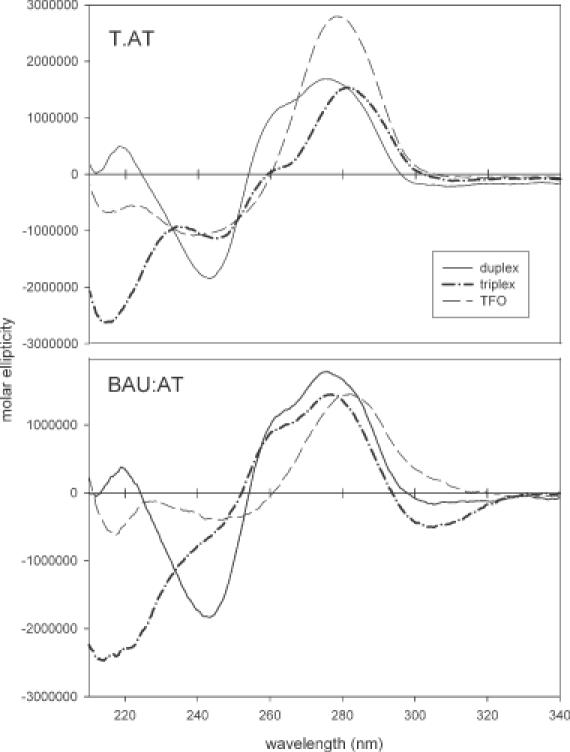
CD spectra of TFO-con2 (upper panel) and TFO-3 (lower panel) together with the target duplex and the triplex as indicated. The spectra were recorded in 10 mM sodium phosphate pH 6.0 containing 1 mM EDTA and 200 mM NaCl.
CONCLUSIONS
The above results clearly show that 2′-aminoethoxy-5-(3-aminoprop-1-ynyl)uridine (BAU) produces very stable triplets with AT base pairs, and that this nucleotide is highly selective for AT relative to all other Watson–Crick DNA base pairs in parallel TFOs. Triplets with pyrimidine:purine base pairs (BAU.TA and BAU.CG) are highly destabilized relative to triplets formed with purine:pyrimidine base pairs. Although BAU has enhanced affinity at RY base pairs, BAU and thymidine produce triplexes with very similar stabilities at YR base pairs (Table 1). The 2′-aminoethoxy and 5-aminopropargyl groups of BAU therefore appear to have no significant influence on the stability of triplets with pyrimidine:purine base pairs. This is especially noteworthy since T.CG is the most stable natural triplet at CG inversions, and this interaction is not enhanced with BAU. It is known that both amino groups are required for maximum triplex stabilization (24) and modelling studies have suggested that the stabilizing interactions occur between the amino groups of BAU and specific phosphates in the BAU.AT triplet (24). It is, therefore, likely that one or both of these specific contacts do not occur in BAU.YR triplets. In conclusion, the high selectivity and the enhanced stability of the BAU.AT triplet relative to T.AT and its discrimination against other base pair indicate that BAU is a very useful base analogue for the sequence-specific creation of stable triple helices at pH 7.0.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at NAR Online.
Acknowledgments
ACKNOWLEDGEMENTS
This work was supported by grants from the European Union and Cancer Research UK. V.E.C.P. and D.A.R. are supported by research studentships from BBSRC and EPRSC, respectively.
REFERENCES
- 1.Chan P.P. and Glazer,P.M. (1997) Triplex DNA: fundamentals, advances and potential applications for gene therapy. J. Mol. Med., 75, 267–282. [DOI] [PubMed] [Google Scholar]
- 2.Gorman L. and Glazer,P.M. (2001) Directed gene modification by triple helix formation. Curr. Mol. Med., 1, 391–399. [DOI] [PubMed] [Google Scholar]
- 3.Praseuth D., Guieysse,A.L. and Hélène,C. (1999) Triple helix formation and the antigene strategy for sequence-specific control of gene expression. Biochim. Biophys. Acta, 1489, 181–206. [DOI] [PubMed] [Google Scholar]
- 4.Neidle S. (1997) Recent developments in triple-helix regulation of gene expression. Anticancer Drug Des., 12, 433–442. [PubMed] [Google Scholar]
- 5.Vasquez K.M. and Wilson,J.H. (1998) Triplex-directed modification of genes and gene activity. Trends Biochem. Sci., 23, 4–9. [DOI] [PubMed] [Google Scholar]
- 6.Knauert M.P. and Glazer,P.M. (2001) Triplex forming oligonucleotides: sequence-specific tools for gene targeting. Hum. Mol. Gen., 10, 2243–2251. [DOI] [PubMed] [Google Scholar]
- 7.Thuong N.T. and Hélène,C. (1993) Sequence specific recognition and modification of double helical DNA by oligonucleotides. Angew. Chem. Int. Ed. Engl., 32, 666–690. [Google Scholar]
- 8.Soyfer V.N. and Potoman,V.N. (1996) Triple-Helical Nucleic Acids. Springer-Verlag, New York. [Google Scholar]
- 9.Fox K.R. (2000) Targeting DNA with triplexes. Curr. Med. Chem., 7, 17–37. [DOI] [PubMed] [Google Scholar]
- 10.Gowers D.M. and Fox,K.R. (1999) Towards mixed sequence recognition by triple helix formation. Nucleic Acids Res., 27, 1569–1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Asensio J.L., Lane,A.N., Dhesi,J., Bergvist,S. and Brown,T. (1998) The contribution of cytosine protonation to the stability of parallel DNA triple helices. J. Mol. Biol., 275, 811–822. [DOI] [PubMed] [Google Scholar]
- 12.Völker J. and Klump,H.K. (1994) Electrostatic effects in DNA triple helices. Biochemistry, 33, 13502–13508. [DOI] [PubMed] [Google Scholar]
- 13.Keppler M.D. and Fox,K.R. (1997) Relative stability of triplexes containing different numbers of T.AT and C+.GC triplets. Nucleic Acids Res., 25, 4644–4649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Soto A.M., Loo,J. and Marky,L.A. (2002) Energetic contributions for the formation of TAT/TAT, TAT/CGC+, and CGC+/CGC+ base triplet stacks. J. Am. Chem. Soc., 124, 14355–14363. [DOI] [PubMed] [Google Scholar]
- 15.Roberts R.W. and Crother,D.M. (1996) Prediction of the stability of DNA triplexes. Proc. Natl Acad. Sci. USA, 93, 4320–4325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.James P.L., Brown,T. and Fox,K.R. (2003) Thermodynamic and kinetic stability of intermolecular triple helices containing different proportions of C+.GC and T.AT triplets. Nucleic Acids Res., 31, 5598–5606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hampel K.J., Crosson,P. and Lee,J.S. (1991) Polyamines favour DNA triplex formation at neutral pH. Biochemistry, 30, 4455–4459. [DOI] [PubMed] [Google Scholar]
- 18.Thomas T. and Thomas,T.J. (1993) Selectivity of polyamines in triplex DNA stabilization. Biochemistry, 32, 14068–14074. [DOI] [PubMed] [Google Scholar]
- 19.Rajeev KG., Jadhav,V.R. and Ganesh,K.N. (1997) Triplex formation at physiological pH: comparative studies on DNA triplexes containing 5-Me-dC tethered at N4 with spermine and tetraethyleneoxyamine. Nucleic Acids Res., 25, 4187–4193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Roig V. and Asseline,U. (2003) Oligo-2′-deoxyribonucleotides containing uracil modified at the 5-position with linkers ending with guanidinium groups. J. Am. Chem. Soc., 125, 4416–4417. [DOI] [PubMed] [Google Scholar]
- 21.Bijapur J., Keppler,M.D., Bergqvist,S., Brown,T. and Fox,K.R. (1999) 5-(1-propargylamino)-2′-deoxyuridine (UP): a novel thymidine analogue for generating DNA triplexes with increased stability. Nucleic Acids Res., 27, 1802–1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Blommers M.J.J., Natt,F., Jahnke,W. and Cuenoud,B. (1998) Dual recognition of double stranded DNA by 2′-aminoethoxy-modified oligonucleotides: the solution structure of an intramolecular triplex obtained by NMR spectroscopy. Biochemistry, 37, 17714–17725. [DOI] [PubMed] [Google Scholar]
- 23.Cuenoud B., Casset,F., Husken,D., Natt,F., Wolf,R.M., Altmann,K.-H., Martin,P. and Moser,H.E. (1998) Dual recognition of double-stranded DNA by 2′-aminoethoxy-modified oligonucleotides. Angew. Chem. Int. Ed., 37, 1288–1291. [DOI] [PubMed] [Google Scholar]
- 24.Sollogoub M., Darby,R.A.J., Cuenoud,B., Brown,T. and Fox,K.R. (2002) Stable DNA triple helix formation using oligonucleotides containing 2′-aminoethoxy,5-propargylamino-U. Biochemistry, 41, 7224–7231. [DOI] [PubMed] [Google Scholar]
- 25.Sollogoub M., Dominguez,B., Fox,K.R. and Brown,T. (2000) Synthesis of a novel bis-amino-modified thymidine monomer for use in DNA triplex stabilisation. Chem. Commun., 2315–2316. [Google Scholar]
- 26.Markiewicz W.T., Padyukova,N.S., Samek,Z. and Smrt,J. (1980) The reaction of 1,3-dichloro-1,1,3,3-tetraisopropyldisiloxane with cytosine arabinoside and 1-(6-deoxy-α-l-talofuranosyl)uracil. Coll. Czech Chem. Commun., 40, 1860–1865. [Google Scholar]
- 27.Mitsunobu O. (1981) The use of diethyl azodicarboxylate and triphenylphosphine in synthesis and transformation of natural-products. Synthesis, 1, 1–28. [Google Scholar]
- 28.Kume A., Sekine,M. and Hata,T. (1982) Phthaloyl group: a new amino protecting group of deoxyadenosine in oligonucleotide synthesis. Tetrahedron Lett., 23, 4365–4368. [Google Scholar]
- 29.Vorbruggen H., Krolikiewicz,K. and Bennua,B. (1981) Nucleoside syntheses. 22. Nucleoside synthesis with trimethylsilyl triflate and perchlorate as catalysts. Chem. Ber. Recl., 114, 1234–1255. [Google Scholar]
- 30.Cruickshank K.A. and Stockwell,D.L. (1988) Oligonucleotide labelling: a concise synthesis of a modified thymidine phosphoramidite. Tetrahedron Lett., 29, 5221–5224. [Google Scholar]
- 31.Hobbs F.W. (1989) Palladium-catalyzed synthesis of alkynylamino nucleosides – a universal linker for nucleic-acids. J. Org. Chem., 54, 3420–3422. [Google Scholar]
- 32.Takahashi S., Kuroyama,K., Sonogashira,N. and Hagihara,N. (1980) A convenient synthesis of ethynylarenes and diethynylarenes. Synthesis, 627–630. [Google Scholar]
- 33.Langley G.J., Herniman,J.M., Davies,N.L. and Brown,T. (1999) Simplified sample preparation for the analysis of oligonucleotides by matrix-assisted laser desorption/ionisation time-of-flight mass spectrometry. Rapid Commun. Mass Spectrom., 13, 1717–1723. [DOI] [PubMed] [Google Scholar]
- 34.Darby R.A.J., Sollogoub,M., McKeen,C., Brown,L., Risitano,A., Brown,N., Barton,C., Brown,T. and Fox,K.R. (2002) High throughput measurement of duplex, triplex and quadruplex melting curves using molecular beacons and a LightCycler. Nucleic Acids Res., 30, e39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brown P.M., Madden,C.A. and Fox,K.R. (1998) Triple helix formation at different positions on nucleosomal DNA. Biochemistry, 37, 16139–16151. [DOI] [PubMed] [Google Scholar]
- 36.Dabrowiak J.C. and Goodisman,J. (1989) Quantitative footprinting analysis of drug-DNA interactions. In: Kallenbach,N.R. (ed.), Chemistry and Physics of DNA-Ligand Interactions. Adenine Press, NY, pp. 143–174. [Google Scholar]
- 37.McDowell J.A. and Turner,D.H. (1996) Investigation of the structural basis for thermodynamic stabilities of tandem GU mismatches: solution structure of (rGAGGUCUC)2 by two-dimensional NMR and simulated annealing. Biochemistry, 35, 14077–14089. [DOI] [PubMed] [Google Scholar]
- 38.Gralla J. and Crothers,D.M. (1973) Free energy of imperfect nucleic acid helices. III. Small internal loops resulting from mismatches. J. Mol. Biol., 78, 301–319. [DOI] [PubMed] [Google Scholar]
- 39.Gray D.M., Hung,S.-H. and Johnson,K.H. (1995) Absorption and circular dichroism spectroscopy of nucleic acid duplexes and triplexes. Meth. Enzymol., 246, 19–34. [DOI] [PubMed] [Google Scholar]
- 40.Gray D.M., Morgan,A.R. and Ratliff,R.L. (1978) A comparison of the circular dichroism spectra of synthetic DNA sequences of the homopurine.homopyrimidine and mixed purine–pyrimidine types. Nucleic Acids Res., 5, 3679–3695. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.