


Abstract
Short interfering RNAs (siRNAs) directed against different regions of genes display marked variation in their potency in mediating mRNA degradation. Various factors have been proposed to affect the efficacy of siRNA. We explored some of the factors by evaluating in cultured human cells 28 randomly selected siRNAs targeting the GPR39 and MGC29643 transcripts derived from the same genetic locus but transcribed in opposite directions. Twenty of the 24 siRNAs targeting the overlapping regions of the transcripts simultaneously reduced the levels of both transcripts. Single nucleotide changes in either of the siRNA strands significantly reduced the gene-silencing efficiency of the siRNA on targeted sense transcript without affecting the antisense transcript. Overall, we observed a greater gene-silencing efficiency on the MGC29643 transcript than on the GPR39 transcript in HeLa cells. Since MGC29643 transcript is more abundant than the GPR39 transcript [0.24 versus 0.008% relative to 100% for glyceraldehyde-3-phosphate dehydrogenase (GAPDH)], the results suggest that the abundance of the mRNA affects the efficiency of silencing. Two additional observations supported this hypothesis. First, GAPDH whose intracellular level is the highest of the three was the most efficiently silenced. Second, a reversal of gene-silencing efficiency was observed in U-138 MG cells in which the relative abundance of the GPR39 and MGC29643 transcripts is also reversed. Our study suggests that low-abundant transcripts are less susceptible to siRNA-mediated degradation than medium- and high-abundant transcripts.
INTRODUCTION
RNA interference (RNAi) is a post-transcriptional gene-silencing process induced in diverse organisms by double-stranded RNAs (dsRNAs) homologous in sequence to the silenced genes (1,2). In mammalian cells, the introduction of long dsRNAs (>30 bp) led to the activation of a global, sequence-nonspecific response resulting in the blockage of protein synthesis and mRNA degradation (3). Small dsRNAs of 21–23 nt in length, however, can bypass the sequence-independent response of mammalian cells and induce sequence-specific degradation of target mRNA (4–6). These small dsRNAs, termed small interfering or short interfering RNAs (siRNAs), act as ‘guides’ within a nuclease complex, the RNA-induced silencing complex (RISC), to direct cleavage and degradation of target mRNA (7–11). Target recognition is a highly sequence-specific process mediated by the siRNA complementary to the target mRNA (12).
Ever since the demonstration that siRNAs mediate sequence-specific gene silencing in cultured mammalian cells, the siRNA-based technology has been successfully used to down-regulate the expression of mammalian genes and various infectious agents (13–20). Gene silencing by siRNA is commonly carried out by transient transfection of cells with synthetic siRNAs or by using expression vectors to produce cells that transiently or stably express siRNAs or short hairpin RNAs. The relative ease of generating synthetic siRNAs, either by chemical synthesis or by in vitro transcription, coupled with the immediate effectiveness makes transient transfection with synthetic siRNAs the preferred method in conducting large-scale analysis of siRNAs in readily transfectable cells.
One difficulty in the application of siRNA-based technology is that siRNAs display a wide range of activities reducing target mRNA or protein expression by 0% to >90%. Only a fraction of siRNAs result in a significant reduction of targets (21,22). Significant progress has been made in recent years toward understanding some of the determinants that affect siRNA efficacy. One group of determinants relates to the nature of the siRNA sequences. For example, it has been reported that nucleotide mismatches created at various positions in the siRNAs have varying effects on the specificity of target recognition (21,23–25) and that siRNA duplex asymmetry and internal stability play a critical role in determining the function and longevity of some siRNAs (26,27). The second group relates to the properties of the target mRNA. It is suggested that extensive secondary structure in the mRNA, the presence of RNA-associated proteins and the specific subcellular localization of the mRNA may render the transcript inaccessible to siRNA-incorporated RISC binding (21). The turnover rate of mRNA is often considered a factor, but how the intracellular level of mRNA affects the effectiveness of an siRNA is unclear. The third group relates to the properties of the target cells: variations in transfection efficiency, growth confluence, passage number, differentiation status and the toxicity of the transfection reagents all play a role in determining the outcome of gene silencing by siRNA.
To eliminate many of the variables, especially those pertaining to the last group, we evaluated the relative gene silencing of a pair of sense and antisense transcripts in the same cells. In the last few years, a number of prokaryotic and eukaryotic genes have been found to have a naturally occurring antisense transcript (28,29). The sense and antisense transcripts of these genes are transcribed from opposite DNA strands of the same locus and their sequences overlap in the protein-coding and/or non-coding regions. Some of the antisense transcripts encode for proteins involved in diverse biological functions. Non-coding antisense transcripts have also been described and their role appears to be associated with the regulation of gene expression (30). The scope of antisense transcription occurring in the human genome has been explored (31,32). A recent study that integrated computational tools with experimental methods estimated that at least 8% of the predicted 40 000 human genes have an antisense partner (32).
In the present study, we evaluated the relative gene-silencing efficiency of a pair of sense and antisense transcripts, GPR39 and MGC29643, in cultured human cells. GPR39 is a member of the G-protein-coupled receptor family and may be involved in the regulation of growth hormone release in the brain (33). MGC29643 encodes a 165 amino acid protein whose function is currently unknown (34). Nucleotide sequence alignment demonstrates that these transcripts are derived from the same genetic locus on chromosome 2q21–2q22 but transcribed in opposite directions. The overlapping region encompasses 1261 nt between the 3′ external exons of these genes. We also evaluated the silencing efficiency of the human glyceraldehyde-3-phosphate dehydrogenase (GAPDH) gene, as an example of a highly expressed transcript (35). Our study demonstrates that siRNAs can induce the degradation of a sense and an antisense transcript simultaneously and that the strand-specific activity of an siRNA can be altered by single-base mismatches. The study also reveals a correlation between the silencing efficiency of siRNA and the intracellular transcript levels.
MATERIALS AND METHODS
siRNA design and synthesis
Sequence information of the target mRNAs was obtained from the NCBI Entrez nucleotide database: GPR39 (accession no. NM_001508, AC079773), MGC29643 (accession no. NM_144586), and GAPDH (accession no. M33197). All siRNAs were designed according to the general guidelines described by Elbashir et al. (36). Briefly, using the siRNA-designing tool (www.ambion.com), the entire length of the mRNA sequences was scanned for the 21 nt sequences that contain the AA(N19) motif (N for any nucleotide). The 21 nt sequences with ∼50% of G/C and with no significant homology to other human genes were selected as the target sequences. The target sites of the siRNAs directed against each of the three transcripts are fairly evenly distributed and span the entire length of the transcripts. The ranges of G/C content of the siRNAs are 38.1–66.7% for GPR39 and 42.9–57.1% for both MGC29643 and GAPDH.
The target sequences of siRNAs begin at the following positions corresponding to each of the three transcripts (counting from the translation initiation codon ATG): GPR39 (siRNAs GPR39-1 through GPR39-14): 196, 616, 895, 925, 1018, 1206, 1230, 1264, 1321, 1362, 1462, 1567, 1632 and 1826; MGC29643 (siRNAs MGC-1 through MGC-14): 86, 170, 280, 397, 576, 641, 655, 747, 885, 903, 1003, 1174, 1324 and1360; GAPDH (siRNAs GAP-1 through GAP-12): 25, 163, 208, 415, 433, 445, 515, 655, 775, 910, 946 and 1033. The target sequence for the nonspecific control siRNA is 5′-AAA CGT GGT CTC GAA CAT GCA. The siRNA duplex was synthesized by a transcription-based method using the Silencer™ siRNA Construction Kit (Ambion, Austin, TX) (37,38). Successful synthesis of the 21mer siRNA duplexes was verified by non-denaturing PAGE and siRNA was quantified by spectrophotometry. The antisense strands of the siRNAs are the reverse complements of the target mRNAs, and the sense siRNA strands have the same sequences as the target mRNAs. A UU dinucleotide sequence was incorporated at the 3′ end of the sense siRNA strands during in vitro transcription. The end product is an RNA duplex consisting of two 21mer RNAs with 19 complementary nucleotides and 3′ terminal non-complementary dimers of uridine. A chemically synthesized siRNA targeting the GAPDH transcript, GAP-Ambion, was obtained from Ambion.
Cell culture and siRNA transfection
Human HeLa cervical cancer cell line and human U-138 MG glioma cell line (ATCC, Manassus, VA) were grown at 37°C in RPMI 1640 complete growth medium (GIBCO/Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum, 1× glutamine and 1% penicillin and streptomycin. Twenty-four hours before siRNA transfection, the cells were trypsinized and seeded in a T25 tissue culture flask in a fresh medium at the following cell densities: 4 × 104 cells/ml for HeLa (2 × 105 cells per flask) and 5 × 104 for U-138 MG (2.5 × 105 cells per flask). After 24 h, when a cell confleunce of 40–50% was reached, three identical T25 flasks of cells were transiently transfected with the sequence-specific siRNAs and the sequence-nonspecific control siRNA using oligofectamine (Invitrogen Inc., Carlsbad, CA) according to the manufacturer's instructions. The final concentration of siRNA in the culture medium was 10 nM. For quantitative real-time RT–PCR, the cells were harvested ∼48 h after transfection at a confleunce of 70–80%. Total cellular RNA was isolated from the cells using the RNeasy Mini Kit (QIAGEN Inc., Valencia, CA), quantified by spectrophotometry and stored in −80°C until use.
Determination of siRNA gene-silencing efficiency
Quantitative real-time RT–PCR (TaqMan assay) was carried out to determine the extent of siRNA-mediated target mRNA degradation. Dual-labeled fluorogenic probes and primers were synthesized at Integrated DNA Technologies, Inc. (Coralville, IA) and their sequences are as follows: 5′-/56-FAM/CCC TGT CCT GCA AGC TGC ACA CTT/3BHQ-1/3′ (GPR39 probe), 5′-GGA ATC CCC TGA CCA CGT CC (GPR39 forward primer), 5′-AGC TGC AGG CCT CGA AGA G (GPR39 reverse primer), 5′-/56-FAM/AAG TGC CGG GAT CAT GTA CCG CAA G/3BHQ-1/3′ (MGC29643 probe), 5′-CAT GTG TCA GAA AGA AGT GAT GGA G (MGC29643 forward primer), 5′-GCC GCT GAT GAT GCA CAG (MGC29643 reverse primer), 5′-/56-FAM/TTG GTC GTA TTG GGC GCC TGG TC/3BHQ-1/3′ (GAPDH probe), 5′-GGT GAA GGT CGG AGT CAA CG (GAPDH forward primer) and 5′-AGA GTT AAA AGC AGC CCT GGT G (GAPDH reverse primer). A one-step real-time RT–PCR was carried out using an ABI PRISM 7700 sequence detection system (Applied Biosystems, Foster City, CA). The 50 μl reaction contained the following components: 1× Taq polymerase buffer A, 0.8 mM of dNTPs, 5.5 mM of MgCl2, 12.5 U of M-MLV reverse transcriptase, 1.25 U of AmpliTaq Gold DNA polymerase, 0.2 μM of probe and each of the primers and 5–500 ng of total cellular RNA. All reagents except the probes and primers were purchased from Applied Biosystems (Foster City, CA). Negative control reactions omitting either RNA or reverse transcriptase were run with all assays. The one-step real-time RT–PCR, performed in a 96-well plate, consists of cycles at 48°C for 30 min (cDNA synthesis), 95°C for 10 min (inactivation of reverse transcriptase and activation of DNA polymerase), and 95°C for 15 s and 60°C for 1 min for 40 cycles (real-time PCR). Each RNA sample was run in triplicates. Real-time PCR data were collected using the ABI PRISM 7700 sequence detection system.
Relative quantification of the target mRNAs was achieved according to the principle of the standard-based quantitative PCR method (39). Briefly, an RNA stock expressing the appropriate target was used as a calibrator to generate a standard curve. The target quantity of test samples was derived from the standard curve through the comparison of the values of fluorescent signal intensity. The amplification of a ‘housekeeping’ gene transcript, 18S rRNA, was carried out to standardize the amount of RNA added to a reaction. The amount of a target transcript was divided by the amount of 18S rRNA to obtain the normalized target quantity.
Regular RT–PCR and gel electrophoresis of amplified products were also carried out to provide visual examples of gene silencing with selected RNA samples. The primers used in the study were synthesized at Integrated DNA Technologies Inc. (Coralville, IA) and their sequences are as follows: 5′-TCC AGT GCT ACC AGT GTG AAG (MGC29643 forward primer), 5′-GGC TGG AAG AAC AAT GCA G (MGC29643 reverse primer), 5′-GCT GAT GAT GCA CAG GAC TTG (GPR39 forward primer), 5′-GTG CCT GTC TCT TTC ACC ATC (GPR39 reverse primer), 5′-CGC TGA GTA CGT CGT GGA GTC (GAPDH forward primer) and 5′-GGT GGC AGT GAT GGC ATG GAC (GAPDH reverse primer). A one-step RT–PCR was carried out using a GeneAmp PCR system 9600 (Applied Biosystems). The 50 μl reaction contained the following components: 1× Taq polymerase buffer A, 0.8 mM of dNTPs, 2.5 mM of MgCl2, 12.5 U of M-MLV reverse transcriptase, 2.5 U of AmpliTaq Gold DNA polymerase, 0.4 μM of each of the primers and 500 ng (for MGC29643 and GPR39) or 50 ng (for GAPDH) of total cellular RNA. All reagents except the primers were purchased from Applied Biosystems (Foster City, CA). The one-step RT–PCR consists of cycles at 48°C for 30 min (cDNA synthesis), 95°C for 10 min (inactivation of reverse transcriptase and activation of DNA polymerase), 95°C for 30 s, 60°C for 1.5 min and 72°C for 30 s for 28 cycles (PCR), and 72°C for 7 min. After RT–PCR, 5 μl of the amplified MGC29643 and GPR39 products were mixed in a tube and were separated by electrophoresis in a 1.5% agarose gel containing ethidium bromide along with a 100 bp DNA ladder (Invitrogen) as the size marker. Used as an internal control, 5 μl of the amplified GAPDH product were also separated by electrophoresis in a 1.5% agarose gel. The gel image was obtained by exposure of the gel to an ultraviolet (UV) transilluminator (302 nm) and recorded using the Kodak EDAS 290 digital camera.
RESULTS
Genomic organization and transcript levels of GPR39 and MGC29643
The cloning of the cDNAs for GPR39 and MGC29643 has been reported previously (33,34). A schematic diagram depicting the genomic organization of these genes is shown in Figure 1. The GPR39 gene is composed of two exons separated by a large intron of ∼228 kb. A bacterial artificial chromosome (BAC) clone that includes exon 2 and the presumptive 3′-untranslated region (3′-UTR) of the gene (accession no. AC079773) was identified by sequence alignment. The gene is localized to chromosome 2q21–q22 (33). The full-length cDNA for MGC29643 was isolated from human cDNA libraries of diverse sources (34). The cDNA clone of MGC29643 contains the 498 nt coding sequence flanked by a 273 nt 5′-UTR and a 1119 nt 3′-UTR. The MGC29643 gene consists of four exons and encodes a 165 amino acids protein of unknown function. Nucleotide sequence alignment of GPR39 and MGC29643 transcripts shows that they are derived from the same genetic locus but transcribed from opposite directions. The transcripts overlap in their 3′ external exons by 1261 nt.
Figure 1.
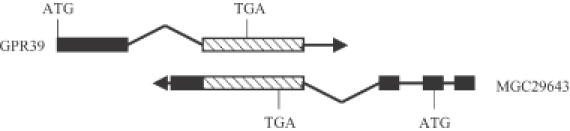
Genomic organization of human GPR39 and MGC29643 genes. The GPR39 and MGC29643 genes are localized to the same site on human chromosome 2q21–q22 but transcribed in opposite directions. The boxes represent the exons and the lines represent the introns. Shaded boxes show the overlapping regions between the 3′ extreme exons of the genes. The locations of translation initiation (ATG) and termination (TGA) codons are indicated.
The relative transcript abundance of GPR39, MGC29643 and GAPDH in HeLa and U-138 MG cells was determined by quantitative real-time RT–PCR. The primers and probes used in the study were designed to hybridize to the non-overlapping regions of the transcripts to ensure the detection of only the corresponding transcript. The relative transcript abundance is expressed as the percentage of the target transcript level to that of the housekeeping gene GAPDH. The average values of five independent RNA preparations were used to calculate the transcript abundance. In HeLa cells, the GPR39 and MGC29643 mRNA levels were 0.008 and 0.24% respectively, relative to GAPDH mRNA (set arbitrarily at 100%). Thus, MGC29643 mRNA is ∼30 times more abundant than the GPR39 mRNA in these cells. In U-138 MG cells, the GPR39 and MGC29643 mRNA levels were 0.07 and 0.005% relative to GAPDH mRNA, respectively. Therefore, GPR39 mRNA is 14 times more abundant than the MGC29643 mRNA in these cells. The GAPDH mRNA level in HeLa cells is ∼60% of the level of GAPDH mRNA in U-138 MG cells.
Gene-silencing efficiency of siRNAs targeting GPR39, MGC29643 and GAPDH in HeLa cells
A schematic map depicting the target sites of siRNAs along the three transcripts is shown in Figure 2. The efficiency of gene silencing is measured by the percentage of target mRNA reduction in siRNA-transfected cells relative to mock-transfected cells. The average transcript levels from six independent siRNA transfection experiments are shown in Figure 3. Consistent with other reports, our study found that siRNAs directed against each of the three endogenous mammalian transcripts vary in their potency, reducing the transcript level from 0 to 90%. As expected, the siRNAs that target the unique (non-overlapping) regions of these transcripts (Figure 3A and B; siRNAs MGC-1, MGC-2, GPR39-1, GPR39-2) only induced the reduction of the corresponding transcripts. Twenty of the 24 siRNAs targeting the overlapping regions of GPR39 and MGC29643 reduced the levels of both transcripts. The MGC29643 transcript was effectively silenced, with 36% (5/14) of the siRNAs reducing the transcript level by at least 70% (Figure 3A). In contrast, only 14% (2/14) of the siRNAs reduced the level of GPR39 transcript by 70% or more (Figure 3B). Surprisingly, the MGC29643 transcript was more efficiently down-regulated even by siRNAs originally selected based on the GPR39 sequence. Six of the 12 (50%) overlapping GPR39-targeted siRNAs reduced the MGC29643 transcript level by 70% or more (Figure 3B). In contrast, none of the 12 overlapping MGC29643-targeted siRNAs reduced GPR39 by 70% (Figure 3A). In total, 18 of the 24 (75%) overlapping siRNAs induced a greater reduction of the MGC29643 transcript than the GPR39 transcript; only 3 of the 24 (13%) siRNAs induced a greater reduction of the GPR39 transcript (Figure 3A and B; siRNAs MGC-4, GPR39-5, GPR39-6). The global silencing efficiency of the siRNAs appears to correlate with the cellular levels of these transcripts, as MGC29643 is expressed at moderate levels, while GPR39 is expressed at very low levels. The siRNAs targeting the highly expressed GAPDH transcript were the most effective, with 58% (7/12) of them reducing the transcript level by 70% or more; the rest of the siRNAs reduced the transcript level by at least 40% (Figure 3C).
Figure 2.
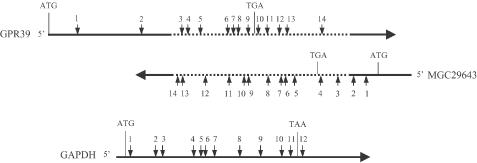
Locations of siRNA target sites on the transcripts of GPR39, MGC29643 and GAPDH genes. The lines represent the three transcripts. Solid lines represent the sequence-unique regions and dotted lines represent the sequence-overlapping regions. Small arrows show the relative positions of siRNA target sites. The numbers above and below the small arrows correlate with the siRNA codes described in the Materials and Methods. The locations of the translation initiation (ATG) and termination (TGA, TAA) codons are indicated.
Figure 3.

Reduction of target mRNA levels in HeLa cells induced by siRNAs targeting MGC29643, GPR39 and GAPDH (A–C). Quantitative real-time RT–PCR was carried out to determine the extent of target mRNA reduction induced by siRNAs. Mean fluorescence is reported from six independent transfection experiments. Error bars show variations among the experiments as the standard deviation of the mean. The extent of target mRNA reduction is measured by the percentage of the transcript level in siRNA-transfected cells relative to the transcript level in mock-transfected cells. (A) Reduction of MGC29643 (solid bars) and GPR39 (empty bars) mRNA levels induced by siRNAs directed against MGC29643. (B) Reduction of MGC29643 (solid bars) and GPR39 (empty bars) mRNA levels induced by siRNAs directed against GPR39. (C) Reduction of GAPDH mRNA level induced by siRNAs directed against GAPDH. (D) RT–PCR and gel electrophoresis of amplified products. The sizes of the amplified products are 402 bp (MGC29643), 220 bp (GPR39) and 280 bp (GAPDH). Lane 1: 100 bp ladder DNA size marker. Lane 2: no-template RT–PCR negative control. Lane 3: HeLa cells transfected with buffer instead of siRNA. Lane 4: HeLa cells transfected with nonspecific siRNA. Lane 5: HeLa cells transfected with MGC-1 siRNA. Lane 6: HeLa cells transfected with MGC-3 siRNA. Lane 7: HeLa cells transfected with MGC-4 siRNA.
To provide visual examples of gene silencing by siRNA, we performed regular RT–PCR to amplify the GPR39 and MGC29643 transcripts from total RNA isolated from HeLa cells transfected with selected siRNAs. The primers used in the study were designed to amplify the non-overlapping regions of the transcripts to ensure the detection of only the corresponding transcript. The amplified products were then separated by electrophoresis in a 1.5% agarose gel containing ethidium bromide (Figure 3D). The intensity of the amplified products correlated well with the degree of transcript reduction determined by real-time RT–PCR (Figure 3A). The amplification of the GAPDH transcript was used as an internal control.
Reversal of siRNA gene-silencing efficiency in U-138 MG cells
To further investigate whether the difference in gene-silencing efficiency might correlate with transcript abundance, we sought to identify other human cell lines that express the opposite levels of the transcripts. A reversal of the relative gene-silencing efficiency of the siRNAs against these transcripts in such cell lines would lend further support to the hypothesis that low-abundant transcripts are less susceptible to siRNA-mediated degradation.
Using quantitative real-time RT–PCR, a human glioma cell line, U-138 MG, was identified that expresses the GPR39 transcript at a level that is 14-fold higher than the level of the MGC29643 transcript (see previous section). We then evaluated in this cell line a subgroup of the siRNAs that induced a greater reduction of the MGC29643 transcript than the GPR39 transcript in HeLa cells. A total of 10 siRNAs were evaluated, including five siRNAs designed for GPR39 and five siRNAs designed for MGC29643. For each transcript, four of the five siRNAs were directed against the overlapping region and the other siRNA was against the non-overlapping region. A partial or a complete reversal of the gene-silencing efficiency of these siRNAs was observed (Figure 4A and B). Four of the eight siRNAs (MGC-8, MGC-13, GPR-4, GPR-13) induced a greater reduction of the GPR39 transcript than the MGC29643 transcript (Figure 3A and B for comparison). The other four siRNAs (MGC-3, MGC-7, GPR-7, GPR12) induced the degradation of both transcripts to a similar extent. As expected, the siRNAs targeting the non-overlapping regions of the transcripts, MGC-1 and GPR-1, induced the reduction of only the corresponding target.
Figure 4.
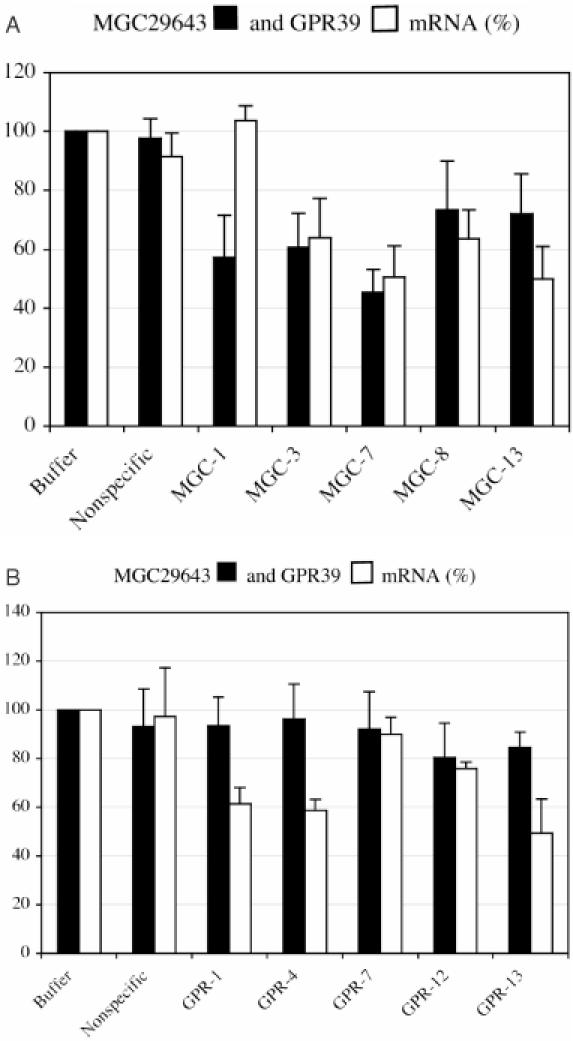
Reduction of target mRNA levels in U-138 MG cells induced by siRNAs targeting MGC29643 and GPR39. Quantitative real-time RT–PCR was carried out to determine the extent of target mRNA reduction induced by siRNAs. Mean fluorescence is reported from six independent transfection experiments. Error bars show variations among the experiments as the standard deviation of the mean. The extent of target mRNA reduction is measured by the percentage of the transcript level in siRNA-transfected cells relative to the transcript level in mock-transfected cells. (A) Reduction of MGC29643 (solid bars) and GPR39 (empty bars) mRNA levels induced by siRNAs directed against MGC29643. (B) Reduction of MGC29643 (solid bars) and GPR39 (empty bars) mRNA levels induced by siRNAs directed against GPR39.
Alteration of strand-specific activity of siRNA by single base mutations
The siRNA MGC-5 induces degradation of both MGC29643 and GPR39 transcripts by ∼50% in HeLa cells (Figure 3A). To evaluate the effect of sequence mismatches on the strand-specific activity of siRNA, we introduced single nucleotide changes near the center of the siRNA in one or both siRNA strands (Figure 5A). The siRNA MGC-5a consists of a C to G change at position 12 in the antisense strand (counting from the 5′ end of this strand) and a wild-type sense strand. When base paired with its targets, the antisense strand forms a G/G mismatch at position 12 with the MGC29643 transcript, while the sense strand perfectly matches with the GPR39 transcript. The siRNA MGC-5b consists of an A to U change at position 12 in the sense strand (counting from the 5′ end of this strand) and a wild-type antisense strand. When base paired with its targets, the sense strand forms a U/U mismatch at position 12 with the GPR39 transcript, while the antisense strand perfectly matches with the MGC29643 transcript. The siRNA MGC-5a/b consists of both mutant strands and should form the same mismatches with the MGC29643 and GPR39 transcripts. Both the parental siRNA and the sequence-modified siRNAs were introduced into HeLa cells and their ability to induce target degradation was evaluated. In contrast to the parental siRNA (MGC-5), MGC-5a and MGC-5b mediated the degradation of only those transcripts that are complementary to the wild-type siRNA strands but not the transcripts complementary to the mutant siRNA strands. As would be expected, siRNA MGC-5a/b did not induce the degradation of either of the transcripts (Figure 5B).
Figure 5.
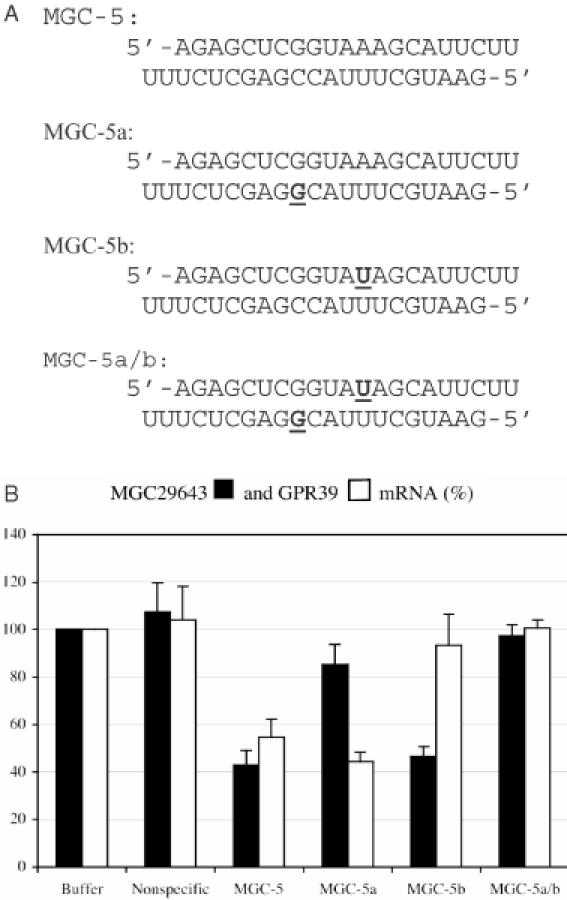
Alteration of siRNA strand-specific activity by mutation (A) Sequences of wild-type (MGC-5) and mutated (MGC-5a, MGC-5b, MGC-5a/b) siRNAs. The top strands are complementary to GPR39 mRNA and the bottom strands are complementary to MGC29643 mRNA. The bold and underlined letters indicate the modified nucleotides. (B) Reduction of MGC29643 (solid bars) and GPR39 (empty bars) mRNA levels induced by siRNAs, determined by quantitative real-time RT–PCR. Mean fluorescence is reported from six independent transfection experiments. Error bars show variations among the experiments as the standard deviation of the mean.
DISCUSSION
Synthetic siRNAs cleaved sense as well as antisense target RNAs in Drosophila melanogaster lysate (5,23). Our study extended this finding to an in vivo system and showed that siRNAs can simultaneously induce sequence-specific degradations of two endogenous mammalian transcripts oriented in opposite directions. Since 8% human genes have been estimated to have an antisense partner (32), our results suggest that when selecting siRNA target sites, it would be useful to examine the possible existence of an antisense mRNA that overlaps with the sense mRNA. Toward this end, it may be desirable to avoid targeting the 5′- and 3′-UTRs of the genes as ∼70% of the overlapped sequences encompass the 5′- and 3′-UTRs (32).
Studies with synthetic siRNAs demonstrate that each siRNA duplex cleaves the target RNA at a single site in the center of the region covered by the siRNA duplex (5,23). Based on the analysis of the effect of single- and double-nucleotide mismatches created at various positions in siRNA duplexes, the RNAi machinery seems to be more discriminative in base pairing between target mRNA and siRNA in the center of the duplex than at the 5′ and 3′ ends (21,22,40,41). Several reports showed that mismatches at or near the center of the siRNA duplex resulted in partial or complete loss of the ability of the siRNAs to reduce mRNA or protein expression levels (21,22,40,41). A careful examination of the sequence modifications in siRNA duplexes, produced either by chemical synthesis (21,22,40) or by in vitro transcription (41), revealed that some of the mutations created on one siRNA strand (antisense strand) were compensated by complementary changes on the other strand creating perfectly matched siRNA duplexes (21,22,40,41). On the other hand, some of the mutations were not compensated for by complementary changes, producing imperfectly matched siRNA duplexes (21,22). In the present study, the sequence changes in siRNAs MGC-5a and MGC-5b were made on only one siRNA strand, resulting in mismatched siRNA duplexes. Our results provide new information that such mismatched siRNA heteroduplexes can induce the degradation of the mRNA complementary to the wild-type siRNA strand, but sparing the mRNA complementary to the mutant siRNA strand. Expression profiling studies show that the sense strand of an siRNA can direct off-target gene silencing (42). Thus, our observation suggests that the use of siRNA heteroduplexes can be useful in gene silencing to confer selectivity of sense or antisense transcripts as well as minimize the off-target effects of siRNA.
Work on Caenorhabditis elegans and Drosophila indicate that the effective concentration of siRNA within cells can be substoichiometric, such that a target mRNA in great excess of the dsRNA is completely destroyed (1,43). Such striking efficacy is thought to be due in part to the ability of the cells to regenerate the trigger RNAs through the catalysis of the enzyme RNA-dependent RNA polymerase (44,45). Till date, evidence suggesting the existence of a similar mechanism in mammals is lacking. In cultured mammalian cells, RNAi induced by synthetic siRNAs is transient and the reduction of target mRNA level lasts for only a few days. Thus, maintaining an effective cellular concentration of siRNA is a key to achieving efficient and sustained gene silencing in mammalian cells. By analogy to siRNA, a similar dependence on the cellular concentration of target RNA would be expected. It has been reported that highly expressed endogenous mammalian genes can be effectively silenced by siRNAs (13). A recent report suggests that a possible difference in mRNA levels between human HeLa cells and mouse SW3T3 cells might be responsible for the differences in lamin A/C silencing by siRNA (46). At present, the overall effectiveness of siRNAs in silencing low-abundant transcripts is unclear. Given the stoichiometric nature of the RNAi process in mammalian cells, it may be envisaged that the efficiency with which siRNA/RISC interacts with target mRNA is dictated by a threshold concentration of mRNA. If this was true, the transcripts that are present at a level below the threshold would be expected to be less susceptible to siRNA-mediated degradation.
In the present study, we explored the presumptive effect of transcript abundance on the effectiveness of siRNA by employing the quantitative real-time RT–PCR method (TaqMan assay) known to be highly sensitive and specific in determining specific mRNA levels (39,47). Our study suggested that low-abundant transcripts are less susceptible to siRNA-mediated gene silencing than intermediate- or high-abundant transcripts, based on two lines of evidence. First, the global gene-silencing efficiency of randomly selected siRNAs correlated with the cellular expression levels of three transcripts in HeLa cells in that the transcript with the highest abundance (GAPDH) was silenced most effectively and the transcript with the lowest abundance (GPR39) was silenced least effectively. Second, the relative gene-silencing efficiency of the siRNAs targeting the overlapping regions of the GPR39 and MGC29643 transcripts also correlated with the cellular expression levels of the transcripts. In particular, regardless of the cell types, the transcript with higher abundance (MGC29643 in HeLa and GPR39 in U-138 MG) was silenced more effectively than the transcript with lower abundance (GPR39 in HeLa and MGC29643 in U-138 MG). It is worth noting that the use of siRNAs that target both a sense and an antisense transcript simultaneously is of particular value as certain experimental and biological variables that could potentially affect the results could be avoided. These variables include the transfection efficiency of siRNA, the toxicity of reagents and the biological status of cultured cells.
Recent studies revealed that siRNA duplexes can be functionally asymmetric, with one of the two strands being able to induce a greater degree of target RNA degradation (26,27). It has been shown that such functional asymmetry was determined mainly by the thermodynamic stabilities at the 5′ ends of the two siRNA strands. In general, the more effective siRNA strand possesses a less stable 5′ end than its less effective counterpart (more A/U base pairs than G/C base pairs). In an attempt to assess the potential contribution of siRNA duplex asymmetry to the relative silencing efficiencies of the sense and antisense siRNA strands targeting the GPR39 and MGC29643 transcripts, we examined the base composition of the first 4 nt at the 5′ end of both siRNA strands. A recent study (26) has indicated that mismatches at positions 1 to 4 of siRNA strands (counting from 5′ end) were most influential to the relative loading of the siRNA strands into RISC and therefore to the relative silencing efficiency of these strands. Our preliminary analysis revealed a fairly random nucleotide composition at the 5′ ends for the majority of the siRNA duplexes, suggesting that duplex asymmetry on the whole did not play a significant role in determining the relative gene-silencing efficiency of the siRNAs. However, the analysis did reveal a strong correlation between duplex asymmetry and the relative silencing efficiency in a pair of ‘outlier’ siRNAs (MGC-4 and GPR-6). These two siRNAs induced a greater reduction of the low abundance GPR39 transcript than the higher abundance MGC29643 transcript in HeLa cells (Figure 3). Sequence analysis indicated that the 5′ ends of the siRNA strands that target the MGC29643 transcript (MGC-4: 5′-UGGC; GPR-6: 5′-GGAG) were more stable than the 5′ ends of the opposite strands that target the GPR39 transcript (MGC-4: 5′-AUUA; GPR-6: 5′-AAAA). Future work is planned to determine if introduction of destabilizing mismatches at the 5′ ends can reverse the relative gene-silencing efficiency of these and other siRNAs, and whether rationally designed siRNAs with asymmetrical duplex ends would still display a relative silencing efficiency that correlates with target mRNA abundance. The latter may provide an indication to the relative importance of target mRNA expression and properties of the siRNA for gene silencing.
Two additional factors may provide alternative explanations to the observation that low-abundant transcripts are less susceptible to siRNA-mediated degradation than medium- and high-abundant transcripts. One is the possibility that the sense and antisense transcripts form hybrids due to their sequence complementarity. If this were the case, most of the lower abundant transcripts may be present in a double-stranded structure, which is more refractory to RNAi than the single-strand transcript. A comparison of the relative abundance and silencing efficiency of transcripts not known to have antisense partners should help address this issue. A second potential factor is the difference in the half-lives of these transcripts. However, we do not have information regarding the half-life of the GPR39 and MGC29643 transcripts currently.
A recent study (21) found that siRNAs with intermediate activities were unable to deplete the transcript of the endogenous human tissue factor gene, but were more effective on depleting the transcript of an exogenous transgene. Although the transcript levels were not determined in this study, it may be assumed that the endogenous transcript is at a lower level than the transcript from the transgene. Our observation would support the general conclusion that low-abundant transcripts are less susceptible to siRNA-mediated degradation. This observation should have important implications in the application of siRNA-based technology. In order to achieve significant silencing of genes that are expressed at low levels, it may be necessary to target multiple sites on the transcripts and/or increase the effective intracellular concentration and stability of siRNA. It should be emphasized that the efficiency of siRNA-mediated gene silencing is affected by a combination of factors. A thorough understanding of these factors should help the design of highly functional siRNAs that can effectively silence all of the transcripts, regardless of their cellular expression levels.
Acknowledgments
ACKNOWLEDGEMENTS
The authors wish to express their gratitude to Drs Nick Paoni and Jed Chatterton for valuable discussions, to Dr Wufang Fan for help in bioinformatics analysis and to Jeremy To, Joseph Siu and Cuiying Wang for technical help and sharing reagents.
REFERENCES
- 1.Fire A., Xu,S., Montgomery,M.K., Kostas,S.A., Driver,S.E. and Mello,C.C. (1998) Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature, 391, 806–811. [DOI] [PubMed] [Google Scholar]
- 2.Hannon G.J. (2002) RNA interference. Nature, 418, 244–251. [DOI] [PubMed] [Google Scholar]
- 3.Bass B.L. (2001) RNA interference. The short answer. Nature, 411, 428–429. [DOI] [PubMed] [Google Scholar]
- 4.Elbashir S.M., Harborth,J., Lendeckel,W., Yalcin,A., Weber,K. and Tuschl,T. (2001) Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature, 411, 494–498. [DOI] [PubMed] [Google Scholar]
- 5.Elbashir S.M., Lendeckel,W. and Tuschl,T. (2001) RNA interference is mediated by 21- and 22-nucleotide RNAs. Genes Dev., 15, 188–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Caplen N.J., Parrish,S., Imani,F., Fire,A. and Morgan,R.A. (2001) Specific inhibition of gene expression by small double-stranded RNAs in invertebrate and vertebrate systems. Proc. Natl Acad. Sci. USA, 98, 9742–9747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hammond S.M., Bernstein,E., Beach,D. and Hannon,G.J. (2000) An RNA-directed nuclease mediates post-transcriptional gene silencing in Drosophila cells. Nature, 404, 293–296. [DOI] [PubMed] [Google Scholar]
- 8.Bernstein E., Caudy,A.A., Hammond,S.M. and Hannon,G.J. (2001) Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature, 409, 363–366. [DOI] [PubMed] [Google Scholar]
- 9.Hammond S.M., Boettcher,S., Caudy,A.A., Kobayashi,R. and Hannon,G.J. (2001) Argonaute2, a link between genetic and biochemical analyses of RNAi. Science, 293, 1146–1150. [DOI] [PubMed] [Google Scholar]
- 10.Hutvagner G., Zamore,P.D. (2002) A microRNA in a multiple-turnover RNAi enzyme complex. Science, 297, 2056–2060. [DOI] [PubMed] [Google Scholar]
- 11.Zeng Y., Yi,R. and Cullen,B.R. (2003) MicroRNAs and small interfering RNAs can inhibit mRNA expression by similar mechanisms. Proc. Natl Acad. Sci. USA, 100, 9779–9784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bass B.L. (2000) Double-stranded RNA as a template for gene silencing. Cell, 101, 235–238. [DOI] [PubMed] [Google Scholar]
- 13.Harborth J., Elbashir,S.M., Bechert,K., Tuschl,T. and Weber,K. (2001) Identification of essential genes in cultured mammalian cells using small interfering RNAs. J. Cell. Sci., 114, 4557–4565. [DOI] [PubMed] [Google Scholar]
- 14.Lee N.S., Dohjima,T., Bauer,G., Li,H., Li,M.J., Ehsani,A., Salvaterra,P. and Rossi,J. (2002) Expression of small interfering RNAs targeted against HIV-1 rev transcripts in human cells. Nat. Biotechnol., 20, 500–505. [DOI] [PubMed] [Google Scholar]
- 15.Jacque J.M., Triques,K. and Stevenson,M. (2002) Modulation of HIV-1 replication by RNA interference. Nature, 418, 435–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Novina C.D., Murray,M.F., Dykxhoorn,D.M., Beresford,P.J., Riess,J., Lee,S.K., Collman,R.G., Lieberman,J., Shankar,P. and Sharp,P.A. (2002) siRNA-directed inhibition of HIV-1 infection. Nature Med., 8, 681–686. [DOI] [PubMed] [Google Scholar]
- 17.Brummelkamp T.R., Bernards,R. and Agami,R. (2002) A system for stable expression of short interfering RNAs in mammalian cells. Science, 296, 550–553. [DOI] [PubMed] [Google Scholar]
- 18.Miyagishi M. and Taira,K. (2002) U6 promoter-driven siRNAs with four uridine 3′ overhangs efficiently suppress targeted gene expression in mammalian cells. Nat. Biotechnol., 20, 497–500. [DOI] [PubMed] [Google Scholar]
- 19.Paul C.P., Good,P.D., Winer,I. and Engelke,D.R. (2002) Effective expression of small interfering RNA in human cells. Nat. Biotechnol., 20, 505–508. [DOI] [PubMed] [Google Scholar]
- 20.Paddison P.J., Caudy,A.A., Bernstein,E., Hannon,G.J. and Conklin,D.S. (2002) Short hairpin RNAs (shRNAs) induce sequence-specific silencing in mammalian cells. Genes Dev., 16, 948–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Holen T., Amarzguioui,M., Wiiger,M.T., Babaie,E. and Prydz,H. (2002). Positional effects of short interfering RNAs targeting the human coagulation trigger tissue factor. Nucleic Acids Res., 30, 1757–1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hamada M., Ohtsuka,T., Kawaida,R., Koizumi,M., Morita,K., Furukawa,H., Imanishi,T., Miyagishi,M. and Taira,K. (2002) Effects on RNA interference in gene expression (RNAi) in cultured mammalian cells of mismatches and the introduction of chemical modifications at the 3′-ends of siRNAs. Antisense Nucleic Acid Drug Dev., 12, 301–309. [DOI] [PubMed] [Google Scholar]
- 23.Elbashir S.M., Martinez,J., Patkaniowska,A., Lendeckel,W. and Tuschl,T. (2001) Functional anatomy of siRNAs for mediating efficient RNAi in Drosophila melanogaster embryo lysate. EMBO J., 20, 6877–6888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Amarzguioui M., Holen,T., Babaie,E. and Prydz,H. (2003) Tolerance for mutations and chemical modifications in a siRNA. Nucleic Acids Res., 31, 589–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Czauderna F., Fechtner,M., Dames,S., Aygun,H., Klippel,A., Pronk,G.J., Giese,K. and Kaufmann,J. (2003) Structural variations and stabilising modifications of synthetic siRNAs in mammalian cells. Nucleic Acids Res., 31, 2705–2716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schwarz D.S., Hutvagner,G., Du,T., Xu,Z., Aronin,N. and Zamore,P.D. (2003) Asymmetry in the assembly of the RNAi enzyme complex. Cell, 115, 199–208. [DOI] [PubMed] [Google Scholar]
- 27.Khvorova A., Reynolds,A. and Jayasena,S.D. (2003) Functional siRNAs and miRNAs exhibit strand bias. Cell, 115, 209–216. [DOI] [PubMed] [Google Scholar]
- 28.Wagner E.G. and Simons,R.W. (1994) Antisense RNA control in bacteria, phages, and plasmids. Annu. Rev. Microbiol., 48, 713–742. [DOI] [PubMed] [Google Scholar]
- 29.Kumar M. and Carmichael,G.G. (1998) Antisense RNA: function and fate of duplex RNA in cells of higher eukaryotes. Microbiol. Mol. Biol. Rev., 62, 1415–1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Knee R. and Murphy,P.R. (1997) Regulation of gene expression by natural antisense RNA transcripts. Neurochem. Int., 31, 379–392. [DOI] [PubMed] [Google Scholar]
- 31.Lehner B., Williams,G., Campbell,R.D. and Sanderson,C.M. (2002) Antisense transcripts in the human genome. Trends Genet., 18, 63–65. [DOI] [PubMed] [Google Scholar]
- 32.Yelin R., Dahary,D., Sorek,R., Levanon,E.Y., Goldstein,O., Shoshan,A., Diber,A., Biton,S., Tamir,Y., Khosravi,R., Nemzer,S., Pinner,E., Walach,S., Bernstein,J., Savistsky,K. and Rotman,G. (2003) Widespread occurrence of antisense transcription in the human genome. Nat. Biotechnol. 21, 379–386. [DOI] [PubMed] [Google Scholar]
- 33.Mckee K.K., Tan,C.P., Palyha,O.C., Liu,J., Feighner,S.D., Hreniuk,D.L., Smith,R.G., Howard,A.D. and Van der Ploeg,L.H. (1997) Cloning and characterization of two human G protein-coupled receptor genes (GPR38 and GPR39) related to the growth hormone secretagogue and neurotensin receptors. Genomics, 46, 426–434. [DOI] [PubMed] [Google Scholar]
- 34.Mammalian Gene Collection (MGC) Program Team. (2002) Generation and initial analysis of more than 15 000 full-length human and mouse cDNA sequences. Proc. Natl Acad. Sci. USA, 99, 16899–16903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tokunaga K., Nakamura,Y., Sakata,K., Fujimori,K., Ohkubo,M., Sawada,K. and Sakiyama,S. (1987) Enhanced expression of a glyceraldehyde-3-phosphate dehydrogenase gene in human lung cancers. Cancer Res., 47, 5616–5619. [PubMed] [Google Scholar]
- 36.Elbashir S.M., Harborth,J., Weber,K. and Tuschl,T. (2002) Analysis of gene function in somatic mammalian cells using small interfering RNAs. Methods, 26, 199–213. [DOI] [PubMed] [Google Scholar]
- 37.Kapadia S.B., Brideau-Anderson,A. and Chisari,F.V. (2003) Interference of hepatitis C virus RNA replication by short interfering RNAs. Proc. Natl Acad. Sci. USA, 100, 2014–2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Splinter P.L., Masyuk,A.I. and LaRusso,N.F. (2003) Specific inhibition of AQP1 water channels in isolated rat intrahepatic bile duct units by small interfering RNAs. J. Biol. Chem., 278, 6268–6274. [DOI] [PubMed] [Google Scholar]
- 39.Ke L.D., Chen,Z. and Yung,W.K. (2000). A reliability test of standard-based quantitative PCR: exogenous vs endogenous standards. Mol. Cell. Probes, 14, 127–135. [DOI] [PubMed] [Google Scholar]
- 40.Vickers T.A., Koo,S., Bennett,C.F., Crooke,S.T., Dean,N.M. and Baker,B.F. (2003) Efficient reduction of target RNAs by small interfering RNA and RNase H- dependent antisense agents. A comparative analysis. J. Biol. Chem, 278, 7108–7118. [DOI] [PubMed] [Google Scholar]
- 41.Miller V.M., Xia,H., Marrs,G.L., Gouvion,C.M., Lee,G., Davidson,B.L. and Paulson,H.L. (2003) Allele-specific silencing of dominant disease genes. Proc. Natl Acad. Sci. USA, 100, 7195–7200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jackson A.L., Bartz,S.R., Schelter,J., Kobayashi,S.V., Burchard,J., Mao,M., Li,B., Cavet,G. and Linsley,P.S. (2003) Expression profiling reveals off-target gene regulation by RNAi. Nat. Biotechnol. 21, 635–637. [DOI] [PubMed] [Google Scholar]
- 43.Kennerdell J.R. and Carthew,R.W. (1998) Use of dsRNA-mediated genetic interference to demonstrate that frizzled and frizzled 2 act in the wingless pathway. Cell, 95, 1017–1026. [DOI] [PubMed] [Google Scholar]
- 44.Lipardi C., Wei,Q. and Paterson,B.M. (2001) RNAi as random degenerative PCR: siRNA primers convert mRNA into dsRNAs that are degraded to generate new siRNAs. Cell, 107, 297–307. [DOI] [PubMed] [Google Scholar]
- 45.Sijen T., Fleenor,J., Simmer,F., Thijssen,K.L., Parrish,S.,Timmons,L., Plasterk,R.H. and Fire,A. (2001) On the role of RNA amplification in dsRNA-triggered gene silencing. Cell, 107, 465–476. [DOI] [PubMed] [Google Scholar]
- 46.Harborth J., Elbashir,S.M., Vandenburgh,K., Manninga,H., Scaringe,S.A., Weber,K. and Tuschl,T. (2003) Sequence, chemical, and structural variation of small interfering RNAs and short hairpin RNAs and the effect on mammalian gene silencing. Antisense Nucleic Acid Drug Dev., 13, 83–105. [DOI] [PubMed] [Google Scholar]
- 47.Sanburn N. and Cornetta,K. (1999) Rapid titer determination using quantitative real-time PCR. Gene Ther., 6, 1340–1345. [DOI] [PubMed] [Google Scholar]