


Abstract
Two competing events, termination and readthrough (or nonsense suppression), can occur when a stop codon reaches the A-site of a translating ribosome. Translation termination results in hydrolysis of the final peptidyl-tRNA bond and release of the completed nascent polypeptide. Alternatively, readthrough, in which the stop codon is erroneously decoded by a suppressor or near cognate transfer RNA (tRNA), results in translation past the stop codon and production of a protein with a C-terminal extension. The relative frequency of termination versus readthrough is determined by parameters such as the stop codon nucleotide context, the activities of termination factors and the abundance of suppressor tRNAs. Using a sensitive and versatile readthrough assay in conjunction with RNA interference technology, we assessed the effects of depleting eukaryotic releases factors 1 and 3 (eRF1 and eRF3) on the termination reaction in human cell lines. Consistent with the established role of eRF1 in triggering peptidyl-tRNA hydrolysis, we found that depletion of eRF1 enhances readthrough at all three stop codons in 293 cells and HeLa cells. The role of eRF3 in eukarytotic translation termination is less well understood as its overexpression has been shown to have anti-suppressor effects in yeast but not mammalian systems. We found that depletion of eRF3 has little or no effect on readthrough in 293 cells but does increase readthrough at all three stop codons in HeLa cells. These results support a direct role for eRF3 in translation termination in higher eukaryotes and also highlight the potential for differences in the abundance or activity of termination factors to modulate the balance of termination to readthrough reactions in a cell-type-specific manner.
INTRODUCTION
Translation termination is a ubiquitous yet incompletely understood step in gene expression during which the final peptidyl-tRNA bond is hydrolyzed, releasing the nascent polypeptide. In higher eukaryotes, it is believed that this reaction is mediated by two proteins, eukaryotic release factor 1 (eRF1) and eRF3. eRF1, a class I release factor, is responsible for stop codon discrimination through direct interaction with the nucleotides of the stop codon triplet (1–4). The eRF1 protein is also directly implicated in triggering peptidyl-tRNA hydrolysis via the highly conserved GGQ motif (5,6). Although the hydrolysis event is catalyzed by the peptidyl-transferase center of the ribosome, eRF1 may make contact with the nascent peptide through the GGQ motif (7). Both in vitro and in vivo, eRF1 is known to interact with the class II release factor eRF3 (8). The eRF3 protein is an eRF1- and ribosome-dependent GTPase that stimulates eRF1 activity in response to GTP hydrolysis (9,10).
In contrast to the apparently direct role of eRF1 in termination, the necessity of eRF3 for the termination reaction is uncertain. In in vitro assays, addition of eRF1 alone is sufficient to trigger peptidyl-tRNA hydrolysis, although addition of eRF3 increases the rate of hydrolysis at low stop codon concentrations (10,11). The prokaryotic eRF3 homolog is dispensable for bacterial growth, but the yeast and Drosophila eRF3 genes are essential (12–14). However, since eRF3 has been implicated in cellular processes other than termination (15,16), it is possible that the essential role of eRF3 is not in termination but for an alternative function. It has also been suggested that instead of directly contributing to termination, the eRF3 interaction may instead link termination to other cellular processes. For example, eRF3 binds to the poly(A) binding protein and may be involved in ribosome recycling through this interaction (17,18). eRF3 also interacts with the Upf proteins, Upf1, Upf2 and Upf3, and thus may mediate the interplay between termination and the nonsense-mediated decay pathway (19–21).
Studies of the role of eRF3 in termination in different eukaryotic systems have yielded discrepant results. Since translational readthrough at stop codons occurs at an increased frequency when termination efficiency is reduced, the observation that yeast with reduced levels of functional eRF3 have an omnipotent nonsense suppressor phenotype (22,23) suggests that eRF3 is needed for efficient termination. Conversely, overexpression of both eRF1 and eRF3 is required to increase termination efficiency in yeast, implicating eRF3 along with eRF1 in the termination reaction (24). In contrast, both in rabbit reticulocyte lysates and in transfected human cells (25,26), increased eRF1 expression alone is sufficient to increase termination efficiency. Increasing eRF3 concentration, either alone or in conjunction with eRF1, produced no further effect on termination efficiency in the mammalian systems (25,26). Collectively, these results raise uncertainties about the necessity of eRF3 for termination in higher eukaryotes.
Since the results of augmented release factor expression differed between the yeast and mammalian systems, the effects of decreasing release factor expression in mammalian systems may or may not correspond to those detected in yeast. To examine this question, we used RNA interference technology to deplete release factor pools in human cell lines, and then examined the effect on termination efficiency using a sensitive quantitative readthrough assay. In agreement with and as an extension of observations reported for the effect at a UAG stop codon (27), we found that depletion of eRF1 results in an omnipotent nonsense suppressor phenotype in human cell lines. Furthermore, we found that depletion of eRF3 also has the potential to decrease termination efficiency at all three stop codons, but in a cell-type-specific manner. Consistent with the observations from studies in yeast, these results imply that eRF3 does play a direct role in translation termination in higher eukaryotes.
MATERIALS AND METHODS
Plasmid construction
The secreted alkaline phosphatase gene was amplified from pcDNAI/SEAP/Neo (provided by Michael Katze, University of Washington) using the forward primer 5′-GGAACTCGAGCTCACCATGCTGCTGCTGCTGCTG-3′ and the degenerate reverse primer 5′-GCGAATTCTGGYYAGCCCGGGTGCGCG-3′, where Y = C or T, followed by ligation of the PCR product into the pCDNA3.1/V5-His TOPO TA expression vector (Invitrogen). The identity of the degenerate codon was determined for individual clones by sequencing, resulting in a TOPO-SEAP vector series. The S-peptide sequence was generated by Taq polymerase fill-in of the overlapping oligonucleotides 5′-GCGAATTCAAAGAAACCGCTGCTGCTAAATTCGAACGCCAG-3′ and 5′-GCTCTAGATCACTATTAGCTGTCCATGTGCTGGCGTTCGAA-3′. The fill-in product was digested with EcoRI and XbaI and ligated into the EcoRI/XbaI sites of the TOPO-SEAP vectors, yielding the SEAP-S-tag series pEQ911 (ochre), pEQ912 (amber), pEQ913 (opal) and pEQ914 (tryptophan). Site-directed mutagenesis using the QuikChange Kit (Stratagene) was performed on pEQ914 to change the tryptophan codon to TGT and create a BsrGI site, using the oligonucleotides 5′-GCCGCGCACCCGGGCTGTACAGAATTCAAAGAAACC-3′ (forward) and 5′-GGTTTCTTTGAATTCTGTACAGCCCGGGTGCGCGGC-3′ (reverse), yielding pEQ961. Oligonucleoides 5′-GTACACAATAACAACTCGAGG-3′ and 5′-AATTCCTCGAGTTGTTATTGT-3′ (ochre), 5′-GTACAC AATAGCAACTCGAGG-3′ and 5′-AATTCCTCGAGTTGCTATTGT-3′ (amber), 5′-GTACACAATGACAACTCGAGG-3′ and 5′-AATTCCTCGAGTTGTCATTGT-3′ (opal), and 5′-GTACACAATGGCAACTCGAGG-3′ and 5′-AATTCCTCGAGTTGCCATTGT-3′ (tryptophan), with noted codon indicated in boldface, were annealed and inserted into the BsrGI/EcoRI sites of pEQ961 to generate the SEAP-Stag series pEQ962 (ochre), pEQ963 (amber), pEQ964 (opal) and pEQ965 (tryptophan), which contains a tobacco mosaic virus (TMV)-like termination codon context. The presence of the desired SEAP-S-tag junction was confirmed by sequencing.
The plasmids pSV-tS su+(ochre) and pSV-tS su−(serine), which express a human suppressor transfer RNA (tRNA) with an ochre anticodon and the parental serine tRNA from which the suppressor was derived were provided by Uttam RajBhandary (Massachusetts Institute of Technology) (28). Plasmids expressing an amber (pEQ933) or opal (pEQ934) suppressor tRNA were derived from pSV-tS su+ by site-directed mutagenesis using the QuikChange Kit (Stratagene) and the primer pairs 5′-GGTTAAGGCGATGGACTCTAAATCCATTGGGGTCTCC-3′ and 5′-GGAGACCCCAATGGATTTAGAGTCCATCGCCTTAACC-3′ (amber) or 5′-GGTTAAGGCGATGGACTTCAAATCCATTGGGGTCTCC-3′ and 5′-GGAGACCCCAATGGATTTGAAGTCCATCGCCTTAACC-3′ (opal), with the nucleotides corresponding to the anticodon indicated in boldface. The presence of the desired anticodon was confirmed by sequencing.
Plasmids directing the synthesis of short hairpin RNA (shRNA) under the control of the human H1 promoter were derived from pBabePuro-p27.1 (provided by Bruce Clurman, Fred Hutchinson Cancer Research Center, Seattle, WA) (29). The control vector pEQ940 (pBp), that does not express shRNA, was made by NheI digestion of pBabePuro-p27.1 to drop out the H1 promoter and associated cassette, followed by religation. Oligonucleotides designed to generate shRNAs targeting eRF1 and eRF3 were annealed, filled-in with Taq polymerase, digested with BglII and EcoRI and ligated to BglII-/EcoRI-digested pBabePuro-p27.1. The oligonucleotides 5′-GCAGATCTGCAATGGCACCAGCATGATATTCAAGAGATATCAT-3′ and 5′-CGGAATTCAAAAAGCAATGGCACCAGCATGATATCTCTTGAAT-3′, targeting nucleotides 221–243 of eRF1 (GenBank accession no. NM_004730), were used to produce pEQ929 (shRNA 1A). Plasmid pEQ941, expressing shRNA 1B directed against nucleotides 1327–1348 of eRF1, was constructed with the oligonucleotides 5′-GCAGATCTGAAGGGTCTCAGTTTGTGAAAGTTCAAGAGACTTTCA-3′ and 5′-GCGAATTCAAAAAGAAGGGTCTCAGTTTGTGAAAGTCTCTTGAAC-3′. Two plasmids were designed to target eRF3 (GenBank accession no. XM_083954). Plasmids pEQ945 expressing shRNA 3A directed against nucleotides 1171–1189 and pEQ946 expressing shRNA 3B directed against nucleotides 1659–1678, were constructed with the oligonucleotide pairs 5′-GCAGATCTGCTGATTTGGCTGTGCTGGTTCAAGAGACCAGCA-3′/5′-GCGAATTCAAAAAGCTGATTTGGCTGTGCTGGTCTCTTGAAC-3′ and 5′-GCAGATCTGCACAACGTGGAAGTTCTTGTTCAAGAGACAAGAA-3′/ 5′-GCGAATTCAAAAAGCACAACGTGGAAGTTCTTGTCTCTTGAAC-3′. The oligonucleotides 5′-CCTTGGATCCGATTCCGCGTGTTCGTGTACGTTCAAGAGACGTA-3′ and 5′-CCTTGAATTCAAAAAGATTCCGCGTGTTCGTGTACGTCTCTTGAACGTA-3′, directed against nucleotides 230 602–230 582 of TRS1 in Towne human cytomegalovirus (HCMV) (GenBank accession no. AY315197), were annealed and filled-in with Taq polymerase, digested with BamHI and EcoRI, and then processed as above to produce pEQ936.
Plasmid pGH5 [provided by Shin-ichi Hoshino, University of Tokyo (30)], which contains eRF3 cDNA, was digested with BamHI and PstI and the 1.9 kb fragment containing the eRF3 gene was ligated into the same sites in pQE10 (Qiagen) to add a histidine (his) tag to the N-terminus of eRF3 and yield pEQ609. PCR amplification of an eRF1 mutant using the pQE-specific primers 5′-GAATTCATTAAAGAGGAGAAA-3′ (forward) and 5′-CTGGATCTATCAACAGGAGTCCAA-3′ (reverse) and insertion of the product into pcDNA3.1/V5-His TOPO TA resulted in pEQ840. EcoRI/BlpI fragments containing his-tagged wild-type eRF1 and eRF3 were moved from pEQ814 (7) and pEQ609, respectively, into the same sites of pEQ840 resulting in plasmids pEQ842 and pEQ863.
Plasmid pEQ909 encodes a truncated (amino acids 1–260), his-tagged version of the human cytomegalovirus TRS1 protein. To produce this plasmid, a plasmid derived from VVCL1 (31) was used as a PCR template with forward primer 5′-GCCTCGACGTCGGATCCGTCCGGCGGCCATGGCC-3′ and reverse primer 5′-CGTCGCGCGCGGGGTGCC-3′, and the product ligated into pcDNA3.1/V5-His TOPO TA.
Transfection and SEAP assay
HeLa, HEK293 (293) or HEK293T (293T) cells were propagated in DMEM supplemented with 10% Nu serum (Collaborative Research, Inc.). Plasmids were transfected into the cells at ∼60% confluence in 24-well plates, unless otherwise specified, using either calcium phosphate or lipofectamine (Invitrogen). After replacement at 16 h post-transfection, media samples were collected at 48–72 h post-transfection, incubated at 65°C for 30 min to inactivate endogenous phosphatases and then clarified by centrifugation at 10 000 g for 10 min.
Readthrough was determined by comparing the bound versus the total secreted alkaline phosphatase (SEAP) activity for each sample, in a manner similar to that described previously (32). Total SEAP activity was measured in 200 μl of SEAP buffer [final concentrations of 1 M diethanolamine, 0.5 mM MgCl2, 10 mM l-homoarginine and 0.5 mM methyl-umbelliferyl phosphate (Sigma)]. The change in fluorescence per minute was determined using an Ascent Fluoroskan (Thermolab systems) at 37°C with λ excitation = 355 and λ emission = 460. Bound SEAP activity was measured by incubating samples overnight at 4°C in S-protein-coated wells. After incubation, the medium was aspirated from the wells and unbound SEAP was removed by three 350 μl washes with 1× phosphate-buffered saline (PBS) containing 0.1% Tween-20 and one 350 μl wash with 1× PBS. After adding 200 μl of SEAP buffer, the activity was measured with the Fluoroskan. At least 10 linear timepoints were used to determine SEAP activity. To generate S-protein-coated plates, Nunc Maxisorp or Costar EIA 96-well plates were incubated overnight at 4°C with 100 μl/well of 10 μg/ml Ribonuclease S-protein (Sigma R-6250)in 1× PBS, followed by a 2 h blocking step at room temperature with 200 μl of 1% non-fat dry milk in1× PBS and then washing the wells 2 times with 350 μl of 1× PBS with 0.1% Tween-20.
For each experiment, a fusion protein control was included to determine the fraction of tagged SEAP that bound to the plate. The percentage readthrough for each sample was calculated by dividing the bound SEAP activity by the total SEAP activity, then normalizing this number by the fraction of SEAP-Stag fusion protein control that was bound and converting the results to a percentage.
Fusion protein binding
293T cells were transfected by calcium phosphate with 0.5 μg pEQ965 (SEAP fusion control vector) per well. After processing of media, total SEAP activity was determined for a 0.2 μl aliquot (20 μl of a 1:100 dilution in 1× PBS). To determine binding, samples were diluted in 1× PBS to a final volume of 100 μl/well and applied to an S-protein-coated plate. Total and bound SEAP were assayed in quadruplicate for each dilution. The amount of bound SEAP activity and the percentage of activity bound were calculated and linear regression was performed to compare the bound activity to the volume of media loaded.
To measure fusion protein binding in the presence of excess untagged SEAP, 293T cells in a 100 mM dish were transfected with 12 μg of pEQ911 (SEAP ochre vector with a poor readthrough context that does not produce detectable levels of S-tagged SEAP) by calcium phosphate. Medium containing the fusion protein from the pEQ965 transfection was diluted 1:100 into medium from the pEQ911 transfection. Total SEAP activity was determined for a 0.2 μl aliquot of both the media from the pEQ911 transfection and the 1:100 dilution of fusion protein into pEQ911 media. Dilutions were added to S-protein-coated plates using 0–70 μl of the 1:100 fusion protein dilution and the final volume was brought to 100 μl/well with pEQ911 media. The amount of bound SEAP activity, as well as the percentage of activity bound, was determined for quadruplicate samples of each dilution. Linear regression was performed to compare the amount of bound activity to the volume of fusion protein loaded.
Readthrough assay with suppressor tRNAs
293T cells were cotransfected by calcium phosphate with combinations of 0.5 μg of SEAP indicator pEQ962 (ochre), pEQ963 (amber), pEQ964 (opal) or pEQ965 (tryptophan), with 0.5 μg of tRNA vector pSV-tS-su− (serine), pSV-tS-su+ (ochre), pEQ933 (amber) or pEQ934 (opal) in duplicate. Total SEAP activity was determined for 0.2 μl aliquots, while bound SEAP activity was determined using 100 μl aliquots for non-cognate or 50 μl (+50 μl 1× PBS) for cognate stop-codon/suppressor tRNA pairs.
To determine the efficiency of readthrough using varying amounts of suppressor tRNAs, 293T cells were transfected by calcium phosphate with 0.5 μg of pEQ962 (SEAP ochre indicator) and 1.0 μg of mixtures of pSV-tS-su− (serine tRNA) and pSV-tS-su+ (ochre tRNA) at varying ratios of the two plasmids, or with 0.5 μg of pEQ965 (SEAP fusion control) and 1.0 μg of pSV-tS-su− (serine tRNA). Total SEAP activity was determined on 0.2 μl aliquots and bound SEAP activity was determined using 50 μl aliquots with the following exceptions; for transfections with pEQ962 with 1.0 μg pSV-tS-su−, 100 μl aliquots were used, while for pEQ965 transfections, bound activity was measured using 0.2 μl aliquots. All bound incubations were brought to a final volume of 100 μl with 1× PBS. Mean (± SD) percentage readthrough was calculated for triplicate transfections.
Readthrough assay with shRNAs
Transfections were performed in triplicate by calcium phosphate for 293 cells or by lipofectamine for HeLa cells, using 0.5 μg of SEAP indicator (pEQ962, pEQ963, pEQ964 or pEQ965) and 0.5 μg of plasmid expressing shRNA (pEQ940, pEQ936, pEQ929, pEQ941, pEQ945 or pEQ946). The total SEAP activity for each sample was determined using a 0.2 μl aliquot of the media, while bound SEAP activity was determined by incubating 100 μl of media per test sample or 0.2 μl of media containing fusion protein diluted to 100 μl with 1× PBS. Statistical analysis was performed using a two-tailed t-test. To calculate the fold increase in readthrough, the percentage readthrough for a specific stop codon context in the presence of an shRNA was divided by the percentage readthrough at the same stop context in the presence of the vector backbone (pBp, pEQ940). Three independent experimental sets were used to determine the average fold increase in readthrough.
Western blot analyses
For assessing transgene knockdown efficiency, 293T or HeLa cells in 6-well plates were transfected with 1.5 μg of each plasmid [eRF1 (pEQ842) or eRF3 (pEQ863), TRS1 (pEQ909) and shRNA (pEQ940, pEQ936, pEQ929, pEQ941, pEQ945, or pEQ946)] using the calcium phosphate method for 293T or lipofectamine for HeLa cells. Forty-eight hours post-transfection, cells were resuspended in 1 ml media, and then pelleted at 2000 g for 5 min at 4°C. After aspirating media, the cells were lysed for 5 min on ice in 20 μl low salt lysis buffer (50 mM Tris pH 8.0, 1% NP-40). Nuclei and cellular debris were pelleted by centrifugation for 5 min at 7500 g at 4°C. The supernatant was sonicated for 30 s in a bath sonicator and stored at −20°C until use. For immunoblots, lysates were separated by SDS–PAGE, electroblotted to nitrocellulose, probed with anti-pentahis antibody (Qiagen) and detected using the CDP-Star Kit (Tropix) according to the manufacturer's specifications. To estimate relative transgene expression, multiple exposures were scanned and NIH Image v1.63 was used to quantify band density. eRF1 and eRF3 levels were normalized to TRS1 expression for each lane.
For assays of endogenous release factor levels, HeLa or 293 cells were washed in 1× PBS and then lysed in low salt lysis buffer (with one Roche mini complete protease inhibitor cocktail tablet per 5 ml) for 5 min on ice. Lysates were centrifuged at 2500 g for 10 min at 4°C, and the supernatant was stored on ice during the determination of protein concentration and then used directly. Equal amounts of protein (20 or 50 μg/well for eRF1 or eRF3 blots, respectively) were fractionated by SDS–PAGE and electroblotted onto nitrocellulose. Rabbit polyclonal antibodies specific for eRF3 were raised to purified his-tagged eRF3 expressed in bacteria using pEQ609. This serum and antiserum to eRF1 (7) was used at 1:10 000 dilution in 1× PBS-T containing 5% BSA. After release factor detection using the CDP-Star Kit, the blots were washed 4 times for 5 min each in 1× PBS-T, stripped by incubation for 30 min at 50°C in stripping buffer (63 mM Tris pH 6.8, 100 mM 2-mercaptoethanol, 2% SDS), then washed 6 times for 5 min each in 1× PBS-T before being reprobed for with a 1:1000 dilution of actin rabbit polyclonal serum (Sigma).
RESULTS
Secreted alkaline phosphatase readthrough assay
In order to measure readthrough levels in cell culture, we utilized a secreted alkaline phosphatase (SEAP)-based assay (32). The SEAP reporter gene was cloned into a plasmid downstream from the CMV major immediate early gene promoter and upstream from the coding sequences for the S-peptide tag (Figure 1A). The S-peptide consists of fifteen N-terminal acids from pancreatic ribonuclease A. The remaining portion of the ribonuclease A, called the S-protein, forms a stable complex with the S-peptide and can be used to isolate S-peptide-tagged proteins (33,34). The S-peptide coding sequence is separated from the SEAP reading frame by an in-frame stop codon (test vectors) or tryptophan codon (fusion vector control) (Figure 1A). The nucleotides flanking the stop codon are similar to those found at the gag–pol junction of TMV (35), a termination codon context known to promote relatively high basal readthrough levels in eukaryotic cells. When the junction contains the tryptophan codon, translation results in a C-terminal fusion of the S-peptide to SEAP. However, if a stop codon is present in the junction, the S-peptide is only fused to SEAP as a result of a readthrough event. Each of the three different stop codons follow the S-peptide sequence to ensure that translation will terminate at this point, if not before. Advantages of this system include the fact that total SEAP serves as an internal control for transfection efficiency and transcript level, and SEAP measurement is non-destructive allowing multiple timepoints to be examined.
Figure 1.
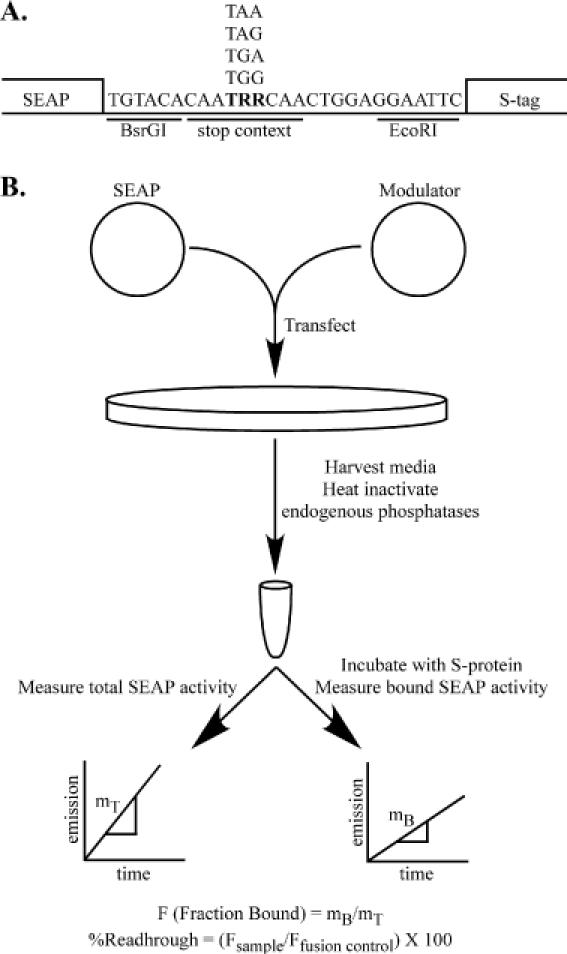
The SEAP readthrough assay. (A) The secreted alkaline phosphatase (SEAP) gene and the S-peptide tag sequence (S-tag) were inserted into a TOPO-TA (Invitrogen) expression vector as described in Materials and Methods. The junction between SEAP and the S-tag contains a unique BsrGI restriction site, followed by either a stop or tryptophan codon embedded in a TMV-like termination context (CAATRRCAA, where R = A or G), and an EcoRI site. The S-tag sequence is followed by each of the three in-frame stop codons to prevent subsequent readthrough events. (B) After cotransfection of a SEAP reporter and a modulator plasmid, the total and S-protein-bound SEAP activity are measured and used to calculate the relative readthrough efficiency. The percentage readthrough is then calculated by normalizing the fraction of SEAP bound per test sample to the binding efficiency of the SEAP–S-tag fusion control.
The general protocol for the assay (Figure 1B) is to cotransfect cells with a SEAP test vector and the termination modulator being tested. For each experiment, cotransfection of the tryptophan fusion vector with the modulator is also performed. At the desired time post-transfection, media is collected and heat-treated to inactivate endogenous phosphatases. The total SEAP activity in a sample aliquot is determined. Following incubation in S-protein-coated wells, untagged SEAP is washed away, and the SEAP activity of the bound fraction is determined. The ratio of the bound versus total SEAP activities per sample aliquot reflects the amount of readthrough in the sample, and normalization of this ratio to the amount of fusion protein bound for the experiment yields the percentage readthrough.
Several experiments were conducted to validate this assay. First, we examined binding of the fusion protein to the S-protein-coated plates. Medium was collected from 293T cells that had been transfected with the SEAP fusion control vector, which should yield exclusively SEAP molecules tagged with the S-peptide. Various amounts of this media diluted in PBS were incubated in S-protein-coated plates, and the amount of SEAP activity bound per well was determined. The bound SEAP activity was linear over two orders of magnitude of applied volume (Figure 2A), indicating that we were not saturating the S-protein binding sites and that binding correlated to the amount of S-peptide fusion protein present.
Figure 2.
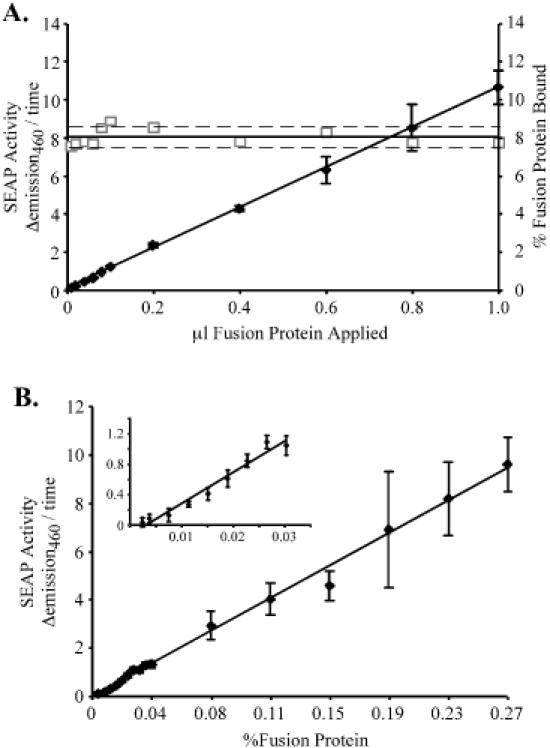
S-tagged SEAP binding to S-protein-coated plates is linear. (A) SEAP bound to S-protein-coated wells was measured using varying amounts of SEAP–S-tagged fusion protein generated by transfection of pEQ965 into 293T cells as described in Materials and Methods. Bound SEAP activity (closed diamonds), was proportional to the amount of fusion protein applied over two orders of magnitude, with a regression coefficient of 0.9995. Measurement of the total SEAP activity in the same dilutions enabled calculation of the fraction bound to the plate (open squares). The solid and broken lines indicate the average percentage bound ± 5% error. (B) As detailed in Materials and Methods, varying amounts of SEAP–S-tagged fusion protein were diluted into a lysate containing a large excess of untagged SEAP, generated by transfection of pEQ911 into 293T cells, and then incubated overnight in an S-protein-coated 96-well plate. Bound SEAP activity correlated to the amount of fusion protein applied over two orders of magnitude, with a regression coefficient of 0.9943. An enlarged view of the low percentage fusion protein data is shown in the inset.
Surprisingly, the percentage of the fusion protein present that bound was only 8.02 ± 0.45%. A number of factors could contribute to the low apparent binding. Appending the S-tag to SEAP does not appear to inhibit SEAP activity (data not shown); however binding of the S-peptide to S-protein may constrain SEAP and reduce its apparent activity. Also, reduction in activity may occur because SEAP substrate must diffuse to SEAP molecules now bound at a surface rather than free in solution. Binding efficiency varied from plate to plate. However, the percentage fusion protein bound was consistent between replicate transfections (data not shown), suggesting that the observed differences were due to variation in the plates and the S-protein coating, and not due to variations in the production of binding competent S-peptide-tagged SEAP. Although the binding efficiency of S-peptide-tagged SEAP was lower than expected, the linearity of the binding and its consistency within each experiment argue that this approach can be used to detect relative changes in readthrough.
To assess the reliability of the assay when tagged SEAP constitutes only a minor fraction of the total SEAP, we diluted the S-tagged SEAP into a constant amount of untagged SEAP, rather than in PBS, such that tagged SEAP comprised ∼1/100 000 to 1/1000 of the total SEAP activity per well. As before, the amount of bound SEAP activity detected correlated with the amount of fusion protein present (Figure 2B). Since each well contained approximately the same amount of total SEAP activity, these results demonstrate that the bound SEAP activity is due to the presence of S-peptide-tagged SEAP and not just to non-specific binding of SEAP. No bound SEAP activity was detected when the binding reaction contained exclusively untagged SEAP. The experiment also shows that excess untagged SEAP does not interfere with fusion protein binding, as we were able to detect fusion protein when it comprised as little as 0.002% of the total SEAP activity present in a binding reaction. Furthermore, the presence of excess untagged SEAP did not significantly affect the percentage of fusion protein that bound per well, which was 9.00 ± 1.44% in this experiment.
Engineered suppressor tRNAs were used to test the specificity of this readthrough assay. All possible combinations of suppressor tRNA plasmids, including the control serine tRNA plasmid from which the others were derived, and SEAP reporter plasmids, were transfected into 293T cells. The basal readthrough detected in the transfections containing the parental serine tRNA was 0.017 ± 0.003% at the ochre (TAA) codon, 0.088 ± 0.005% at the amber (TAG) codon, and 0.063 ± 0.002% at the opal (TGA) codons. Large increases in readthrough were observed only in transfections that contained the cognate SEAP stop codon and suppressor tRNA pairing (Figure 3A). The presence of the ochre suppressor tRNA increased readthrough from the ochre SEAP construct by ∼700-fold. Readthrough increases were 400- and 500-fold for the amber and opal pairings, respectively. Although the opal suppressor had no effect on non-cognate stop codons, we did detect a very small cross-response between the ochre and amber stop codons and anticodons. Expression of the amber suppressor tRNA increased readthrough at the ochre stop codon ∼2-fold and a similar response was observed at the amber termination signal in the presence of the ochre suppressor tRNA. However, >99% of the impact of the tRNA suppressors was limited to cognate stop codons, validating the stop codon specificity of the assay.
Figure 3.
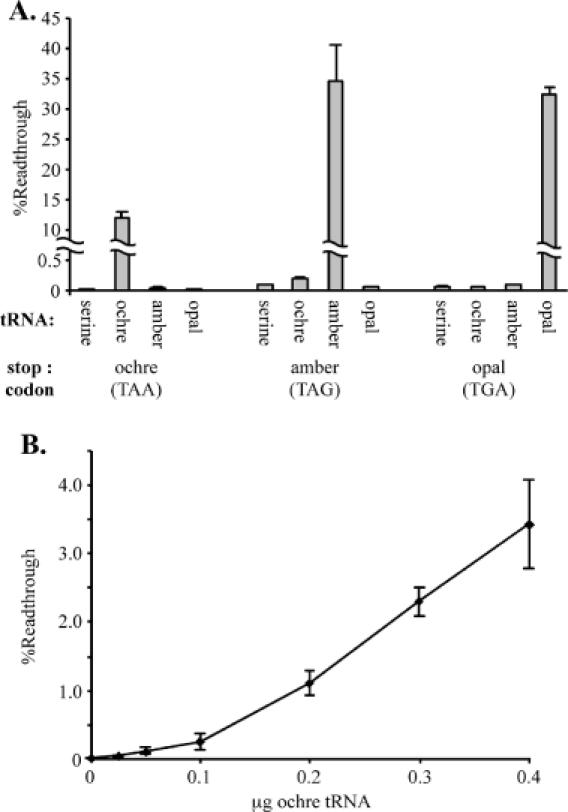
Detection of stop codon-specific increases in readthrough with the SEAP assay system. (A) SEAP indicator vectors [pEQ962 (ochre), pEQ963 (amber), pEQ964 (opal)] were cotransfected into 293T cells along with suppressor tRNA constructs [pSV-tS-su+ (ochre), pEQ933 (amber) or pEQ934 (opal)], or the parental serine tRNA plasmid [pSV-tS-su−(serine)] from which they were derived, in all possible combinations. The readthrough levels were determined as described in Materials and Methods. Increases in readthrough were detected only in combinations containing the cognate suppressor tRNA and stop codon. (B) Increasing amounts of the ochre suppressor tRNA construct [pSV-tS-su+ (ochre)] were cotransfected into 293T cells with a constant amount of ochre stop codon junction SEAP indicator vector (pEQ962), while the plasmid encoding the parental serine tRNA [pSV-tS-su−(serine)] was used to equalize DNA mass among transfections. The percentage readthrough increased with the amount of ochre suppressor tRNA transfected.
Since the changes in readthrough observed in the previous experiment were much larger than the alterations of termination efficiency expected when using other termination modulators, we examined the sensitivity of the assay by cotransfecting 293T cells with varying amounts of the ochre suppressor tRNA plasmid and a constant amount of ochre stop codon junction SEAP indicator. Increasing amounts of suppressor tRNA enhanced the readthrough level (Figure 3B), with as little as a 2-fold increase in readthrough level detected at the lowest suppressor tRNA concentration, suggesting that the assay is quite sensitive in detecting changes in suppression frequency.
The previous experiment was performed with a SEAP vector that has the ochre stop codon surrounded by a TMV-like context nucleotide that is known to allow a relatively high basal readthrough level. To further examine the utility of this assay, we repeated the experiment using a SEAP indicator construct that contains the ochre stop codon surrounded by an alternative context of nucleotides not predicted to allow a high level of readthrough (pEQ911). In this experiment (data not shown) basal readthrough was undetectable, but the addition of the cognate suppressor tRNA enhanced readthrough from this construct to detectable levels. Although the overall level of readthrough that could be achieved was lower with this construct, the increases observed in readthrough levels correlated linearly with the amount of suppressor tRNA expression vector used (data not shown). These results suggest that the SEAP system should be useful for detecting the alterations in readthrough that would result from perturbation of termination efficiency at stop codons in alternative contexts.
Release factor knockdown in 293 cells
Because the effects of release factor overexpression on termination differ between yeast and human cells (24–26), release factor depletion might also affect termination efficiency differently in these two systems. To assess the impact of release factor depletion in mammalian cells, we employed RNA interference, using a plasmid-based system (29) to generate shRNAs. Two target sequences were chosen for each release factor. For eRF1, the vectors we used expressed shRNA 1A, corresponding to sequences near the 5′ end and 1B, matching a sequence located near the 3′ end of the eRF1 reading frame. Translation of eRF3 may initiate at any one of three alternate start codons, with the short- and long forms of the protein predominating (36). Therefore, sequences near the 5′ end of the coding region were avoided in selecting eRF3 RNA interference targets. Instead, we used shRNAs 3A and 3B, corresponding to the middle and 3′ end of the eRF3 coding region, respectively. The sequences in shRNA 3A but not 3B should also target the recently described eRF3-related protein eRF3b (37–39).
Since the release factors are essential proteins (13,14,40), we attempted to deplete their expression by transient transfection of the shRNA constructs rather than by trying to generate stable cell lines. Because DNA will enter only a fraction of the cells in a typical transfection, we cotransfected his-tagged eRF1 or eRF3 transgenes and assessed the efficacy of the RNA interference constructs by immunoblot assays using anti-His antibodies. A his-tagged non-cellular protein, TRS1, was included in these cotransfection experiments to allow comparison of release factor levels among samples. While cotransfection of the shRNA construct backbone did not change the expression level of the eRF1 transgene (data not shown), shRNA constructs 1A and 1B resulted in decreased transgene expression (Figure 4A). RNA interference against target 1B was substantially more effective at knocking down eRF1 expression, reducing the level of his-tagged eRF1 expression ∼10-fold, as opposed to the 2-fold reduction in transgene expression observed with shRNA 1A. Not surprisingly, the shRNAs that target eRF3 had no effect on expression of eRF1.
Figure 4.
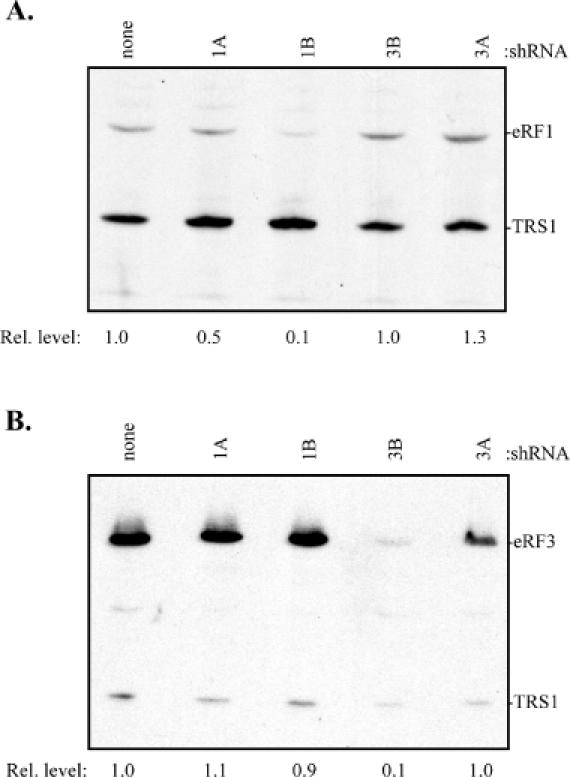
Release factor transgene knockdown by RNA interference. (A) 293T cells were cotransfected with an expression plasmid for his-tagged eRF1 (pEQ842) and no shRNA construct (none) or the indicated shRNA construct [1A (pEQ929), 1B (pEQ941), 3B (pEQ946), or 3A (pEQ945)]. A his-tagged non-cellular gene, TRS1 (pEQ909)—derived from the HCMV TRS1 coding sequence—was included to allow comparison between transfections. Transgene expression was visualized by immunoblot with antibody specific for the his tag. Positions of eRF1 and TRS1 migration are shown. Transgene expression relative to transfections containing no shRNA vector is indicated below each lane. (B) The transgene knockdown experiment described above was repeated with a his-tagged eRF3 (pEQ863) construct.
Next, the experiment was repeated using his-tagged eRF3 in place of his-tagged eRF1. As expected shRNAs directed against eRF1 had no effect on eRF3 transgene expression (Figure 4B). While the shRNA construct containing the 3A target did not result in any knockdown of eRF3 transgene expression, the construct containing the 3B construct effectively decreased transgene expression by ∼10-fold.
The effect of release factor depletion on termination efficiency was then examined in 293 cells using the SEAP readthrough assay. In order to determine the termination response to the various shRNA constructs, we first examined the effects of eRF1 and eRF3 depletion at the amber stop codon junction. The basal readthrough level, determined from cotransfection of the shRNA vector backbone (pBp) with the SEAP indicator (Figure 5), was not significantly different from that obtained with transfection of the readthrough indicator alone or with cotransfection of an irrelevant plasmid (pBS—pBluescript SK+, Stratagene) (data not shown). Cotransfection of the shRNA TRS1 construct, which targets a non-cellular gene, also did not alter the observed readthrough level. Cotransfection of the shRNA construct 1A, which caused only minimal reduction of eRF1 transgene expression, caused a slight but not statistically significant increase in the readthrough level. In contrast, the more efficient eRF1 knockdown caused by cotransfection of the shRNA 1B construct caused a ∼2.5-fold increase in readthrough, reflecting a reduction in termination efficiency. Cotransfection with shRNA 3A did not affect readthrough. Although the 3B shRNA construct was quite effective at depleting eRF3 transgene expression, it caused only a slight increase in readthrough that was not significantly different from the basal level.
Figure 5.
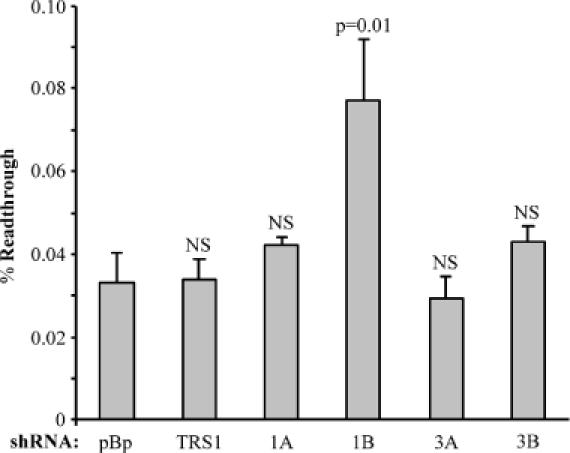
Effects of release factor depletion on termination efficiency at the amber stop codon in 293 cells. 293 cells were cotransfected with equal amounts of amber (UAG) stop codon junction SEAP indicator vector (pEQ963) and the shRNA constructs or control plasmids as described in Materials and Methods. At 72 h post-transfection, the percentage readthrough for each sample was determined. Each column represents the average readthrough level determined from triplicate transfections, with error bars signifying one sample standard deviation. The constructs used for cotransfection were pBp (pEQ940)—the RNA interference construct backbone, TRS1 (pEQ936)—an shRNA construct targeting the non-cellular gene TRS1, and 1A (pEQ929), 1B (pEQ941), 3A (pEQ945) or 3B (pEQ946)—RNA interference constructs targeting eRF1 or eRF3. The significance with respect to the pBp basal readthrough value, as indicated by the P-value derived from a two-tailed t-test, is listed above each sample (NS, not significant, P > 0.05). Only eRF1 depletion by the 1B RNA interference construct resulted in substantial changes in readthrough.
If release factor depletion affects termination efficiency, we expect it to do so at all three stop codons. Therefore, we examined the impact of shRNA-mediated knockdown of eRF1 and eRF3 at each stop codon using the full set of SEAP vectors. In these experiments we used the shRNA constructs 1B and 3B since they were more effective than 1A and 3A at knocking down their respective targets. Depletion of eRF1 decreased termination efficiency at each stop codon as indicated by a ∼3-fold increase in readthrough in all cases (Figure 6A). A marginal increase in the mean readthrough level was noted at each stop codon when eRF3 was depleted, but this increase was only statistically significant at the ochre stop codon. We also examined the effects of simultaneous depletion of both release factors. The combined effect of cotransfecting plasmids expressing 1B and 3B shRNAs was a slight increase in readthrough at amber and opal stop codons compared to eRF1 depletion alone; however, this increase was only significant at the amber stop codon. No measurable effect beyond the results from eRF1 depletion alone was detected at the ochre stop codon. The experiments in which the release factors were depleted individually were repeated 3 times, with triplicate transfections for each readthrough measurement. Only depletion of eRF1 consistently exhibited a statistically significant decrease in termination efficiency at all stop codons. Depletion of eRF3 had a significant effect on termination in only one other instance (two out of nine total measurements), in this case at the opal stop codon. The results are summarized in Figure 6B, with the fold change in readthrough calculated relative to readthrough levels in the presence of the parental shRNA construct pBp. In 293 cells, depletion of eRF1 consistently caused a decrease in termination efficiency, increasing readthrough levels of 2.5- to 3-fold at all stop codons tested. While depletion of eRF3 appeared to cause a slight increase in readthrough, this effect was not observed in all experiments and was not statistically significant.
Figure 6.
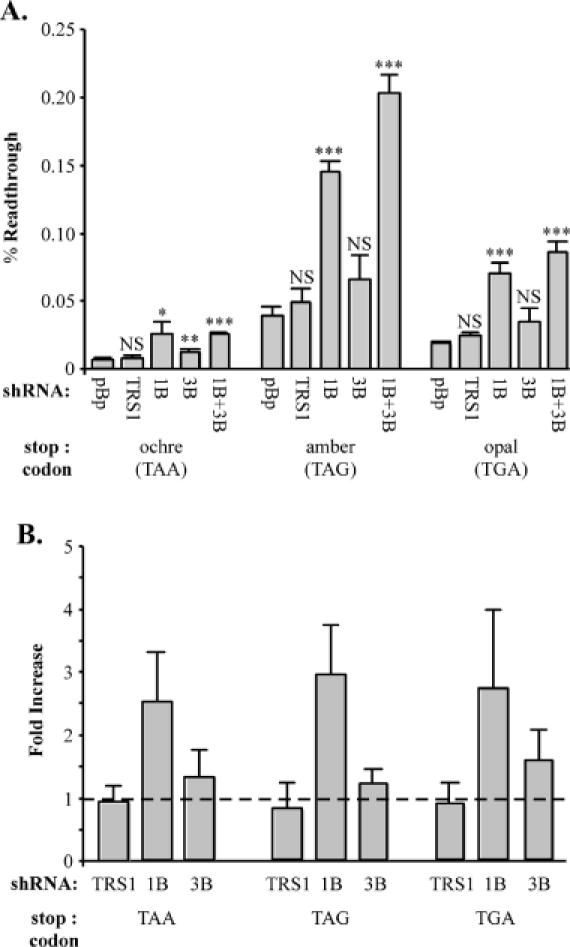
Depletion of eRF1 decreases termination in 293 cells. (A) The effects of release factor depletion were examined at each stop codon. Equal amounts of SEAP readthrough vectors with the indicated stop codon [ochre (pEQ962), amber (pEQ963), or opal (pEQ964)] and shRNA constructs were cotransfected into 293 cells as described in Materials and Methods. In transfections where both release factors were depleted simultaneously, equal amounts of all three plasmids were used. Bound and total alkaline phosphatase activity was measured and used to calculate the mean (± SD) percentage readthrough from triplicate transfections for each RNA interference construct. RNA interference constructs used were pBp (pEQ940), TRS1 (pEQ936), 1B (pEQ941) or 3B (pEQ946). The significance of the test samples with respect to the pBp basal value is shown above the each bar. The P-values, as determined by a two-tailed t-test, are indicated by NS (P > 0.05), asterisk (0.05 ≥ P > 0.01), double asterisks (0.01 ≥ P > 0.001) and triple asterisks (0.001 ≥ P). (B) The single experiment shown in (A) was repeated on three separate occasions for each SEAP indicator. The results, expressed as the fold increase in percentage readthrough in the presence of the indicated shRNA construct with respect to the basal readthrough level determined with pBp, are summarized with error bars representing one sample standard deviation. The dashed line represents the theoretical value when readthrough is unaffected by the test shRNA.
Release factor knockdown in HeLa cells
To determine whether the effects of release factor depletion were similar in other cell types, we repeated the readthrough experiments using HeLa cells. The basal readthrough levels detected were substantially lower in HeLa cells compared to 293 cells (Figure 7A) and were similar among transfections containing just the SEAP indicator, or with cotransfection of the controls pBS, pBp or pTRS1 (data not shown). Consistent with the data from assays in 293 cells, depletion of eRF1 decreased termination efficiency in HeLa cells as indicated by an increase of ∼2- to 3-fold in readthrough at each of the three stop codons (Figure 7A). However, in contrast to the results in 293 cells, depletion of eRF3 caused a clear reduction in termination efficiency in the HeLa cells. The effect was generally similar in magnitude to that caused by depletion of eRF1. Dual depletion of eRF1 and eRF3 caused a slightly higher level of readthrough than depletion of either eRF alone at amber and opal but not ochre codons; however, this effect was not consistently significantly different from either eRF1 or eRF3 depletion. The fact that the effect of dual release factor depletion was generally similar to that of single depletion suggests that the two proteins may act at the same step in the termination reaction. The effect of release factor depletion was determined at each stop codon in three separate experiments (Figure 7B). Depletion of either release factor caused a statistically significant decrease in termination efficiency in all cases. The fold increase in readthrough in response to release factor depletion was consistently 2- to 3-fold at each stop codon regardless of which release factor was depleted. These results suggest that eRF3 is directly involved in the termination process.
Figure 7.
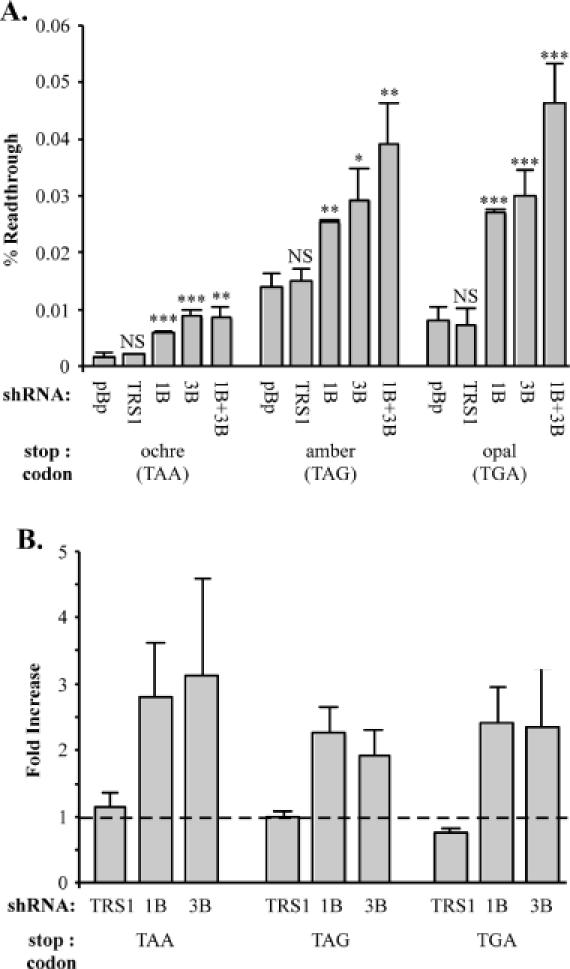
Increase in termination efficiency in HeLa cells by depletion of either eRF1 or eRF3. (A) The effects of release factor depletion were determined by transfection, in triplicate, in HeLa cells as described in Materials and Methods. RNA interference constructs used were pBp (pEQ940), TRS1 (pEQ936), 1B (pEQ941) or 3B (pEQ946). The significance of the test samples with respect to the pBp basal value, determined by a two-tailed t-test is shown above each bar: NS (P > 0.05), asterisk (0.05 ≥ P > 0.01), double asterisks (0.01 ≥ P > 0.001) and triple asterisks (0.001 ≥ P). (B) Summary of results from experiments performed on three separate occasions. Results are expressed as the mean fold (± SD) increase in percentage readthrough as compared to transfections with pBp. The dashed line represents the theoretical value when readthrough is unaffected by the test shRNA.
It was surprising that eRF3 depletion had differing effects in the two cell types. One possible explanation was that RNA interference was more effective in the HeLa cells. However, we found that knockdown of eRF3 was actually somewhat less efficient in HeLa than in 293 cells (data not shown). Another possible explanation was that the level of endogenous release factor pools may cause the HeLa cells to be more sensitive to eRF3 knockdown. In fact, immunoblot assays revealed that HeLa cells do appear to have less endogenous eRF3 but similar levels of eRF1 compared to 293 cells (Figure 8). Thus, it is possible that a further reduction of eRF3 in HeLa cells results in it becoming rate-limiting for termination. These results highlight the potential for variation in release factor levels or activities among cell types.
Figure 8.
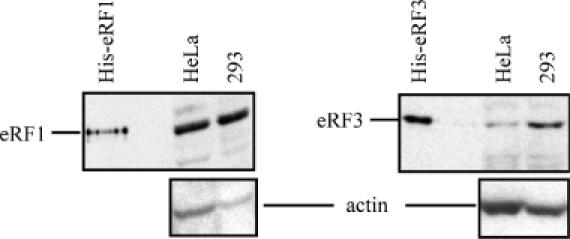
Relative expression levels of eRF1 and eRF3 in 293 and HeLa cells. Fresh lysates of confluent HeLa or 293 cells were analyzed by immunoblot to determine the relative levels of endogenous release factors. Equal amounts of total protein (20 or 50 μg/well for eRF1 or eRF3 blots, respectively) were fractionated by SDS–PAGE and electroblotted onto nitrocellulose. Blots were first probed with antibody directed against eRF1 (left) or eRF3 (right), and then reprobed with antibody specific for actin.
DISCUSSION
We adapted a SEAP assay system, first described by Sogaard et al. (32) to examine the effects of release factor depletion on termination efficiency in human cell lines. Since this assay has not been widely used, we examined its performance in a series of control experiments. The measurement of S-peptide-tagged SEAP was linear over a wide range of enzyme concentrations. However, the binding efficiency of the tagged SEAP molecules was approximately one-tenth of the expected value. Although it is possible that S-peptide binding constrains the activity of the SEAP molecule, causing an artificially low apparent binding efficiency, we detected a substantial amount of tagged-SEAP protein in the medium after incubation of the SEAP–S-tag fusion protein in S-peptide-coated wells (data not shown). This result suggests that the apparently low binding efficiency is not simply due to alteration of SEAP activity by virtue of the physical constraints caused by binding to the plastic surface. Although it has been reported that an S-peptide fusion protein had a dissociation constant only slightly higher than that of S-peptide alone (34), it is possible that fusion to the SEAP molecule decreased the affinity of the S-peptide for S-protein. We have observed a 2-fold increase in binding with a SEAP construct possessing a longer spacer region between the SEAP and S-tag moieties (data not shown), suggesting that low binding efficiency may be due in part to steric hindrance of S-tag binding by SEAP. It may be possible to improve binding efficiency by multimerizing the tag. Regardless, the low binding efficiency of the fusion protein affects the detection limit of the SEAP system, but not its ability to detect changes in readthrough. With this system, we were able to detect changes in readthrough level as small as 2-fold from the basal levels of readthrough as low as 0.002%.
We found basal readthrough values of ∼0.01, 0.04 and 0.03% at the ochre (UAA), amber (UAG) and opal (UGA) stop codons, respectively, in 293 cells. Our absolute readthrough level is lower than the 2% reported at the amber TMV stop codon context in NIH 3T3 cells (41). Although some of the differences may be due to assay methods, in the original report of the SEAP–S-tag assay higher readthrough levels were reported albeit with a corresponding high assay background (32). Part of the variation in basal readthrough is likely due to cell-type-specific differences. In fact, basal readthrough levels were 2.5-fold lower in HeLa cells than in 293 cells in our experiments. The low level of basal readthrough may also be due in part to subtle differences in the nucleotide context surrounding the termination codons. Our SEAP vectors contained a modified TMV readthrough context, CAA TRR CAA CTC. Even relatively minor alterations in the nucleotide context of the termination codon can have major effects. For example, mutation of the bona fide TMV stop context CAA TAG CAA TTA to CAA TAG CAA CTT caused a 10-fold decrease in readthrough in yeast (42). In a study with 3T3 cells, subtle differences in stop codon context caused up to a 100-fold alteration in termination efficiency (41). We were unable to detect any basal readthrough when we used the context GGC TAG CCA GAA (data not shown). Thus, differences in the translational termination fidelity in various cells and in the termination codon context may result in a large effect on the basal level of readthrough, making exact comparisons between systems difficult.
RNA interference using transfection of synthetic 21–23mer dsRNA molecules has been tested previously as a method for inhibiting termination. As in our study, knockdown of eRF1 promoted readthrough by 2- to 3-fold (27), but the study by Carnes et al. only tested termination at an amber codon and did not examine eRF3 depletion. Using in situ generation of the shRNAs, we observed that eRF1 knockdown augments readthrough at all three stop codons. The particular eRF1 shRNAs and siRNAs used varied in the efficacy of eRF1 depletion, possibly due to the precise RNA targets or to the method of delivery. Nonetheless, the effects of eRF1 depletion on readthrough were strikingly similar. In the previous study, eRF1 depletion with the more effective siRNA caused a 2.8-fold increase in readthrough at the UAG stop codon, while our shRNA 1B caused a 3-fold increase in readthrough. In our study, the magnitude of the effect on termination efficiency was similar at all three stop codons, and was observed in HeLa as well as 293 cells. A potential advantage of the in situ generation of shRNAs is that it can be used to generate stable cell lines. However, in an attempt to make such lines using shRNA 1B, we found that an initial increase in readthrough disappeared after repeated cell passage (data not shown), possibly reflecting selective pressure against depletion of an essential gene product.
Since promoting readthrough has potential therapeutic applications for a variety of genetic diseases in which nonsense codons prevent expression of the full-length protein, several other approaches for modulating termination have been investigated. Aminoglycoside antibiotics increase readthrough by promoting ribosomal misreading. The degree to which aminoglycosides increase readthrough depends upon the exact drug and stop codon context (43). Gentamicin increased readthrough 5- to 10-fold in cell-free assays and in 293 cells (44,45), and increased protein expression 8-fold in a Hurler cell line (46). A pilot test with gentamicin in our system yielded a 2- to 3-fold increase from the amber SEAP construct (data not shown), suggesting that aminoglycoside-promoted readthrough may be lower with our test construct stop context. Other methods of interfering with termination include the use of RNA aptamers and suppressor tRNAs. Aptamers that bind to and inhibit eRF1 function caused a 1.5- to 2.5-fold increase in readthrough when added to in vitro translation reactions (27). When the reactions were supplemented with eRF1 prior to aptamer addition, readthrough increased 3- to 4-fold relative to no aptamer addition. The efficacy of these RNA aptamers in cell culture has not been reported. Since nonsense suppression by suppressor tRNAs allows cognate base pairing at the termination codon, whereas readthrough in release factor depletion experiments relies on non-cognate binding, the suppressor tRNA is much more effective at increasing readthrough levels. Suppressor tRNAs may have an advantage for therapeutic use in that they act in a stop-codon-dependent manner. However, each of these methods designed to promote readthrough will undoubtedly increase readthrough events at many termination sites, generating unintended C-terminally extended proteins that will complicate use of this approach for treatment of genetic diseases.
Since no link has been found between eRF3 overexpression and increased termination efficiency in mammalian systems (25,26), we were unsure whether decreased eRF3 expression would reduce termination efficiency in human cell lines. We found that eRF3 depletion promoted readthrough to a similar extent as eRF1 depletion in HeLa cells. This result agrees with observations in yeast cells and argues that eRF3 plays a direct role in termination in higher eukaryotes. However, eRF3 depletion did not detectably decrease termination efficiency in 293 cells. This difference was not due to a difference in the effectiveness of RNA interference between the two cell types (Figure 4 and data not shown), nor does it appear to be caused by a general destabilization of eRF1 due to eRF3 depletion (Figure 4A). Rather our data demonstrating a relatively lower abundance of eRF3 in HeLa cells than in 293 cells (Figure 8) suggest that following depletion by shRNAs, eRF3 levels might drop below a critical level in HeLa cells but not 293 cells. Relatively high levels of eRF3 expression could explain why overexpression of eRF1, but not eRF3 increased termination efficiency in 293 cells (26). However, we do not know whether the abundance of eRF1 and eRF3 is proportional to their activities nor do we know whether the two cell lines express similar levels eRF3b (38,39). Since the shRNA 3B targets eRF3 but not eRF3b, high eRF3b levels might functionally substitute for eRF3 upon depletion. Recent studies in Drosophilia (14) have shown that the progeny of maternal eRF3 haploinsufficient flies exhibit increased readthrough during early development at certain premature termination codons, and results from one study suggest that overexpression of eRF3 in HeLa cells may decrease nonsense suppression (32). These results bolster our findings that depletion of eRF3 in HeLa cells decreases termination efficiency and further support a direct role for eRF3 in termination in higher eukaryotes.
Acknowledgments
ACKNOWLEDGEMENTS
We would like to thank Drs Uttam RajBandary (Massachusetts Institute for Technology), Shin-ichi Hoshino (University of Tokyo), Michael Katze (University of Washington) and Bruce Clurman, Michael Emerman, Ted Gooley, Alana Ruddell (Fred Hutchinson Cancer Research Center), members of the Geballe laboratory and the Biotechnology Resource of the Fred Hutchinson Cancer Research Center for advice, technical assistance, and reagents. This work was supported by NIH grants AI26672 and CA91329. D.M.J. was supported in part by a training grant from the National Cancer Institute, CA 09229.
REFERENCES
- 1.Seit-Nebi A., Frolova,L. and Kisselev,L. (2002) Conversion of omnipotent translation termination factor eRF1 into ciliate-like UGA-only unipotent eRF1. EMBO Rep., 3, 881–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ito K., Frolova,L., Seit-Nebi,A., Karamyshev,A., Kisselev,L. and Nakamura,Y. (2002) Omnipotent decoding potential resides in eukaryotic translation termination factor eRF1 of variant-code organisms and is modulated by the interactions of amino acid sequences within domain 1. Proc. Natl Acad. Sci. USA, 99, 8494–8499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chavatte L., Frolova,L., Kisselev,L. and Favre,A. (2001) The polypeptide chain release factor eRF1 specifically contacts the s(4)UGA stop codon located in the A site of eukaryotic ribosomes. Eur. J. Biochem., 268, 2896–2904. [DOI] [PubMed] [Google Scholar]
- 4.Chavatte L., Seit-Nebi,A., Dubovaya,V. and Favre,A. (2002) The invariant uridine of stop codons contacts the conserved NIKSR loop of human eRF1 in the ribosome. EMBO J., 21, 5302–5311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Frolova L.Y., Tsivkovskii,R.Y., Sivolobova,G.F., Oparina,N.Y., Serpinsky,O.I., Blinov,V.M., Tatkov,S.I. and Kisselev,L.L. (1999) Mutations in the highly conserved GGQ motif of class 1 polypeptide release factors abolish ability of human eRF1 to trigger peptidyl-tRNA hydrolysis. RNA, 5, 1014–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Seit-Nebi A., Frolova,L., Justesen,J. and Kisselev,L. (2001) Class-1 translation termination factors: invariant GGQ minidomain is essential for release activity and ribosome binding but not for stop codon recognition. Nucleic Acids Res., 29, 3982–3987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Janzen D.M., Frolova,L. and Geballe,A.P. (2002) Inhibition of translation termination mediated by an interaction of eukaryotic release factor 1 with a nascent peptidyl-tRNA. Mol. Cell. Biol., 22, 8562–8570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Frolova L.Y., Simonsen,J.L., Merkulova,T.I., Litvinov,D.Y., Martensen,P.M., Rechinsky,V.O., Camonis,J.H., Kisselev,L.L. and Justesen,J. (1998) Functional expression of eukaryotic polypeptide chain release factors 1 and 3 by means of baculovirus/insect cells and complex formation between the factors. Eur. J. Biochem., 256, 36–44. [DOI] [PubMed] [Google Scholar]
- 9.Frolova L., Le Goff,X., Zhouravleva,G., Davydova,E., Philippe,M. and Kisselev,L. (1996) Eukaryotic polypeptide chain release factor eRF3 is an eRF1- and ribosome-dependent guanosine triphosphatase. RNA, 2, 334–341. [PMC free article] [PubMed] [Google Scholar]
- 10.Zhouravleva G., Frolova,L., Le Goff,X., Le Guellec,R., Inge-Vechtomov,S., Kisselev,L. and Philippe,M. (1995) Termination of translation in eukaryotes is governed by two interacting polypeptide chain release factors, eRF1 and eRF3. EMBO J., 14, 4065–4072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Frolova L., Le Goff,X., Rasmussen,H.H., Cheperegin,S., Drugeon,G., Kress,M., Arman,I., Haenni,A.L., Celis,J.E., Philippe,M. et al. (1994) A highly conserved eukaryotic protein family possessing properties of polypeptide chain release factor. Nature, 372, 701–703. [DOI] [PubMed] [Google Scholar]
- 12.Grentzmann G., Brechemier-Baey,D., Heurgue,V., Mora,L. and Buckingham,R.H. (1994) Localization and characterization of the gene encoding release factor RF3 in Escherichia coli. Proc. Natl Acad. Sci. USA, 91, 5848–5852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stansfield I. and Tuite,M.F. (1994) Polypeptide chain termination in Saccharomyces cerevisiae. Curr. Genet., 25, 385–395. [DOI] [PubMed] [Google Scholar]
- 14.Chao A.T., Dierick,H.A., Addy,T.M. and Bejsovec,A. (2003) Mutations in eukaryotic release factors 1 and 3 act as general nonsense suppressors in Drosophila. Genetics, 165, 601–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Valouev I.A., Kushnirov,V.V. and Ter-Avanesyan,M.D. (2002) Yeast polypeptide chain release factors eRF1 and eRF3 are involved in cytoskeleton organization and cell cycle regulation. Cell Motil. Cytoskeleton, 52, 161–173. [DOI] [PubMed] [Google Scholar]
- 16.Basu J., Williams,B.C., Li,Z., Williams,E.V. and Goldberg,M.L. (1998) Depletion of a Drosophila homolog of yeast Sup35p disrupts spindle assembly, chromosome segregation, and cytokinesis during male meiosis. Cell Motil. Cytoskeleton, 39, 286–302. [DOI] [PubMed] [Google Scholar]
- 17.Hoshino S., Imai,M., Kobayashi,T., Uchida,N. and Katada,T. (1999) The eukaryotic polypeptide chain releasing factor (eRF3/GSPT) carrying the translation termination signal to the 3′-poly(A) tail of mRNA. Direct association of erf3/GSPT with polyadenylate-binding protein. J. Biol. Chem., 274, 16677–16680. [DOI] [PubMed] [Google Scholar]
- 18.Uchida N., Hoshino,S., Imataka,H., Sonenberg,N. and Katada,T. (2002) A novel role of the mammalian GSPT/eRF3 associating with poly(A)-binding protein in Cap/Poly(A)-dependent translation. J. Biol. Chem., 277, 50286–50292. [DOI] [PubMed] [Google Scholar]
- 19.Wang W., Czaplinski,K., Rao,Y. and Peltz,S.W. (2001) The role of Upf proteins in modulating the translation read-through of nonsense-containing transcripts. EMBO J., 20, 880–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Czaplinski K., Ruiz-Echevarria,M.J., Paushkin,S.V., Han,X., Weng,Y., Perlick,H.A., Dietz,H.C., Ter-Avanesyan,M.D. and Peltz,S.W. (1998) The surveillance complex interacts with the translation release factors to enhance termination and degrade aberrant mRNAs. Genes Dev., 12, 1665–1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Weng Y., Czaplinski,K. and Peltz,S.W. (1996) Identification and characterization of mutations in the UPF1 gene that affect nonsense suppression and the formation of the Upf protein complex but not mRNA turnover. Mol. Cell. Biol., 16, 5491–5506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Inge-Vechtomov S., Zhouravleva,G. and Philippe,M. (2003) Eukaryotic release factors (eRFs) history. Biol. Cell, 95, 195–209. [DOI] [PubMed] [Google Scholar]
- 23.Hawthorne D.C. and Leupold,U. (1974) Suppressors in yeast. Curr. Top. Microbiol. Immunol., 64, 1–47. [DOI] [PubMed] [Google Scholar]
- 24.Stansfield I., Jones,K.M., Kushnirov,V.V., Dagkesamanskaya,A.R., Poznyakovski,A.I., Paushkin,S.V., Nierras,C.R., Cox,B.S., Ter-Avanesyan,M.D. and Tuite,M.F. (1995) The products of the SUP45 (eRF1) and SUP35 genes interact to mediate translation termination in Saccharomyces cerevisiae. EMBO J., 14, 4365–4373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Drugeon G., Jean-Jean,O., Frolova,L., Le Goff,X., Philippe,M., Kisselev,L. and Haenni,A.L. (1997) Eukaryotic release factor 1 (eRF1) abolishes readthrough and competes with suppressor tRNAs at all three termination codons in messenger RNA. Nucleic Acids Res., 25, 2254–2258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Le Goff X., Philippe,M. and Jean-Jean,O. (1997) Overexpression of human release factor 1 alone has an antisuppressor effect in human cells. Mol. Cell. Biol., 17, 3164–3172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Carnes J., Jacobson,M., Leinwand,L. and Yarus,M. (2003) Stop codon suppression via inhibition of eRF1 expression. RNA, 9, 648–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Capone J.P., Sharp,P.A. and RajBhandary,U.L. (1985) Amber, ochre and opal suppressor tRNA genes derived from a human serine tRNA gene. EMBO J., 4, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Grandori C., Wu,K.J., Fernandez,P., Ngouenet,C., Grim,J., Clurman,B.E., Moser,M.J., Oshima,J., Russell,D.W., Swisshelm,K. et al. (2003) Werner syndrome protein limits MYC-induced cellular senescence. Genes Dev., 17, 1569–1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hoshino S., Miyazawa,H., Enomoto,T., Hanaoka,F., Kikuchi,Y., Kikuchi,A. and Ui,M. (1989) A human homologue of the yeast GST1 gene codes for a GTP-binding protein and is expressed in a proliferation-dependent manner in mammalian cells. EMBO J, 8, 3807–3814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Child S.J., Hakki,M., De Niro,K.L. and Geballe,A.P. (2004) Evasion of cellular antiviral responses by human cytomegalovirus TRS1 and IRS1. J Virol., 78, 197–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sogaard T.M., Jakobsen,C.G. and Justesen,J. (1999) A sensitive assay of translational fidelity (readthrough and termination) in eukaryotic cells. Biochemistry (Mosc), 64, 1408–1417. [PubMed] [Google Scholar]
- 33.Kim J.S. and Raines,R.T. (1993) Ribonuclease S-peptide as a carrier in fusion proteins. Protein Sci., 2, 348–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Karpeisky M., Senchenko,V.N., Dianova,M.V. and Kanevsky,V. (1994) Formation and properties of S-protein complex with S-peptide-containing fusion protein. FEBS Lett., 339, 209–212. [DOI] [PubMed] [Google Scholar]
- 35.Skuzeski J.M., Nichols,L.M., Gesteland,R.F. and Atkins,J.F. (1991) The signal for a leaky UAG stop codon in several plant viruses includes the two downstream codons. J. Mol. Biol., 218, 365–373. [DOI] [PubMed] [Google Scholar]
- 36.Jean-Jean O., Le Goff,X. and Philippe,M. (1996) Is there a human [psi]? C R Acad. Sci. III, 319, 487–492. [PubMed] [Google Scholar]
- 37.Hoshino S., Imai,M., Mizutani,M., Kikuchi,Y., Hanaoka,F., Ui,M. and Katada,T. (1998) Molecular cloning of a novel member of the eukaryotic polypeptide chain-releasing factors (eRF). Its identification as eRF3 interacting with eRF1. J. Biol. Chem., 273, 22254–22259. [DOI] [PubMed] [Google Scholar]
- 38.Le Goff C., Zemlyanko,O., Moskalenko,S., Berkova,N., Inge-Vechtomov,S., Philippe,M. and Zhouravleva,G. (2002) Mouse GSPT2, but not GSPT1, can substitute for yeast eRF3 in vivo. Genes Cells, 7, 1043–1057. [DOI] [PubMed] [Google Scholar]
- 39.Jakobsen C.G., Segaard,T.M., Jean-Jean,O., Frolova,L. and Justesen,J. (2001) [Identification of a novel termination release factor eRF3b expressing the eRF3 activity in vitro and in vivo]. Mol. Biol. (Mosk), 35, 672–681. [PubMed] [Google Scholar]
- 40.Kisselev L.L. and Buckingham,R.H. (2000) Translational termination comes of age. Trends Biochem. Sci., 25, 561–566. [DOI] [PubMed] [Google Scholar]
- 41.Cassan M. and Rousset,J.P. (2001) UAG readthrough in mammalian cells: effect of upstream and downstream stop codon contexts reveal different signals. BMC Mol. Biol., 2, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Namy O., Hatin,I. and Rousset,J.P. (2001) Impact of the six nucleotides downstream of the stop codon on translation termination. EMBO Rep., 2, 787–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Manuvakhova M., Keeling,K. and Bedwell,D.M. (2000) Aminoglycoside antibiotics mediate context-dependent suppression of termination codons in a mammalian translation system. RNA, 6, 1044–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Keeling K.M. and Bedwell,D.M. (2002) Clinically relevant aminoglycosides can suppress disease-associated premature stop mutations in the IDUA and P53 cDNAs in a mammalian translation system. J. Mol. Med., 80, 367–376. [DOI] [PubMed] [Google Scholar]
- 45.Howard M.T., Shirts,B.H., Petros,L.M., Flanigan,K.M., Gesteland,R.F. and Atkins,J.F. (2000) Sequence specificity of aminoglycoside-induced stop condon readthrough: potential implications for treatment of Duchenne muscular dystrophy. Ann. Neurol., 48, 164–169. [PubMed] [Google Scholar]
- 46.Keeling K.M., Brooks,D.A., Hopwood,J.J., Li,P., Thompson,J.N. and Bedwell,D.M. (2001) Gentamicin-mediated suppression of Hurler syndrome stop mutations restores a low level of alpha-L-iduronidase activity and reduces lysosomal glycosaminoglycan accumulation. Hum. Mol. Genet., 10, 291–299. [DOI] [PubMed] [Google Scholar]