


Abstract
During retroviral replication, full-length viral RNAs are encapsidated into new virus particles, while spliced RNAs are excluded. The Retroviridae are unique among viruses in that infectious viral particles contain a dimer of two identical genomic RNA strands. A variety of experimental data has suggested that dimerization and encapsidation of full-length viral RNAs are linked processes, although whether dimerization is a prerequisite for encapsidation, or conversely, dimerization follows encapsidation, has not been firmly established. If dimerization was the sole determinant for encapsidation, then spliced viral RNAs might be expected to display a reduced capacity for dimerization, resulting in their exclusion from the dimerization pool. Here, we studied the in vitro dimerization properties of unspliced and spliced HIV-2 RNA. We find that the rate and yield of dimerization of Nef, Rev and Tat spliced RNAs exceeded those of unspliced RNA. Although these data do not support a simple correlation between dimerization efficiency and the presence of introns, they establish that splicing affects the presentation of dimerization signal(s), which we corroborate with structure probing. This change in RNA conformation likely affects the RNA's suitability for packaging. Furthermore, the presence of upstream and downstream elements that affect the conformation of the packaging signal represents a potentially efficient viral strategy for correctly sorting spliced versus unspliced RNAs.
INTRODUCTION
Retroviruses encapsidate two copies of their RNA genome per viral particle. After extraction and deproteinization of the viral RNA from mature particles, electron microscopy studies have revealed that the two strands of genomic RNA are associated with a region located close to the 5′ end, called the dimer linkage structure (DLS) (1,2). The DLS overlaps with various structural and functional elements, especially elements of the RNA encapsidation signal [for a review see (3,4)]. Synthetic RNA transcripts spanning the DLS have been shown to dimerize in vitro in the absence of any viral proteins, indicating that direct RNA–RNA intermolecular interaction(s) could play a role in the dimerization process (5). Further analytical studies using synthetic transcripts led to the identification of the mechanisms and structural elements involved in dimerization of the leader region of HIV-1 genomic RNA.
In HIV-1, a short sequence called the dimerization initiation site (DIS) (6,7) or stem–loop 1 (SL1) (8) promotes dimerization of leader RNA fragments. This element is capable of maintaining two viral RNA fragments in a loose or tight dimer state, depending on the dimerization protocol. Loose dimers are thought to be associated via an intermolecular kissing loop interaction (9–13). In general, they can be observed on a native gel [Tris-borate magnesium (TBM) buffer at 4°C] but not on a semidenaturing gel in Tris-borate EDTA (TBE) buffer at 28°C or a TBM gel at 28°C, presumably because the loose dimers dissociate under these electrophoresis conditions (14). In contrast, when HIV-1 RNA is incubated at high temperature (55°C) or in the presence of viral nucleocapsid protein at 37°C, it forms stable dimers called tight dimers. Tight dimers withstand semidenaturing electrophoresis in TBE buffer at 28°C, presumably forming an extended duplex base pairing arrangement.
In HIV-2, a homologous SL1 structure was identified by sequence alignment and secondary structure prediction, but it does not readily mediate loose or tight dimerization of a complete leader RNA construct (residues 1–561; Figure 1) (15–17). However, shorter RNA fragments are capable of forming SL1-dependent tight dimers upon incubation at 55°C suggesting that the role of SL1 in dimerization can be regulated (16,18). In addition, both short and long RNA constructs readily form loose dimers through a second dimerization element located at the 5′ end of the primer-binding site (PBS; Figure 1) (15,16).
Figure 1.
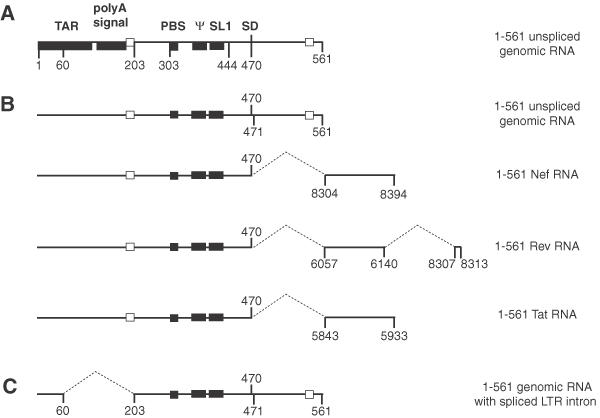
Representation of the HIV-2 ROD RNA constructs used in this study. (A) The landmark sequences with known functions for the first 561 nt of HIV-2 RNA are indicated by boxes. TAR, polyA signal, PBS, ψ, SL1 and SD represent the trans-activation region, the poly(A) signal domain, the primer binding site, the encapsidation signal, the stem–loop 1 and the major splice donor site, respectively. (B) RNAs used in this study. The closed boxes represent the three in vitro characterized dimerization elements, the 5′ end of the PBS, the palindromic sequence in the encapsidation signal ψ and the SL1. The open boxes represent the cores of the two elements forming the long distance interaction, 189–196 and 543–550 (19). The numbers to the right indicate the origin of the nucleotides located downstream of the SD. Sequences removed in spliced RNAs are indicated by dotted lines. (C) The 5′ LTR intron missing in the 1–561 genomic RNA is represented by dotted lines. The splice acceptor and donor sites shown here were originally described in HIV-2 Rev transcripts (23).
The ability of HIV-2 SL1 to mediate tight dimer formation is controlled in vitro by sequences located upstream and downstream of the major splice donor site (SD) (17,19). We identified two structural elements, 189–196 and 543–550 nt, that when base-paired, prevent the use of SL1 as a dimerization signal by sequestering it in a stable intramolecular arrangement with the encapsidation signal ψ that is located just upstream of SL1 (19,20). A similar sequestration of SL1 by base pairing was proposed by Berkhout and co-workers, although there are minor differences in the details of the base pairing that result in sequestration of SL1 (17). A recent detailed structural analysis of HIV-1 5′ region RNA based on phylogenetic, structure probing and crosslinking data by Kjems and co-workers strongly supports the existence of this long distance interaction and its conservation among diverse viruses (21).
To further analyze the functional relationships between structural elements mediating or regulating dimerization, we noted the location of dimerization regulatory elements relative to known splice donor and acceptor sites. Two different naturally occurring splicing events have been described within this region. First, use of the SD at position 470 removes the downstream 543–550 element (Figure 1B). Second, a less well-characterized splicing event leads to the deletion of nt 60–202, and thus the upstream 189–196 element is removed (Figure 1C). This event, called 5′ long terminal repeat (5′ LTR) intron splicing, was first described in simian immunodeficiency virus (SIV) (22), and affects HIV-2 RNAs as well (23). Thus, usage of these splicing sites necessarily disrupts the long-range interaction between the regulatory elements (Figure 1).
The present study is a comparative analysis of the in vitro dimerization properties of HIV-2 leader region RNA harboring splicing-dependent sequence rearrangements. We found that the absence of one or both dimerization regulatory elements in the Nef, Rev, Tat or LTR-spliced constructs enables the formation of tight dimers upon incubation at 55°C. Antisense oligonucleotide binding indicated that the ψ-SL1 region is involved in tight dimerization and that the encapsidation signal ψ and SL1 interact with each other. The dimerization regulatory elements are located in intronic sequences flanking the signals for encapsidation and dimerization. Our present dimerization and structure probing results show that splicing events that occur normally during virus replication alter the presentation of RNA sequences known to be involved in dimerization and encapsidation. This is suggestive of a mechanism by which spliced versus unspliced RNAs might be differentiated in vivo so that only full-length RNAs are packaged into virions.
MATERIALS AND METHODS
Template construction for in vitro transcription
The clones of the first 561 nt of Nef, Rev and Tat leader region were built using a partial sub-cloning protocol. We took advantage of the close proximity between a StuI site and the major SD, located respectively at 455–460 and 470. The numbering is based on the genomic RNA sequence of HIV-2 ROD isolate (24). A sense primer containing the 454–470 nt followed by Nef, Rev or Tat exonic sequences and an antisense primer containing an EcoRI site were used to amplify the first 91 nt of either Nef, Rev or Tat coding regions (Figure 1 and Table 1). We used the exonic sequence of the most representative species of Nef, Rev or Tat mRNA from HIV-2 and SIV (22,23,25,26). After PCR using the modified pROD10 plasmid as template, the PCR products were gel-purified and digested using StuI and EcoRI. This fragment was ligated in the plasmid containing the first 561 nt of HIV-2 ROD in which the StuI–EcoRI 455–561 fragment had been removed. The HIV-2 ROD DNA template (modified plasmid pROD10) was provided by the EU Programme EVA/MRC Centralized Facility for AIDS Reagents (NIBSC, UK; grant numbers QLK2-CT-1999-00609 and GP828102). All constructs were checked by DNA sequencing.
Table 1. Oligonucleotides used in this study.
| sBAMT7R | 5′-TAG GAT CCT AAT ACG ACT CAC TAT AGG TCG CTC TGC GGA GAG-3′ |
| asECO561 | 5′-AAG AAT TCA GTT TCT CGC GCC CAT CTC CC-3′ |
| sNEF454 | 5′-GAG GCC TCC GGG TGA AGC AGA TCC ATA TCC ACA AGG-3′ |
| sREV454 | 5′-GAG GCC TCC GGG TGA AGG CTC GGG ATA TGT TAT GAA CG-3′ |
| sTAT454 | 5′-GAG GCC TCC GGG TGA AGA CAT GGA GAC ACC CTT GAA G-3′ |
| asNEF561 | 5′-AAG AAT TCA GGG CCA GTA TCT GTC TCC-3′ |
| asREV561 | 5′-ATG AAT TCT ATG GAT TTG TCT GGT GTA GGA GAC GG-3′ |
| asTAT561 | 5′-TTG AAT TCT TGA GTG GCC ACA TCC-3′ |
| asLTR60 | 5′-CTG CTA GTG CTG GAG AGA ACC-3′ |
| sLTR203 | 5′-TCG CCG CCT GGT CAT TCG GTG-3′ |
| asPBS | 5′-GGC GCC AAC CTG CTA GGG ATT-3′ |
| asΨ | 5′-CTA GGA GCA CTC CGT CGT GGT TTG-3′ |
| asSL1 | 5′-TGG TAC CTC GGC CCG CGC CT-3′ |
| asDIM | 5′-TGG TAC CTC GGC CCG CGC CTT TCT AGG AGC-3′ |
| asTAT | 5′-GAG TGG CCA CAT CCT GCT CTG AAG-3′ |
To build the 1–561 constructs with the LTR intron removed, a two step PCR-ligation was used. A sense primer containing a BamHI site and the promoter for the T7 RNA polymerase and a 5′-phosphorylated antisense primer ending at nucleotide 60 were used to amplify the first 60 nt of the common leader RNA region from an HIV-2 ROD isolate DNA template. Similarly, a 5′-phosphorylated sense primer starting at nucleotide 203 and an antisense primer containing a HIV-2 sequence ending at nucleotide 561 and an EcoRI site were used to amplify 203–561 nt of the leader region of the genomic RNA (Figure 1 and Table 1). The two PCR products were purified on an agarose gel and then ligated together using the T4 DNA ligase (New England Biolabs). A second gel purification was used to purify the correct ligation product, i.e. one 1–60 fragment ligated to one 203–561 fragment. The ligation product was digested using BamHI and EcoRI and cloned into the BamHI and EcoRI sites of the pUC18 plasmid.
Nef, Rev or Tat constructs without the LTR intron were built by removing the BamHI–BbsI fragment encompassing the LTR intron from these plasmids and ligating the BamHI–BbsI fragment isolated from the LTR-spliced 1–561 construct described above.
RNA synthesis and purification
The different plasmids were linearized with EcoRI prior to in vitro transcription. RNAs were synthesized by in vitro transcription of the EcoRI-digested plasmids with the AmpliScribe™ T7 transcription kit (Epicentre). After transcription, the DNA was digested with the supplied RNase-free DNase, and the RNA was purified by ammonium acetate precipitation followed by size exclusion chromatography (Bio-Gel® P-4; Bio-Rad).
In vitro dimerization of HIV-2 RNA
An aliquot of 5–7 pmol of RNA was denatured in 8 μl water for 2 min at 90°C and quench cooled on ice for 2 min. After the addition of 2 μl monomer buffer (final concentrations: 50 mM Tris–HCl, pH 7.5 at 37°C, 40 mM KCl and 0.1 mM MgCl2) or dimer buffer (final concentrations: 50 mM Tris–HCl, pH 7.5 at 37°C, 300 mM KCl and 5 mM MgCl2), dimerization was allowed to proceed for 15 or 30 min at 37 or 55°C, respectively. We used 55°C since the incubation of the 1–444 RNA at 55°C allows a maximal yield of TBE-resistant SL1-dependent dimers (16). The samples were then cooled on ice to stabilize dimers formed during incubation and loaded onto a 0.8% agarose gel with 2 μl of glycerol loading dye 6× (40% glycerol, 44 mM Tris-borate, pH 8.3, 0.25% bromophenol blue). Electrophoresis was carried out for 90 min at a monitored temperature of 4 or 28°C in 44 mM Tris-borate, pH 8.3, 0.1 mM MgCl2 (TBM) or 44 mM Tris-borate, pH 8.3 and 1 mM EDTA (TBE), respectively. After electrophoresis, the ethidium bromide-stained gel was scanned on a Fluorescent Image Analyzer FLA-3000 (Fujifilm).
Kinetics of tight dimer formation
An aliquot of 100 pmol of RNA was denatured in 160 μl water for 2 min at 90°C and quench cooled on ice for 2 min. After the addition of 2 μl dimer buffer under the lid of 20 tubes, 8 μl of denatured RNA was aliquoted to each tube. The dimerization was then initiated by a 5 s spin in a bench top centrifuge and immediate loading of the tubes in a heating block at 55°C. Dimerization was allowed to proceed for 2–30 min. At each time point a tube containing 10 μl reaction mixture was removed from the heating block, mixed with 2 μl of glycerol loading dye 6× and loaded onto a 0.8% agarose TBE gel. Electrophoresis was carried out at 28°C and 3 V/cm. After electrophoresis, the ethidium bromide-stained gel was scanned on a Fluorescent Image Analyzer FLA-3000 (Fujifilm). Quantification of the extent of dimerization was done using Fujifilm Image Gauge V3.3 software. The data were fitted using a second order conformation model (27):
![]()
where Mt is the concentration of monomer at time t, M0 is the initial concentration of dimerization-competent monomer, and kdim is the second-order rate constant of dimerization (μM−1 min−1).
Antisense oligonucleotides
Antisense oligonucleotides served two purposes in this study. First, the relative availability of an RNA sequence can be monitored by how efficiently it binds free oligonucleotide, with the assumption that nominally single-stranded RNA binds oligonucleotide more efficiently than base paired RNA. Second, oligonucleotides can be used to alter the conformation and, thus, the behavior of RNAs. Because binding of an oligonucleotide can alter the conformation of the larger RNA, interpretation of results when they are used as structure probes must be carried out carefully and with corroboration by other methods, such as structure probing and mutagenesis or deletion analysis, as we have done here.
An aliquot of 5 pmol of RNA with or without 100 pmol of antisense oligonucleotide was denatured in 8 μl water for 2 min at 90°C and quench cooled on ice for 2 min. After the addition of 2 μl of 5-fold concentrated dimer buffer, dimerization was allowed to proceed for 15 min at 37°C or 30 min at 55°C. The samples were then cooled on ice to stabilize dimers formed during incubation and loaded onto a 0.8% agarose gel in TBM or TBE buffer. Electrophoresis was carried out for 90 min at 4°C (for TBM gel) or 28°C (for TBE gel). After electrophoresis, the ethidium bromide-stained gel was scanned on a Fluorescent Image Analyzer FLA-3000 (Fujifilm).
RNA solution structure probing
In a standard experiment, 5 pmol of 1–561 genomic RNA or Rev RNA was heated in water for 2 min at 90°C, and then quench cooled on ice. The dimerization was started by the addition of dimer buffer (final concentrations: 50 mM Tris–HCl, pH 7.5 at 37°C, 300 mM KCl and 5 mM MgCl2). After 0, 4 or 20 min incubation at 55°C, dimethylsulfate (DMS; Aldrich) or kethoxal (ICN Biomedicals) was added to the RNA and the mixture incubated for 2 min at room temperature (27°C). The final concentrations of DMS and kethoxal were 0.5 and 0.1%, respectively. Chemical probing was stopped by the addition of glycogen, EDTA, sodium acetate and ethanol. After a 30-min precipitation at −20°C, the samples were pelleted by centrifugation at 15 000 r.p.m. for 30 min, ethanol washed, vacuum dried and resuspended in water (DMS-treated samples) or borate-KOH, pH 7.5 and 50 mM (kethoxal-treated samples). One-fourth of the resuspended material was then reverse-transcribed with the avian myeloblastosis virus reverse transcriptase (Promega) and a 5′ end-32P-labeled radioactive DNA oligonucleotide primer. After a 30-min primer extension at 42°C, the samples were precipitated as described above and the dried pellet was resuspended in formamide loading and tracking dye and analyzed using denaturing PAGE. A DNA sequencing of the plasmid DNA used for the transcription was loaded together with the primer extension to identify the modified nucleotides.
RESULTS
The first 561 nt of HIV-2 Nef, Rev and Tat mRNAs efficiently form loose dimers
The influence of sequences downstream of the SD on dimerization was analyzed using RNA fragments encompassing the first 561 nt of HIV-2 Nef, Rev or Tat mRNA (Figure 1A and B). The mRNA sequences correspond to representative viral transcript sequences isolated from HIV-2 or SIV-infected cells (22,23,25). The numbering is based on the corrected genomic sequence, nucleotide +1 being the first nucleotide of the viral genomic RNA.
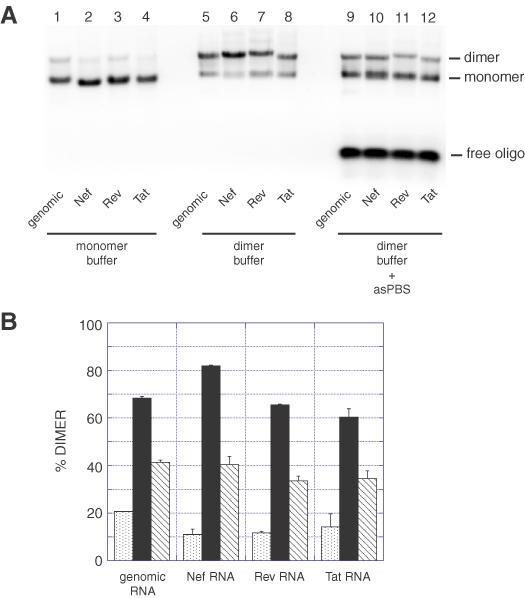
The dimerization of Nef, Rev and Tat RNAs was first assayed by incubating the RNAs in monomer or dimer buffer for 15 min at 37°C and loading the samples on an agarose gel in TBM buffer run at 4°C (Figure 2A). The yields were similar to that of the 1–561 genomic RNA fragment (∼17% in monomer buffer and 60% in dimer buffer; Figure 2B). Since the 5′ end of the PBS was shown to mediate loose dimerization at 37°C (15,16), each RNA was co-incubated in the presence of a 20-fold excess of antisense oligonucleotide asPBS to test the involvement of the PBS as a dimerization element. In the presence of asPBS, the dimerization of all RNAs was inhibited relative to the incubation without oligonucleotide (Figure 2B, compare closed and hatched bars). However, the inhibition by asPBS of Nef, Rev and Tat RNA dimers was less effective than that of genomic RNA, which may suggest that the spliced RNA constructs were capable of using another dimerization site (Figure 2B, compare hatched bars).
Figure 2.
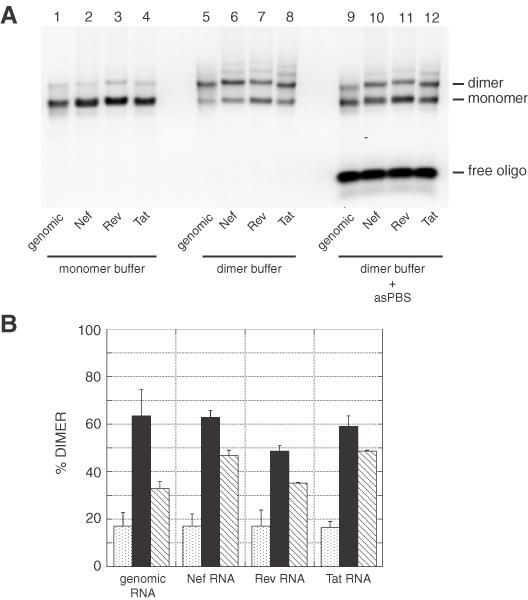
Dimerization of HIV-2 RNAs. (A) RNA fragments corresponding to the first 561 nt of genomic, Nef, Rev or Tat HIV-2 RNAs were incubated for 15 min in monomer (lanes 1–4) or dimer (lanes 5–12) buffer and loaded onto a TBM gel run at 4°C. In lanes 9–12, the RNAs were dimerized in presence of a 20-fold excess of the asPBS antisense oligonucleotide. asPBS binds to the 5′ end of the PBS and nucleotides upstream and inhibits the PBS-dependent dimerization of HIV-2 RNA fragments (15,16). (B) Plot of dimerization as a function of incubation buffer and the presence of asPBS oligonucleotide at 37°C. The y-axis error bars represent the standard deviation of two experiments as shown in (A).
Nef, Rev and Tat RNA fragments efficiently form tight dimers while genomic RNA does not
Tight dimers are exceptionally stable and withstand semidenaturing electrophoresis in TBE buffer at 28°C. As shown previously (15,16), the 1–561 leader genomic RNA yielded a low level of tight dimers (Figure 3A, lane 1). On the other hand, Nef, Rev and Tat RNAs formed a high level of tight dimers (Figure 3A, compare lanes 2–4 with lane 1). To further characterize their dimerization properties, we performed a kinetic analysis of tight dimerization at 55°C (Figure 3B). The data were fitted using a second-order reaction model to obtain rate constants (see Materials and Methods). The dimerization rates of 1–561 Nef, Rev and Tat RNAs were 0.072 ± 0.014, 0.053 ± 0.001 and 0.075 ± 0.005 μM−1 min−1, respectively (Figure 3C). The dimerization rate of 1–561 genomic RNA was much slower (0.005 ± 0.001 μM−1min−1). Thus, the presence of different nucleotides at the 3′ end of the spliced Nef, Rev and Tat RNA constructs, relative to the unspliced genomic RNA, facilitated the formation of tight dimers.
Figure 3.
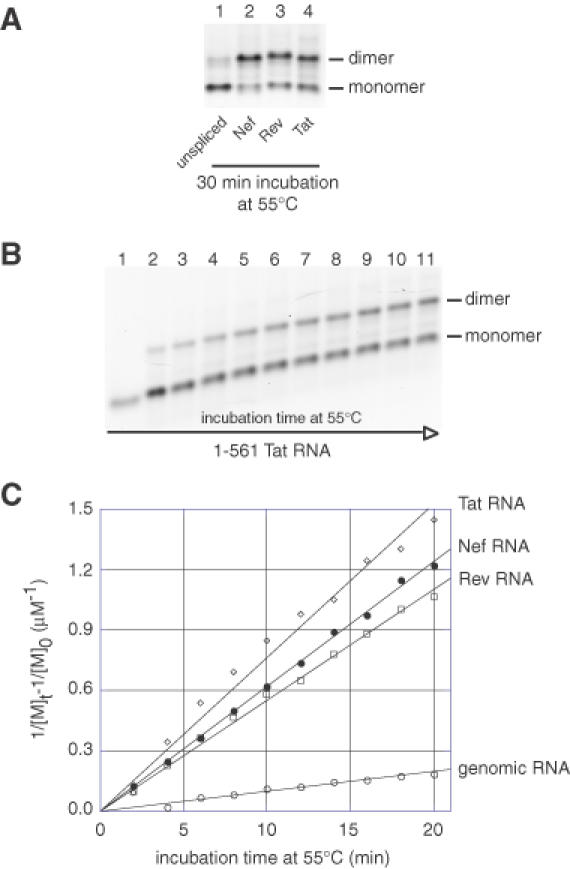
HIV-2 Nef, Rev and Tat RNAs form more tight dimers than the unspliced genomic leader RNA. (A) Genomic, Nef, Rev or Tat HIV-2 1–561 RNAs were incubated in dimer buffer at 55°C. After incubation for 30 min, RNAs were subjected to electrophoresis on a TBE agarose gel run at 28°C. (B) Kinetics of tight dimerization of 1–561 Tat RNA at 55°C. The time-dependent yield of dimerization of 1–561 Tat RNA at 55°C was monitored on a TBE/28°C agarose gel. Lanes 1–11 represent aliquots loaded after incubation for 0, 2, 4, 6, 8, 10, 12, 14, 16, 18 and 20 min, respectively. (C) Plots of the kinetic data for genomic, Nef, Rev or Tat HIV-2 1–561 RNAs (represented by open circles, closed circles, open squares and open diamonds, respectively). [M]t is the concentration of monomer at time t and [M]0 is the initial concentration of dimerization-competent monomer. Kinetic constants, kdim, were obtained by fitting the data to a second-order reaction model (20).
Analysis of dimerization site usage and interactions in the ψ-SL1 domain
To determine more precisely which element in the ψ-SL1 domain mediates the formation of tight dimers, we incubated the different 1–561 RNA fragments in the presence of antisense oligonucleotides. The asψ and asSL1 oligonucleotides bind to the encapsidation signal (ψ) and SL1, respectively (Figure 4A). We have previously showed that both elements can modulate tight dimerization of the 1–561 genomic RNA fragment (28). The binding of asSL1 to the HIV-2 leader 1–561 RNA fragment induced tight dimerization through the ψ domain, and reciprocally, the binding of asψ induced tight dimerization through the SL1 domain. Oligonucleotide binding could either activate or inhibit dimerization of spliced RNAs relative to the level of dimers in the absence of any oligonucleotide, and the effects of asψ and asSL1 binding differed for each RNA (Figure 4B and C). asψ binding activated Nef and Tat RNAs, but inhibited Rev RNA dimerization (Figure 4C, stippled bars). asSL1 binding activated Tat RNA dimerization, but inhibited Nef and Rev RNA dimerization (Figure 4C, hatched bars). It is noteworthy that tight dimerization was inhibited in the presence of both oligonucleotides, completely for Nef and Rev RNAs, and to a low level for Tat RNA (Figure 4C, open bars). These results suggest that the region downstream of SD in spliced mRNA fragments influences the usage of dimerization elements in the ψ-SL1 domain.
Figure 4.
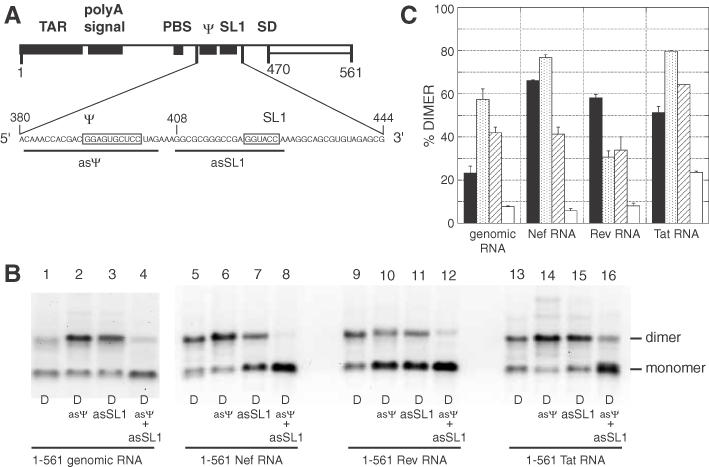
Effects of antisense oligonucleotides directed against the ψ-SL1 region on tight dimerization of Nef, Rev or Tat RNAs. (A) Representation of the ψ-SL1 region and antisense oligonucleotides used in this experiment. The two palindromic sequences with tight dimerization properties are boxed (28). asψ and asSL1 antisense oligonucleotides binding sites are represented by thick lines. (B) 1–561 wild-type (unspliced), Nef, Rev or Tat RNAs were incubated in dimer buffer at 55°C without or with a 20-fold molar excess of asψ, asSL1 or both oligonucleotides, and subjected to TBE electrophoresis. (C) Percentage dimerization for each RNA alone or with asψ, asSL1, or both oligonucleotides (represented by closed, stippled, hatched and open bars, respectively). The y-axis error bars represent the standard deviation of duplicate experiments.
PBS-dependent dimerization of RNAs lacking the 5′ LTR intron
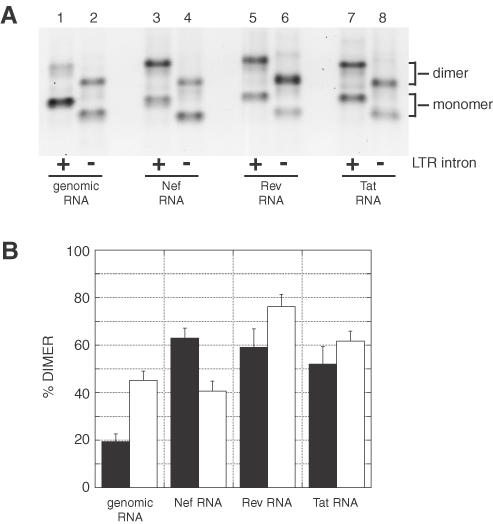
Some SIV and HIV-2 transcripts undergo a specific splicing event in the region upstream of SD (22,23,25,29–31). This event, called 5′ LTR splicing, removes a part of the transactivation element (TAR) and the polyadenylation signal structure (Figure 1C). The LTR-spliced RNAs also lack the 189–196 element, which was shown to influence tight dimerization through a long-range interaction with the gag translation start site (19). We thus assayed dimerization at 37°C for the genomic, Nef, Rev and Tat RNA constructs that lack the LTR intron (LTR-spliced RNAs). Upon incubation in dimer buffer, the four LTR-spliced RNA fragments showed a high yield of dimers when assayed on a TBM gel (from 60 to 80%; Figure 5). Incubation in the presence of asPBS indicated that at least half of these salt-induced dimers occurred through a PBS–PBS interaction (Figure 5B, compare closed and hatched bars). Thus, the absence of the nucleotides forming the LTR intron does not prevent the PBS-dependent dimerization of all tested RNA fragments.
Figure 5.
Dimerization of LTR-spliced HIV-2 RNAs at 37°C. (A) Genomic, Nef, Rev or Tat HIV-2 1–561 RNAs without the LTR intron (nt 61–202) were incubated for 15 min in monomer (lanes 1–4) or dimer (lanes 5–12) buffer and loaded onto a TBM gel run at 4°C. In lanes 9–12, the RNAs were dimerized in the presence of a 20-fold excess of the asPBS antisense oligonucleotide. (B) Plot of dimerization as a function of incubation buffer and the presence of asPBS oligonucleotide at 37°C. Stippled, closed and hatched bars represent the percentage of dimers in monomer buffer or in dimer buffer without or with asPBS, respectively. The y-axis error bars represent the standard deviation of two experiments shown in (A).
The absence of the 5′ LTR intron differentially influences tight dimerization of genomic, Nef, Rev and Tat RNAs
We tested the LTR-spliced RNAs for their ability to form tight dimers upon incubation at 55°C. All LTR-spliced constructs except Nef showed an increase in the tight dimerization yield, relative to their unspliced counterpart (Figure 6A). The increase ranged 1.2–2.5-fold, the highest relative increase was evident in the genomic RNA species (Figure 6B). The lack of the LTR intron sequence in Nef RNA decreased the tight dimer yield from 60 to 40% (Figure 6B). The lack of the LTR intron sequence thus influenced the use of the tight dimerization element positively for the genomic, Rev and Tat RNA species, and negatively for the Nef RNA species.
Figure 6.
Effect of the absence of the LTR intron on tight dimerization of genomic, Nef, Rev and Tat RNAs. (A) Genomic, Nef, Rev or Tat HIV-2 1–561 RNAs with (odd-numbered lanes) or without (even-numbered lanes) the sequence 61–202 corresponding to the LTR intron, were incubated in dimer buffer at 55°C. After incubation for 30 min, RNAs were subjected to electrophoresis on a TBE agarose gel run at 28°C. (B) Plot of the percentage of tight dimerization at 55°C. Closed bars represent the dimer level of LTR-unspliced RNAs and open bars represent the dimer level of LTR-spliced constructs. The y-axis error bars represent the standard deviation of two experiments shown in (A).
Analysis of dimerization element usage in the ψ-SL1 domain
Since all LTR-spliced RNA fragments were capable of forming tight dimers, we tested the effects of asψ and asSL1 oligonucleotides. As described for the LTR-unspliced RNA fragments, the effects of oligonucleotide binding were diverse (Figure 7) asψ binding induced an increase in the tight dimer yield (a few percentage increase for Rev, but a doubling for Nef; Figure 7B, stippled bars). The binding of asSL1 increased the dimer yield for the genomic, Nef and Tat LTR-spliced RNAs, but a decrease in Rev RNA (Figure 7B, hatched bars). Incubation of the RNAs in the presence of both oligonucleotides completely inhibited the dimerization of the LTR-spliced genomic, Nef and Rev RNA fragments, but did not affect the dimerization of Tat RNA (Figure 7B, open bars). The Tat RNA contains a palindromic sequence at the extreme 3′ end that promotes dimerization, as annealing an oligonucleotide to this sequence in combination with asSL1 and asψ completely inhibits dimerization (Figure 7, lanes 18 and 19). These results indicated that splicing of the LTR intron influences the presentation of the ψ-SL1 domain. In turn, the identity of nucleotides downstream of the major SD qualitatively and quantitatively influences the effect of 5′ LTR splicing on dimerization.
Figure 7.
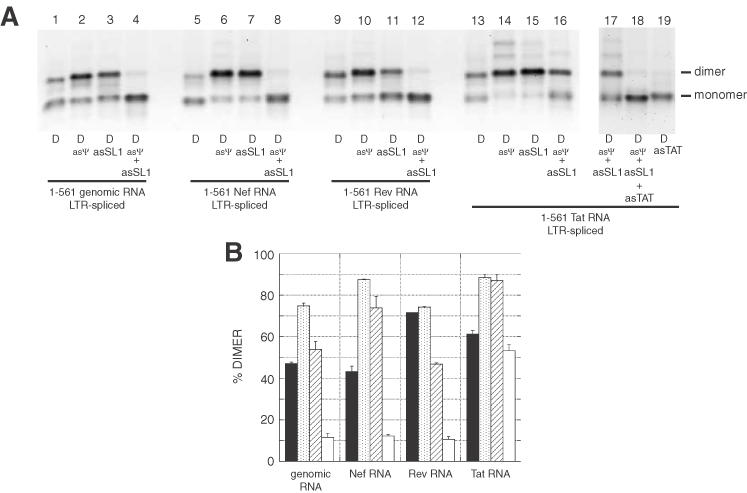
Effects of antisense oligonucleotides against the ψ-SL1 region on tight dimerization of LTR-spliced RNAs. (A) LTR-spliced 1–561 RNAs were incubated in dimer buffer at 55°C without or with a 20-fold molar excess of asψ, asSL1 or both oligonucleotides, and subjected to TBE electrophoresis. The gel is visualized with ethidium bromide. The data indicate that the removal of the LTR intron activated the ψ-SL1 region for dimerization in each of the constructs except Tat. The resistance of Tat LTR-spliced RNA dimers to inhibition by asSL1 and asψ oligonucleotides (lanes 16 and 17) was investigated further by using the additional oligonucleotide asTAT targeted against an autocomplementary sequence near the 3′ end of this construct. Lanes 18 and 19 indicate that this autocomplementary sequence was responsible for this dimerization. (B) Plot of the percentage of tight dimerization at 55°C as a function of the presence of oligonucleotides. The y-axis error bars represent the standard deviation of two experiments shown in (A).
Structure probing of unspliced and spliced RNAs shows conformational differences
The behavioral differences described above for the spliced versus unspliced RNA strongly suggested that the conformational differences affected presentation of dimerization sequences. We therefore probed unspliced wild-type and Rev-spliced RNA with the structure-specific chemical probes kethoxal and DMS, which selectively modify the unpaired guanines and adenines (and to a lesser extent, cytosines), respectively. Although structure probing in these conditions could be complicated by the existence of multiple intramolecular conformations and the formation of dimers with increasing incubation time, the results were surprisingly straightforward.
First, we examined the reactivity of nucleotides in the upstream element of the long distance interaction in the spliced and unspliced RNAs. Figure 8 shows that when nucleotides in the downstream element of the long distance interaction are removed by splicing, the reactivity of A192 and adjacent cytosines in the upstream element to DMS is distinctly higher. This strongly supports the basepairing model between the upstream and downstream elements of the long distance interaction, and is the first example of chemical structure probing in support of the long distance interaction in HIV-2.
Figure 8.
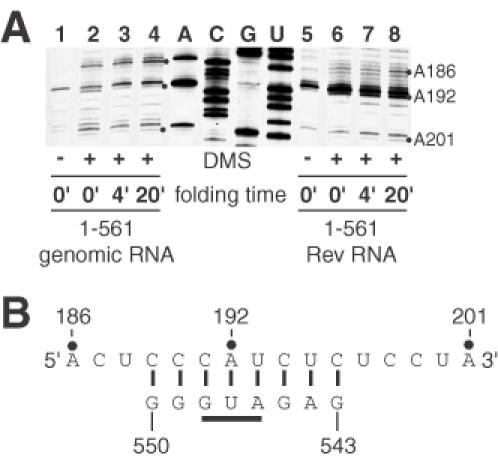
DMS probing of the upstream element of the long distance interaction. (A) Wild-type unspliced and Rev-spliced RNAs were denatured, then incubated in the presence of dimerization buffer at 55°C for the times indicated. DMS was then added (0.5% final concentration) and incubation was continued at 27°C for 2 min. Reactions were stopped and primer extensions were used to visualize the sites of modification as described in Materials and Methods. (B) The increased modification of A192 and flanking cytosines in the Rev RNA samples indicates that this region is less protected when the complementary sequence (543–550 nt in the unspliced RNA) is removed by splicing.
Next, we examined the structure of RNA in the vicinity of the known dimerization signals, namely SL1, the palindrome that resides in the ψ packaging signal upstream of SL1, and the PBS. Remarkably, using both DMS and kethoxal, the structural differences observed were relatively subtle. Figure 9 shows the results of kethoxal probing in the vicinity of ψ-SL1, where the most significant differences were observed. G389 (the lower band of the observed doublet in Figure 8A) flanks the packaging signal, and is seen to be more reactive in the unspliced RNA at short time intervals, but retains higher reactivity at longer times in the spliced RNA. In the stem of SL1 G413, and to a lesser extent G414, were found to be much more reactive in the unspliced versus the spliced RNA, while in the loop G420 and G421 differed significantly in their reactivity between unspliced and spliced RNAs. These localized alterations in reactivity of SL1 nucleotides probably represent incremental adjustments of the overall RNA conformation, since widespread differences were not observed. Such differences in conformation could nevertheless mean the difference between productive and non-productive binding of a partner RNA or viral protein.
Figure 9.
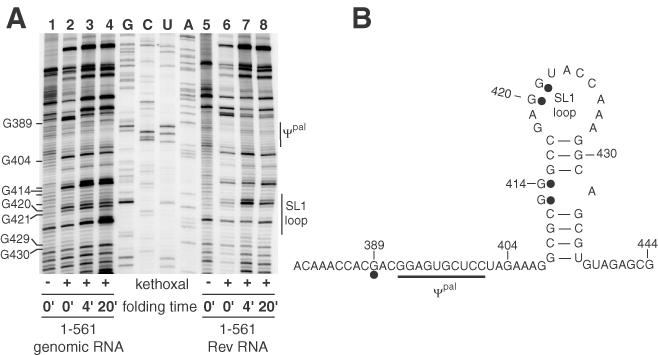
Kethoxal probing of the ψ-SL1 in spliced and unspliced RNAs. (A) Primer extension reactions of kethoxal modified wild-type (lanes 1–4) or Rev-spliced (lanes 5–8) RNA were carried out as described in Materials and Methods. Overall, the reactivities in this region and elsewhere in the RNA (data not shown) were similar, indicating that gross rearrangements of the RNA did not occur upon splicing. However, several nucleotides in the ψ-SL1 region did display reactivity differences as indicated. (B) Summary of the principal reactivity differences between unspliced and spliced RNAs superimposed on a secondary structure model of ψ-SL1.
DISCUSSION
In this study, we analyzed the behavior of several HIV-2 RNA fragments representing the 5′ end of spliced mRNAs coding for the viral proteins Nef, Rev and Tat. This research was inspired by the observation that although both spliced and unspliced viral RNAs contain signals for dimerization and encapsidation, only unspliced RNAs are efficiently packaged. We hypothesized, therefore, that it is the conformation of these signals, rather than their simple presence, that directs encapsidation in vivo. Here, we used RNA dimerization in vitro as an assay for conformational changes that could affect the presentation of known dimerization signals in this region in vivo. Subtle differences in conformation in the two types of RNA were verified by chemical probing.
We previously demonstrated that a long distance base pairing interaction inhibits tight dimerization of the leader fragment of HIV-2 genomic RNA in vitro (19). The long distance interaction involves base pairing between 189–196 and 543–550 nt (Figure 1A). Studies with truncated mutants of HIV-2 leader RNA indicated that the lack of one or both elements allows efficient tight dimerization. In agreement with the proposed model, the lack of the 189–196 element in LTR-spliced constructs and/or the lack of the 543–550 element in Nef, Rev and Tat constructs increased the tight dimerization yield in a way similar to the effects of 5′ or 3′ truncations in the 1–561 genomic RNA (19,28). Thus, tight dimerization of HIV-2 RNAs is modulated by elements located in introns flanking the dimerization signals, suggesting that the deletion of intronic sequence during splicing can significantly alter the conformation of neighboring sequences, including the packaging signal, ψ.
Although Nef, Rev and Tat RNA fragments can form tight dimers, the nature of the interaction may differ. In unspliced leader region RNA, ψ and SL1 sequences form an intramolecular interaction that precludes their involvement in dimerization (28). Disruption of this intramolecular interaction (using oligonucleotides or through disruption of the long distance interaction) promotes dimerization in full-length unspliced constructs. However, the spliced RNAs used in this study display differential behavior upon binding of the asψ and asSL1 antisense oligonucleotides (Figure 4). These results suggest that in spliced RNAs, the absence of the long distance interaction leads to a different conformation of the ψ-SL1 region, as evidenced by an increase in tight dimerization. Discrete differences in conformation in the ψ-SL1 region were observed in the probing experiments of spliced versus unspliced RNA.
Clearly, if the present results are extrapolated to the in vivo context, they indicate that dimerization is not the primary determinant for encapsidation, since subgenomic RNAs dimerize more efficiently than unspliced RNAs in vitro. However, because dimerization and encapsidation in vivo involve a complex interaction of RNA and host and viral proteins, we emphasize here that the difference in dimerization properties of naked RNAs demonstrates that the signals for dimerization and encapsidation can be differentially displayed, depending on the state of flanking intronic sequences. One of these conformational states may represent the cognate structure for recognition by a protein to promote encapsidation while another state (i.e. the conformation adopted by the spliced RNAs) occludes the three-dimensional encapsidation signal. The structure probing results (Figures 8 and 9) suggest that the difference in conformations is relatively small and localized, since no wholesale rearrangement of the RNA was observed between the two types of RNA.
During the formation of the retroviral particle in vivo, unspliced genomic-length RNA molecules are preferentially encapsidated despite the presence of other cytoplasmic spliced RNA species. In most retroviruses, it is believed that the spliced retroviral RNA species are not encapsidated because the RNA encapsidation signal is located downstream of the major SD and thus disappear upon splicing [for a review see (4)]. However, the HIV-2 and SIV RNA encapsidation signal, as defined by the analysis of deletion mutants, is located upstream of the major SD (20,32), suggesting that other structures or mechanisms help to ensure the preferential encapsidation of the genomic RNA over the spliced sub-genomic species. The results presented in this study suggest that interactions between elements upstream and downstream of the major SD may help to select for encapsidation of the unspliced genomic RNA preferentially over other spliced species. It is presently not clear whether the ψ-SL1 region is the primary dimerization element in vivo or how the encapsidation signal ψ is activated. Lever and co-workers have shown that the activation of ψ is a co-translational event mediated by the nucleocapsid domain of the Gag polyprotein (20,33), and Aldovini and co-workers have demonstrated that packaging in SIV depends on elements upstream and downstream of the major splice donor site (34). The present results suggest that long-range interactions spanning the splice sites provide a potential structural mechanism to differentiate the leader region of the unspliced genomic RNA from spliced RNAs by regulating the conformation of the nested encapsidation and dimerization elements. It will be of interest to characterize the discrete conformational changes in the ψ-SL1 region as a function of NC/Gag binding, splicing, translation and dimerization.
Acknowledgments
ACKNOWLEDGEMENTS
We acknowledge Patrick Watson for critical reading of the manuscript and helpful suggestions. The pROD10 plasmids from Dr J.-M. Bechet and Dr A. M. L. Lever were obtained from the Centralised Facility for AIDS Reagents supported by EU Programme EVA (contract QLK2-CT-1999-00609) and the UK Medical Research Council. Q.N.S. was supported by NIH grant number P20RR-16455-03 from the BRIN Program of the National Center for Research Resources. This research is supported by the National Institutes of Health (grant number AI45388) to J.S.L.
REFERENCES
- 1.Bender W. and Davidson,N. (1976) Mapping of poly(A) sequences in the electron microscope reveals unusual structure of type C on cornavirus RNA molecules. Cell, 7, 595–607. [DOI] [PubMed] [Google Scholar]
- 2.Kung H.J., Hu,S., Bender,W., Bailey,J.M., Davidson,N., Nicolson,M.O. and McAllister,R.M. (1976) RD-114, baboon, and woolly monkey viral RNA's compared in size and structure. Cell, 7, 609–620. [DOI] [PubMed] [Google Scholar]
- 3.Greatorex J. and Lever,A. (1998) Retroviral RNA dimer linkage. J. Gen. Virol., 79, 2877–2882. [DOI] [PubMed] [Google Scholar]
- 4.Jewell N.A. and Mansky,L.M. (2000) In the beginning: genome recognition, RNA encapsidation and the initiation of complex retrovirus assembly. J. Gen. Virol., 81, 1889–1899. [DOI] [PubMed] [Google Scholar]
- 5.Darlix J.L., Gabus,C., Nugeyre,M.T., Clavel,F. and Barre-Sinoussi,F. (1990) Cis elements and trans-acting factors involved in the RNA dimerization of the human immunodeficiency virus HIV-1. J. Mol. Biol., 216, 689–699. [DOI] [PubMed] [Google Scholar]
- 6.Laughrea M. and Jette,L. (1994) A 19-nucleotide sequence upstream of the 5′ major splice donor is part of the dimerization domain of human immunodeficiency virus 1 genomic RNA. Biochemistry, 33, 13464–13474. [DOI] [PubMed] [Google Scholar]
- 7.Skripkin E., Paillart,J.C., Marquet,R., Ehresmann,B. and Ehresmann,C. (1994) Identification of the primary site of the human immunodeficiency virus type 1 RNA dimerization in vitro. Proc. Natl Acad. Sci. USA, 91, 4945–4949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McBride M.S. and Panganiban,A.T. (1996) The human immunodeficiency virus type 1 encapsidation site is a multipartite RNA element composed of functional hairpin structures. J. Virol., 70, 2963–2973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Paillart J.C., Skripkin,E., Ehresmann,B., Ehresmann,C. and Marquet,R. (1996) A loop–loop ‘kissing’ complex is the essential part of the dimer linkage of genomic HIV-1 RNA. Proc. Natl Acad. Sci. USA, 93, 5572–5577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Muriaux D., Fosse,P. and Paoletti,J. (1996) A kissing complex together with a stable dimer is involved in the HIV-1Lai RNA dimerization process in vitro. Biochemistry, 35, 5075–5082. [DOI] [PubMed] [Google Scholar]
- 11.Laughrea M. and Jette,L. (1996) Kissing-loop model of HIV-1 genome dimerization: HIV-1 RNAs can assume alternative dimeric forms, and all sequences upstream or downstream of hairpin 248–271 are dispensable for dimer formation. Biochemistry, 35, 1589–1598. [DOI] [PubMed] [Google Scholar]
- 12.Haddrick M., Lear,A.L., Cann,A.J. and Heaphy,S. (1996) Evidence that a kissing loop structure facilitates genomic RNA dimerisation in HIV-1. J. Mol. Biol., 259, 58–68. [DOI] [PubMed] [Google Scholar]
- 13.Clever J.L., Wong,M.L. and Parslow,T.G. (1996) Requirements for kissing-loop-mediated dimerization of human immunodeficiency virus RNA. J. Virol., 70, 5902–5908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shubsda M.F., McPike,M.P., Goodisman,J. and Dabrowiak,J.C. (1999) Monomer–dimer equilibrium constants of RNA in the dimer initiation site of human immunodeficiency virus type 1. Biochemistry, 38, 10147–10157. [DOI] [PubMed] [Google Scholar]
- 15.Jossinet F., Lodmell,J.S., Ehresmann,C., Ehresmann,B. and Marquet,R. (2001) Identification of the in vitro HIV-2/SIV RNA dimerization site reveals striking differences with HIV-1. J. Biol. Chem., 276, 5598–5604. [DOI] [PubMed] [Google Scholar]
- 16.Lanchy J.M. and Lodmell,J.S. (2002) Alternate usage of two dimerization initiation sites in HIV-2 viral RNA in vitro. J. Mol. Biol., 319, 637–648. [DOI] [PubMed] [Google Scholar]
- 17.Dirac A.M., Huthoff,H., Kjems,J. and Berkhout,B. (2002) Regulated HIV-2 RNA dimerization by means of alternative RNA conformations. Nucleic Acids Res., 30, 2647–2655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dirac A.M., Huthoff,H., Kjems,J. and Berkhout,B. (2001) The dimer initiation site hairpin mediates dimerization of the human immunodeficiency virus, type 2 RNA genome. J. Biol. Chem., 276, 32345–32352. [DOI] [PubMed] [Google Scholar]
- 19.Lanchy J.M., Rentz,C.A., Ivanovitch,J.D. and Lodmell,J.S. (2003) Elements located upstream and downstream of the major splice donor site influence the ability of HIV-2 leader RNA to dimerize in vitro. Biochemistry, 42, 2634–2642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Griffin S.D., Allen,J.F. and Lever,A.M. (2001) The major human immunodeficiency virus type 2 (HIV-2) packaging signal is present on all HIV-2 RNA species: cotranslational RNA encapsidation and limitation of Gag protein confer specificity. J. Virol., 75, 12058–12069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Damgaard C.K., Andersen,E.S., Knudsen,B., Gorodkin,J. and Kjems,J. (2004) RNA interactions in the 5′ region of the HIV-1 genome. J. Mol. Biol., 336, 369–379. [DOI] [PubMed] [Google Scholar]
- 22.Viglianti G.A., Sharma,P.L. and Mullins,J.I. (1990) Simian immunodeficiency virus displays complex patterns of RNA splicing. J. Virol., 64, 4207–4216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chatterjee P., Garzino-Demo,A., Swinney,P. and Arya,S.K. (1993) Human immunodeficiency virus type 2 multiply spliced transcripts. AIDS Res. Hum. Retroviruses, 9, 331–335. [DOI] [PubMed] [Google Scholar]
- 24.Guyader M., Emerman,M., Sonigo,P., Clavel,F., Montagnier,L. and Alizon,M. (1987) Genome organization and transactivation of the human immunodeficiency virus type 2. Nature, 326, 662–669. [DOI] [PubMed] [Google Scholar]
- 25.Reinhart T.A., Rogan,M.J. and Haase,A.T. (1996) RNA splice site utilization by simian immunodeficiency viruses derived from sooty mangabey monkeys. Virology, 224, 338–344. [DOI] [PubMed] [Google Scholar]
- 26.Kuiken C., Foley,B., Hahn,B., Marx,P., McCutchan,F., Mellors,J., Wolinsky,S. and Korber,B. (2001) HIV Sequence Compendium 2001. Theoretical Biology and Biophysics Group, Los Alamos National Laboratory, Los Alamos, NM. [Google Scholar]
- 27.Marquet R., Paillart,J.C., Skripkin,E., Ehresmann,C. and Ehresmann,B. (1994) Dimerization of human immunodeficiency virus type 1 RNA involves sequences located upstream of the splice donor site. Nucleic Acids Res., 22, 145–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lanchy J.M., Ivanovitch,J.D. and Lodmell,J.S. (2003) A structural linkage between the dimerization and encapsidation signals in HIV-2 leader RNA. RNA, 9, 1007–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Viglianti G.A., Rubinstein,E.P. and Graves,K.L. (1992) Role of TAR RNA splicing in translational regulation of simian immunodeficiency virus from rhesus macaques. J. Virol., 66, 4824–4833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Unger R.E., Stout,M.W. and Luciw,P.A. (1991) Simian immunodeficiency virus (SIVmac) exhibits complex splicing for tat, rev, and env mRNA. Virology, 182, 177–185. [DOI] [PubMed] [Google Scholar]
- 31.Bibollet-Ruche F., Cuny,G., Pourrut,X., Brengues,C., Galat-Luong,A., Galat,G. and Delaporte,E. (1998) Multiply spliced env and nef transcripts of simian immunodeficiency virus from West African green monkey (SIVagm-sab). AIDS Res. Hum. Retroviruses, 14, 515–519. [DOI] [PubMed] [Google Scholar]
- 32.Strappe P.M., Greatorex,J., Thomas,J., Biswas,P., McCann,E. and Lever,A.M. (2003) The packaging signal of simian immunodeficiency virus is upstream of the major splice donor at a distance from the RNA cap site similar to that of human immunodeficiency virus types 1 and 2. J. Gen. Virol., 84, 2423–2430. [DOI] [PubMed] [Google Scholar]
- 33.Kaye J.F. and Lever,A.M. (1999) Human immunodeficiency virus types 1 and 2 differ in the predominant mechanism used for selection of genomic RNA for encapsidation. J. Virol., 73, 3023–3031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Patel J., Wang,S.W., Izmailova,E. and Aldovini,A. (2003) The simian immunodeficiency virus 5′ untranslated leader sequence plays a role in intracellular viral protein accumulation and in RNA packaging. J. Virol., 77, 6284–6292. [DOI] [PMC free article] [PubMed] [Google Scholar]