


Abstract
A novel microarray system that utilizes a porous aluminum-oxide substrate and flow-through incubation has been developed for rapid molecular biological testing. To assess its utility in gene expression analysis, we determined hybridization kinetics, variability, sensitivity and dynamic range of the system using amplified RNA. To show the feasibility with complex biological RNA, we subjected Jurkat cells to heat-shock treatment and analyzed the transcriptional regulation of 23 genes. We found that trends (regulation or no change) acquired on this platform are in good agreement with data obtained from real-time quantitative PCR and Affymetrix GeneChips. Additionally, the system demonstrates a linear dynamic range of 3 orders of magnitude and at least 10-fold decreased hybridization time compared to conventional microarrays. The minimum amount of transcript that could be detected in 20 μl volume is 2–5 amol, which enables the detection of 1 in 300 000 copies of a transcript in 1 μg of amplified RNA. Hybridization and subsequent analysis are completed within 2 h. Replicate hybridizations on 24 identical arrays with two complex biological samples revealed a mean coefficient of variation of 11.6%. This study shows the potential of flow-through porous microarrays for the rapid analysis of gene expression profiles in clinical applications.
INTRODUCTION
With the effective completion of the human genome sequence and other genome sequences (1), DNA microarrays have been widely adopted in genomics because of their ability to simultaneously examine the expression levels of multiple genes. The analysis of transcriptional regulation in the entire genome has facilitated progress in more complete characterization of molecular pathways that are fundamental to cellular behavior. This has yielded new classes of molecular targets that may be amenable to therapeutic intervention (2). It has also identified subsets of genes that could be useful markers for diagnosis and prediction of clinical outcome (3–6). The prognostic and diagnostic power of DNA microarrays promises to increase the reliability of diagnostic classifications and optimize effective use of drugs (7). A main challenge, however, is how best to apply microarray analysis to routine clinical practice. In the clinical setting, implementation of a diagnostic tool demands not only speed and small starting samples but also a high level of data quality. Limitations of conventional microarrays include slow hybridization kinetics requiring incubation times of 14–18 h. Therefore, conventional microarrays may not represent the most cost-effective option for routine applications in gene expression profiling.
We have developed a novel flow-through porous microarray (porous array) that can be used for rapid molecular biological testing (8). This technology involves the use of a porous three-dimensional aluminum-oxide substrate (9) and flow-through incubation. The substrate contains millions of pores (0.2 × 60 μm2) in parallel orientation connecting the top and bottom surfaces (Figure 1A–D). In comparison with the two-dimensional geometry, the reactive surface in the substrate is increased ∼500-fold. As samples are actively pumped back and forth through the porous structure by moderate air pressure, they react with the capture molecules that are immobilized on the surface. The depleted solution near the spots is replenished by the continuous flow-through incubation (Figure 1E) and the diffusion distance is dramatically reduced (maximum 100 nm). Therefore, the binding kinetics can be significantly accelerated. Using a 4-array system (10) or a newly developed 96-array system, 4 or 96 samples can be analyzed in parallel. When the sample is pumped to the underside of the substrate, an image is recorded through the entire porous structure (Figure 1F), allowing real-time measurement as the hybridization reaction is progressing. The differences between the porous array system and other existing flow-through microarray techniques (11) are as follows:
The porous substrate has very small pores. The diameter of an individual pore is ∼0.2 μm (Figure 1D). As a consequence, the diffusion time in the pores is 2500 times smaller than with the existing flow-through technology with typical pore sizes of 10 μm (11,12).
The large capillary action of the substrate enables the simple but effective sample pumping/mixing scheme (cycling up and down).
Figure 1.
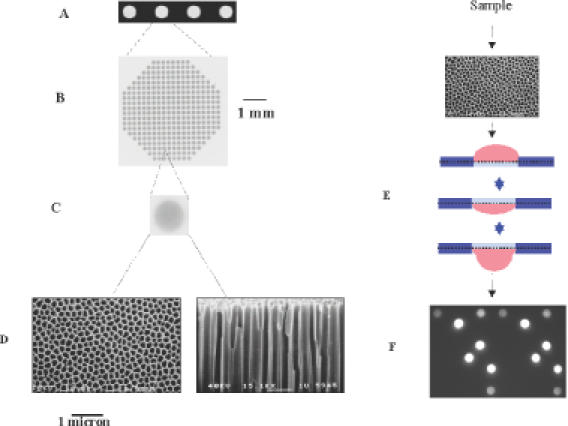
Schematic depiction of porous array technology. (A) Four arrays in a chip. (B) 400 spots on an array (scale given by the bar). (C) A single spot. (D) Porous structure of an aluminum-oxide substrate (100 000 pores per spot, scale given by the bar). Left, top view of the substrate. The diameter of an individual pore is ∼200 nm. Right, partial cross section of the substrate. The substrate has a thickness of 60 μm with capillary pores. (E) Flow-through incubation. The sample is pumped back and forth by air pressure through the porous substrate during the incubation. (F) A raw image acquired on porous array. A Tiff image is recorded through the entire porous structure by an epi-fluorescent CCD imaging system. The image information is converted into spot intensity values using ArrayPro Analyzer software.
The mechanism of cellular heat-shock response, which is conserved among many different organisms, is well studied. To verify the gene expression profile acquired on our platform, we performed a heat-shock experiment on cultured Jurkat cells and analyzed the transcriptional regulation of 23 genes after heat-shock treatment. Of the 23 genes, 10 were selected from the report of Schena et al. (13), and another 13 were chosen from a database of genes with a known function in stress response or protein biosynthesis (http://www.hugeindex.org).
The results obtained with the arrays were compared with real-time quantitative PCR and Affymetrix GeneChips. In addition, we assessed the technical specifications of the system with individually labeled exogenous transcripts. We present here the use of this system for fast expression profiling.
MATERIALS AND METHODS
Heat-shock array
Preparation of the porous arrays was performed as described previously (8). The heat-shock array used in this study contained 48 features that were spotted in duplicate (96 spots). The 48 features consisted of 60mer oligonucleotides (BioSpring) corresponding to 23 human genes, 10 oligonucleotides corresponding to exogenous transcripts from the SpotReport Alien Oligo Array Validation System (Stratagene), 8 oligonucleotides corresponding to ArrayControl RNA Spikes (Ambion), 2 Cy3/Cy5 reference oligonucleotides, 2 human Cot-1 DNA, 2 poly(dA) and 1 water. The 10 oligonucleotides from Stratagene were used as positive controls for normalization during data analysis. The Cot-1 DNA, poly(dA) and oligonucleotides from Ambion were used as negative controls.
Oligonucleotides corresponding to the 23 genes in sense orientation were selected using ArrayDesigner 2.0 software (Premier Biosoft). These oligonucleotides were chosen from within 1000 bases of the 3′ end of the transcript and had a calculated Tm between 75 and 80°C. A threshold for hairpins and self dimers of the oligonucleotides was set to be less than three base hybrids. The sequences of the oligonucleotides are available from the authors upon request.
Cell culture and RNA extraction
Jurkat cells were grown in RPMI-1640 media supplemented with 10% fetal calf serum, streptomycin (100 μg/ml), penicillin (500 U/ml) and l-glutamine (300 μg/ml). Jurkat cells were grown at 37°C in a humidified atmosphere of 5% CO2. Heat-shocked Jurkat cells were incubated at 43°C for 4 h (13). Subsequently, the cells were collected and RNA extraction was performed using Trizol reagent according to the manufacturer's instructions (GibcoBRL). Total RNA was then treated with DNase I and purified using RNeasy spin columns (Qiagen). The concentration of the RNA was estimated by measuring the optical density (OD) at 260 nm using a SpectraMax Plus 384 instrument (Molecular Devices).
Sample preparation for hybridization
Preparation of exogenous transcripts
Individually labeled transcripts of 525 bp were generated for 5 (spike 1, 2, 5, 6 and 8) of 10 exogenous mRNAs (Stratagene). From each of the 5 transcripts, 10 ng of mRNA were used as input for linear amplification and labeling with MessageAmp aRNA kit (Ambion). In vitro transcription (IVT) reactions with incorporation of cyanine-5-UTP (Cy5, Perkin Elmer) or biotin-16-UTP (Roche) were performed for 6.5 h. The labeled transcript was further purified using RNeasy spin columns. The concentration of the transcript was determined by OD260.
Preparation of complex background amplified RNA
Five micrograms of total human reference RNA (Stratagene) was used as input for linear amplification and labeling with the MessageAmp aRNA kit. The IVT reactions with and without the incorporation of biotin-16-UTP were performed as described above.
Preparation of complex biological amplified RNA
Five micrograms of total RNA derived from either control or heat-shocked Jurkat cells were used as input for linear amplification and labeling. Prior to cDNA synthesis, 50 pg of each of the 10 exogenous mRNAs (Stratagene) was added to each of the RNA samples. The IVT reactions with incorporation of biotin-11-CTP (Perkin Elmer) and biotin-16-UTP (Roche) were performed according to the protocol in the Affymetrix GeneChip Expression Manual.
Preparation of complex biological cDNA
Five micrograms of total RNA prepared from either control or heat-shocked Jurkat cells were labeled with biotin-11-dUTP (Roche) during reverse transcription with the cDNA labeling module in the LabelStar Array kit (Qiagen). Prior to the cDNA synthesis, 50 pg of each of the 10 exogenous mRNAs (Stratagene) was added to each of the RNA samples. The cDNA was purified using QIAquick PCR purification kit (Qiagen). The cDNA labeling reaction for each of the two RNA samples was performed in quadruplicate.
Hybridization and detection
General protocol
Hybridization, washing and detection were performed using a 4-array system (FD10, Olympus) as described previously (10). The FD10 integrated fluidics and epi-fluorescent CCD imaging system allows for automated analysis of up to four samples in parallel. Furthermore, the system is capable of providing kinetic read-out and changing the temperature during incubation.
Each sample was dried in a Speed-Vac and dissolved in 20 μl of hybridization buffer (50% formamide, 3× SSPE, 1% SDS and 5× Denhardt's solution, pH 7.0). The sample was denatured at 95°C for 5 min and kept at 42°C until hybridization. The sample was pumped back and forth through the porous substrate by air pressure (0.1 bar) (5 cycles/min) during incubation. Images were recorded after the final washing step. For kinetic measurements, images were taken during the hybridization between the pumping cycles.
Biotinylated samples were hybridized in 20 μl of solution at 42°C for 60 min. After removing the hybridization solutions, arrays were washed three times with staining buffer [2× phosphate-buffered saline (PBS), 2 μg/μl acetylated BSA and 0.05% Tween-20] at 37°C. After washing, the hybridized RNA was stained by flow-through incubation with 10 μg/ml streptavidin labeled with R-phycoerythrin (Kirkegaard & Perry Laboratories) in the staining buffer for 5 min at 37°C. After removing unbound streptavidin–phycoerythrin by three times washing with 2× PBS buffer at 37°C, images were taken using a Cy3 filter set. The hybridization conditions for each assessment are described below.
Hybridization kinetics
Prior to hybridization, 4 μg of unlabeled human reference aRNA was fragmented randomly to a length of 100–200 nt by incubating at 70°C for 10 min in 1× fragmentation buffer (Ambion). One microgram of the aRNA was added to each of the three mixtures for 75, 300 and 600 pM concentrations of five individually Cy5-labeled exogenous transcripts. Hybridizations were carried out at 42°C for 300 min without washing. Images were automatically taken every 5 min using a Cy5 filter set during the incubation.
Sensitivity and dynamic range
Biotinylated human reference aRNA (60 μg) was fragmented as described above. Four micrograms of the aRNA was added to each of the 14 mixtures for a concentration range of 0.1 pM to 50 nM (0.1 pM, 0.25 pM, 0.5 pM, 1 pM, 5 pM, 10 pM, 50 pM, 100 pM, 500 pM, 1 nM, 5 nM, 10 nM, 25 nM and 50 nM) of five biotinylated exogenous transcripts and one negative control. The triplicate experiments were performed using 12 chips (4 arrays per chip).
Gene expression profiling
Each of the two biotinylated aRNA samples (13 μg per sample) derived from control or heat-shocked Jurkat cells was fragmented and dissolved in 260 μl of hybridization buffer. Each of the two RNA samples (1 μg per hybridization) was hybridized to 12 identical arrays. In addition, two biotinylated cDNA samples from control and heat-shocked RNA were hybridized to 8 identical arrays (4 arrays per sample).
Data analysis
The image information was converted into spot intensity values using a customized version of ArrayPro Analyzer software (Media Cybernetics). Median signal intensity and local background measurements were obtained for each spot on the hybridized array. Local background was subtracted from the value of each spot on the array. The median signal intensity after background subtraction was used for further analysis. The mean signal intensity of duplicate spots on each array was normalized using the mean signal values of the 10 exogenous spikes. A cut-off value for a positive signal was defined as three times above the SD of the background. The coefficients of variation (CV) for all genes from different arrays were determined to assess the variability of hybridizations.
For expression profiles, the ratios of control and heat-shocked RNA samples and their corresponding SD were calculated from the normalized values. The 95% confidence intervals of these ratios were used to identify differentially expressed genes. Genes were considered as being up-regulated if the ratio exceeded 1.2 or as being down-regulated if the ratio was below 0.8.
Real-time quantitative PCR
Real-time quantitative PCR of the 23 genes investigated was performed on the ABI PRISM 7000 Sequence Detection System (Applied Biosystems) using the SYBR Green PCR Master Mix kit (Applied Biosystems) according to the manufacturer's protocol. The primer sequences are available from the authors upon request. Five micrograms of total RNA from control and heat-shocked Jurkat cells were used to generate cDNA as described above. The concentration of complex cDNA was measured by OD260. PCR was carried out in triplicate on 5 μl (10 ng/μl) of complex cDNA in a total volume of 50 μl. The amplification of single-size products was further verified by gel electrophoresis. The changes in fluorescence of SYBR Green I were monitored at 72°C in every cycle and the threshold cycle (Ct) was calculated. The samples were quantified using Ct values and a calibration curve of known concentrations was generated for each gene (14).
Affymetrix GeneChip
Aliquots of the same biotinylated aRNAs analyzed on the porous arrays were fragmented and hybridized to four Affymetrix U133A GeneChips (2 chips per sample) at the Leiden Genome Technology Center in The Netherlands. Ten micrograms of each aRNA sample were hybridized to the GeneChip in a final volume of 200 μl according to the protocol in the Affymetrix GeneChip Expression Manual. Analysis of raw data files was performed using the original .DAT file with Affymetrix GeneChip Operating Software (GCOS version 1.1.1.052).
RESULTS
Hybridization kinetics
Hybridization kinetics were monitored by real-time measurements over 300 min using 75, 300 and 600 pM concentrations of spiking mixtures. A set of successive raw images obtained on a single array during hybridization with a 600 pM spiking mixture is shown in Figure 2A. Signal intensities obtained from three concentrations of one transcript over time are displayed in Figure 2B. A similar pattern of hybridization kinetics was observed among the three concentrations of the other four exogenous transcripts. The kinetics data obtained from the other four spikes can be found in Supplementary Figure 4. The hybridization signals were detected within 5 min and reached a plateau between 90 and 120 min for all samples. During 300 min of hybridization, the ratio of signals between 300 and 600 pM concentrations of the respective transcript was constant at 2.1 with a CV of 5.9%, while the ratio between 75 and 600 pM remained constant at 7.9 with a CV of 4.6%. On average, 65% of the maximum signal at 90 min was reached after 60 min. Typically, a hybridization time of 60 min was used for gene expression analysis with a flow rate of 5 cycles/min.
Figure 2.
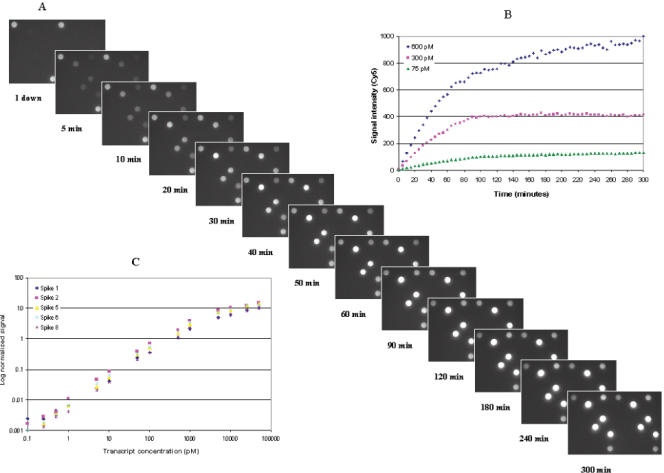
Hybridization kinetics on porous arrays. (A) Raw images displaying hybridization kinetics, acquired on a single array from hybridization of five Cy5-labeled exogenous transcripts at a 600 pM concentration. (B) Hybridization kinetics signal intensity versus hybridization time for an exogenous transcript 2. The signal intensities were obtained from three hybridizations using 75, 300 and 600 pM concentrations of the transcript 2. (C) Dynamic range of detection on porous arrays. Log–log plot of normalized signal intensity versus concentration for five exogenous transcripts (spike 1, 2, 5, 6 and 8). The concentrations of the five exogenous transcripts ranged from 0.1 pM to 50 nM. The dynamic range experiment was performed by using the streptavidin–phycoerythrin detection process.
We performed additional experiments to determine the effect of the hybridization kinetics by varying the flow rate from 5 cycles/min to 2 cycles/min to 1 cycle every 3 min. Hybridization kinetics were monitored over 60 min using a 1 nM concentration of five individually Cy5-labeled exogenous transcripts (spike 1, 2, 5, 7 and 8). One microgram of an unlabeled human reference aRNA was added to each of the three mixtures for hybridizations with a flow speed from 5 cycles/min to 1 cycle every 3 min. The results on the effect of the hybridization kinetics by varying the flow rate can be found in Supplementary Figure 5. Different patterns of hybridization kinetics on the flow rates were observed among the five exogenous transcripts. The signal increase per time unit was considerably lower for the slow flow rate of 1 cycle every 3 min than for the flow rates of 2 and 5 cycles/min. The overall signal intensities for the flow rate of 5 cycles/min were higher than the for the flow rate of 2 cycles/min.
Sensitivity and dynamic range of detection
The minimum detectable amount and dynamic range were monitored using the streptavidin–phycoerythrin detection process. Triplicate experiments were performed using five biotinylated exogenous transcripts with concentrations ranging from 0.1 pM to 50 nM in a background of biotinylated human aRNA. The signal intensities measured at three different exposure times were used for data analysis. The signal intensities of the exogenous transcripts were normalized against β-actin gene. When only the human aRNA was hybridized to the array, no hybridization signal was detected for the exogenous genes, indicating the absence of cross hybridization.
Positive signals were detected in three (60%) and five (100%) of the five exogenous transcripts at a concentration as low as 0.1 and 0.25 pM, respectively. The minimum detectable amount was therefore determined to be in the range of 2–5 amol in 20 μl hybridization solution. This enables the detection of 1 in 300 000 copies of a transcript in 1 μg of amplified RNA.
As indicated in Figure 2C, the dynamic range of detection exceeded 4 orders of magnitude. Saturation occurred between 5 and 10 nM, depending on the transcript tested. When the signal intensity was plotted as a function of spiking concentration on a linear scale, linearity (R2 > 0.99) was observed for 3 orders of magnitude from 1 to 1000 pM for all of the five transcripts. The average variability (CV) of signal intensity for triplicate hybridizations over the concentration range examined was 11.2%.
Variability and specificity
Replicate experiments were performed to determine the variability and specificity of hybridization using two biological RNA samples. The two samples were biotinylated aRNAs derived from control and heat-shocked Jurkat cells. Aliquots of the same aRNA sample were subsequently hybridized to 12 identical arrays. The CV for each individual spot was calculated based on the normalized signals across the 12 replicates. Of the 48 features, positive signals could be detected in 10 exogenous spikes and all of the human genes, with the exception of one transcript (NM_012266). Specificity of hybridization was determined by the absence of positive signal in all negative controls spotted on the array. The intra- and inter-array variability of signal intensities for hybridizations are summarized in Table 1. The average intra-array variability (CV) between duplicate spots over 12 arrays was 6.2%. The average inter-array variability of signal intensity across 4 arrays within a chip was 8.9%, while the variability across 12 arrays was 11.6%. Variability was increased at low signal values (Table 1).
Table 1. Variability of hybridization.
| Heat-shocked RNA | Control RNA | |||
|---|---|---|---|---|
| Numbera | CV% | Numbera | CV% | |
| Duplicate spots within an array | ||||
| Average | 32 | 5.7 | 32 | 6.6 |
| Normalized signal < 1.0 | 19 | 5.9 | 20 | 7.2 |
| Normalized signal > 1.0 | 13 | 5.4 | 12 | 5.8 |
| Four arrays within a chip | ||||
| Average | 32 | 8.7 | 32 | 9.0 |
| Normalized signal < 1.0 | 19 | 9.7 | 20 | 10.5 |
| Normalized signal > 1.0 | 13 | 7.9 | 12 | 7.4 |
| Twelve arrays among three chips | ||||
| Average | 32 | 10.3 | 32 | 12.8 |
| Normalized signal < 1.0 | 19 | 11.8 | 20 | 15.5 |
| Normalized signal > 1.0 | 13 | 9.1 | 12 | 9.3 |
aThe number of genes scored as positive signals on the array.
CV, coefficient of variation.
Differential gene expression analysis
To assess the performance of the system for profiling of complex biological RNA samples, experiments were performed using two complex aRNA and cDNA samples derived from control and heat-shocked Jurkat cells. The same total RNA preparations were used in the aRNA and cDNA experiments. Of the 23 genes, 22 gave rise to positive signals in both control and heat-shocked samples. Detectable expression for all 22 genes was seen in hybridizations using 0.05–5 μg aRNAs and with direct cDNA labeling starting from 2 to 20 μg total RNA (data not shown). The input materials for hybridization were typically either 1 μg of aRNA, or cDNA directly labeled from 5 μg of total RNA. The average normalized signal from the replicated hybridizations was used for comparative expression analysis. The fold-change trends (regulation or no change) after heat-shock induction are shown in Table 2. Of the 22 genes, 11 were identified as up-regulated (P < 0.05) in the aRNA experiments, and 13 (P < 0.05) in the cDNA experiments. The remaining genes did not show differential expression. The results were subsequently confirmed by real-time quantitative PCR. Of the 22 genes detected on the arrays, trends could be confirmed for 17 (77%) cases in both aRNA and cDNA samples by quantitative PCR (Table 2).
Table 2. Correlation of fold-change trends among platforms.
| Accession number | Blast ID | cDNA | aRNA | Affymetrix | QPCR | cDNA versus QPCR | aRNA versus QPCR | Affymetrix versus QPCR | cDNA versus aRNA | aRNA versus Affymetrix |
|---|---|---|---|---|---|---|---|---|---|---|
| X15183 | HSP90α | Up | Up | 0 | Up | + | + | − | + | − |
| M16660 | HSP90β | Up | Up | Up | Up | + | + | + | + | + |
| M17597 | Polyubiquitin | Up | Up | Up | Up | + | + | + | + | + |
| X52882 | TCP-1 | Up | Up | Up | 0 | − | − | − | + | + |
| U56655 | Novel | Up | 0 | np | 0 | − | + | nd | − | nd |
| X00351 | b-Actin | 0 | 0 | 0 | 0 | + | + | + | + | + |
| L11329 | PAC-1 | Up | Up | Up | Up | + | + | + | + | + |
| D13388 | DNAJA1 | Up | Up | Up | Up | + | + | + | + | + |
| L00160 | PGK | 0 | 0 | 0 | Up | − | − | − | + | + |
| M55643 | NF-kB1 | Up | 0 | 0 | Up | + | − | − | − | + |
| X68277 | DUSP1 | Up | Up | Up | Up | + | + | + | + | + |
| X02317 | SOD1 | Up | Up | 0 | 0 | − | − | + | + | − |
| U90878 | PDLIM1 | 0 | 0 | 0 | 0 | + | + | + | + | + |
| M88279 | FKBP4 | Up | Up | Up | Up | + | + | + | + | + |
| NM_002156 | HSPD1 | Up | Up | Up | Up | + | + | + | + | + |
| NM_004134 | HSPA9B | 0 | 0 | 0 | 0 | + | + | + | + | + |
| NM_012266 | DNAJB5 | nd | nd | nd | 0 | nd | nd | nd | nd | nd |
| NM_003334 | UBE1 | 0 | 0 | 0 | 0 | + | + | + | + | + |
| NM_022739 | SMURF2 | 0 | Up | 0 | 0 | + | − | + | − | − |
| NM_003342 | UBE2G | 0 | 0 | 0 | 0 | + | + | + | + | + |
| L06499 | RPL37A | Up | 0 | Up | 0 | − | + | − | − | − |
| NM_000994 | RPL32 | 0 | 0 | 0 | 0 | + | + | + | + | + |
| NM_000991 | RPL28 | 0 | 0 | Up | 0 | + | + | − | + | − |
| Correlation | 77% | 77% | 71% | 82% | 76% |
Up, Up-regulated; 0, unchanged; +, correlated; −, uncorrelated; nd, not detected; np, not presented.
To further examine consistency with other platforms, data from Affymetrix GeneChips were generated from control and heat-shocked Jurkat cells. Biotinylated aRNA samples (identical to those analyzed on the porous arrays) were hybridized to Affymetrix GeneChips. Of the 23 genes, one (U56655) was not represented and another (NM_012266) was not detectable on the U133A GeneChip. The transcript that could not be detected on the porous array was the same as the one that was not detectable on Affymetrix GeneChips (NM_012266). Of the 21 genes detected on the GeneChip, trends could be confirmed for 15 (71%) cases by quantitative PCR (Table 2). The gene regulations acquired on the porous arrays from the aRNA samples were in 76% correlation with data obtained from the Affymetrix GeneChips (Table 2).
Figure 3 compares the expression profiles obtained with the three different platforms. The quantitative correlation of fold-change between the cDNA data and quantitative PCR (R = 0.90) was greater than the correlation found between aRNA and quantitative PCR (R = 0.71). The correlation of the fold-change obtained with the porous arrays and quantitative PCR was higher than the correlation found between the data obtained with Affymetrix GeneChips and quantitative PCR (R = 0.45). The overall fold-change ratios obtained on Affymetrix GeneChips were lower than on porous arrays and those determined by quantitative PCR.
Figure 3.

Multi-platform comparisons of quantitative fold-change ratios. Comparison of the fold-change values determined by real-time quantitative PCR, by porous arrays using complex cDNA samples [porous array (cDNA)] and complex aRNA samples [porous array (aRNA)] and by Affymetrix GeneChips [GeneChip (U133A)]. Error bars indicate the SD of the fold-change.
DISCUSSION
The flow-through porous microarray described here is a rapid, low-density system in which up to 400 features can be analyzed on a single array. The array area with a diameter of 4.45 mm, in combination with the relatively large spots with a diameter of 120 μm and a typical pitch distance of 200 μm, results in a maximum number of 400 spots per array. For future clinical applications, we consider it of great value to have the basic specifications of our system determined with well-controlled samples and to compare the system with widely used technologies such as real-time quantitative PCR and Affymetrix GeneChips. To assess hybridization reactions quantitatively on the platform, we determined hybridization kinetics in real time using exogenous transcripts. We show that signal can be detected within 5 min and approaches a plateau after 90 min. We recommend a hybridization time of 60 min for gene expression analysis, since this is sufficient for the detection of genes expressed at low levels. In contrast to overnight incubation for conventional microarrays, the hybridization time is thus reduced >10-fold (3–7). A flow rate of 5 cycles/min is used here, since this pumping condition showed a higher signal increase per time unit than the other tested pumping conditions. By comparing the ratios of signal intensities for different concentrations of transcripts, we found that these are constant with a CV of 4.6–5.9%. This indicates that the expression levels can be quantitatively detected. When analyzing the signal intensities of the five exogenous transcripts, we found that different transcripts display different signal intensities at the same concentration and show the different patterns of hybridization kinetics on the flow rates. This is probably due to variable hybridization efficiency induced by the base composition of oligonucleotides spotted on the array and/or by secondary structure of mRNA molecules (15,16). The availability of kinetic measurement on our platform offers experimental flexibility for assay development; however, real-time detection may not be applicable for most gene expression analyses. Complex biological samples often have high background fluorescence, and require washing of the array before intensity measurement if very sensitive detection is required.
Typically, hybridization and subsequent analysis are completed within 2 h. Using the current protocols for RNA extraction and direct cDNA labeling, gene expression experiments starting from a biological sample can be completed within one day. This is a significant advantage in clinical situations where the information needs to be provided rapidly for therapeutic intervention. In addition, the data acquired on the porous arrays are highly reproducible with a variation (CV) of 11.6% across 24 arrays. The minimum detectable concentration (0.1–0.25 pM) is better than other existing flow-through microarray techniques (11) but is similar to values obtained with Affymetrix GeneChips (17). However, the minimum detectable amount (2–5 amol) is 10 times less, since we use a 10 times smaller sample volume than on Affymetrix GeneChips. Therefore, our protocol requires only 1 μg of aRNA instead of the 10 μg of aRNA used in the protocol of Affmetrix GeneChips (6,7). We found that a linear dynamic range of 3 orders of magnitude can be reached, which is broader than on Affymetrix GeneChip (17) and similar to that obtained on other three-dimensional microarray platforms (11,18). The broad dynamic range is due to the large binding capacity provided by the internal surface area of the substrate (8,19).
We subjected Jurkat cells to heat-shock treatment and analyzed the transcriptional regulation of 23 genes. Of the 22 genes detected on our platform, 10 could be identified as up-regulated with both heat-shocked cDNA and aRNA samples. All of the 10 genes have a known function in stress response (http://www.hugeindex.org). We applied real-time quantitative PCR (14,20) to cross-validate the expression patterns. The results were also compared to data obtained with Affymetrix GeneChips. Although the expression profiles obtained with the three different platforms did not display identical quantitative changes, they were well correlated with the gene regulations. This observation is consistent with reports comparing array-based profiling with quantitative PCR (21,22). When comparing the quantitative fold change ratios among the three platforms, we found that the overall fold-change ratios obtained on the Affymetrix GeneChips are lower than those determined using both porous arrays and quantitative PCR. This could be due to the method used for averaging of signal intensities on the 11 probes per gene on Affymetrix GeneChips, which may decrease the measured ratio between the two conditions (17).
We selected Affymetrix GeneChips for cross-platform comparison since they have been widely used for global gene expression profiling (6,7). While 60mer oligonucleotide arrays are closer to our platform in terms of probe length, they are less well standardized and do not necessarily use the same programs for probe design, or the same hybridization protocols. Classical hybridization studies show that the rate constant for hybridization reactions that are not diffusion-limited is proportional to the square root of the length of the shortest strand participating in duplex formation. Therefore, only a 1.5-fold increase in hybridization velocity is expected when switching from 25 to 60mers (23). Obviously, diffusion limitation determines hybridization kinetics in solid phase conditions such as array hybridizations much more than does fragment length. It has been reported that accurate gene expression measurement on DNA microarrays can be achieved with both multiple short probes and a long probe per gene (24–26). Although our approach differs significantly from Affymetrix GeneChips, the profiles of gene regulation have a good correlation with each other. This suggests that our platform could reproduce the major findings acquired from Affymetrix GeneChips.
We compared the heat-shock data from the report of Schena et al. (13) with those collected on our platform. Of the seven genes monitored on the glass cDNA arrays after heat-shock induction, trends were confirmed in six cases by our platform with the cDNA samples and five cases with the aRNA samples. Although the total RNA source and labeling procedures used for both experiments were not identical, there was a significant concordance of the findings between the two platforms.
In summary, our results show good correlation between results obtained with porous arrays, real-time quantitative PCR and Affymetrix GeneChips. They are also in agreement with data from glass cDNA arrays obtained from the literature (13). The results demonstrate that our platform provides a capability for quantitative expression analysis in a novel format. We also show that the porous array is associated with speed, wide dynamic range, good sensitivity and reproducibility. Therefore, we expect that the flow-through porous microarray will serve as a diagnostic tool for the rapid analysis of gene expression profiles.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at NAR Online.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Riet Hilhorst, Rob Ruijtenbeek and Savithri Rangarajan for discussions, and Bertrand Jordan for helpful comments. Part of the work presented here was supported by a grant (BTS01002) from the Dutch BTS.
REFERENCES
- 1.Venter J.C., Adams,M.D., Myers,E.W., Li,P.P., Mural,R.J., Sutton,G.G., Smith,H.O., Yandell,M., Evans,C.A., Holt,R.A. et al. (2001) The sequence of the human genome. Science, 291, 1304–1351. [DOI] [PubMed] [Google Scholar]
- 2.Dan S., Tsunoda,T., Kitahara,O., Yanagawa,R., Zembutsu,H., Katagiri,T., Yamazaki,K., Nakamura,Y. and Yamori,T. (2002) An integrated database of chemosensitivity to 55 anticancer drugs and gene expression profiles of 39 human cancer cell lines. Cancer Res., 62, 1139–1147. [PubMed] [Google Scholar]
- 3.Golub T.R., Slonim,D.K., Tamayo,P., Huard,C., Gaasenbeek,M., Mesirov,J.P., Coller,H., Loh,M.L., Downing,J.R., Caligiuri,M.A. et al. (1999) Molecular classification of cancer: class discovery and class prediction by gene expression profiling. Science, 286, 531–537. [DOI] [PubMed] [Google Scholar]
- 4.Perou C.M., Sorlie,T., Eisen,M.B., van de Rijn,M., Jeffrey,S.S., Rees,C.A., Pollack,J.R., Ross,D.T., Hohnsen,H., Akslen,L.A. et al. (2000) Molecular portraits of human breast tumors. Nature, 406, 747–752. [DOI] [PubMed] [Google Scholar]
- 5.Dhanasekaran S.M., Barrette,T.R., Ghosh,D., Shah,R., Varambally,S., Kurachi,K., Pienta,K.J., Rubin,M.A. and Chinnaiyan,A.M. (2001) Delineation of prognostic biomarkers in prostate cancer. Nature, 412, 822–826. [DOI] [PubMed] [Google Scholar]
- 6.Dyrskjot L., Thykjaer,T., Kruhoffer,M., Jensen,J.L., Marcussen,N., Hamilton-Dutoit,S., Wolf,H. and Orntoft,T.F. (2003) Identifying distinct classes of bladder carcinoma using microarrays. Nature Genet., 33, 90–96. [DOI] [PubMed] [Google Scholar]
- 7.Valk P.J., Verhaak,R.G., Beijen,M.A., Erpelinck,C.A.J, Barjesteh van Waalwijk van Doorn-Khosrovani,S., Boer,J.M., Beverloo,H.B., Moorhouse,M.J., van der Spek,P.J., Lowenberg,B. et al. (2004) Prognostically useful gene-expression profiles in acute myeloid leukemia. N. Engl. J. Med., 15, 1617–1628. [DOI] [PubMed] [Google Scholar]
- 8.Van Beuningen R., van Damme,H., Boender,P., Bastiaensen,N., Chan,A. and Kievits,T. (2001) Fast and specific hybridisation using flow-through microarrays on porous metal oxide. Clin. Chem., 47, 1931–1933. [Google Scholar]
- 9.Rigby W.R., Cowieson,D.R., Davies,N.C. and Furneaux,R.C. (1990) An anodising process for production of inorganic microfiltration membranes. Trans. Inst. Met. Finish., 68, 95–108. [Google Scholar]
- 10.Hokaiwado N., Asamoto,M., Tsujimura,K., Hirota,T., Ichihara,T., Satoh,T. and Shirai,T. (2004) Rapid analysis of gene expression changes caused by liver carcinogens and chemopreventive agents using a newly developed three-dimensional microarray system. Cancer Sci., 95, 123–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cheek B.J., Steel,A.B., Torres,M.P., Yu,Y.Y. and Yang.H. (2001) Chemiluminescence detection for hybridization assays on the flow-thru chip, a three-dimensional microchannel biochip. Anal. Chem., 15, 5777–5783. [DOI] [PubMed] [Google Scholar]
- 12.Benoit V., Steel,A., Torres,M., Yu,Y.Y., Yang,H. and Cooper,J. (2001) Evaluation of three-dimensional microchannel glass biochips for multiplexed nucleic acid fluorescence hybridization assays. Anal. Chem., 73, 2412–2420. [DOI] [PubMed] [Google Scholar]
- 13.Schena M., Shalon,D., Heller,R., Chai,A., Brown,P.O. and Davis,R.W. (1996) Parallel human genome analysis: microarray-based expression monitoring of 1000 genes. Proc. Natl Acad. Sci. USA, 93, 10614–10619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rajeevan M.S., Vernon,S.D., Taysavang,N. and Unger,E.R. (2001) Validation of array-based gene expression profiles by real-time (kinetic) RT–PCR. J. Mol. Diagn., 3, 26–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang L., Miles,M.F. and Aldape,K.D. (2003) A model of molecular interactions on short oligonucleotide microarrays. Nat. Biotechnol., 21, 818–821. [DOI] [PubMed] [Google Scholar]
- 16.Hekstra D., Taussig,A.R., Magnasco,M. and Naef,F. (2003) Absolute mRNA concentrations from sequence-specific calibration of oligonucleotide arrays. Nucleic Acids Res., 31, 1962–1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chudin E., Walker,R., Kosaka,A., Wu,S.X., Rabert,D., Chang,T.K. and Kreder,D.E. (2001) Assessment of the relationship between signal intensities and transcript concentration for Affymetrix GeneChip arrays. Genome Biol., 3, research 0005.1–0005.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ramakrishnan R., Dorris,D., Lublinsky,A., Nguyen,A., Domanus,M., Prokhorova,A., Gieser,L., Touma,E., Lockner,R., Tata,M. et al. (2001) An assessment of Motorola CodeLink microarray performance for gene expression profiling applications. Nucleic Acids Res., 30, e30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xu Y. and Bao,G. (2003) A filtration-based protein microarray technique. Anal. Chem., 15, 5345–5351. [DOI] [PubMed] [Google Scholar]
- 20.Schmittgen T.D., Zakrajsek,B.A., Mills,A.G., Gorn,V., Singer,M.J. and Reed,M.W. (2000) Quantitative reverse transcription-polymerase chain reaction to study mRNA decay: comparison of endpoint and real-time methods. Anal. Biochem., 285, 194–204. [DOI] [PubMed] [Google Scholar]
- 21.Chuaqui R.F., Bonner,R.F., Best,C.J., Gillespie,J.W., Flaig,M.J., Hewitt,S.M., Phillips,J.L., Krizman,D.B., Tangrea,M.A., Ahram,M. et al. (2002) Post-analysis follow-up and validation of microarray experiments. Nature Genet., 32, Suppl, 509–514. [DOI] [PubMed] [Google Scholar]
- 22.Tsukasaki K., Tanosaki,S., DeVos,S., Hofmann,W.K., Wachsman,W., Gombart,A.F., Krebs,J., Jauch,A., Bartram,C.R., Nagai,K. et al. (2004) Identifying progression-associated genes in adult T-cell leukemia/lymphoma by using oligonucleotide microarrays. Int. J. Cancer., 109, 875–881. [DOI] [PubMed] [Google Scholar]
- 23.Wetmur J.G. (1991) DNA probes: applications of the principles of nucleic acid hybridization. Crit. Rev. Biochem. Mol. Biol., 26, 227–259. [DOI] [PubMed] [Google Scholar]
- 24.Kane M.D., Jatkoe,T.A., Stumpf,C.R., Lu,J., Thomas,J.D. and Madore,S.J. (2000) Assessment of the sensitivity and specificity of oligonucleotide (50mer) microarrays. Nucleic Acids Res., 28, 4552–4557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Barczak A., Rodriguez,M.W., Hanspers,K., Koth,L.L., Tai,Y.C., Bolstad,B.M., Speed,T.P. and Erle,D.J. (2003) Spotted long oligonucleotide arrays for human gene expression analysis. Genome Res., 13, 1775–1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chou C.C., Chen,C.H., Lee,T.T. and Peck,K. (2004) Optimization of probe length and the number of probes per gene for optimal microarray analysis of gene expression. Nucleic Acids Res., 32, e99. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.