

Abstract
Cutaneous flushing is a common presenting complaint in endocrine disorders. The pathophysiology of flushing involves changes in cutaneous blood flow triggered by multiple intrinsic factors that are either related to physiology or disease. Flushing can be divided into episodic or persistent causes. Episodic flushing is mediated by the release of endogenous vasoactive mediators or medications, while persistent flushing results in a fixed facial erythema with telangiectasia and a cyanotic tinge owing to the large cutaneous blood vessels that contain slow-flowing deoxygenated blood. The differential diagnosis of cutaneous flushing in neuroendocrine disorders is limited, yet encompasses a broad spectrum of benign and malignant entities, including carcinoid syndrome, pheochromocytoma, Cushing syndrome, medullary thyroid cancer, and pancreatic neuroendocrine tumors. In this review, we provide a concise and up-to-date discussion on the differential diagnosis and approach of flushing in neuroendocrinology.
Keywords: Flushing, Neuroendocrine tumor, Carcinoid, Pheochromocytoma, Histamine, Substance P
Introduction
Flushing is a subjective sensation of warmth that is accompanied by reddening of the skin anywhere on the body but favors the face, neck, and upper torso (1). This bodily predilection is primarily due to increased relative volume of visible superficial cutaneous vasculature, as well as qualitative differences in skin vascular response and vascular regulation, as compared with other body areas (1–3). Flushing can be broadly divided into episodic or persistent. Episodic flushing is mediated by the release of endogenous vasoactive mediators or medications, while persistent flushing results in a fixed facial erythema with telangiectasia’s and a cyanotic tinge owing to the large cutaneous blood vessels that contain slow-flowing deoxygenated blood (2). Flushing should be differentiated from facial plethora, which is more chronic and develops insidiously, although the physiology of the two phenomena is similar.
The differential diagnosis of a patient with flushing is extensive and includes a broad range of benign and malignant conditions. Benign causes of flushing include rosacea, climacterium, fever, benign cutaneous flushing (as seen in emotional distress), and medication-induced. To the contrary, the following causes of flushing are associated with increased morbidity and mortality: carcinoid syndrome, pheochromocytoma, mastocytosis, neuroendocrine tumors, anaphylaxis, medullary thyroid cancer, renal cell carcinoma, inborn errors of metabolism such as Fabry disease, and autonomic dysfunction.
With the exception of carcinoids, flushing due to tumors is rare and tends to occur in advanced stages or as a manifestation of paraneoplastic syndrome. The signs and symptoms of benign cutaneous flushing include abdominal complaints and flushing of the blush area, which may overlap with those of carcinoid syndrome and pheochromocytoma (4,5). Thus, clinicians should obtain a careful history and perform a thorough physical examination in all cases of flushing as an early diagnosis of an endocrine tumor, such as a pheochromocytoma, may lead to decreased morbidity and mortality. In this review, we provide a concise and up-to-date discussion on the differential diagnosis and approach of flushing in neuroendocrinology (Table 1).
Table 1.
Neuroendocrine causes of flushing.
| Diagnosis | Estimates | Clinical findings | Triggers | Substances |
|---|---|---|---|---|
| Carcinoid syndrome | 10% of patients with carcinoid tumors (incidence rate of 1.9 per 100,000) | Flushing Diarrhea Abdominal cramping Fatigue |
|
Serotonin Substance P Histamine Catecholamines Prostaglandins |
| Pheochromocytoma and Paraganglioma | 1 in 2500–6500 individuals (500–1600 cases diagnosed annually in the United States) | Hypertension is the most frequent finding (sustained or paroxysmal, or associated with flushing or pallor) The typical PPGL attack, which is seen in approximately 30% of patients, presents headaches, sweating, palpitations, and with or without flushing. |
|
Pheochromocytomas: catecholamines (epinephrine, norepinephrine, and dopamine) Paragangliomas: does not produce catecholamines Both may be biochemically silent |
| Medullary Thyroid Cancer | Rare | Most patients are asymptomatic; in the symptomatic patient, secretory diarrhea is the most prominent hormone-mediated clinical finding, with or without flushing | None | Calcitonin Prostaglandins Histamine Substance P Ketacalcin Levodopa, Adrenocorticotropic hormone Corticotropin-releasing hormone |
| Pancreatic Neuroendocrine Tumors | <1 per 100 000 persons per year | Most individuals usually only have symptoms relating to the hormone that is chiefly produced. | None | Vasoactive intestinal peptide Gastric inhibitory polypeptide Prostaglandin Insulin Gastrin Glucagon Adrenocorticotropic hormone Corticotropin-releasing hormone Somatostatin Growth hormone–releasing factor Neurotensin Parathyroid hormone-related peptide Pancreatic polypeptide Melanocyte-Stimulating Hormone |
| Endogenous Cushing syndrome | 0.7–2.4 per million population per year | Facial plethora Central body weight gain with limb thinning Acanthosis nigricans Proximal muscle weakness Easy bruising Striae Flushing (rare) |
None | Cortisol Adrenocorticotropic hormone and/or Corticotropin-releasing hormone (in Cushing disease or ectopic Cushing syndrome) |
Initial evaluation
The initial evaluation of a patient with flushing includes a thorough history and physical examination. A directed history should focus on the type of a flush and its associated symptoms, including a detailed description of the type of flush and its distribution, associations (i.e.: lightheadedness, low blood pressure, bronchospasm, tachycardia) and triggers (i.e.: emotion, food, drugs, physical exertion, alcohol, stress). Vague complaints may be associated with a psychiatric comorbidity, such as anxiety, depression, or somatization disorder. Elevation of both arms can lead to marked facial plethora, indicative of compressed jugular veins, in patients with large sub/retrosternal goiter or mediastinal mass. This phenomenon is called Pemberton’s sign (6,7).
Certain clinical characteristics may be elicited to assist in narrowing the differential diagnosis of flushing. Associated sweating (“wet flushes”) indicates autonomic hyperactivation, while lack of (“dry flushes”) is usually the result of a substance(s) that lead to vascular smooth muscle activation. Accompanying symptoms may further point to the cause; antidromic sensorineural-mediated flushing is accompanied by pain or burning sensation in the affected areas, while the presence of urticaria and pruritus is seen in histamine-mediated reactions, including mastocytosis, or as a side effect to vancomycin therapy. However, the majority of conditions that manifest with flushing do overlap in symptoms (Table 2).
Table 2.
The characteristics of flushing in neuroendocrine tumors.
| Syndrome | Characteristics |
|---|---|
| Carcinoid syndrome |
|
| Pancreatic Neuroendocrine Tumors |
|
| Endogenous Cushing syndrome |
|
Neuroendocrine causes of flushing
1. Carcinoid syndrome
Carcinoid syndrome (CS) is a life-threatening medical condition that affects approximately 10% of patients with carcinoid tumors (8,9). Carcinoid tumors are slow-growing benign lesions of enterochromaffin or Kulchitsky cells that are derived from the neuroendocrine lineage and have a low incidence rate of 1.9 per 100,000 (10–16). Patients with carcinoid tumors are usually asymptomatic or may have vague gastrointestinal complaints. However, patients with CS typically present with flushing that is often times accompanied by diarrhea, abdominal cramping, and fatigue. In the majority of patients (approximately 50%), particularly males, tumors are located in the small bowel or proximal colon, and rarely in the stomach, bronchus or appendix (3,8,17). CS may rarely arise from ovarian teratomas, tumors of the uterine cervix, glomus jugulare, and thyroid gland or present with right-sided cardiac failure from valvular disease or severe bronchoconstriction (8,17,18).
Aggressive carcinoid tumors tend to be functional and secrete several biologically active substances. The clinical hallmark of functional carcinoids in over 90% of cases is flushing, which is often episodic (3,10). Generally, vasoactive substances, such as serotonin (5-HT), substance P, histamine, catecholamines, prostaglandins, among others, that escape hepatocyte inactivation, provoke flushing (12,19,20). These substances are usually secreted by carcinoid tumors that are located distal to the portal vein or downstream of functioning hepatocytes. The release of these substances is triggered by amine-rich foods, such as sherry, beer, fermented foods, and chocolate, pharmacologic triggers such as catecholamines, dopamine, pentagastrin and isoproterenol (also seen in patients with mastocytosis and benign cutaneous flushing), and increase in adrenergic activity, as seen in pain, anger, embarrassment, or exertion. Niacin-induced flushing, which can only be reduced by ~ 30% by taking aspirin, is influenced by 9alpha, 11beta-prostaglandin F (2) combined with an imbalance of the sympathetic and parasympathetic nervous system (21). Facial flushing, sometimes seen when 1-desamino-8-D-arginine vasopressin (DDAVP) is administered intravenously, appears to be mediated by prostacyclin production (22).
The flush in CS may be distinguished from other causes and can help point out the location of the tumor. Midgut tumors cause a rapid cyanotic flush that last for less than a minute and commonly associated with a mild burning sensation, while foregut tumors produce pruritic wheals that are reddish-brown and occur over the entire body (Image 1), and those with bronchoconstriction are usually bright red and confluent, and associated with chemosis, facial edema, or hypotension. Accompanying symptoms are common, including diarrhea, dyspnea, abdominal pain or wheezing. The chronicity of symptoms will dictate the length of episodes, development of bluish coloration of bodily areas, such as the malar area or nose, or thick skin changes with venous telangiectasia.
Image 1.
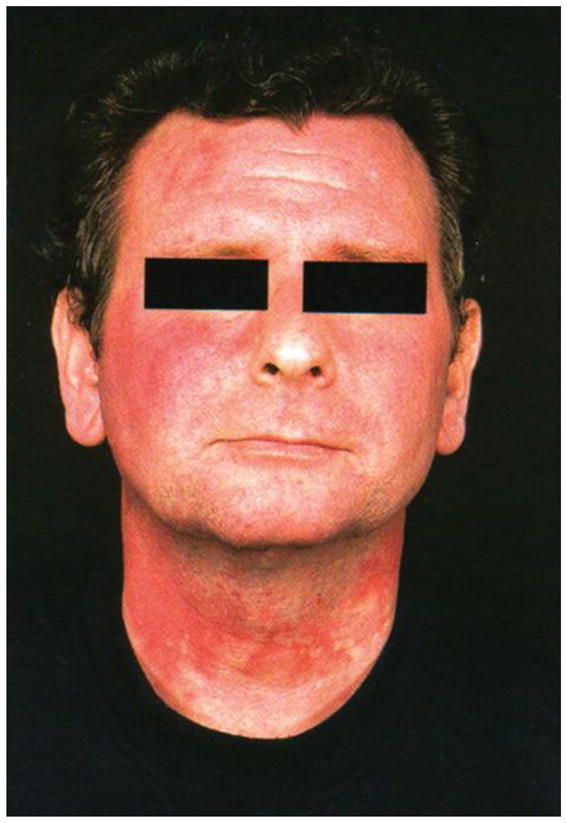
Flushing from a carcinoid tumour in the foregut due to histamine release. With permission from John Wiley and Sons
The diagnosis of CS is established by measuring the 24-hour urine levels of 5-hydroxyindolacetic acid (5-HIAA) or chromogranin A (CgA). 5-HIAA is a major urinary metabolite of 5-HT, with a sensitivity of 73% and a specificity of 100% for diagnosing carcinoids (23). Urinary 5-HIAA is not elevated in mastocytosis, because 5-HT is not made by human mast cells or in idiopathic anaphylaxis or idiopathic flushing. Moreover, some patients with carcinoid tumors have symptoms of flushing with low or normal levels of 5-HIAA (24). Unlike 5-HIAA, CgA levels are independent of symptoms and are elevated to 100–1000 times normal in 85%–100% of patients with a carcinoid tumor regardless of whether the tumor is functional or nonfunctional. The specificity and sensitivity of CgA is 98.4% and 62.9%, respectively (25).
The main goals of CS management are symptom control. This is best achieved with somatostatin analogs (e.g.: octreotide or lanreotide) by reducing the secretion of vasoactive mediators (10,12,15,26,27,28). Histamine-induced flushing may be treated with H1 and H2 receptor blockers (10). Alternatives in management include various combinations of isotope therapy, addition of interferon, 5-HT antagonist ketanserin, and use of chemotherapy (10).
2. Pheochromocytoma and Paraganglioma
Pheochromocytoma and paraganglioma (PPGL) are neuroendocrine tumors that arise from the chromaffin cells of the adrenal glands, and ganglia along the sympathetic and parasympathetic chain, respectively (29). PPGL affect about 1 in 2500–6500 individuals, with 500–1600 cases diagnosed annually in the United States (30). Pheochromocytomas typically produce catecholamines (epinephrine, norepinephrine, and dopamine) and can lead to secondary diabetes mellitus or hypertension, while paragangliomas that arise from the parasympathetic ganglia do not produce catecholamines, and rarely are PPGL’s biochemically silent. Hypertension is the most frequent finding, and can be sustained or paroxysmal, or associated with flushing or pallor. The typical PPGL attack, which is seen in approximately 30% of patients (29,31,32), presents with headaches, sweating, palpitations, and with or without flushing. Such paroxysms may be precipitated by medications such as glucocorticoids or β-adrenergic receptor blockers, which may be used in the treatment of flushing before a diagnosis is established.
Several factors may be responsible for the generation of flushing in PPGL. The most convincing mechanism is the production of catecholamines that lead to thermal vasodilation of the face and other bodily areas (33,34). Other recognized factors include general blood pressure lability and episodes of increased cardiac output, and the production of flushing mediators, such as calcitonin gene-related peptide (35), vasoactive intestinal polypeptide (VIP) (36–38), and adrenomedullin (39,40), a potent vasodilatory peptide with significant vasodilatory effects on skin.
Syndromic PPGL may also present with flushing and their frequency is unknown (29). These conditions include; multiple endocrine neoplasia type 2A (MEN2A; medullary thyroid cancer, primary hyperparathyroidism, and cutaneous lichen amyloidosis); multiple endocrine neoplasia type 2B (MEN2B; medullary thyroid cancer, mucocutaneous neuromas, skeletal deformities (e.g., kyphoscoliosis or lordosis), joint laxity, myelinated corneal nerves, and intestinal ganglioneuromas (Hirschsprung disease); von Hippel-Lindau syndrome (VHL; hemangioblastoma that involve the cerebellum, spinal cord, or brainstem, retinal angioma, clear cell renal cell carcinoma, pancreatic neuroendocrine tumors and serous cystadenomas, endolymphatic sac tumors of the middle ear, papillary cystadenomas of the epididymis and broad ligament) and Neurofibromatosis type 1 (neurofibromas, multiple café-au-lait spots, axillary and inguinal freckling, iris hamartomas (Lisch nodules), bony abnormalities, central nervous system gliomas, macrocephaly, and cognitive deficits). Plethora can develop when neurogenic polyglobulia occurs via hemangioblastomas expressing erythropoietin or via other VHL-associated tumors secreting erythropoietin (41). Plethora is a sign of other endocrine conditions, like Cushing syndrome (see below).
The diagnosis of PPGL is established by measurements of plasma free or 24-hour urinary fractionated metanephrines (29). These tests are superior to other tests of catecholamine excess, as free metanephrines are produced continuously within adrenal chromaffin cells (or the tumors derived from these cells) and are specific markers of chromaffin tumors.
3. Medullary Thyroid Cancer
Medullary thyroid cancer (MTC) is a rare malignant tumor of the parafollicular C cells of the thyroid gland (42). MTC is derived from the neural crest and secretes a variety of biologically active substances including calcitonin, prostaglandins, histamine, substance P, ketacalcin, levodopa, adrenocorticotropic hormone (ACTH), and corticotropin-releasing hormone, that can cause flushing and sweating. MCT is caused by mutations in the RET proto-oncogene, and may be sporadic or inherited in an autosomal dominant pattern in approximately 25% of cases as part of multiple endocrine neoplasia (MEN2A and MEN2B) or familial MTC. Most patients with MTC are asymptomatic; in the symptomatic patient, secretory diarrhea is the most prominent hormone-mediated clinical feature, while protracted flushing of the face and upper extremities, discoloration, and telangiectasias, is seen less frequently (42). Examining a fine needle aspirate from a thyroid nodule initially makes the diagnosis of MTC with 95–98% accuracy (42). Serum calcitonin levels are usually elevated and often establish the diagnosis of MTC. Carcinoembryonic antigen (CEA) may be elevated in advanced MTC and could be used as a marker for possible later recurrence of disease. Total thyroidectomy with central lymph node dissection is the minimum recommended operation for MTC. Genetic testing of the proband and all first-degree relatives should include RET mutation analysis (43,44).
4. Pancreatic Neuroendocrine Tumors
Pancreatic neuroendocrine tumors (PNETs) are rare neuroendocrine neoplasms from pluripotent cells of the pancreas with an incidence of les than 1 per 100 000 persons per year (45,46). Their prevalence ranges from 0.8% to 10% in patients undergoing a postmortem examination, suggesting that people frequently harbor asymptomatic PNETs (47). When PNETs are malignant, they are called pancreatic endocrine cancer or islet cell carcinoma.
PNETs secrete several hormones and peptides, including VIP, gastric inhibitory polypeptide, prostaglandin, insulin, gastrin, glucagon, ACTH, somatostatin, growth hormone–releasing factor (GRF), neurotensin, parathyroid hormone-related peptide, pancreatic polypeptide, and melanocyte-stimulating hormone (48,49). Most individuals usually only have symptoms relating to the hormone that is chiefly produced. Flushing can be seen in all forms of PNETs and resembles that of gastric carcinoid syndrome (pruritic reddish-brown with variegated margination over the entire body). As with CS, chronic flushing from PNETs may develop thick skin changes with venous telangiectasia and bluish coloration of the chin, nose, and malar area.
PNETs are divided into several subtypes. VIPomas classically present with Verner-Morrison syndrome that manifest with watery diarrhea, hypokalemia, achlorhydria, and rarely flushing. A VIP tumor is diagnosed by demonstrating a high plasma VIP level in the setting of stool volume greater than 1 L per day. Neurotensinomas are rare forms of PNETs that are usually malignant and manifest with hypotension, hypokalemia, weight loss, flushing, and hyperglycemia, while calcitoninomas, another rare form, cause watery diarrhea and facial flushing, as one would see with MTC. Flushing is a rare clinical finding in: gastrinomas which are usually malignant and form in the head of the pancreas (often referred to as Zollinger-Ellison syndrome); insulinomas which are usually benign tumors in the head, body, or tail of the pancreas that may produce flushing during hypoglycemic episodes; glucagonomas which are usually malignant and located in the tail of the pancreas and present with hyperglycemia and a characteristic necrolytic migratory erythema rash; and somatostatinomas which lead to hyperglycemia, gallstones, and steatorrhea.
Evaluation of PNETs requires biochemical workup of the various secretory peptides and amines, and advanced imaging techniques. Abdominal and pancreatic ultrasound, as well as computed tomography and aortography, could be performed to localize the tumor or metastases (50). Therapy for PNETs is primarily surgical resection for localized disease and selected patients with metastatic disease. Although somatostatin analogues have proven to be very effective in ameliorating symptoms of hormone overproduction, options regarding systemic therapy for advanced disease continue to be limited (10,12,49).
5. Endogenous Cushing syndrome
Endogenous Cushing syndrome is the result of chronic glucocorticoid excess from either an ACTH-secreting pituitary tumor, cortisol producing adenoma, adrenal hyperplasia, or ectopic production of ACTH and/or CRH from a neuroendocrine tumor (51,52). Endogenous Cushing syndrome is rare with an overall incidence of approximately 0.7–2.4 per million population per year (53,54). Patients classically present with facial plethora (Image 2), central body weight gain with limb thinning, glucose intolerance or diabetes with extensive acanthosis nigricans, hypertension, proximal muscle weakness, easy bruising and striae. Flushing is rarely a presenting complaint and is usually subjective. Facial plethora is consistent and unlike flushing rarely episodic; it is also decreased after surgery, due to reduction of blood volume perfusion (Image 2) (55).
Image 2.
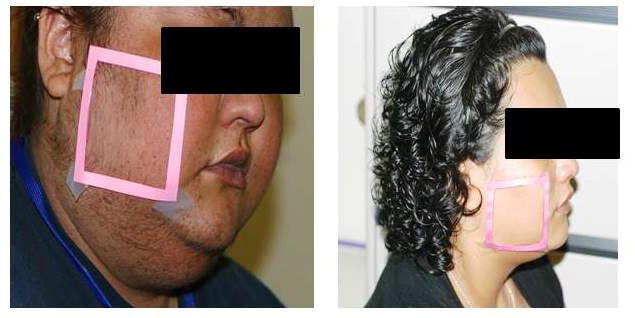
Facial plethora in a patient with Cushing syndrome before and after surgery. With permission from Ali Afshari and Constantine A. Stratakis, NIH
6. Other causes
Several comorbidities may be associated with neuroendocrine disorders and are known to cause flushing, including; central hypogonadism in men (56); POEMS syndrome (polyneuropathy, organomegaly, endocrinopathy, monoclonal proteins, and skin changes) (57); malignant histiocytoma, neuroblastoma, and ganglioneuroma, from increased production of VIP; TSH-secreting pituitary adenoma causing hyperthyroidism (58); dysautonomia, orthostatic hypotension, migraines; anxiety and panic attacks presenting with hot flashes and sweating; and vascular pulsation in inflammatory skin diseases, examples of which are spider arterial angiomas, acquired pulsating arteriovenous angiomas and arteriovenous capillary shunt in inflamed lesions or in local trauma (only visible in inflamed skin implicating the roles of neuropeptides) (59, 60). More than two thirds of perimenopausal women experience hot flashes which may represent a marker of underlying cardiovascular disease (61); a recent study examined the role of genetic variation in the tachykinin receptor 3 (TACR3) as a contributor to hot flashes (62). Treatment should be individually tailored and includes use of systemic hormone therapy (estrogen +/− progestogen), selective serotonin reuptake inhibitors, gabapentin, clonidine, and other modalities (63,64).
Conclusion
The differential diagnosis of cutaneous flushing in neuroendocrine disorders is limited, yet encompasses a broad spectrum of benign and malignant entities. A thorough history and physical examination to elicit the characteristics of the underlying syndrome associated with flushing, and laboratory and radiographic investigations to ascertain the cause, are mandatory. Several neuroendocrine disorders are associated with flushing, including CS, PPGL, MTC, and endogenous Cushing syndrome, among others. It is imperative for the clinician to separate benign from potentially life-threatening conditions associated with flushing and to provide an appropriate workup and treatment to decrease morbidity and mortality that are associated with neuroendocrine disorders.
Footnotes
Conflict of Interest Statement:
The authors declare that they have no conflict of interest.
References
- 1.Ikizoglu G. Red face revisited: flushing. Clinic in Dermatology. 2014;32:800–808. doi: 10.1016/j.clindermatol.2014.02.019. [DOI] [PubMed] [Google Scholar]
- 2.Ray D, Williams G. Pathophysiological causes and clinical significance of flushing. Br J Hosp Med. 1993;50:594–8. [PubMed] [Google Scholar]
- 3.Aldrich LB, Moattari AR, Vinik AI. Distinguishing features of idiopathic flushing and carcinoid syndrome. Arch Intern Med. 1988;148:2614–8. [PubMed] [Google Scholar]
- 4.Metcalfe DD. Differential diagnosis of the patient with unexplained flushing/anaphylaxis. Allergy Asthma Proc. 2000;21:21–4. doi: 10.2500/108854100778249006. [DOI] [PubMed] [Google Scholar]
- 5.Hartmann K, Escribano L, Grattan C, Brockow K, Carter MC, Alvarez-Twose I, et al. Cutaneous manifestations in patients with mastocytosis: consensus report of the European Competence Network on Mastocytosis, the American Academy of Allergy, Asthma & Immunology, and the European Academy of Allergology and Clinical Immunology. J Allergy Clin Immunol. 2016 Jan;137(1):35–45. doi: 10.1016/j.jaci.2015.08.034. [DOI] [PubMed] [Google Scholar]
- 6.De Filippis EA, Sabet A, Sun MR, Garber JR. Pemberton’s sign: explained nearly 70 years later. J Clin Endocrinol Metab. 2014;99(6):1949–1954. doi: 10.1210/jc.2013-4240. [DOI] [PubMed] [Google Scholar]
- 7.Jukic T, Kusic Z. Pemberton’s sign in patient with substernal goiter. J Clin Endocrinol Metab. 2010;95(9):4175. doi: 10.1210/jc.2010-0944. [DOI] [PubMed] [Google Scholar]
- 8.Schnirer II, Yao JC, Ajani JA. Carcinoid--a comprehensive review. Acta Oncol. 2003;42:672–92. doi: 10.1080/02841860310010547. [DOI] [PubMed] [Google Scholar]
- 9.Feldman JM. Carcinoid tumors and the carcinoid syndrome. Curr Probl Surg. 1989;26(12):835–85. doi: 10.1016/0011-3840(89)90010-5. [DOI] [PubMed] [Google Scholar]
- 10.Singh S, Asa SL, Dey C, Kennecke H, Laidley D, Law C, Asmis T, Chan D, et al. Diagnosis and management of gastrointestinal neuroendocrine tumors: an evidence-based Canadian consensus. Cancer Treat Rev. 2016;47:32–45. doi: 10.1016/j.ctrv.2016.05.003. [DOI] [PubMed] [Google Scholar]
- 11.Boudreaux JP1, Klimstra DS, Hassan MM, Woltering EA, Jensen RT, Goldsmith SJ, Nutting C, Bushnell DL, Caplin ME, Yao JC North American Neuroendocrine Tumor Society (NANETS) The NANETS consensus guideline for the diagnosis and management of neuroendocrine tumors: well-differentiated neuroendocrine tumors of the Jejunum, Ileum, Appendix, and Cecum. Pancreas. 2010 Aug;39(6):753–66. doi: 10.1097/MPA.0b013e3181ebb2a5. [DOI] [PubMed] [Google Scholar]
- 12.Liu EH, Solorzano CC, Katznelson L, Vinik AI, Wong R, Randolph G. AACE/ACE disease state clinical review: diagnosis and management of midgut carcinoids. Endocr Pract. 2015;21:534–545. doi: 10.4158/EP14464.DSC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.O’Toole D, Kianmanesh R, Caplin M. ENETS 2016 consensus guidelines for the management of patients with digestive neuroendocrine tumors: an update. Neuroendocrinology. 2016;103(2):117–118. doi: 10.1159/000443169. [DOI] [PubMed] [Google Scholar]
- 14.Kulke MH, Shah MH, Benson AB, 3rd, Bergsland E, Berlin JD, Blaszkowsky LS, Emerson L, et al. Neuroendocrine tumors, version 1. 2015. J Natl Compr Canc Netw. 2015;13(1):78–108. doi: 10.6004/jnccn.2015.0011. [DOI] [PubMed] [Google Scholar]
- 15.Kunz PL, Reidy-Lagunes D, Anthony LB, Bertino EM, Brendto K, Chan JA, Chen H, et al. Consensus guidelines for the management and treatment of neuroendocrine tumors. Pancreas. 2013;42(4):557–577. doi: 10.1097/MPA.0b013e31828e34a4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Crocetti E, Paci E. Malignant carcinoids in the USA, SEER 1992–1999. An epidemiological study with 6830 cases. Eur J Cancer Prev. 2003;12:191–4. doi: 10.1097/00008469-200306000-00004. [DOI] [PubMed] [Google Scholar]
- 17.Ganim RB, Norton JA. Recent advances in carcinoid pathogenesis, diagnosis and management. Surgical Oncol. 2000;9:173–9. doi: 10.1016/s0960-7404(01)00007-x. [DOI] [PubMed] [Google Scholar]
- 18.Koch CA, Azumi N, Furlong MA, Jha RC, Kehoe TE, Trowbridge CH, O’Dorisio TM, Chrousos GP, Clement SC. Carcinoid syndrome caused by an atypical carcinoid of the uterine cervix. J Clin Endocrinol Metab. 1999;84(11):4209–4213. doi: 10.1210/jcem.84.11.6126. [DOI] [PubMed] [Google Scholar]
- 19.Vinik AI, Gonin J, England BG, Jackson T, McLeod MK, Cho K. Plasma substance-P in neuroendocrine tumors and idiopathic flushing: the value of pentagastrin stimulation tests and the effects of somatostatin analog. J Clin Endocrinol Metab. 1990;70(6):1702–9. doi: 10.1210/jcem-70-6-1702. [DOI] [PubMed] [Google Scholar]
- 20.Balks HJ, Conlon JM, Creutzfeldt W, Stockmann F. Circulating bradykinin-like immunoreactivity and the pentagastrin-induced carcinoid flush. Clin Endocrinol. 1988;29(2):141–151. doi: 10.1111/j.1365-2265.1988.tb00256.x. [DOI] [PubMed] [Google Scholar]
- 21.Parson HK, Harati H, Cooper D, Vinik AI. Role of prostaglandin D2 and the autonomous nervous system in niacin-induced flushing. J Diabetes. 2013;5(1):59–67. doi: 10.1111/j.1753-0407.2012.00216.x. [DOI] [PubMed] [Google Scholar]
- 22.Belch JJ, Small M, McKenzie F, Hill PA, Lowe GD, McIntyre DE, Forbes CD, Prentice CR. DDAVP stimulates prostacyclin production. Thromb Haemost. 1982 Apr 30;47(2):122–3. [PubMed] [Google Scholar]
- 23.Feldman JM, O’Dorisio TM. Role of neuropeptides and serotonin in the diagnosis of carcinoid tumors. The American journal of medicine. 1986;81:41–8. doi: 10.1016/0002-9343(86)90583-8. [DOI] [PubMed] [Google Scholar]
- 24.Eriksson B, Oberg K, Stridsberg M. Tumor markers in neuroendocrine tumors. Digestion. 2000;62(Suppl 1):33–8. doi: 10.1159/000051853. [DOI] [PubMed] [Google Scholar]
- 25.Nehar D, Lombard-Bohas C, Olivieri S, et al. Interest of Chromogranin A for diagnosis and follow-up of endocrine tumours. Clinical Endocrinol. 2004;60:644–52. doi: 10.1111/j.1365-2265.2004.02030.x. [DOI] [PubMed] [Google Scholar]
- 26.Ruszniewski P, Ish-Shalom S, Wymenga M, O’Toole D, Arnold R, Tomassetti P, Bax N, et al. Rapid and sustained relief from the symptoms of carcinoid syndrome: results from an open 6-month study of the 28-day prolonged-release formulation of lanreotide. Neuroendocrinology. 2004;80:244–251. doi: 10.1159/000082875. [DOI] [PubMed] [Google Scholar]
- 27.Pokuri VK, Fong MK, Iyer R. Octreotide and lanreotide in gastroenteropancreatic neuroendocrine tumors. Curr Oncol Rep. 2016;18(1):7. doi: 10.1007/s11912-015-0492-7. [DOI] [PubMed] [Google Scholar]
- 28.Woltering EA, Wright AE, Stevens MA, Wang YZ, Boudreaux JP, Mamikunian G, Riopelle JM, Kaye AD. Development of effective prophylaxis against intraoperative carcinoid crisis. J Clin Anesth. 2016 Aug;32:189–93. doi: 10.1016/j.jclinane.2016.03.008. [DOI] [PubMed] [Google Scholar]
- 29.Lenders JW, Duh QY, Eisenhofer G, et al. Pheochromocytoma and paraganglioma: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. 2014;99:1915–42. doi: 10.1210/jc.2014-1498. [DOI] [PubMed] [Google Scholar]
- 30.Chen H, Sippel RS, O’Dorisio MS, et al. The North American Neuroendocrine Tumor Society consensus guideline for the diagnosis and management of neuroendocrine tumors: pheochromocytoma, paraganglioma, and medullary thyroid cancer. Pancreas. 2010;39:775–83. doi: 10.1097/MPA.0b013e3181ebb4f0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kopetschke R, Slisko M, Kilisli A, Tuschy U, Wallaschofski H, Fassnacht M, Ventz M, Beuschlein F, et al. Frequent incidental discovery of phaeochromocytoma: data from a German cohort of 201 phaeochromocytoma. Eur J Endocrinol. 2009;161(2):355–361. doi: 10.1530/EJE-09-0384. [DOI] [PubMed] [Google Scholar]
- 32.Plouin PF, Degoulet P, Tugaye A, Ducrocq MB, Menard J. Screening for phaeochromocytoma: in which hypertensive patients ? A semiological study of 2585 patients, including 11 with phaeochromocytoma. Nouv Presse Med. 1981;10(11):869–872. [PubMed] [Google Scholar]
- 33.Metz SA, Halter JB, Porte D, Jr, Robertson RP. Suppression of plasma catecholamines and flushing by clonidine in man. J Clin Endocrinol Metab. 1978;46:83–90. doi: 10.1210/jcem-46-1-83. [DOI] [PubMed] [Google Scholar]
- 34.McGuinness ME, Talbert RL. Pharmacologic stress testing: experience with dipyridamole, adenosine, and dobutamine. Am J Hosp Pharm. 1994;51:328–46. quiz 404–5. [PubMed] [Google Scholar]
- 35.Mouri T, Takahashi K, Sone M, et al. Calcitonin gene-related peptide-like immunoreactivities in pheochromocytomas. Peptides. 1989;10:201–4. doi: 10.1016/0196-9781(89)90097-1. [DOI] [PubMed] [Google Scholar]
- 36.Herrera MF, Stone E, Deitel M, Asa SL. Pheochromocytoma producing multiple vasoactive peptides. Arch Surg. 1992;127:105–8. doi: 10.1001/archsurg.1992.01420010123020. [DOI] [PubMed] [Google Scholar]
- 37.Smith SL, Slappy AL, Fox TP, Scolapio JS. Pheochromocytoma producing vasoactive intestinal peptide. Mayo Clin Proc. 2002;77:97–100. doi: 10.4065/77.1.97. [DOI] [PubMed] [Google Scholar]
- 38.Kitamura K, Kangawa K, Kawamoto M, Ichiki Y, Matsuo H, Eto T. Isolation and characterization of peptides which act on rat platelets, from a pheochromocytoma. Biochem Biophys Res Commun. 1992;185:134–41. doi: 10.1016/s0006-291x(05)80966-0. [DOI] [PubMed] [Google Scholar]
- 39.Letizia C, Rossi G, Cerci S. Adrenomedullin and endocrine disorders. Panminerva medica. 2003;45:241–51. [PubMed] [Google Scholar]
- 40.Nicholls MG, Lainchbury JG, Lewis LK, et al. Bioactivity of adrenomedullin and proadrenomedullin N-terminal 20 peptide in man. Peptides. 2001;22:1745–52. doi: 10.1016/s0196-9781(01)00508-3. [DOI] [PubMed] [Google Scholar]
- 41.Gläsker S, Neumann HPH, Koch CA, Vortmeyer AO. Von Hippel-Lindau Disease. In: De Groot LJ, Beck-Peccoz P, Chrousos G, Dungan K, Grossman A, Hershman JM, Koch C, McLachlan R, New M, Rebar R, Singer F, Vinik A, Weickert MO, editors. Endotext [Internet] Vol. 2000. South Dartmouth (MA): MDText.com, Inc; 2015. Jul 11, [Google Scholar]
- 42.Wells SA, Jr, Asa SL, Dralle H, et al. Revised American Thyroid Association guidelines for the management of medullary thyroid carcinoma. Thyroid. 2015;25:567–610. doi: 10.1089/thy.2014.0335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Koch CA. Molecular pathogenesis of MEN2-associated tumors. Fam Cancer. 2005;4(1):3–7. doi: 10.1007/s10689-004-7022-3. [DOI] [PubMed] [Google Scholar]
- 44.Brauer VF, Scholz GH, Neumann S, Lohmann T, Paschke R, Koch CA. RET germline mutation in codon 791 in a family representing 3 generations from age 5 to age 70 years: should thyroidectomy be performed? Endocr Pract. 2004 Jan-Feb;10(1):5–9. doi: 10.4158/EP.10.1.5. [DOI] [PubMed] [Google Scholar]
- 45.Lepage C, Bouvier AM, Phelip JM, Hatem C, Vernet C, Faivre J. Incidence and management of malignant digestive endocrine tumours in a well defined French population. Gut. 2004;53:549–53. doi: 10.1136/gut.2003.026401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Halfdanarson TR, Rabe KG, Rubin J, Petersen GM. Pancreatic neuroendocrine tumors (PNETs): incidence, prognosis and recent trend toward improved survival. Ann Oncol. 2008;19:1727–33. doi: 10.1093/annonc/mdn351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kimura W, Kuroda A, Morioka Y. Clinical pathology of endocrine tumors of the pancreas. Analysis of autopsy cases. Dig Dis Sci. 1991;36:933–42. doi: 10.1007/BF01297144. [DOI] [PubMed] [Google Scholar]
- 48.Miehle K, Tannapfel A, Lamesch P, Borte G, Schenker E, Kluge R, Ott RA, Wiechmann V, Koch M, Kassahun W, Paschke R, Koch CA. Pancreatic neuroendocrine tumor with ectopic adrenocorticotropin production upon second recurrence. J Clin Endocrinol Metab. 2004;89(8):3731–3736. doi: 10.1210/jc.2003-032164. [DOI] [PubMed] [Google Scholar]
- 49.Nilubol N, Freedman EM, Quezado MM, Patel D, Kebebew E. Pancreatic neuroendocrine tumor secreting vasoactive intestinal peptide and dopamine with pulmonary emboli: a case report. J Clin Endocrinol Metab. 2016 Oct;101(10):3564–3567. doi: 10.1210/jc.2016-2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Baur AD, Pavel M, Prasad V, Denecke T. Diagnostic imaging of pancreatic neuroendocrine neoplasms (pNEN): tumor detection, staging, prognosis, and response to treatment. Acta Radiol. 2016 Mar;57(3):260–70. doi: 10.1177/0284185115579932. [DOI] [PubMed] [Google Scholar]
- 51.Stratakis CA. Diagnosis and Clinical Genetics of Cushing Syndrome in Pediatrics. Endocrinol Clin N Am. 2016;45:311–28. doi: 10.1016/j.ecl.2016.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Robyn JA, Koch CA, Montalto J, Yong A, Warne GL, Batch JA. Cushing’s syndrome in childhood and adolescence. J Paediatr Child Health. 1997;33(6):522–527. doi: 10.1111/j.1440-1754.1997.tb01663.x. [DOI] [PubMed] [Google Scholar]
- 53.Lindholm J, Juul S, Jorgensen JO, et al. Incidence and late prognosis of cushing’s syndrome: a population-based study. J Clin Endocrinol Metab. 2001;86:117–23. doi: 10.1210/jcem.86.1.7093. [DOI] [PubMed] [Google Scholar]
- 54.Etxabe J, Vazquez JA. Morbidity and mortality in Cushing’s disease: an epidemiological approach. Clinical Endocrinol. 1994;40:479–84. doi: 10.1111/j.1365-2265.1994.tb02486.x. [DOI] [PubMed] [Google Scholar]
- 55.Afshari A, Ardeshirpour Y, Lodish MB, et al. Facial Plethora: Modern Technology for Quantifying an Ancient Clinical Sign and Its Use in Cushing Syndrome. J Clin Endocrinol Metab. 2015;100:3928–33. doi: 10.1210/jc.2015-2497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.McReynolds SM, Freidberg SR, Guay AT, Lee AK, Pazianos AG, Hussain SF. Hot flushes in men with pituitary adenoma. Surg Neurol. 1995;44:14–7. doi: 10.1016/0090-3019(95)00163-8. discussion 7–8. [DOI] [PubMed] [Google Scholar]
- 57.Myers BM, Miralles GD, Taylor CA, Gastineau DA, Pisani RJ, Talley NJ. POEMS syndrome with idiopathic flushing mimicking carcinoid syndrome. Am J Med. 1991;90:646–8. [PubMed] [Google Scholar]
- 58.Niepomniszcze H, Amad RH. Skin disorders and thyroid diseases. J Endocrinol Invest. 2001;24:628–38. doi: 10.1007/BF03343905. [DOI] [PubMed] [Google Scholar]
- 59.Zouboulis CC, Liakou AI. Images in clinical medicine. Flashing, pulsating angioma. N Engl J Med. 2012 Jun 14;366(24):e36. doi: 10.1056/NEJMicm1110901. [DOI] [PubMed] [Google Scholar]
- 60.Seo I, Bargo PR, Kollias N. Simultaneous assessment of pulsating and total blood in inflammatory skin lesions using functional diffuse reflectance spectroscopy in the visible range. J Biomed Opt. 2010;15(6):060507. doi: 10.1117/1.3524191. [DOI] [PubMed] [Google Scholar]
- 61.Sassarini J, Lumsden MA. Vascular function and cardiovascular risk factors in women with severe flushing. Maturitas. 2015;80:379–383. doi: 10.1016/j.maturitas.2015.01.007. [DOI] [PubMed] [Google Scholar]
- 62.Crandall CJ, Manson JE, Hohensee CM, Horvath S, Wactawski-Wende J, LeBlanc E, et al. Association of genetic variation in the tachykinin receptor 3 locus with hot flashes and night sweats in the Women’s Health Initiative Study. Menopause. doi: 10.1097/GME.0000000000000763. Published ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Krause MS, Nakaijma ST. Hormonal and nonhormonal treatment of vasomotor symptoms. Obstet Gyncecol Clin N Am. 2015;42:163–179. doi: 10.1016/j.ogc.2014.09.008. [DOI] [PubMed] [Google Scholar]
- 64.Stuenkel CA, Davis SR, Gompel A, Lumsden MA, Murad MH, Pinkerton JV, Santen RJ. Treatment of symptoms of the menopause: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. 2015 Nov;100(11):3975–4011. doi: 10.1210/jc.2015-2236. [DOI] [PubMed] [Google Scholar]