

Abstract
Histone deacetylase 8 (HDAC8) catalyzes the hydrolysis of acetyl-L-lysine to yield products L-lysine and acetate through a mechanism in which a nucleophilic water molecule is activated by a histidine general base and a catalytic metal ion (Zn2+ or Fe2+). Acetyl-L-lysine also requires activation by metal coordination and a hydrogen bond with catalytic tyrosine Y306, which also functions in transition state stabilization. Interestingly, Y306 is located in the conserved glycine-rich loop G302GGGY. The potential flexibility afforded by the tetraglycine segment may facilitate induced-fit conformational changes of Y306 between “in” and “out” positions, as observed in related deacetylases. To probe the catalytic importance of the glycine-rich loop in HDAC8, we rigidified this loop by preparing the G302A, G303A, G304A, and G305A mutants, and we measured their steady-state kinetics and determined their X-ray crystal structures. Substantial losses of catalytic efficiency are observed (10–500-fold based on kcat/KM), particularly for G304A HDAC8 and G305A HDAC8. These mutants also exhibit the greatest structural changes for catalytic tyrosine Y306 (1.3–1.7 Å shifts of the phenolic hydroxyl group). Molecular dynamics simulations further indicate that G304 and G305 undergo pronounced structural changes as residue 306 transitions between “in” and “out” conformations. Thus, the G304A and G305A substitutions likely compromise the position and conformational changes of Y306 required for substrate activation and transition state stabilization. The G302A and G303A substitutions have less severe catalytic consequences, and these substitutions may influence an internal channel through which product acetate is believed to exit.
Graphical abstract
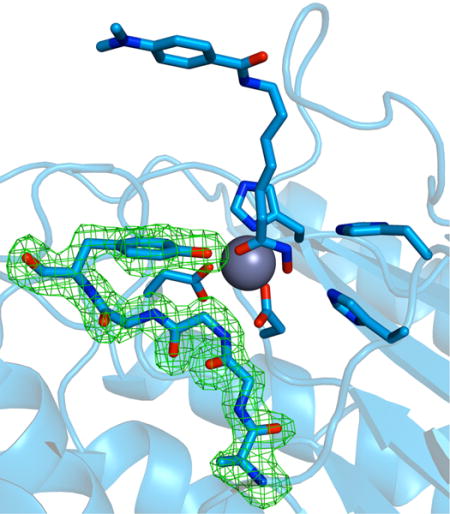
Introduction
Histone deacetylases (HDACs) catalyze the hydrolysis of posttranslationally modified acetyl-L-lysine side chains to yield acetate and free L-lysine side chains in histone and non-histone protein substrates.1–3 Notably, certain HDAC isozymes have been identified as critical targets for cancer chemotherapy due to their biological functions throughout the cell cycle.4–8 Class I, II, and IV HDACs are metal-dependent enzymes and require a divalent metal ion, either Zn2+ or Fe2+, for the chemistry of catalysis in vivo.9 The scissile carbonyl of acetyl-L-lysine coordinates to the metal ion and accepts a hydrogen bond from a catalytic tyrosine in the precatalytic enzyme-substrate complex;10,11 in human HDAC8, this catalytic tyrosine is Y306. Both of these interactions are required to polarize the carbonyl for nucleophilic attack by a metal-bound solvent molecule, and both interactions are important for transition state stabilization.9,12,13 Additionally, a tandem histidine pair is required for general acid-general base catalysis and transition state stabilization.12,13 In HDAC8, H143 serves both general base and general acid functions, with H142 serving as an electrostatic catalyst (Figure 1).13 In HDAC6, however, structural studies in which the intact substrate and tetrahedral intermediate are trapped in the active site suggest that the tandem histidine residues may serve separate general base and general acid functions.14
Figure 1.
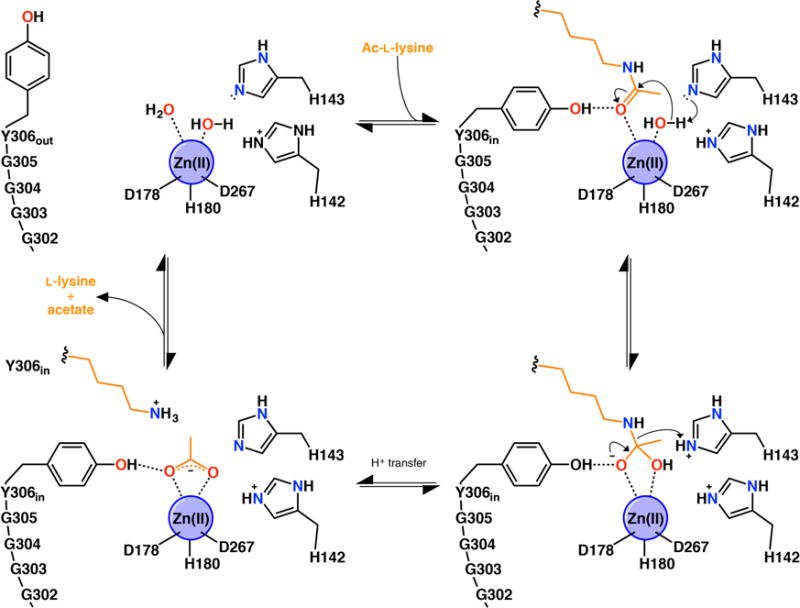
Summary of the HDAC8 mechanism, including possible induced-fit conformational changes of catalytic tyrosine Y306.
It has been suggested that the side chain of Y306 in HDAC8 may undergo conformational changes between “in” and “out” positions as part of an induced-fit substrate binding mechanism,15 reminiscent of the classic induced-fit mechanism of carboxypeptidase A.16 Even though crystal structures of wild-type HDAC8 reveal exclusively “in” conformations for Y306,17,18 the possibility of conformational flexibility for the catalytic tyrosine is suggested by experimental and computational studies of related deacetylases. For example, both “in” and “out” conformations of the catalytic tyrosine are observed in the crystal structure of H159A acetylpolyamine amidohydrolase (APAH) complexed with substrate N8-acetylspermidine.15 Additionally, the crystal structures of H976Y HDAC4-inhibitor complexes reveal “in” and “out” conformations for the newly-introduced catalytic tyrosine.19 Interestingly, molecular dynamics simulations of HDAC3 in the presence and absence of binding partners indicate conformational mobility for the catalytic tyrosine, which adopts a predominantly “out” conformation in the apo state and an “in” conformation upon activator binding.20 Finally, molecular dynamics simulations of certain HDAC8 mutants indicate modest conformational flexibility for Y306 involving ~2 Å shifts of the phenolic hydroxyl group.21 The “in” conformation of the catalytic tyrosine facilitates binding and activation of the scissile carbonyl of acetyl-L-lysine for hydrolysis, whereas the “out” conformation would facilitate dissociation of the deacetylated L-lysine product.
HDAC8 contains a glycine-rich loop, G302GGGY, immediately preceding catalytic tyrosine Y306 in the polypeptide segment between β-strand 8 and α-helix H1. This tetraglycine segment is conserved in class I deacetylases (HDAC1, HDAC2, HDAC3, and HDAC8) as well as the histone deacetylase-like protein from Aquifex aeolicus; moreover, G304 and G305 are strictly conserved in all metal-dependent HDACs and related deacetylases. In HDAC8, G302 and G304 adopt backbone conformations accessible to chiral L-amino acids, but G303 and G305 adopt backbone conformations accessible only to glycine. These conformations orient all of the glycine backbone carbonyl groups toward the protein interior. The carbonyl oxygens of G303 and G305 accept hydrogen bonds from R37, a crucial residue proposed to serve as a gatekeeper for an internal channel through which product acetate is believed to exit based on computational and enzymological studies.22,23 These hydrogen bonds, along with a van der Waals contact between the carbonyl of G139 and the amide nitrogen of G303, are believed to lock the channel gate closed.22 Additionally, a water molecule, referred to as the “bridging water molecule”, forms hydrogen bonds with R37, W137, G139, and G303 in wild-type HDAC8. Since opening the gate would require reorganization of this hydrogen bond network, it is possible that the potential flexibility afforded by the glycine-rich loop enables egress of the acetate product as well as dissociation of the L-lysine product.
To probe the functional significance of the glycine-rich loop in the active site of HDAC8, we rigidified this loop by preparing the G302A, G303A, G304A, and G305A mutants. We measured the catalytic activities of these mutants and determined their X-ray crystal structures. Surprisingly, these modest amino acid substitutions substantially compromise catalytic activity. Even though X-ray crystal structures of enzyme-inhibitor complexes reveal that Y306 predominantly adopts the “in” conformation required for inhibitor binding and catalysis, molecular dynamics (MD) simulations of HDAC8 indicate that residue 306 is capable of conformational transitions between “in” and “out” positions; the potential flexibility afforded by the glycine-rich G302GGGY loop may facilitate these transitions. Thus, rigidification of the glycine-rich loop may hinder conformational changes that facilitate substrate binding and product dissociation.
Materials and Methods
Reagents
Most chemicals used in buffers and crystallization were purchased from Fisher, Sigma-Aldrich, or Hampton Research and used without further purification.
Mutagenesis and protein preparation
The G302A, G303A, G304A, and G305A mutations were introduced into the human HDAC8-His6-pET20b construct11 using Quickchange site-directed mutagenesis (Agilent Technologies, Inc.). Forward and reverse primers were synthesized by Integrated DNA Technologies (Table 1). PCR plasmid products were sequenced to confirm mutations. HDAC8 constructs were expressed in BL21(DE3) Escherichia coli cells as previously described,11 with minor modifications. Briefly, 50 mL cultures (Lysogeny Broth (LB) at 100 μg/mL ampicillin) were grown overnight and used to inoculate 12 × 1-L M9 minimal media supplemented with 100 μg/mL ampicillin. Cells were grown at 37 °C until OD600 reached ~1.0, at which point the temperature was lowered to 16 °C. After cooling for 30 min, expression was induced with 100 μM isopropyl-β-thiogalactopyranoside and 100 μM ZnCl2. Protein was expressed overnight. Cells were pelleted via centrifugation and stored at −80 °C until purification. Wild-type and mutant constructs of human HDAC8 were purified as previously described.11 All protein was concentrated via centrifugation using 10k MWCO centrifugal filters (Millipore) to 10–20 mg/mL. Protein concentrations were determined from the absorbance at 280 nm using the calculated extinction coefficient 50,240 M−1cm−1. Aliquots were flash-cooled in liquid nitrogen and stored at −80 °C until further use.
Table 1.
PCR primers used to generate HDAC8 mutants
| Mutation | Direction | Nucleotide Sequencea |
|---|---|---|
| G302A | forward | 5′–CT ACC CTG ATC CTG GCT GGC GGT GGT TAC AAC–3′ |
| reverse | 5′–GTT GTA ACC ACC GCC AGC CAG GAT CAG GGT AG–3′ | |
| G303A | forward | 5′–CTG ATC CTG GGT GCC GGT GGT TAC AAC C–3′ |
| reverse | 5′–G GTT GTA ACC ACC GGC ACC CAG GAT CAG–3′ | |
| G304A | forward | 5′–G ATC CTG GGT GGC GCT GGT TAC AAC CT–3′ |
| reverse | 5′–CAG GTT GTA ACC AGC GCC ACC CAG GAT C–3′ | |
| G305A | forward | 5′–CTG GGT GGC GGT GCT TAC AAC CTG GCG AAC–3′ |
| reverse | 5′–GTT CGC CAG GTT GTA AGC ACC GCC ACC CAG–3′ |
Boldface letters indicate the introduced mutant codon.
Activity assays
Michaelis-Menten kinetics were measured using the Fluor de Lys HDAC8 tetrapeptide assay substrate Ac-Arg-His-Lys(Ac)-Lys(Ac)-aminomethylcoumarin (BML-KI178-0005, Enzo Life Sciences). Substrate deacetylation by HDAC8 was followed by trypsin cleavage of the amide bond with the 7-amino-4-methylcoumarin moiety. The resultant fluorescence shift was analyzed using a standard curve to quantify product generation. Assays were performed in triplicate at room temperature in 50 μL assay buffer [25 mM Tris (pH 8.2), 137 mM NaCl, 2.7 mM KCl, and 1 mM MgCl2] containing 100 μM–2 mM assay substrate with the following enzyme concentrations: 0.1 μM (wild-type HDAC8), 2 μM (G302A HDAC8, G303A HDAC8, and G305A HDAC8), or 10 μM (G304A HDAC8). The reaction was quenched after 5 min (wild-type HDAC8) or 20 min (all HDAC8 mutants) by the addition of 50 μL assay buffer containing trypsin and 200 μM of the hydroxamate inhibitor 4-(dimethylamino)-N-[7-(hydroxyamino)-7-oxoheptyl]-benzamide (M344). After 30 min, fluorescence was recorded using a Tecan Infinite M1000Pro plate reader (λex = 380 nm, λem = 480 nm). Data were analyzed using non-linear least-squares regression and kcat and KM were calculated based on the Michaelis-Menten equation, v0 = kcat[E][S] / (KM + [S]); v0 = initial rate, [E] = enzyme concentration, [S] = substrate concentration.24
Crystallization
All enzyme-inhibitor complexes were crystallized in sitting drops by the vapor diffusion method. Complexes with G302A HDAC8, G303A HDAC8, and G305A HDAC8 were crystallized at 21 °C, and the complex with G304A HDAC8 was crystallized at 4 °C.
For cocrystallization of the G302A HDAC8-M344 complex, a 500 nL drop containing 5 mg/mL G302A HDAC8, 50 mM Tris (pH 8.0), 150 mM KCl, 5% glycerol, 1 mM tris(2-carboxyethyl)phosphine (TCEP), 2 mM M344, and 30 mM glycylglycylglycine (GGG) was added to a 500 nL drop of precipitant solution [50 mM imidazole (pH 7.0), 15% (w/v) PEG 35,000 (Fluka), and 4 mM TCEP] and equilibrated against a 100 μL reservoir of precipitant solution. The use of GGG as a crystallization additive was first reported by Vannini and colleagues.17
For cocrystallization of the G303A HDAC8-M344 complex, a 2 μL drop containing 5 mg/mL G303A HDAC8, 50 mM Tris (pH 8.0), 150 mM KCl, 5% glycerol, 1 mM TCEP, 2 mM M344, and 30 mM GGG was added to a 2 μL drop of precipitant solution [100 mM HEPES (pH 7.5), 22% (w/v) PEG 6,000, and 4 mM TCEP] and equilibrated against a 1 mL reservoir of precipitant solution. Additionally, heterogeneous streak seeding was performed using a seed stock derived from crystals of the G304A HDAC8-M344 complex.
For cocrystallization of the G304A HDAC8-M344 complex, a 500 nL drop containing 5 mg/mL G304A HDAC8, 50 mM Tris (pH 8.0), 150 mM KCl, 5% glycerol, 1 mM TCEP, 2 mM M344, and 30 mM GGG was added to a 500 nL drop of precipitant solution [100 mM BisTris (pH 6.5), 10% (w/v) PEG 35,000 (Fluka), and 4 mM TCEP] and equilibrated against a 100 μL reservoir of precipitant solution.
For cocrystallization of the G305A HDAC8-M344 complex, a 2 μL drop containing 5 mg/mL G305A HDAC8, 50 mM Tris (pH 8.0), 150 mM KCl, 5% glycerol, 1 mM TCEP, 2 mM M344, and 30 mM GGG was added to a 2 μL drop of precipitant solution [100 mM BisTris (pH 6.5), 13% (w/v) PEG 4,000, and 4 mM TCEP] and equilibrated against a 1 mL reservoir of precipitant solution. Additionally, heterogeneous streak seeding was performed using a seed stock derived from crystals of the G304A HDAC8-M344 complex.
All crystals formed within 1–2 days and were soaked in a cryoprotectant solution comprised of mother liquor supplemented with 17% ethylene glycol (G302A HDAC8), 25% ethylene glycol (G303A HDAC8 and G305A HDAC8), or 25% glycerol (G304A HDAC8) prior to flash-cooling in liquid nitrogen.
Data collection and structure determination
X-ray diffraction data were collected on NE-CAT beamline 24-ID-E at the Advanced Photon Source, Argonne National Laboratory, from crystals of G302A HDAC8, G303A HDAC8, and G305A HDAC8 complexes with M344. X-ray diffraction data were collected on beamline 14-1 at the Stanford Synchrotron Radiation Lightsource from crystals of the G304A HDAC8-M344 complex. Data were integrated and indexed with iMosflm25 and scaled using Aimless in the CCP4 program suite.26 Data collection statistics are recorded in Table 2.
Table 2.
Crystallographic data collection and refinement statistics
| HDAC8 mutant | G302A | G303A | G304A | G305A |
|---|---|---|---|---|
|
Unit Cell
| ||||
| Space group | P21 | P21 | P21 | P21 |
| a, b, c (Å) | 53.1, 84.5, 94.6 | 97.7, 83.8, 99.8 | 53.5, 83.9, 98.4 | 53.0, 82.9, 94.6 |
| α,β, γ (°) | 90.0, 99.0, 90.0 | 90.0, 91.7, 90.0 | 90.0, 101.8, 90.0 | 90, 99.1, 90 |
|
Data Collection | ||||
| Wavelength (Å) | 0.97919 | 0.97923 | 1.18076 | 0.97923 |
| Resolution (Å) | 52.52–1.90 | 97.66–2.41 | 52.36–1.95 | 82.95–1.87 |
| Total/unique no. of reflections | 469621/65162 | 219182/62588 | 189166/62024 | 238947/65132 |
| Rmerge a,b | 0.141 (0.951) | 0.068 (0.313) | 0.078 (0.473) | 0.045 (0.306) |
| CC(1/2)a,c | 0.995 (0.745) | 0.996 (0.836) | 0.991 (0.785) | 0.998 (0.867) |
| I/σ (I)a | 9.8 (2.7) | 12.5 (3.5) | 8.0 (2.5) | 15.4 (3.5) |
| Redundancya | 7.3 (7.3) | 3.5 (2.9) | 3.0 (3.0) | 3.7 (3.1) |
| Completeness (%)a | 100 (100) | 99.9 (99.4) | 99.8 (100) | 97.2 (93.7) |
|
Refinement | ||||
| No. of reflections used in refinement/test set | 65134/3260 | 62558/2928 | 61982/3219 | 65094/6273 |
| Rworka,d | 0.176 (0.244) | 0.180 (0.223) | 0.175 (0.224) | 0.172 (0.231) |
| Rfreea,d | 0.208 (0.308) | 0.212 (0.281) | 0.200 (0.258) | 0.210 (0.283) |
| No. of nonhydrogen atoms: | ||||
| protein | 5649 | 11050 | 5534 | 5549 |
| ligand | 102 | 208 | 63 | 154 |
| solvent | 303 | 211 | 391 | 349 |
|
Average B-factors (Å2) |
||||
| protein | 23 | 26 | 29 | 29 |
| ligand | 30 | 37 | 40 | 35 |
| solvent | 28 | 24 | 34 | 34 |
|
Root-mean-square deviation from ideal geometry |
||||
| bonds (Å) | 0.007 | 0.003 | 0.006 | 0.006 |
| angles (°) | 0.84 | 0.51 | 0.80 | 0.74 |
| Ramachandran plot (%)e | ||||
| favored | 97.0 | 97.0 | 98.4 | 98.9 |
| allowed | 2.9 | 2.9 | 1.6 | 1.1 |
| generously allowed | 0.1 | 0.1 | 0.0 | 0.0 |
|
| ||||
| PDB accession code | 5THS | 5THT | 5THU | 5THV |
Values in parentheses refer to the data from the highest shell.
Rmerge = Σ|Ih − 〈I〉hI|/ΣIh, where 〈I〉h is the average intensity calculated for reflection h from replicate measurements.
Pearson correlation coefficient between random half-datasets.
Rwork = Σ||Fo| − |Fc||/Σ|Fo| for reflections contained in the working set. |Fo| and |Fc| are the observed and calculated structure factor amplitudes, respectively. Rfree is calculated using the same expression for reflections contained in the test set held aside during refinement.
Calculated with PROCHECK.31
All crystal structures were solved by molecular replacement using the atomic coordinates of H143A HDAC8 (PDB ID: 3EWF)11 as a search model for rotation and translation function calculations using the program Phaser.27 Model building was accomplished using the graphics program Coot28 and crystallographic refinement was executed using Phenix.29 The inhibitor M344 and water molecules were included in the later stages of refinement. Occasionally, spurious electron density peaks were observed that could not be satisfactorily modeled by ordered solvent, so these peaks were left uninterpreted. Such peaks possibly corresponded to partially disordered crystallization buffer components such as polyethylene glycol fragments. The quality of each final model was evaluated through MolProbity30 and PROCHECK.31 Final refinement statistics are recorded in Table 2.
Molecular Dynamics Simulations
The free, open-source program GROMACS (GROningen MAchine for Chemical Simulation)32–34 was utilized for molecular dynamics (MD) simulations of wild-type and Y306F HDAC8. The starting points for MD simulations were the crystal structures of wild-type HDAC8 (PDB accession code 3RQD)35 and Y306F HDAC8 (PDB accession code 2V5W).10 Protein structures were stripped of all water and inhibitor atoms and the pdb2gmx command was used to generate a Gromacs-compatible topology, position restraint, and structure file using the AMBER99SB-ILDN force field.36 Protein structures were placed in a cubic box of water molecules (protein surfaces were 10–17 Å away from box edges), ions were added to make the net charge of the system = 0, and the protein-solvent system was subject to energy minimization. To probe the effects of anion binding in the active site, simulations were run in the presence and absence of a chloride ion located in a potential acetate release channel near W141. All MD simulations were run for 10 ns with a step size of 2 fs; frames were generated every 2 ps to sample each trajectory.
Results
G302A HDAC8
Of the four glycine residues in the glycine-rich loop, G302 is most distant from the active site (Cα-Zn2+ distance = 9.6 Å). G302 appears as leucine in class IIa and IIb HDACs and threonine in the class IV enzyme, HDAC11. Of the four glycine-to-alanine HDAC8 mutants studied here, G302A HDAC8 retains the highest levels of catalytic activity and exhibits 13% residual activity based on kcat/KM (Table 3, Figure 2).
Table 3.
Steady-state kinetic parameters for HDAC8 mutants
| HDAC8 | kcat (s−1) | KM (μM) | kcat/KM (M−1s−1) | Rel. Efficiency (%) |
|---|---|---|---|---|
| wild-type | 0.50 ± 0.01 | 460 ± 40 | 1100 ± 100 | 100 |
| G302A | 0.030 ± 0.001 | 220 ± 40 | 140 ± 30 | 13 ± 1 |
| G303A | 0.020 ± 0.001 | 340 ± 50 | 60 ± 10 | 5.0 ± 0.8 |
| G304A | 0.0016 ± 0.0001 | 1000 ± 70 | 2.0 ± 0.2 | 0.20 ± 0.03 |
| G305A | 0.013 ± 0.001 | 680 ± 80 | 20 ± 3 | 2.0 ± 0.3 |
Figure 2.
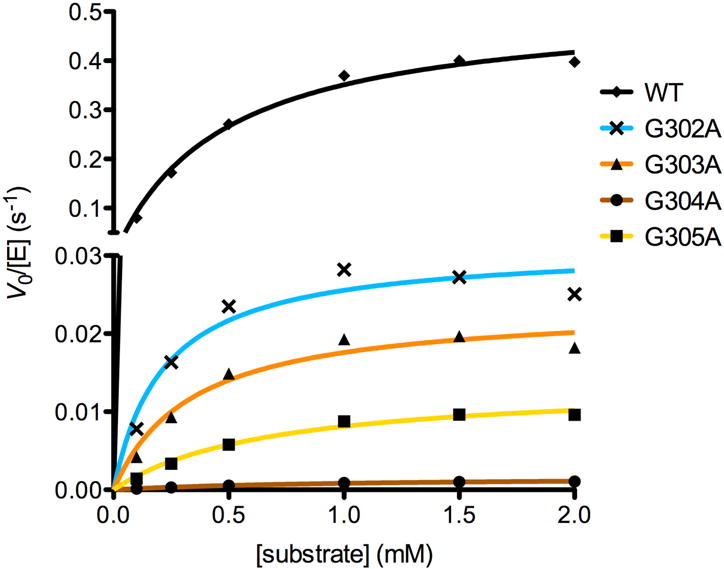
Steady state kinetics of wild-type and mutant HDAC8 enzymes. Activity was measured using the tetrapeptide substrate Arg-His-Lys(Ac)-Lys(Ac)-AMC (AMC = 7-amino-4-methylcoumarin). V0 = initial velocity, [E] = enzyme concentration. Error bars are present (mean ± s.e.m., n = 3) but are sufficiently small that they are masked by the data points.
The crystal structure of the G302A HDAC8-M344 complex was determined at 1.9 Å resolution (Figure 3a). The overall structure is similar to that of the wild-type HDAC8-M344 complex (PDB ID: 1T67)18 with a root-mean-square deviation (rmsd) of 0.51 Å for 357 Cα atoms and 0.55 Å for 352 Cα atoms for monomers A and B, respectively. The methyl side chain of A302 resides in a small hydrophobic pocket between W137 and W315. Steric interactions with the A302 methyl group cause a 1.3 Å shift in the backbone carbonyl of W137 (Figure 4). Since the backbone conformation of G302 in wild-type HDAC8 is close to the generally allowed region of the Ramachandran plot, the G302A substitution causes only subtle shifts in the ϕ and ψ angles of each residue in the loop (Figure 5). Additionally, the bridging water molecule that hydrogen bonds with R37, W137, G139, and G303 in wild-type HDAC8 shifts approximately 1.6 Å in G302A HDAC8 due to the shift in the backbone carbonyl of W137. Consequently, the bridging water molecule only forms hydrogen bonds with R37 and G139. It also shares a hydrogen bond with another water molecule that in turn forms a hydrogen bond with the backbone carbonyl of S138.
Figure 3.
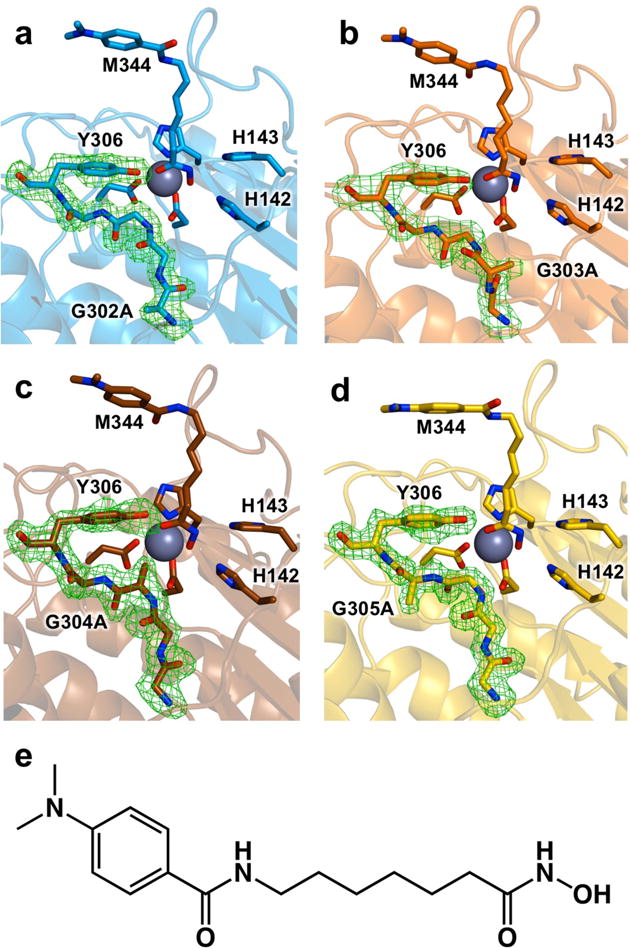
Simulated annealing omit maps of the glycine-rich loop contoured at 3.0σ in the crystal structures of (a) G302A HDAC8, (b) G303A HDAC8, (c) G304A HDAC8, and (d) G305A HDAC8 complexed with the hydroxamate inhibitor M344 (e). For reference, metal-binding residues, M344, and catalytic residues H142, H143, and Y306 are indicated. The catalytic Zn2+ ion is a large grey sphere.
Figure 4.
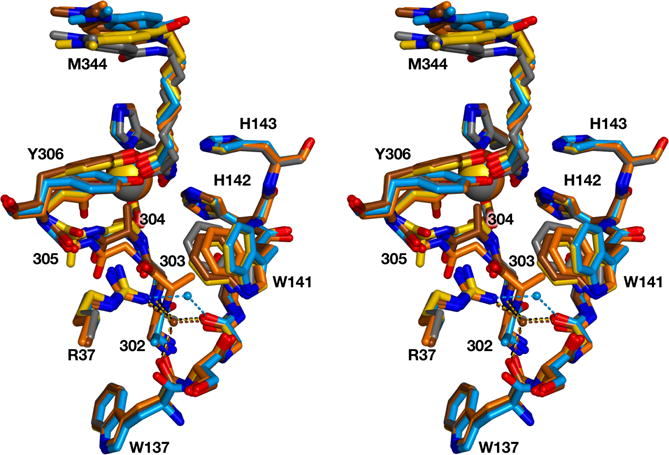
Stereoview of the glycine-rich loop of each structure superimposed on that of wild-type HDAC8 complexed with M344 (grey; PDB ID: 1T67). Each structure is color-coded as in Figure 3: G302A HDAC8, blue; G303A HDAC8, orange; G304A HDAC8, brown; G305A HDAC8, yellow. For reference, R37 and the W137–W141 segment are also illustrated, and the catalytic Zn2+ ion is a large sphere. The bridging water molecule near R37 is shown as a small sphere and hydrogen bonds are shown as dotted lines.
Figure 5.
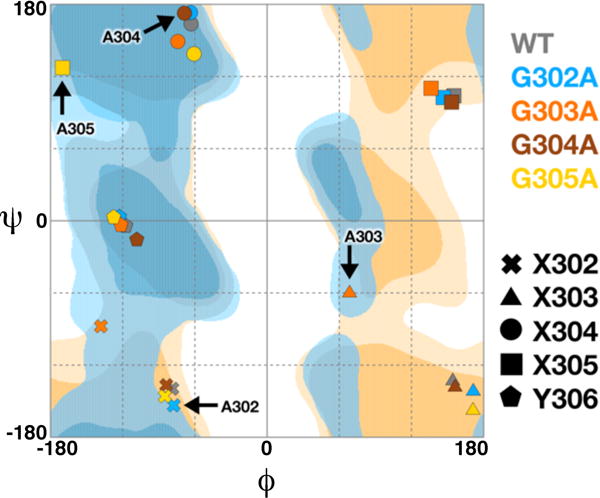
Ramachandran plot for the glycine-rich loop G302GGGY in wild-type and mutant HDAC8 enzymes. Symbols are defined in the key and colors are consistent with those in Figures 3 and 4: G302A HDAC8, blue; G303A HDAC8, orange; G304A HDAC8, brown; G305A HDAC8, yellow. Regions of the Ramachandran plot are indicated as follows: generally favored (dark blue), generally allowed (light blue), glycine favored (dark yellow), and glycine allowed (light yellow). Plot generated using RAMPAGE.43
Eleven ethylene glycol molecules are observed in the structure of G302A HDAC8, as this was the cryoprotectant utilized in data collection. In both monomers, the side chain of W141 is in the “out” conformation with flanking ethylene glycol molecule binding on each side of the indole ring. Another ethylene glycol molecule is observed in a large internal pocket connected to the acetate release channel in monomer B, where it donates a hydrogen bond to the backbone carbonyl of W137 and accepts a hydrogen bond from R37. Insofar that the binding of ethylene glycol (C2H6O2, Mr = 62.07) could mimic the binding of product acetate (C2H3O2−, Mr = 59.04), it is notable that ethylene glycol binds in the acetate release channel. The L1 loop (residues S30–K36) adjacent to the exit portal of the acetate release channel is generally not well-defined in electron density maps. However, satisfactory electron density is observed for the side chain of L31 which packs against Y111, thereby forcing Y111 into the “out” conformation previously described by Dowling and colleagues.11
G303A HDAC8
G303 is closer to the active site (Cα-Zn2+ distance = 6.2 Å) and appears as glutamate in class IIa and IIb HDACs and serine in HDAC11. G303A HDAC8 exhibits 5% residual catalytic activity compared with wild-type HDAC8 based on kcat/KM (Table 3, Figure 2).
The crystal structure of the G303A HDAC8-M344 complex was determined at 2.4 Å resolution (Figure 3b). There is very little overall difference between this structure and the structure of the wild-type HDAC8-M344 complex, since monomers A, B, C, and D exhibit rmsd values ranging from 0.55 Å–0.58 Å for 352–357 Cα atoms. The introduction of the A303 methyl group causes the most significant structural changes in the glycine-rich loop of all the mutants studied (Figure 4). In the structure of wild-type HDAC8, the backbone dihedral angles of G303 are accessible only to glycine and not a chiral L-amino acid. Accordingly, the backbone conformation of A303 in G303A HDAC8 changes to accommodate L-alanine at this position (Figure 5). This conformational change also propagates to adjacent residue G302. As a result, G139 appears to make stronger interactions with the backbone amides of G302 and A303. However, G139 and A303 are still poorly oriented for hydrogen bonding, despite an O—N separation of 2.9 Å; in fact, the A303 amide is more favorably oriented to donate a bond to the backbone carbonyl of W137 with an N—O separation of 3.1 Å. Hydrogen bond interactions among R37, A303, and G305 are also significantly shorter in G303A HDAC8. Additionally, the A303 side chain sterically displaces the bridging water molecule, which is completely absent in this structure.
The overall conformational change of the glycine-rich loop triggers a 1.3 Å shift in the side chain of W141, which also regulates access to the acetate release channel37 (Figure 4). Nevertheless, W141 still adopts a predominantly “in” conformation in all four monomers despite the newly introduced steric bulk of A303. On the opposite side of the W141 indole ring from A303, an ethylene glycol molecule resides in the acetate release channel. A total of 27 ethylene glycol molecules are observed in this structure, most of which interact at the protein surface. The only ethylene glycol molecules observed on the protein interior are those that pack against the indole ring of W141.
G304A HDAC8
G304 is strictly conserved in all metal-dependent deacetylases and it is closest to the active site (Cα-Zn2+ distance = 3.9 Å). G304A HDAC8 exhibits the most severely compromised activity of all mutants studied, retaining only 0.2% residual activity compared with wild-type HDAC8 based on kcat/KM (Table 3, Figure 2). This represents a 500-fold loss of catalytic efficiency, which is comparable to the 150-fold loss of catalytic efficiency observed for Y306F HDAC8 in which the phenolic hydroxyl group is deleted altogether from the aromatic side chain at position 306.13
The crystal structure of the G304A HDAC8-M344 complex was determined at 1.95 Å resolution (Figure 3c). Overall, the structure of this mutant is generally similar to that of the wild-type HDAC8-M344 complex, with rmsd values of 0.44 Å and 0.43 Å for 357 and 346 Cα atoms in monomers A and B, respectively. The methyl group of A304 protrudes into the active site and is 3.4 Å away from the catalytic Zn2+ ion. Compared to the wild-type HDAC8 structure, the Zn2+ ion shifts 0.5 Å and the M344 C=O—Zn2+ coordination interaction lengthens by 0.4 Å. The conformation of the Y306 side chain is slightly altered in G304A HDAC8, such that the phenolic hydroxyl group shifts 1.7 Å relative to its position in wild-type HDAC8 (Figure 4). All of the hydrogen bond interactions in the acetate release channel are similar to those observed in the wild-type enzyme, and the bridging water molecule remains in place with its hydrogen bonds to R37, W137, G139, and G303 intact. Despite the fact that G304A HDAC8 is the most severely catalytically compromised among all the mutants studied, the backbone conformations of the G302GGGY loop in G304A HDAC8 are most similar to those of wild-type HDAC8 (Figure 4). However, G304A HDAC8 also exhibits the greatest conformational shift for Y306 among all four mutants.
Glycerol was used for cryoprotection of this crystalline mutant rather than ethylene glycol, but no ordered glycerol molecules are observed in the structure. The internal cavities and acetate release channel of G304A HDAC8 most closely resemble those of the wild-type enzyme in comparison with all mutants studied.
G305A HDAC8
G305 is strictly conserved in all metal-dependent deacetylases and is immediately adjacent to catalytic tyrosine Y306. The Cα-Zn2+ distance of 6.7 Å for G305 in wild-type HDAC8 indicates that this residue is approximately equidistant from the active site in comparison with G303. G305A HDAC8 exhibits 2% residual activity compared with wild-type HDAC8 based on kcat/KM (Table 3, Figure 2).
The crystal structure of the G305A HDAC8-M344 complex was determined at 1.87 Å resolution (Figure 3d). There is very little difference between the global structures of G305A HDAC8 and wild-type HDAC8 as indicated by rmsd values of 0.53 Å and 0.52 Å for 348 and 345 Cα atoms in monomers A and B, respectively. The A305 side chain protrudes into a small pocket formed by T311 and W315, the former of which shifts slightly to accommodate the newly introduced methyl group. The G305A substitution pushes the glycine loop closer to the active site, which triggers a 1.4 Å shift in the position of the phenolic hydroxyl group of Y306 (Figure 4). Backbone conformations are slightly different for all residues in the glycine-rich loop, but the greatest deviations are observed for G304 and A305 since, in wild-type HDAC8, G305 adopts torsion angles that could only accommodate glycine and not a chiral L-amino acid (Figure 5). Therefore the introduction of L-alanine at this position necessitates a substantial shift to a generally allowed conformation. The bridging water remains in place, making hydrogen bond interactions with R37, W137, G139, and G303.
Intriguingly, the side chain of W141 adopts both “in” and “out” conformations (each with approximately 50% occupancy), in both monomers of G305A HDAC8. In monomer A, two ethylene glycol molecules flank W141, one on the outer side of the indole ring (i.e., in the acetate release channel), and one closer to the active site with an occupancy of 0.66. There are also two ethylene glycol molecules bound in the large internal pocket, near the binding site of the ethylene glycol molecule observed here in the structure of the G302A HDAC8–M344 complex. The more deeply located ethylene glycol molecule in the pocket forms hydrogen bonds with the phenolic oxygen of Y18 and the backbone carbonyl of A38, while the second ethylene glycol molecule in the pocket interacts with solvent. In monomer B, only one ethylene glycol molecule binds against the indole ring face of W141. A total of 26 ethylene glycol molecules are observed in this structure.
Molecular dynamics of the glycine-rich loop
The 10-ns MD trajectories of wild-type HDAC8 and Y306F HDAC8 do not reveal substantial conformational mobility for Y306. However, the 10-ns MD trajectory of Y306F HDAC8 containing a chloride ion in the acetate release channel near W141 reveals that F306 oscillates between “in” and “out” conformations (Figure 6). Possibly, anion binding in or passage through the channel weakens interactions that otherwise stabilize the “in” conformation of residue 306. These results suggest that residue 306 of HDAC8 is capable of making conformational transitions between “in” and “out” positions, as observed in crystallographic and computational studies of related zinc deacetylases.15,19,20 The side chain χ1 angle in Y306F HDAC8 oscillates within a ~110° range in these simulations, which compares well with ~120° conformational changes observed for the catalytic tyrosine in crystal structures of the polyamine deacetylase APAH15 and H976Y HDAC4,19 respectively. Thus, the agreement between independent computational and experimental studies strongly suggests that the full range of motion for the catalytic tyrosine is reflected in MD simulations of HDAC8.
Figure 6.
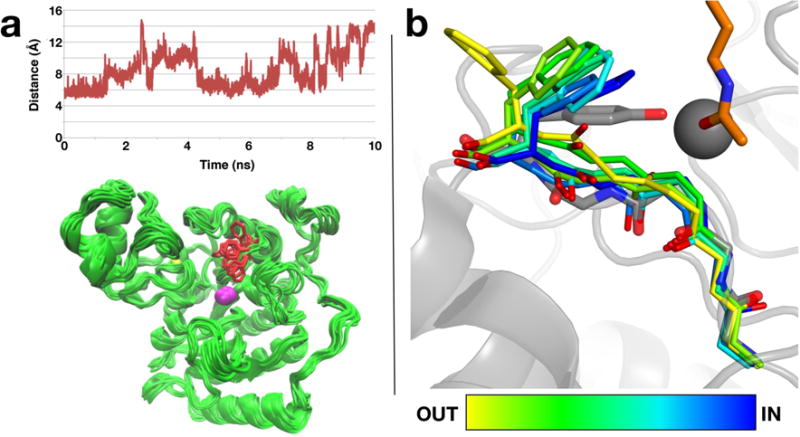
(a) Time-dependent changes in the Cζ-Zn2+ distance in a 10-ns MD simulation of Y306F HDAC8. The side chain of residue 306 oscillates between “in” and “out” conformations in the absence of any bound ligands in the enzyme active site. (b) Superposition of intermediate conformations sampled in one of the conformational transitions of F306 HDAC8 in the 10-ns MD simulation (the orientation differs from that shown in (a)). Conformations are colored from yellow to blue according to their position between the “out” and “in” conformations. The conformation of Y306 in the substrate bound state in H143A HDAC8 (PDB ID: 3EWF) is shown in grey. The active site zinc ion is shown as a grey sphere and the substrate acetyllysine is shown in orange.
Discussion
The current study demonstrates that the glycine-rich loop preceding catalytic tyrosine Y306 is vital for efficient HDAC8 catalysis. Introduction of the minimal steric bulk of a methyl group in place of any glycine hydrogen atom in this loop substantially compromises catalytic efficiency with approximately 10–500-fold reductions in kcat/KM (Figure 2, Table 3). These glycine-to-alanine substitutions could influence the postulated interconversion of Y306 between the “in” and “out” conformations, consistent with an induced-fit model of enzyme catalysis. Moreover, even a slight 1–2 Å shift of Y306 could perturb its hydrogen bond with the scissile carbonyl of the acetyllysine substrate and the developing oxyanion of the tetrahedral intermediate, which in turn would compromise substrate activation and transition state stabilization. Accordingly, it is notable that in the most severely compromised mutants, G304A HDAC8 and G305A HDAC8, the Y306 hydroxyl group shifts 1.7 Å and 1.3 Å, respectively, from its position in wild-type HDAC8. In contrast, the Y306 hydroxyl group shifts only 0.3 Å in G302A HDAC8 and G303A HDAC8, and these mutants retain higher levels of catalytic activity.
Our results suggest that the first pair of glycine residues in the G302GGGY loop may influence catalytic function differently in comparison with the second pair of glycine residues. The first pair of glycine residues, G302 and G303, are conserved in class I HDACs but are not strictly conserved in other HDACs and related deacetylases. Thus, while these glycine residues presumably afford some flexibility to the glycine-rich loop, they are not universally required for catalytic function. In wild-type HDAC8, G303 forms a hydrogen bond with the bridging water molecule in the acetate release channel, which also forms hydrogen bonds with R37, W137, and G139. In both G302A HDAC8 and G303A HDAC8, the position of this water molecule is perturbed and the hydrogen bond network is reorganized. Thus, the detrimental effects on catalysis observed for the G302A and G303A mutants may arise from structural changes in the acetate release channel.
In contrast, there are fewer structural changes in the acetate release channel in G304A HDAC8 and G305A HDAC8; indeed, the hydrogen bond network between the bridging water molecule and R37, W137, G139, and G303 is preserved in both mutants. Instead, the primary structural change common to the structures of both of these mutants is a slight (~1.3–1.7 Å) outward movement of the Y306 side chain compared to its position in the wild-type enzyme. This is consistent with the hypothesis that G304 and G305, which are strictly conserved in all metal-dependent deacetylases, influence the structural dynamics of catalysis: these residues may confer flexibility to the polypeptide backbone that enables the interconversion of Y306 between “in” and “out” conformations. If so, this could facilitate substrate association and product dissociation through an induced-fit mechanism. Snapshots of the molecular dynamics trajectory (Figure 6) show that the G304–G305 segment is more conformationally mobile than the G302–G303 segment when residue 306 transitions between “in” and “out” conformations. Thus, the rigidification of the G302GGGY loop with glycine-to-alanine substitutions may increase the free energy barrier for conformational changes of Y306 required for catalysis.
Interestingly, the first mutation observed in the glycine-rich loop of HDAC8, G304R, was identified through genomic sequencing data acquired from a child diagnosed with Cornelia de Lange Syndrome.38–41 The G304R substitution renders HDAC8 catalytically inactive.21,39 Molecular modeling and MD simulations of G304R HDAC8 indicate that the bulky R304 side chain sterically blocks the acetyllysine binding groove, thereby preventing substrate and inhibitor binding.21 Additionally, MD simulations show that the G304R substitution causes the side chain of Y306 to move approximately 2 Å away from the “in” conformation required for catalysis. The movement of Y306 predicted by MD simulations of G304R HDAC8 is consistent with the crystal structure of G304A HDAC8, in which Y306 shifts approximately 2 Å away from the “in” conformation even though the substrate binding groove is not blocked (Figure 3). G304A HDAC8 also exhibits the lowest activity of all the mutants studied (Table 3, Figure 2), suggesting that the flexibility afforded by G304 is critical for catalysis.
In addition to the conformational flexibility of the G302GGGY loop, the current work highlights conformational transitions for other aromatic residues in the HDAC8 active site. The “in” and “out” conformations of W141 and the “out” conformation of Y111 observed in the structure of the G302A HDAC8–M344 complex were previously observed in the structure of the D101L HDAC8–M344 complex.11 Conformational mobility for W141 is implied by alternate positions observed in the crystal structure of G305A HDAC8 (Figure 3), as well as a slight shift triggered by the newly introduced methyl group in G303A HDAC8 (Figure 4). Notably, a substantial conformational change for W141 is observed in the crystal structure of C153F HDAC8, identified in a child diagnosed with Cornelia de Lange Syndrome.37,39 Here, the steric bulk of the F153 side chain locks W141 in the “in” conformation, thereby blocking the acetate release channel and accounting for a 50-fold loss of catalytic activity. Thus, conformational changes of W141 may additionally compromise catalysis in G303A HDAC8 due to the blockage or partial blockage of the acetate release channel.
It is notable that the crystal structures of G302A HDAC8, G303A HDAC8, and G305A HDAC8 are the first HDAC8 structures to be determined using ethylene glycol as a cryoprotectant, because each of these structures reveals a number of ethylene glycol molecules bound in the acetate release channel. Insofar that the binding sites observed for ethylene glycol could represent possible binding sites of the acetate anion, potential trajectories of acetate binding and egress from the HDAC8 active site are illuminated. For example, W141 is sandwiched by two ethylene glycol molecules in the structure of G302A HDAC8, consistent with the proposed role of this residue as a channel gate. A second ethylene glycol molecule interacts with R37 and the backbone carbonyl of W137. In the structure of G303A HDAC8, only one ethylene glycol molecule is observed adjacent to W141, on the outer face of the indole side chain. Additionally, ethylene glycol molecules bind on both sides of W141 in G305A HDAC8, and W141 additionally adopts two conformations. Finally, two ethylene glycol molecules bind in the large internal cavity of G305A HDAC8. Thus, among these glycine loop mutants, ethylene glycol molecules bind to four different sites in the acetate release channel and an adjacent internal cavity (Figure 7). Interestingly, potential acetate trajectories suggested by ethylene glycol binding sites are in general agreement with acetate trajectories observed in recent MD simulations.42
Figure 7.
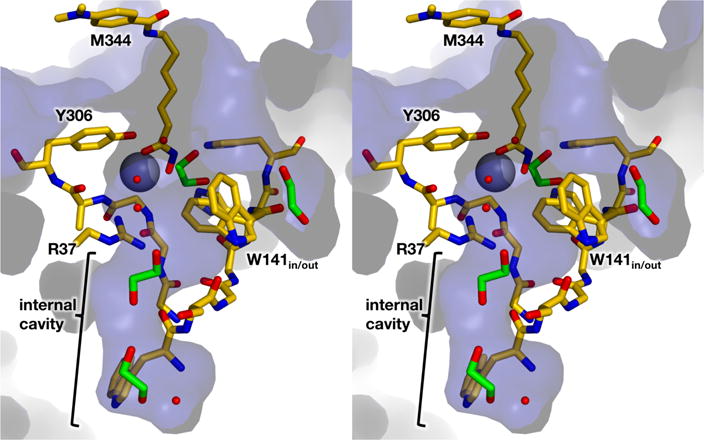
Solvent-accessible surface (blue) of the G305A HDAC8-M344 complex (C = yellow, N = blue, O = red) showing M344, ethylene glycol molecules (C = green), and solvent molecules (small red spheres). Internal cavities and channels accommodate four ethylene glycol molecules and solvent molecules; these cavities and channels could similarly accommodate the acetate product.
Finally, the crystal structure of the high-activity catalytic domain (CD2) of human HDAC6 has recently been reported,14 which allows for the direct structural comparison of class IIb and class I human HDACs. Notable conformational differences are observed between the L778EGGY loop of HDAC6 CD2 and the G302GGGY loop of HDAC8, and between the L3 loops of HDAC6 CD2 (V605RPPGHH, conserved in all class II HDACs) and HDAC8 (W137SGGWHH; the L3 loops contain tandem histidine residues required for catalysis) (Figure 8). Hydrogen bond interactions between the L3 loop and the glycine-rich loop also differ between HDAC6 and HDAC8. Curiously, the gate residue of the acetate release channel, W141 in HDAC8, is not conserved in HDAC6 CD2. Moreover, there are fewer internal pockets and channels in the structure of HDAC6 CD2, so this isozyme may lack an acetate release channel altogether. Although R37 is conserved as R506 in HDAC6 CD2, making similar contacts to the loop preceding the catalytic tyrosine as in HDAC8, it does not appear to serve as a channel gate in HDAC6 CD2, as proposed for HDAC8.22 Instead, R506 simply stabilizes the conformation of the glycine-rich loop of HDAC6 CD2 through hydrogen bonding. In doing so, this conserved arginine may simply function to stabilize the position of the catalytic tyrosine.
Figure 8.
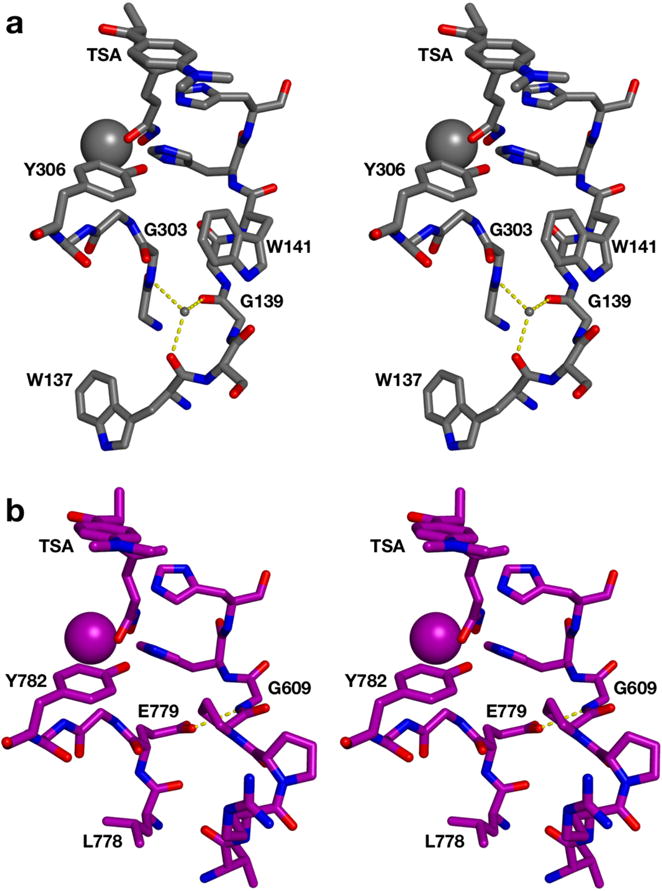
Stereo view of (a) the L3 loop and the glycine-rich loop (including the catalytic tyrosine) in HDAC8 (C = grey, N = blue, O = red; PDB 1T64) and (b) the corresponding loops in human HDAC6 (C = purple; PDB 5EDU). Active site zinc ions are large spheres and the bridging water molecule in the internal cavity of HDAC8 is a small sphere. Both enzymes are complexed with the hydroxamate inhibitor TSA. Hydrogen bonds are indicated by dashed yellow lines. Hydrogen bonds with the bridging water molecule link the L3 loop and the glycine-rich loop in HDAC8. In contrast, there is no bridging water molecule in HDAC6, but the loops are linked by a hydrogen bond between the side chain of E779 and the backbone NH group of G609.
Summary and Conclusions
In conclusion, site-directed mutagenesis, steady-state kinetics, molecular dynamics simulations, and X-ray crystal structures establish the structural and functional importance of the conserved glycine-rich loop G302GGGY in the active site of HDAC8. The catalytic importance of this loop was first suspected following the identification of catalytically-inactive G304R HDAC8 in a child diagnosed with Cornelia de Lange Syndrome.37,39 We show that glycine residues are required in the G302GGGY loop to hold Y306 in the proper position required for catalysis, since individual glycine-to-alanine substitutions cause up to a 1.7 Å shift in the position of the Y306 hydroxyl group and significantly compromise catalysis. The backbone conformations of G303 and G305 are only accessible to glycine, so the substitution of these residues with a chiral L-amino acid requires backbone conformational changes. Additionally, MD simulations indicate that all four glycines, but especially G304 and G305, confer flexibility on the G302GGGY segment that would enable the transition of Y306 between “in” and “out” conformations (only the “in” conformation is catalytically competent). Conformational mobility for the catalytic tyrosine in related deacetylases has been observed in structural and computational studies,15,19–21 so this mobility may reflect an induced-fit substrate binding mechanism as well as a general strategy for the regulation of catalysis in this enzyme family. For example, the conformational transition of the catalytic tyrosine in HDAC3 is modulated by protein binding partners,20 and it is possible that cellular HDAC8 activity could be regulated in similar fashion. Future work in our laboratory will continue to probe and clarify these structure-function relationships in HDAC8 and related deacetylases.
Acknowledgments
For access to synchrotron data collection facilities, we thank the Northeastern Collaborative Access Team beamlines, which are funded by NIH grant P41 GM103403. This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated by Argonne National Laboratory under contract DE-AC02-06CH11357. Additionally, we thank the NSLS-SSRL user transition program supported jointly by the LSBR, NSLSII and SMB, SSRL under NIH grants P41GM111244 and P41GM103393, and DOE BER contract DE-SC0012704. SSRL is operated under DOE BES contract DE-AC02-76SF00515.
Funding
Supported by National Institutes of Health (NIH) grant GM49758. N.J.P was supported by NIH Structural Biology and Molecular Biophysics Training Grant T32 GM008275.
Abbreviations
- APAH
acetylpolyamine amidohydrolase
- HDAC
histone deacetylase
- M344
4-(dimethylamino)-N-[7-(hydroxyamino)-7-oxoheptyl]-benzamide
- MD
molecular dynamics
- rmsd
root-mean-square deviation
- TCEP
tris(2-carboxyethyl)phosphine
Footnotes
Accession Codes
The atomic coordinates and structure factors of G302A HDAC8, G303A HDAC8, G304A HDAC8, and G305A HDAC8 have been deposited in the Protein Data Bank (www.rcsb.org) with accession codes 5THS, 5THT, 5THU, and 5THV, respectively.
Notes
The authors declare no competing financial interests.
References
- 1.De Ruijter AJM, Van Gennip AH, Caron HN, Kemp S, Van Kuilenburg ABP. Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem J. 2003;370:737–749. doi: 10.1042/BJ20021321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lombardi PM, Cole KE, Dowling DP, Christianson DW. Structure, mechanism, and inhibition of histone deacetylases and related metalloenzymes. Curr Op Struct Biol. 2011;21:735–743. doi: 10.1016/j.sbi.2011.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wolfson NA, Pitcairn CA, Fierke CA. HDAC8 substrates: Histones and beyond. Biopolymers. 2013;99:112–126. doi: 10.1002/bip.22135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Haberland M, Montgomery RL, Olson EN. The many roles of histone deacetylases in development and physiology: Implications for disease and therapy. Nat Rev Genet. 2009;10:32–42. doi: 10.1038/nrg2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Emanuele S, Lauricella M, Tesoriere G. Histone deacetylase inhibitors: Apoptotic effects and clinical implications. Int J Oncol. 2008;33:637–646. [PubMed] [Google Scholar]
- 6.Marks PA. Histone deacetylase inhibitors: A chemical genetics approach to understanding cellular functions. Biochim Biophys Acta. 2010;1799:717–725. doi: 10.1016/j.bbagrm.2010.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.New M, Olzscha H, La Thangue NB. HDAC inhibitor-based therapies: Can we interpret the code? Mol Oncol. 2012;6:637–656. doi: 10.1016/j.molonc.2012.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Delcuve GP, Khan DH, Davie JR. Roles of histone deacetylases in epigenetic regulation: Emerging paradigms from studies with inhibitors. Clin Epigenet. 2012;4:5. doi: 10.1186/1868-7083-4-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gantt SL, Gattis SG, Fierke CA. Catalytic activity and inhibition of human histone deacetylase 8 is dependent on the identity of the active site metal ion. Biochemistry. 2006;45:6170–6178. doi: 10.1021/bi060212u. [DOI] [PubMed] [Google Scholar]
- 10.Vannini A, Volpari C, Galinari P, Jones P, Mattu M, Carfí A, De Francesco R, Steinkühler C, Di Marco S. Substrate binding to histone deacetylases as shown by the crystal structure of the HDAC8-substrate complex. EMBO Rep. 2007;8:879–884. doi: 10.1038/sj.embor.7401047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dowling DP, Gantt SL, Gattis SG, Fierke CA, Christianson DW. Structural studies of human histone deacetylase 8 and its site-specific variants complexed with substrate and inhibitors. Biochemistry. 2008;47:13554–13563. doi: 10.1021/bi801610c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gantt SL, Joseph CG, Fierke CA. Activation and inhibition of histone deacetylase 8 by monovalent cations. J Biol Chem. 2010;285:6036–6043. doi: 10.1074/jbc.M109.033399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gantt SL, Decroos C, Lee MS, Gullet LE, Bowman CM, Christianson DW, Fierke CA. General base-general acid catalysis in human histone deacetylase 8. Biochemistry. 2016;55:820–832. doi: 10.1021/acs.biochem.5b01327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hai Y, Christianson DW. Histone deacetylase 6 structure and molecular basis of catalysis and inhibition. Nat Chem Bio. 2016;12:741–747. doi: 10.1038/nchembio.2134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lombardi PM, Angell HD, Whittington DA, Flynn EF, Rajashankar KR, Christianson DW. Structure of a prokaryotic polyamine deacetylase reveals evolutionary functional relationships with eukaryotic histone deacetylases. Biochemistry. 2011;50:1808–1817. doi: 10.1021/bi101859k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Christianson DW, Lipscomb WN. Carboxypeptidase A. Acc Chem Res. 1989;22:62–69. [Google Scholar]
- 17.Vannini A, Volpari C, Filocamo G, Casavola EC, Brunetti M, Renzoni D, Chakravarty P, Paolini C, De Francesco R, Gallinari P, Steinkühler C, Di Marco S. Crystal structure of a eukaryotic zinc-dependent histone deacetylase, human HDAC8, complexed with a hydroxamic acid inhibitor. Proc Natl Acad Sci USA. 2004;101:15064–15069. doi: 10.1073/pnas.0404603101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Somoza JR, Skene RJ, Katz BA, Mol C, Ho JD, Jennings AJ, Luong C, Arvai A, Buggy JJ, Chi E, Tang J, Sang BC, Verner E, Wynands R, Leahy EM, Dougan DR, Snell G, Navre M, Knuth MW, Swanson RV, McRee DE, Tari LW. Structural snapshots of human HDAC8 provide insights into the class I histone deacetylases. Structure. 2004;12:1325–1334. doi: 10.1016/j.str.2004.04.012. [DOI] [PubMed] [Google Scholar]
- 19.Bottomley MJ, Lo Surdo P, Di Giovine P, Cirillo A, Scarpelli R, Ferrigno F, Jones P, Nedderman P, De Francesco R, Steinkühler C, Gallinari P, Carfí A. Structural and functional analysis of the human HDAC4 catalytic domain reveals a regulatory structural zinc-binding domain. J Biol Chem. 2008;283:26694–26704. doi: 10.1074/jbc.M803514200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Arrar M, Turnham R, Pierce L, de Oliviera CAF, McCammon JA. Structural insight into the separate roles of inositol tetraphosphate and deacetylase-activating domain in activation of histone deacetylase 3. Prot Sci. 2013;22:83–92. doi: 10.1002/pro.2190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Decroos C, Christianson NH, Gullet LE, Bowman CM, Christianson KE, Deardorff MA, Christianson DW. Biochemical and structural characterization of HDAC8 mutants associated with Cornelia de Lange Syndrome spectrum disorders. Biochemistry. 2015;54:6501–6513. doi: 10.1021/acs.biochem.5b00881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Haider S, Joseph CG, Neidle S, Fierke CA, Fuchter MJ. On the function of the internal cavity of histone deacetylase protein 8: R37 is a crucial residue for catalysis. Bioorg Med Chem Lett. 2011;21:2129–2132. doi: 10.1016/j.bmcl.2011.01.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang DF, Wiest O, Helquist P, Lan-Hargest HY, Wiech NL. On the function of the 14 Å long internal cavity of histone deacetylase-like protein: implications for the design of histone deacetylase inhibitors. J Med Chem. 2004;47:3409–3417. doi: 10.1021/jm0498497. [DOI] [PubMed] [Google Scholar]
- 24.Michaelis L, Menten ML. Die Kinetik der Invertinwirkung. Biochem Z. 1913;49:333–369. [Google Scholar]
- 25.Battye TGG, Kontogiannis L, Johnson O, Powell HR, Leslie AGW. iMosflm: a new graphical interface for diffraction-image processing with Mosflm. Acta Crystallogr D Biol Crystallogr. 2011;67:271–281. doi: 10.1107/S0907444910048675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AGW, McCoy A, McNicholas SJ, Murshudov GN, Pannu NS, Potterton EA, Powell HR, Read RJ, Vagin A, Wilson KS. Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr. 2011;67:235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Cryst. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung L, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwillinger TC, Zwart PH. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen VB, Arendal WB, III, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: A program to check the stereochemical quality of protein structures. J Appl Cryst. 1993;26:283–291. [Google Scholar]
- 32.Berendsen HJC, van der Spoel D, van Drunen R. GROMACS: A message-passing parallel molecular dynamics implementation. Comput Phys Commun. 1995;91:43–56. [Google Scholar]
- 33.van der Spoel D, Lindahl E, Hess B, Groenhof G, Mark AE, Berendsen HJ. GROMACS: Fast, flexible, and free. J Comput Chem. 2005;26:1701–1718. doi: 10.1002/jcc.20291. [DOI] [PubMed] [Google Scholar]
- 34.Pronk S, Páll S, Schulz R, Larsson P, Bjelkmar P, Apostolov R, Shirts MR, Smith JC, Kasson PM, van der Spoel D, Hess B, Lindahl E. GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics. 2013;29:845–854. doi: 10.1093/bioinformatics/btt055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cole KE, Dowling DP, Boone MA, Phillips AJ, Christianson DW. Structural basis of the antiproliferative activity of largazole, a depsipeptide inhibitor of the histone deacetylases. J Am Chem Soc. 2011;133:12474–12477. doi: 10.1021/ja205972n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lindorff-Larsen K, Piana S, Palmo K, Maragakis P, Klepeis JL, Dror RO, Shaw DE. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins: Struct, Funct, Bioinf. 2010;78:1950–1958. doi: 10.1002/prot.22711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Decroos C, Bowman CM, Moser JS, Christianson KE, Deardorff MA, Christianson DW. Compromised structure and function of HDAC8 mutants identified in Cornelia de Lange Syndrome spectrum disorders. ACS Chem Biol. 2014;9:2157–2164. doi: 10.1021/cb5003762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Deardorff MA, Bando M, Nakato R, Watrin E, Itoh T, Minamino M, Saitoh K, Komata M, Katou Y, Clark D, Cole KE, De Baere E, Decroos C, Di Donato N, Ernst S, Francey LJ, Gyftodimou Y, Hirashima K, Hullings M, Ishikawa Y, Jaulin C, Kaur M, Kiyono T, Lombardi PM, Magnaghi-Jaulin L, Mortier GR, Nozaki N, Petersen MB, Seimiya H, Siu VM, Suzuki Y, Takagaki K, Wilde JJ, Willems PJ, Prigent C, Gillessen-Kaesbach G, Christianson DW, Kaiser FJ, Jackson LG, Hirota T, Krantz ID, Shirahige K. HDAC8 mutations in Cornelia de Lange syndrome affect the cohesin acetylation cycle. Nature. 2012;489:313–317. doi: 10.1038/nature11316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kaiser FJ, Ansari M, Braunholz D, Gil-Rodríguez MC, Decroos C, Wilde JJ, Fincher CT, Kaur M, Bando M, Amor DJ, Atwal PS, Bahlo M, Bowman CM, Bradley JJ, Brunner HG, Clark D, Del Campo M, Di Donato N, Diakumis P, Dubbs H, Dyment DA, Eckhold J, Ernst S, Ferreira JC, Francey LJ, Gehlken U, Guillen-Navarro E, Gyftodimou Y, Hall BD, Hennekam R, Hudgins L, Hullings M, Hunter JM, Yntema H, Innes AM, Kline AD, Krumina Z, Lee H, Leppig K, Lynch SA, Mallozzi MB, Mannini L, McKee S, Mehta SG, Micule I, Care4Rare Canada Consortium. Mohammed S, Moran E, Mortier GR, Moser J-AS, Noon SE, Nozaki N, Nunes L, Pappas JG, Penney LS, Pérez-Aytés A, Petersen MB, Puisac B, Revencu N, Roeder E, Saitta S, Scheuerle AE, Schindeler KL, Siu VM, Stark Z, Strom SP, Thiese H, Vater I, Willems P, Williamson K, Wilson LC, University of Washington Center for Mendelian Genomics. Hakonarson H, Quintero-Rivera F, Wierzba J, Musio A, Gillessen-Kaesbach G, Ramos FJ, Jackson LG, Shirahige K, Pie J, Christianson DW, Krantz ID, Fitzpatrick DR, Deardorff MA. Loss of function HDAC8 mutations cause a phenotypic spectrum of Cornelia de Lange Syndrome-like features, ocular hypertelorism, large fontanelle and X-linked inheritance. Hum Mol Genet. 2014;23:2888–2900. doi: 10.1093/hmg/ddu002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Parenti I, Gervasini C, Pozojevic J, Wendt KS, Watrin E, Azzollini J, Braunholz D, Buiting K, Cereda A, Engels H, Garavelli L, Glazar R, Graffmann B, Larizza L, Lüdecke HJ, Mariani M, Masciadri M, Pié J, Ramos FJ, Russo S, Selicorni A, Stefanova M, Strom TM, Werner D, Wierzba J, Zampino G, Gillessen-Kaesbach G, Wieczorek D, Kaiser FJ. Expanding the clinical spectrum of the “HDAC8-phenotype” – implications for molecular diagnostics, counseling and risk prediction. Clin Genet. 2016;89:564–573. doi: 10.1111/cge.12717. [DOI] [PubMed] [Google Scholar]
- 41.Deardorff MA, Porter NJ, Christianson DW. Structural aspects of HDAC8 mechanism and dysfunction in Cornelia de Lange Syndrome spectrum disorders. Prot Sci. 2016;25:1965–1976. doi: 10.1002/pro.3030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pietra F. Restoring histone deacetylase activity by waste product release. A view from molecular mechanics simulations with mammalian HDAC. Chem Biodivers. 2015;12:503–512. doi: 10.1002/cbdv.201400314. [DOI] [PubMed] [Google Scholar]
- 43.Lovell SC, Davis IW, Arendall WB, III, de Bakker PIW, Word JM, Prisant MG, Richardson JS, Richardson DC. Structure validation by Cα geometry: ϕ, ψ, and Cβ deviation. Proteins: Struct, Funct, Genet. 2002;50:437–450. doi: 10.1002/prot.10286. [DOI] [PubMed] [Google Scholar]