

Abstract
Life-sustaining responses to low oxygen, or hypoxia, depend on signal transduction by HIFs, but the underlying mechanisms by which cells sense hypoxia are not completely understood. Based on prior studies suggesting a link between the β-adrenergic receptor (β-AR) and hypoxia responses, we hypothesized that the β-AR mediates hypoxia sensing and is necessary for HIF-1α accumulation. Beta blocker treatment of mice suppressed hypoxia induction of renal HIF-1α accumulation, erythropoietin production, and erythropoiesis in vivo. Likewise, beta blocker treatment of primary human endothelial cells in vitro decreased hypoxia-mediated HIF-1α accumulation and binding to target genes and the downstream hypoxia-inducible gene expression. In mechanistic studies, cAMP-activated PKA and/or GPCR kinases (GRK), which both participate in β-AR signal transduction, were investigated. Direct activation of cAMP/PKA pathways did not induce HIF-1α accumulation, and inhibition of PKA did not blunt HIF-1α induction by hypoxia. In contrast, pharmacological inhibition of GRK, or expression of a GRK phosphorylation–deficient β-AR mutant in cells, blocked hypoxia-mediated HIF-1α accumulation. Mass spectrometry–based quantitative analyses revealed a hypoxia-mediated β-AR phosphorylation barcode that was different from the classical agonist phosphorylation barcode. These findings indicate that the β-AR is fundamental to the molecular and physiological responses to hypoxia.
Hypoxia activates a unique βAR phosphorylation barcode that allows for distinct downstream signals, which are essential for HIF-1α accumulation.
Introduction
Oxygen delivery to tissues is essential for life. Under hypoxia, adaptive responses take place over the course of hours and days to generate erythrocytes and create new blood vessels. These responses have been identified as the downstream effects of HIFs (1–4). HIF-1 is a master transcription factor for oxygen sensing. It is composed of an oxygen-regulated α subunit (HIF-1α) and a constitutively expressed β subunit (HIF-1β) (5–7). Under hypoxia, HIF-1α is stabilized, dimerizes with HIF-1β, and translocates to the nucleus to promote transcription of genes, such as erythropoietin (EPO) and vascular endothelial growth factors (VEGF), to orchestrate greater capacity of oxygen delivery to tissues (1). However, the determinants of HIF-1α regulation are incompletely understood in the complex physiologic environment of cells (8–10).
More than 40 years ago, long before the discovery of HIF and erythropoietin, β-adrenergic receptor (β-AR) blockade was shown to diminish the erythropoietic response to hypoxia in animal models, but the mechanisms of these effects have been unexplored (11, 12). Since that time, there have been a wealth of studies that have detailed the downstream signal transduction events that result from activation of the β-ARs. β-ARs belong to a large family of GPCRs. β-ARs are ubiquitously expressed throughout the body, with β1-AR as the major subtype expressed in the heart and β2-AR in the lung, vascular endothelium, and kidneys (13–15). When an agonist, such as the endogenous ligands noradrenaline and adrenaline, binds to β-AR, the receptor is stabilized in an active conformation and couples to the heterotrimeric G proteins, resulting in dissociation of Gα and Gβγ subunits. These molecular events rapidly propagate complex intracellular signals. For instance, the dissociation of the Gα subunit initiates the release of cAMP-dependent PKA (16), while Gβγ subunits recruit GPCR kinases (GRKs). Although both PKA and GRK participate in β-AR phosphorylation, the level of phosphorylation at distinct PKA and GRK sites depends on the type and concentrations of agonists (17).
Phosphorylation ultimately diminishes β-AR responsiveness to agonist stimulation, also known as receptor desensitization, and modulates receptor trafficking and turnover. The diminished responsiveness to an overload of noradrenaline is the cardinal feature of congestive heart failure (18–20). Effective therapies with beta blockers, such as carvedilol, have resulted in this class of drug being widely prescribed for left and right heart failure (21, 22). Elevation of HIF-1α levels is also implicated in the pathophysiology of the failing heart (23). We hypothesized that the β-AR is part of the pathway for hypoxia sensing and necessary for HIF-1α accumulation. We tested this hypothesis using in vivo models; genomic, pharmacologic, and molecular approaches; and detailed barcoding of the phosphorylation status of the β-AR.
Results
Decreased hypoxia responses in mice with beta blocker treatment.
To determine whether β-AR blockade prevents hypoxia-mediated erythropoietic responses via HIF-1α in vivo, mice were orally administered vehicle or the beta blocker carvedilol, followed by exposure to hypoxia (10% oxygen) (Figure 1A). Under hypoxia, HIF-1α accumulated in kidneys and was significantly decreased by carvedilol (Figure 1B). In addition, HIF-1α–specific binding to the Epo gene increased and was blocked by carvedilol (Figure 1C). Moreover, hypoxia increased erythropoietin mRNA and protein (24), both of which were suppressed by β-AR blockade (Figure 1, D and E). Since erythropoietin is known to induce the proliferation of erythroid progenitor cells (25), we quantified the percentages of proerythroblasts and basophilic and polychromatic erythroblasts in the bone marrow with flow cytometry (26). Under hypoxia, the erythroid progenitor cell population increased by 35%, and the effect was blunted by carvedilol (Figure 1F). These findings indicate that β-AR blockade by carvedilol suppresses the classical hypoxia erythropoietic responses through HIF-1α.
Figure 1. Beta blocker blunts HIF-mediated erythropoiesis under hypoxia in vivo.

(A) Mice were orally administered vehicle or carvedilol (beta blocker), followed by exposure to 21% or 10% oxygen. Kidneys and serum were harvested after 2 hours, and bone marrow was collected at 24 hours (n = 5/group). (B) Expression of HIF-1α and Lamin B in nuclear extracts of kidneys, as detected by Western blot. Samples were run on the same gel but were noncontiguous. (C) HIF-1α occupancy at the erythropoietin gene Epo, detected by ChIP with control IgG or HIF-1α antibody and quantitative PCR. Fold enrichment was percentage of input (treatment) divided by average percentage of input (normoxia). (D) Expression of Epo and Actin mRNA in kidneys, as detected by reverse-transcription quantitative PCR. (E) Serum erythropoietin by ELISA. (F) Flow cytometry for erythroid progenitor populations. Proerythroblasts (I) were defined by CD44hiTER119lo and basophilic (II) and polychromatic erythroblasts (III) were defined by TER119hi as well as size and CD44 expression. Data are mean ± SD. *P < 0.05; **P < 0.005, ANOVA.
Decreased HIF responses in hypoxic cells with beta blocker treatment.
Next, we tested the in vitro effects of β-AR blockade on HIF-1α accumulation in hypoxic human endothelial cells isolated from umbilical cord veins (HUVECs). HUVECs were exposed to escalating doses of propranolol and then exposed to hypoxia (2% oxygen). Western blot analyses of nuclear proteins revealed that hypoxia increased HIF-1α levels when compared with normoxia and propranolol dose-dependently decreased HIF-1α (Figure 2A). To confirm β-AR blockade inhibitory effects on HIF-1α, ChIP studies were performed to test whether β-AR blockade decreases the binding of HIF-1α to the promoters of its classical target genes, such as chemokine stromal cell–derived factor-1 (SDF-1 or CXCL12), hexokinase 2 (HK2), and VEGFA. Quantitative PCR revealed that hypoxia significantly increased HIF-1α–specific binding to promoter sequences, an effect that was blocked by propranolol (Figure 2B).
Figure 2. Beta blocker attenuates hypoxia responses in vitro.

(A) Expression of HIF-1α and Lamin B in HUVECs treated with escalating doses of propranolol, followed by 5-hour hypoxia (2% oxygen) (n = 2–7/condition). Data are mean ± SEM. *P < 0.05, Student’s t test. (B) HIF-1α occupancy at target genes in HUVECs treated with diluent or propranolol, followed by 12-hour hypoxia. ChIP with control IgG or HIF-1α antibody and quantification of promoter regions of CXCL12, HK2, and VEGFA by quantitative PCR. Fold enrichment was percentage of input (hypoxia with or without beta blocker) divided by average percentage of input (normoxia) (n = 3). *P < 0.05, ANOVA. (C) Heatmap showing the levels of differentially expressed transcripts of HUVECs treated with diluent (–) or propranolol (+), followed by 24-hour hypoxia. The levels of expression relative to normoxia are represented on a continuous scale from blue (lowest) to pink (highest) (n = 5 biological replicates). FDR = 0.05. (D) Kyoto Encyclopedia of Genes and Genomes pathway analysis of transcripts reversed by propranolol under hypoxia in HUVECs. Enrichment score is computed by –log (P value).
To evaluate the entire spectrum of signaling pathways affected by beta blockers under hypoxia, we performed transcriptome-profiling studies that tested 47,000 probes representing well-characterized genes, gene candidates, and splice variants. HUVECs were exposed to diluent or propranolol alone and then exposed to 24-hour hypoxia (data are accessible at GEO, accession GSE86793). Hypoxia induced changes in RNA expression levels relative to normoxia, corresponding to 1,247 probes, 434 of which were reversed by propranolol (at a FDR of 0.05) (Figure 2C and Supplemental Table 1). Pathway analyses and functional annotation clustering of the identified transcripts revealed the biological significance of β-AR in regulating hypoxia-inducible phenotypes, including focal adhesion, glycolysis, p53 signaling pathways, angiogenesis, and cell migration (Figure 2D, Supplemental Table 2, and refs. 27, 28).
β-Agonist increases HIF-1α accumulation under normoxia.
To investigate if β-AR activation can increase HIF-1α levels, dose-response and time-course studies with the β-agonist isoproterenol were performed in HUVECs under normoxia. Isoproterenol increased nuclear HIF-1α in a dose- and time-dependent manner, with a maximum response at 2 hours (Figure 3, A and B). To test the possibility that stabilization of HIF-1α is mediated via a specific β-AR subtype (β1-AR or β2-AR), HUVECs were exposed to CGP-20712A (a beta 1 blocker) or ICI-118551 (a beta 2 blocker), followed by isoproterenol stimulation. Isoproterenol stabilization of HIF-1α was dose-dependently decreased by both beta 1 and beta 2 blockers (Figure 3C). This suggests that isoproterenol stabilization of HIF-1α can be mediated by β1-AR or β2-AR and excludes off-target effects.
Figure 3. β-Agonist promotes HIF-1α accumulation under normoxia.

(A) Expression of HIF-1α and Lamin B in HUVECs treated with diluent or isoproterenol (33–900 μM) for 2 hours (n = 3–6/condition). Data are mean ± SEM. (B) Expression of HIF-1α and Lamin B in HUVECs treated with 300 μM isoproterenol for 0.5 to 24 hours (n = 3–4/condition). Data are mean ± SD. (C) Expression of HIF-1α and Lamin B in HUVECs treated with beta 1 blocker CGP-20712A or beta 2 blocker ICI-118551 (1–1,000 nM) for 45 minutes, followed by 2-hour isoproterenol (n = 3). Data are mean ± SD. *P < 0.05; **P < 0.005; ***P < 0.0005, ANOVA.
HIF-1α accumulation under hypoxia relies on GRK phosphorylation of β-AR.
To establish the mechanistic link between β-AR and HIF-1α, we investigated classical downstream signal pathways of β-ARs. Agonist binding to β-AR stabilizes the receptor in an active conformation and leads to coupling to the heterotrimeric G proteins. This results in production of cAMP by adenylate cyclase that activates PKA, which, in turn, phosphorylates the β-AR (13). To test if hypoxia activates the β-AR canonical downstream signal transduction, we measured the intracellular cAMP level in HUVECs exposed to hypoxia (2% oxygen) for 10 to 15 minutes. Similar to isoproterenol, forskolin (which activates adenylate cyclase), and dibutyryl (a cAMP analog), hypoxia increased cAMP levels (Figure 4A). However, forskolin and dibutyryl did not lead to HIF-1α accumulation (Figure 4B). Further, the PKA inhibitor H89 did not affect HIF-1α accumulation by isoproterenol (Figure 4B). Thus, hypoxia can activate β-AR signaling, but activation of the PKA/cAMP pathway does not lead to HIF-1α accumulation.
Figure 4. Hypoxia or β-agonist–mediated HIF-1α accumulation depends on phosphorylation of β-adrenergic receptor (β-AR) by GPCR kinase (GRK).

(A) Intracellular cAMP in HUVECs 10–15 minutes after stimulation with factors shown (n = 3–6). (B) Expression of HIF-1α and Lamin B in HUVECs treated with 300 μM β-agonist isoproterenol with or without 10 μM of the PKA inhibitor H89 or forskolin or dibutyryl cAMP (dbcAMP) for 2 hours. Cobalt chloride (CoCl2) shown as positive control (n = 3). (C) Expression of HIF-1α and Lamin B in HUVECs exposed to GRK inhibitor (1–125 μM) for 45 minutes followed by 2-hour isoproterenol. The blot is representative of 2 independent experiments. (D) Expression of HIF-1α and Lamin B in HUVECs exposed to a GRK (125 μM) or PKA inhibitor H89 (10 μM), followed by 5-hour hypoxia (2% oxygen) (n = 3). (E) Map of β-AR serine and threonine residues that are phosphorylated by PKA and GRK, mutated to alanine. (F) Expression of HIF-1α and Lamin B in human embryonic kidney cells (HEK293) overexpressing β1-AR (WTβ1-AR) or mutants lacking PKA (PKA–β1-AR) or GRK (GRK–β1-AR) sites, as shown in E, exposed to normoxia or 5-hour hypoxia (n = 3). Data are mean ± SD. *P < 0.05; **P < 0.005; ***P < 0.0005, Student’s t test.
GRKs are the other classical pathway known to phosphorylate β-ARs following receptor activation (13, 29–31). Evaluation of the GRK phosphorylation pathway using pharmacological inhibition of GRK2 in HUVECs revealed a dose-dependent decrease in β-agonist–mediated HIF-1α accumulation (Figure 4C). Likewise, GRK inhibition, but not PKA, significantly attenuated hypoxia-induced HIF-1α levels (Figure 4D). The results suggest that hypoxia-mediated HIF-1α accumulation is dependent on GRK phosphorylation of β-AR. To test this idea, we evaluated the hypoxia response of human embryonic kidney cells (HEK293), which overexpress wild-type β1-AR (WTβ1-AR) or mutant β1-ARs lacking either the phosphorylation target sites of PKA (PKA–β1-AR) or GRK (GRK–β1-AR) (Figure 4E and ref. 32). Hypoxia significantly induced HIF-1α levels in WTβ1-AR and PKA–β1-AR cells but not in GRK–β1-AR cells (Figure 4F). These results indicate that HIF-1α accumulation occurs in response to hypoxia in a manner that requires β-AR–GRK signal transduction.
Hypoxia-specific β-AR phosphorylation barcode.
Because agonist binding to β-AR results in unique receptor phosphorylation patterns that correlate with distinct downstream signals (33), we investigated whether hypoxia imparts a specific receptor phosphorylation signature, i.e., a unique barcode. HEK293 cells overexpressing β2-AR were exposed to the β-agonist isoproterenol (10 μM) or hypoxia (2% oxygen). β2-ARs from the plasma membranes were enriched via binding to alprenolol and subjected to mass spectrometry analyses, which successfully identified 8 key phospho-peptide species of the receptor (Table 1 and Supplemental Table 3).
Table 1. Quantitative analyses of β2-AR phosphorylation sites.

Similar to canonical phosphorylation seen with isoproterenol, hypoxia decreased β2-AR phosphorylation at S246 by 2-fold and increased phosphorylation at S396 by 6.7-fold (Table 1 and Figure 5, A and B). S246 on β2-AR is known to be phosphorylated by ataxia telangiectasia mutated protein kinase (ATM) and S396 is known to be phosphorylated by GRK2. In contrast to isoproterenol, hypoxia did not alter β2-AR phosphorylation at S261 and S262, known PKA sites (Figure 5, C and D). Although isoproterenol and hypoxia increased S355 and S356 phosphorylation, the level of increase was substantially different between the two (hypoxia, 1.6-fold versus isoproterenol, 153-fold) (Figure 5, E and F). These findings are consistent with Western blot analysis of protein extracts from hypoxic HUVECs. Hypoxia increased S355 and S356 phosphorylation, an effect that was blunted by GRK inhibition (Figure 5, G and H). Altogether, these findings identify a distinct hypoxia-specific β2-AR phosphorylation signature, which provides a definitive biochemical link between hypoxia sensing and activation of the β-AR (Figure 5I).
Figure 5. Hypoxia induces a phosphorylation barcode in the absence of agonist binding.

Human embryonic kidney cells (HEK293) overexpressing β2-AR were exposed to the β-agonist isoproterenol or 5-hour hypoxia. β2-ARs were enriched by alprenolol for quantitative mass spectrometry analysis of phosphorylated peptides. 21% and 2% O2 (n = 3); β-agonist (n = 2). (A) Spectrum for the pS246-containing peptide. The mass difference between the y7 and y8 ions is consistent with phosphorylation at S246. (C) Spectrum for the pS261- and pS262-containing peptide. The masses of the y5, y6, and y7 ions are consistent with phosphorylation at S261 and S262. (E) Spectrum for the pS355- and pS356-containing peptide. The masses of the y16, y17, and y18 ions are consistent with phosphorylation at S355 and S356. (B, D, and F) Dot plots showing abundance of each phosphorylated peptide at 21% or 2% oxygen or with β-agonist. (G and H) Expression of phosphorylated β2-AR at S355/S356 and total β2-AR in HUVECs with 21% or 2% oxygen (vehicle or GRK inhibitor at 125 μM). Replicate samples run on parallel gels are presented (n = 2). (I) Hypoxia-specific β-AR phosphorylation barcode with increased (pink) and decreased (blue) phosphorylation at unique sites.
Discussion
Physiologic responses are rapidly initiated within minutes of hypoxia. Catecholamines activate the β1-AR in the cardiac muscles, resulting in increased cardiac output (34–36), while the peripheral vasculature dilates to increase the perfusion of blood to oxygen-limited tissues (37). Secondary to these immediate events, adaptive responses take place over the course of hours and days to generate erythrocytes and create new blood vessels in order to provide greater oxygen delivery to tissues. These late responses have been identified as the downstream effects of HIFs (1, 2). HIF-1 is the primary intracellular mediator of the myriad physiological processes for adaptation. This study provides a biochemical connection between the catecholamine pathway and hypoxia responses. Here, we indicate that hypoxia, like catecholamine agonists, can activate the β-AR and lead to HIF accumulation and adaptive responses. Prior work has suggested a close relationship between HIF and β-AR, i.e., both are hydroxylated by prolyl hydroxylase 3 under hypoxia (38). However, to our knowledge, this is the first connection of hypoxia to activation of β-AR.
In early studies of beta blockers in the 1970s, intraperitoneal injection of rabbits with the subtype-nonspecific beta blocker propranolol prior to an 18-hour exposure to hypobaric hypoxia resulted in decreased erythropoietic responses (11). In another study, rats preinjected with metipranolol (another subtype-nonspecific beta blocker) had less erythropoiesis (12). There were some discrepant findings based on type of beta blocker used, but the studies suggested the possibility that β-AR may regulate hypoxia-inducible erythropoiesis. Many years after these studies, Semenza et al. identified HIF-1 in the molecular mechanisms that lead to production of erythropoietin and the subsequent erythropoiesis associated with hypoxia (2). However, the effect of β-AR on HIF-1 accumulation under hypoxia has been untested until now.
In this study, mice were orally administered the readily absorbed beta blocker carvedilol, which has a longer half-life (6–10 hours) as compared with propranolol (3–6 hours) (39). The mice given carvedilol had lower levels of renal HIF-1α and consequently less binding of the transcription factor to Epo in vivo. Erythropoietin mRNA in the kidneys, the principal site of production, and serum erythropoietin levels were significantly lower with carvedilol treatment. Consistent with these results, mice with carvedilol treatment had fewer erythroid progenitor cells in the bone marrow. Importantly, the beta blocker decreased HIF-1α levels and its binding to target genes and reversed approximately 30% (equivalent to 434 identified transcripts) of the hypoxia-inducible changes in expression. These transcripts mapped to known signal transduction pathways that contribute to cancer biology and dilated cardiomyopathy. In this context, HIF-1α levels are increased in heart tissues obtained from patients with ischemic cardiomyopathy (23). It is interesting to speculate that benefits of beta blockers in heart failure may be related to suppression of HIF-1α.
To define the molecular mechanisms that link β-AR and HIF-1α, the cAMP/PKA pathway was evaluated using pharmacologic approaches and genetic mutants. Under normoxia, simply increasing the intracellular cAMP levels in cells could not cause HIF-1α accumulation. These findings are slightly in variance from a recent study that suggests that isoproterenol can increase HIF-1α protein levels (40). In that study, treatment of human cardiomyocytes with the β-agonist isoproterenol or cervical cancer cells with forskolin increased HIF-1α protein abundance, but forskolin did not increase HIF-1α transcriptional activity (40). In this study, pharmacological inhibition of PKA in primary human endothelial cells, or the expression of a PKA phosphorylation–deficient β-AR mutant in human kidney embryonic cells, does not alter hypoxia-mediated HIF-1α accumulation. This is consistent with the study in which a stable knockdown of the catalytic subunit of PKA did not decrease hypoxic accumulation of HIF-1α (40). Unlike PKA, pharmacological inhibition of GRK, or genetic mutation of the β-AR that impairs GRK phosphorylation, profoundly decreased hypoxia-mediated HIF-1α accumulation in this study. These results support the idea that hypoxia-mediated HIF-1α accumulation is dependent on GRK phosphorylation of β-AR. In a study by Baloğlu, et al., preexposure to 24 hours of hypoxia decreased β-agonist induction of cAMP in alveolar epithelial cells (41). Here, 10-minute hypoxia exposure of human endothelial cells rapidly increased cAMP. These findings suggest that under hypoxia the β-AR produces higher basal levels of cAMP but is overall less responsive to agonist stimulation. Another possibility for the different findings in this study is that cAMP responses to hypoxia are dependent on specific cell types and/or times of exposure.
Carvedilol and propranolol were both employed in experiments performed in this study, and distinctions should be noted in these beta blockers. Carvedilol, but not propranolol, induces phosphorylation at GRK6 sites within β2-AR and initiates β-arrestin–mediated signaling (33, 42). Thus, carvedilol might exert some of its effects independent of receptor antagonism. Shenoy et al. previously found that β-arrestin 1 interacts with HIF-1α, which could be one mechanistic link between the hypoxia barcode and HIF-1α stabilization (43). Studies are needed to test the possible roles of β-arrestins, receptor internalization, and other mechanisms through which GRK phosphorylation may induce HIF.
Human β1-AR and β2-AR protein sequences share 54% sequence identity and contain phosphorylation sites that are targeted by PKA and GRK; but they have different expression patterns and activate different signaling pathways (18, 32). One limitation of the current study is that β1-AR mutants were used in evaluation of hypoxia signal transduction, but β2-AR was used to investigate effects of hypoxia on the phosphorylation barcode. Effects of β2-AR PKA and GRK mutants were not studied. Likewise, the phosphorylation barcodes of β1-AR or its GRK/PKA mutant forms were not studied. In addition, expression levels of β1-AR or β2-AR were not quantitated in vitro or in vivo. Studies delineating differences and similarities between β1-AR and β2-AR under hypoxia will be important to understanding mechanisms and cell-specific effects. Abundance and membrane localization of the β1-AR and β2-AR under hypoxia will also be important to understanding regulatory mechanisms of the receptors under hypoxia.
Quantitative mass spectrometry analyses of β2-AR peptides, enriched from alprenolol binding, indicate highly differential percentages of phosphorylation of known serine and threonine amino acid residues, ranging from 0.045% at S396 to 99% at S262 (Supplemental Table 3). Our method identified 9 amino acids in the β2-AR phosphorylated with isoproterenol treatment, compared with 13 amino acids that were previously reported using stable isotope labeling with amino acids in cell culture with phosphopeptide enrichment (33). Despite differences in methodologies, we identified an overall similar level of phosphorylation change from isoproterenol stimulation at S246 (1.7-fold decrease compared with previously reported 2-fold), at S396 (6.7-fold increase compared with previously reported 3.5-fold), and at S261 and S262 (4.9-fold compared with 15.5-fold). Strikingly, hypoxia alone induces decreased phosphorylation at S246 and increased phosphorylation at S396 at a comparable level to isoproterenol. No change in the phosphorylation level at PKA sites is observed with hypoxia. These findings point to a unique hypoxia-specific β2-AR phosphorylation signature, perhaps mediating a different pattern of signal transduction for HIF-1α regulation.
Our results collectively support a model in which hypoxia activates a β-AR phosphorylation barcode that allows for distinct downstream signals, which are essential for HIF-1α accumulation (Figure 6). The agonist-independent hypoxia-driven β-AR activation is mediated by GRK phosphorylation, which results in a unique phosphorylation signature that mediates HIF-1α stabilization.
Figure 6. Working model.
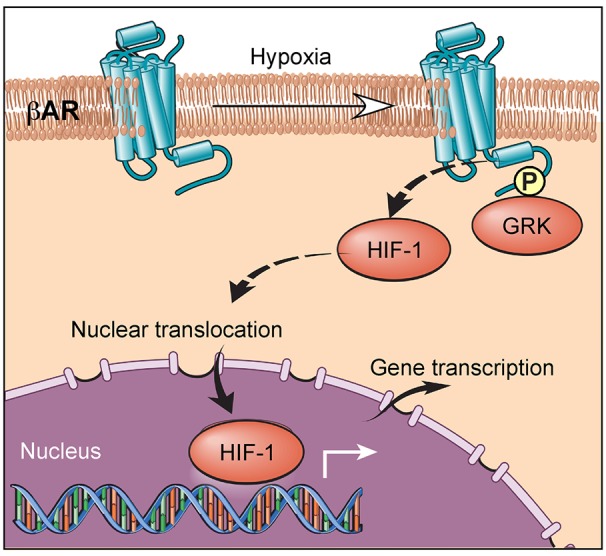
Hypoxia induces a unique β-adrenergic receptor (β-AR) phosphorylation barcode that is GPCR kinase–dependent (GRK-dependent), which drives HIF-1α accumulation.
Although β-ARs were first identified for their roles in the fight-or-flight response as part of the sympathetic nervous system, this study identifies an unsuspected essential role of β-AR in hypoxia sensing in renal hypoxia-induced accumulation of HIF in vivo and in endothelial and epithelial cells in vitro. However, beta blockers are used principally in the context of cardiovascular disease, such as heart failure. Further work is needed to define whether β-AR–dependent hypoxic signaling is operative in the heart and in a clinically relevant model, such as ischemic heart failure.
Methods
Animal model.
Eight-week-old female balb/c mice from The Jackson Laboratory were orally administered with vehicle or carvedilol (C3993, Sigma-Aldrich) dissolved in 20% DMSO/water at 15 mg/kg/d for 7 days, followed by exposure to hypoxia (10% O2). Heart rates were monitored consciously with the MouseOx pulse oximeter.
Flow cytometry.
Bone marrow was isolated from the femur, tibia, and vertebrae. Nonspecific binding sites were blocked by incubation of cells with 10% normal goat serum in PBS for 15 minutes at room temperature. Cells were washed with 1% BSA in PBS and incubated with APC-conjugated anti-CD45 (eBioscience) at 1:100 (2 μg/ml), PE-conjugated TER119 (eBioscience) at 1:50 (4 μg/ml), and FITC-conjugated anti-CD44 (eBioscience) at 1:100 (5 μg/ml) for 30 minutes on ice. All antibodies were diluted in 1% BSA in PBS. Cells were washed twice with 1% BSA in PBS and resuspended in FACS Flow containing the dead cell marker 7-AAD (BD Biosciences) at 1:200. Samples were run on a LSRII flow cytometry (Becton Dickinson), and data were analyzed using Flowjo vX.0.7 (Tree Star Inc.). Aggregates, cell debris, and dead cells were gated out. Leukocytes were excluded using CD45. Three populations were defined based on size and CD44 and TER119 levels. Proerythroblasts were defined as CD44hiTER119lo, and basophilic and polychromatic erythroblasts were defined as TER119hi and selected based on decreasing size and CD44 expression. Proerythroblasts and basophilic and polychromatic erythroblasts were defined such that the percentage of cells in each population fit the expected ratio of 1:2:4 (26).
ELISA.
Blood obtained from cardiac puncture was clotted at 37°C for at least 30 minutes. Serum was isolated after centrifugation at 10,000 g for 10 minutes. The Mouse Erythropoietin Quantikine ELISA Kit (MEP00B, R&D Systems) was used to quantify serum erythropoietin concentration.
Cell culture.
HUVECs were cultured in Endothelial Basal Medium-2 basal medium, supplemented with the Endothelial Cell Grow Medium-2 bulletkit (Lonza). They were cultured up to passage 6 for experiments. All kidney embryonic cells (HEK293) were cultured in minimum essential media with 10% heat-inactivated fetal bovine serum and 1% penicillin/streptomycin. Cells were serum starved for 4 hours or overnight prior to any experiment. For hypoxia studies, cells were incubated in a sealed chamber at 37°C with 2% O2, 5% CO2, balanced with 93% N2. For treatment of cells, propranolol (P0884, Sigma-Aldrich), β-ARK1/GRK2 inhibitor (125 μM) (182200, EMD Millipore), or PKA inhibitor H89 (10 μM) (B1427, Sigma-Aldrich) was added to cells for 45 minutes prior to normoxia or hypoxia. HUVECs were gifts from F. Terenzi and P. Fox at Cleveland Clinic; HEK293 cells overexpressing β1-AR (WTβ1-AR) or mutants lacking PKA (PKA–β1-AR) or GRK (GRK–β1-AR) sites were from Howard Rockman at Duke University, Durham, North Carolina, USA; and HEK293 cells overexpressing β2-AR were from Robert Lefkowitz at Duke University.
Western blot.
For HIF-1α protein quantification, nuclear protein extracts were prepared according to the manufacturing protocol of the nuclear extraction kit (Affymetrix). Proteins were subjected to SDS-PAGE in 4%–15% or 18% Tris-HCl gel (Bio-Rad) and transferred to polyvinylidene difluoride membranes. Antibodies for human HIF-1α (610959, BD Biosciences), mouse HIF-1α (sc-10790, Santa Cruz Biotechnology Inc.), phosphorylated β2-AR (sc-22191-R, Santa Cruz Biotechnology Inc.), β2-AR (sc-569, Santa Cruz Biotechnology Inc.), and Lamin B (sc6216, Santa Cruz Biotechnology Inc.) were used for detection.
Isolation of RNA.
Cells were lysed using QIAzol buffer. RNA was isolated according to the manufacturing protocol of the miRNeasy Mini Kit (Qiagen). The quality of RNA was assessed with the 2100 BioAnalyzer (Agilent Technologies). Samples with an RNA Integrity number higher than 7 were used for genome-wide expression assay.
Genome-wide expression assay.
The HumanHT-12 v4 Expression BeadChip (BD-103-0204, Illumina) was used. The experiment was performed on 5 biological replicates. Data have been deposited in the GEO repository (accession GSE86793).
ChIP — quantitative PCR.
HUVECs were treated with propranolol, isoproterenol, or hypoxia for 12 hours. Chromatins were prepared according to the manufacturing protocol of the Chromatin Immunoprecipitation Assay Kit (17295, EMD Millipore). Rabbit anti-HIF-1α antibody (sc10790, Santa Cruz Biotechnology Inc.) was used to pull down chromatin bound by HIF-1α; rabbit IgG (sc2027, Santa Cruz Biotechnology Inc.) was used as control. DNA primers used for quantitative PCR to quantify HIF-1α–bound promoters are as follows: SDF-1 forward, 5′-TCTAACGGCCAAAGTGGTTT-3′; SDF-1 reverse, 5′-GCCACCTCTCTGTGTCCTTC-3′; HK2 forward, 5′-CACATTGTTGCATGAAACTCC-3′; HK2 reverse, 5′-GACCTCTCCGATTCACAGG-3′; VEGFA forward, 5′-TCTTCGAGAGTGAGGACGTGTGT-3′; VEGFA reverse, 5′-AAGGCGGAGAGCCGGAC-3′ (44); Epo forward, 5′-TCTCTTCATGACTGTACACACC-3′; Epo reverse, 5′-TGACAGCCACATAGAATAAAAG-3′ (45). Quantitative PCR was performed using iQ SYBR Green Supermix (170-8880, Bio-Rad).
Measurements of cAMP.
cAMP was measured according to the manufacturing protocol of the CatchPoint cAMP Fluorescent Assay Kit (R8089, Molecular Devices).
Reverse transcription–quantitative PCR.
mRNA was prepared according to manufacturing protocol of the miRNeasy kit (217004, Qiagen). mRNA was reverse transcribed using the SuperScript First-Strand Synthesis System (11907, Invitrogen). DNA primers used for quantitative PCR to quantify EPO and ACTIN cDNA were as follows: EPO forward, 5′-GAGGCAGAAAATGTCACGATG-3′; EPO reverse, 5′-CTTCCACCTCCATTCTTTTCC-3′; ACTIN forward, 5′-CTAGGCACCAAGGTGTGAT-3′; ACTIN reverse, 5′-CAAACATGATCTGGGTCATC-3′.
Pathway analysis and annotation clustering.
DAVID v6.7 (Database for Annotation, Visualization and Integrated Discovery) was used for Kyoto Encyclopedia of Genes and Genomes pathways analysis and annotation clustering of genes that show significant differential expression in cells treated with hypoxia, isoproterenol, and hypoxia with propranolol pretreatment.
β2-AR alprenolol bead purification.
Kidney embryonic kidney cells which overexpress β2-AR (HEK293 β2-AR) were serum starved for at least 4 hours prior to exposure to isoproterenol (10 μM) for 5 minutes or hypoxia (2% oxygen) for 5 hours. After cell lysis with hypotonic solution, plasma membranes were solubilized with 1% n-Dodecyl β-D-maltoside supplemented with Halt Protease and Phosphatase Inhibitor Cocktail (78440, Thermo Fisher Scientific). Alprenolol beads (CM29101, CellMosaic Inc.) (46) were incubated with plasma membranes at 4°C overnight. Receptors were eluted with loading buffer for gel electrophoresis.
Mass spectrometry.
The protein samples were subjected to in-gel digestion in which the bands were cut from the gels and washed in 50% ethanol, 5% acetic acid. The gel pieces were then dehydrated in acetonitrile, dried in a Savant Speedvac (ThermoFisher Scientific), and digested by adding 5 μl 10 ng/μl of trypsin or chymotrypsin, in 50 mM ammonium bicarbonate, followed by incubation overnight. The peptides were extracted into two portions of 30 μL aliquots of 50% acetonitrile, 5% formic acid. The combined extracts were evaporated to <10 μl in a Speed-vac and then resuspended in 1% acetic acid to make up a final volume of approximately 30 μl for liquid chromatography–mass spectrometry (LC-MS) analysis. The LC-MS system was a Finnigan LTQ-Obitrap Elite hybrid mass spectrometer system. The HPLC column was a Dionex 15 cm × 75 μm id Acclaim Pepmap C18, 2 μm, 100 Å reversed-phase capillary chromatography column. Five μl volumes of the extract were injected, and the peptides — eluted from the column in an acetonitrile, 0.1% formic acid gradient at a flow rate of 0.25 μl/min — were introduced online into the ion source of the mass spectrometer.
The digest was analyzed in both a survey manner and a targeted manner. The survey experiments were performed using the data-dependent multitask capability of the Orbitrap Elite mass spectrometry system (ThermoFisher Scientific) acquiring full-scan mass spectra to determine peptide molecular weights and product ion spectra to determine amino acid sequence in successive instrument scans. The LC-MS/MS data were searched with the programs Mascot and Sequest against both the full human reference sequence database and specifically against the sequence of β2-AR. The parameters used in this search include a peptide mass accuracy of 10 ppm, fragment ion mass accuracy of 0.6 Da, carbamidomethylated cysteines as a constant modification, and oxidized methionine and phosphorylation at S, T, and Y as a dynamic modification. The results were filtered based on Mascot ion scores and Sequest XCorr scores. All positively identified phosphopeptides were manually validated. The targeted experiments involve the analysis of specific β2-AR peptides, including the phosphorylated and unmodified forms of several tryptic and chymotryptic peptides. The chromatograms for these peptides were plotted based on known fragmentation patterns and the peak areas of these chromatograms were used to determine the extent of phosphorylation.
Statistics.
JMP PRO 10 was used for analysis. ANOVA or Student’s 1-tailed t test was used to test the statistical significance for the observed differences, as described in figure legends. The P value threshold chosen for statistical significance was 0.05. For mouse kidney ChIP, data outside 95% confidence interval were excluded from analysis. For genome-wide expression array, the raw expression data were log2 transformed with force-positive background correction and were then quantile normalized. Differential expression analysis was carried out between controls and the hypoxia, propranolol, or hypoxia with propranolol pretreatment groups. For each 2-group comparison, unexpressed and unannotated probes were first excluded from differential analysis. Probes with differential expression levels were identified using the “limma” package in R. FDR was used to account for multiple comparisons. Significant probes were identified as those with a FDR of less than 0.05. All analyses were conducted using R-3.1.3 (https://cran.r-project.org/).
Study approval.
All animal experiments were approved by the Cleveland Clinic Institutional Animal Care and Use Committee at the Lerner Research Institute in Cleveland, Ohio.
Author contributions
HIC performed research; collected, analyzed and interpreted data; and wrote the manuscript. KA, ORS, and KAQ collected data and analyzed data for the animal model. BW collected and analyzed data for mass spectrometry. BH contributed to statistical analysis. WX and JKTD contributed to data collection and analysis. GRS and SVNP provided research design and directions. SCE designed and performed research; analyzed and interpreted data; and wrote the manuscript.
Supplementary Material
Acknowledgments
This work is supported by NIH grants (HL115008, HL103453, HL081064, and HL60917). HIC is in the Molecular Medicine PhD Program of the Cleveland Clinic and Case Western Reserve University, funded in part, by the Med-into-Grad initiative of the Howard Hughes Medical Institute. SCE is supported in part by the Alfred Lerner Memorial Chair in Innovative Biomedical Research. The Perinatal Clinical Research Center at the Cleveland Metrohealth Hospital (UL1TR000439) supported this work through the harvest of HUVECs. The Orbitrap Elite instrument was purchased via an NIH-shared instrument grant (1S10RR031537-01). We thank H. Rockman for the generous gift of HEK293 β1-AR wild-type and mutant cells; R. Lefkowitz for HEK293 β2-AR cells; M. Aldred for the detailed methods of RNA isolation; C. Beall, K. Strohl, and M. Gupta for their scientific input; F. Terenzi, S. Comhair, and D. Mavrakis for providing HUVECs; G. Semenza for the detailed methods for nuclear extract preparation; and David Schumick for the graphical illustration of the working model.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information:JCI Insight. 2016;1(21):e90240. doi:10.1172/jci.insight.90240.
Contributor Information
Kewal Asosingh, Email: asosink@ccf.org.
Weiling Xu, Email: xuw@ccf.org.
Belinda Willard, Email: willarb@ccf.org.
Bo Hu, Email: hub@ccf.org.
George R. Stark, Email: starkg@ccf.org.
Sathyamangla V. Naga Prasad, Email: prasads2@ccf.org.
References
- 1.Liu Y, Cox SR, Morita T, Kourembanas S. Hypoxia regulates vascular endothelial growth factor gene expression in endothelial cells. Identification of a 5′ enhancer. Circ Res. 1995;77(3):638–643. doi: 10.1161/01.RES.77.3.638. [DOI] [PubMed] [Google Scholar]
- 2.Semenza GL, Wang GL. A nuclear factor induced by hypoxia via de novo protein synthesis binds to the human erythropoietin gene enhancer at a site required for transcriptional activation. Mol Cell Biol. 1992;12(12):5447–5454. doi: 10.1128/MCB.12.12.5447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kapitsinou PP, et al. Hepatic HIF-2 regulates erythropoietic responses to hypoxia in renal anemia. Blood. 2010;116(16):3039–3048. doi: 10.1182/blood-2010-02-270322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beall CM, et al. Natural selection on EPAS1 (HIF2α) associated with low hemoglobin concentration in Tibetan highlanders. Proc Natl Acad Sci U S A. 2010;107(25):11459–11464. doi: 10.1073/pnas.1002443107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Huang LE, Gu J, Schau M, Bunn HF. Regulation of hypoxia-inducible factor 1α is mediated by an O2-dependent degradation domain via the ubiquitin-proteasome pathway. Proc Natl Acad Sci U S A. 1998;95(14):7987–7992. doi: 10.1073/pnas.95.14.7987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jaakkola P, et al. Targeting of HIF-α to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science. 2001;292(5516):468–472. doi: 10.1126/science.1059796. [DOI] [PubMed] [Google Scholar]
- 7.Wallace DC. Colloquium paper: bioenergetics, the origins of complexity, and the ascent of man. Proc Natl Acad Sci U S A. 2010;107(suppl 2):8947–8953. doi: 10.1073/pnas.0914635107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Semenza GL. Hypoxia-inducible factors in physiology and medicine. Cell. 2012;148(3):399–408. doi: 10.1016/j.cell.2012.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cash TP, Pan Y, Simon MC. Reactive oxygen species and cellular oxygen sensing. Free Radic Biol Med. 2007;43(9):1219–1225. doi: 10.1016/j.freeradbiomed.2007.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schumacker PT. Lung cell hypoxia: role of mitochondrial reactive oxygen species signaling in triggering responses. Proc Am Thorac Soc. 2011;8(6):477–484. doi: 10.1513/pats.201103-032MW. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fink GD, Paulo LG, Fisher JW. Effects of β adrenergic blocking agents on erythropoietin production in rabbits exposed to hypoxia. J Pharmacol Exp Ther. 1975;193(1):176–181. [PubMed] [Google Scholar]
- 12.Zivný J, Ostádal B, Neuwirt J, Procházka J, Pelouch V. Effect of beta adrenergic blocking agents on erythropoiesis in rats. J Pharmacol Exp Ther. 1983;226(1):222–225. [PubMed] [Google Scholar]
- 13.Rockman HA, Koch WJ, Lefkowitz RJ. Seven-transmembrane-spanning receptors and heart function. Nature. 2002;415(6868):206–212. doi: 10.1038/415206a. [DOI] [PubMed] [Google Scholar]
- 14.Hedberg A, Minneman KP, Molinoff PB. Differential distribution of β-1 and β-2 adrenergic receptors in cat and guinea-pig heart. J Pharmacol Exp Ther. 1980;212(3):503–508. [PubMed] [Google Scholar]
- 15.Krief S, et al. Tissue distribution of β3-adrenergic receptor mRNA in man. J Clin Invest. 1993;91(1):344–349. doi: 10.1172/JCI116191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Daaka Y, Luttrell LM, Lefkowitz RJ. Switching of the coupling of the β2-adrenergic receptor to different G proteins by protein kinase A. Nature. 1997;390(6655):88–91. doi: 10.1038/36362. [DOI] [PubMed] [Google Scholar]
- 17.Hausdorff WP, Bouvier M, O’Dowd BF, Irons GP, Caron MG, Lefkowitz RJ. Phosphorylation sites on two domains of the β2-adrenergic receptor are involved in distinct pathways of receptor desensitization. J Biol Chem. 1989;264(21):12657–12665. [PubMed] [Google Scholar]
- 18.Lefkowitz RJ, Rockman HA, Koch WJ. Catecholamines, cardiac β-adrenergic receptors, and heart failure. Circulation. 2000;101(14):1634–1637. doi: 10.1161/01.CIR.101.14.1634. [DOI] [PubMed] [Google Scholar]
- 19.Thomas JA, Marks BH. Plasma norepinephrine in congestive heart failure. Am J Cardiol. 1978;41(2):233–243. doi: 10.1016/0002-9149(78)90162-5. [DOI] [PubMed] [Google Scholar]
- 20.Bristow MR, et al. Decreased catecholamine sensitivity and β-adrenergic-receptor density in failing human hearts. N Engl J Med. 1982;307(4):205–211. doi: 10.1056/NEJM198207223070401. [DOI] [PubMed] [Google Scholar]
- 21.Bristow MR, et al. Carvedilol produces dose-related improvements in left ventricular function and survival in subjects with chronic heart failure. MOCHA Investigators. Circulation. 1996;94(11):2807–2816. doi: 10.1161/01.CIR.94.11.2807. [DOI] [PubMed] [Google Scholar]
- 22.Quaife RA, Christian PE, Gilbert EM, Datz FL, Volkman K, Bristow MR. Effects of carvedilol on right ventricular function in chronic heart failure. Am J Cardiol. 1998;81(2):247–250. doi: 10.1016/S0002-9149(97)00874-6. [DOI] [PubMed] [Google Scholar]
- 23.Moslehi J, et al. Loss of hypoxia-inducible factor prolyl hydroxylase activity in cardiomyocytes phenocopies ischemic cardiomyopathy. Circulation. 2010;122(10):1004–1016. doi: 10.1161/CIRCULATIONAHA.109.922427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cahan C, Hoekje PL, Goldwasser E, Decker MJ, Strohl KP. Assessing the characteristic between length of hypoxic exposure and serum erythropoietin levels. Am J Physiol. 1990;258(4 pt 2):R1016–R1021. doi: 10.1152/ajpregu.1990.258.4.R1016. [DOI] [PubMed] [Google Scholar]
- 25.Malik J, Kim AR, Tyre KA, Cherukuri AR, Palis J. Erythropoietin critically regulates the terminal maturation of murine and human primitive erythroblasts. Haematologica. 2013;98(11):1778–1787. doi: 10.3324/haematol.2013.087361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu J, et al. Quantitative analysis of murine terminal erythroid differentiation in vivo: novel method to study normal and disordered erythropoiesis. Blood. 2013;121(8):e43–e49. doi: 10.1182/blood-2012-09-456079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4(1):44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 28.Huang da W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37(1):1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Luttrell LM, et al. β-Arrestin-dependent formation of beta2 adrenergic receptor-Src protein kinase complexes. Science. 1999;283(5402):655–661. doi: 10.1126/science.283.5402.655. [DOI] [PubMed] [Google Scholar]
- 30.Rapacciuolo A, et al. Protein kinase A and G protein-coupled receptor kinase phosphorylation mediates β-1 adrenergic receptor endocytosis through different pathways. J Biol Chem. 2003;278(37):35403–35411. doi: 10.1074/jbc.M305675200. [DOI] [PubMed] [Google Scholar]
- 31.Belmonte SL, Blaxall BC. G protein coupled receptor kinases as therapeutic targets in cardiovascular disease. Circ Res. 2011;109(3):309–319. doi: 10.1161/CIRCRESAHA.110.231233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rapacciuolo A, et al. Protein kinase A and G protein-coupled receptor kinase phosphorylation mediates β-1 adrenergic receptor endocytosis through different pathways. J Biol Chem. 2003;278(37):35403–35411. doi: 10.1074/jbc.M305675200. [DOI] [PubMed] [Google Scholar]
- 33.Nobles KN, et al. Distinct phosphorylation sites on the β(2)-adrenergic receptor establish a barcode that encodes differential functions of β-arrestin. Sci Signal. 2011;4(185):ra51. doi: 10.1126/scisignal.2001707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Adachi H, Strauss W, Ochi H, Wagner HN. The effect of hypoxia on the regional distribution of cardiac output in the dog. Circ Res. 1976;39(3):314–319. doi: 10.1161/01.RES.39.3.314. [DOI] [PubMed] [Google Scholar]
- 35.Calbet JA. Chronic hypoxia increases blood pressure and noradrenaline spillover in healthy humans. J Physiol (Lond) 2003;551(pt 1):379–386. doi: 10.1113/jphysiol.2003.045112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hansen J, Sander M. Sympathetic neural overactivity in healthy humans after prolonged exposure to hypobaric hypoxia. J Physiol (Lond) 2003;546(pt 3):921–929. doi: 10.1113/jphysiol.2002.031765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Michiels C. Physiological and pathological responses to hypoxia. Am J Pathol. 2004;164(6):1875–1882. doi: 10.1016/S0002-9440(10)63747-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xie L, et al. Oxygen-regulated β(2)-adrenergic receptor hydroxylation by EGLN3 and ubiquitylation by pVHL. Sci Signal. 2009;2(78):ra33. doi: 10.1126/scisignal.2000444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.De BK, et al. Acute and 7-day portal pressure response to carvedilol and propranolol in cirrhotics. J Gastroenterol Hepatol. 2002;17(2):183–189. doi: 10.1046/j.1440-1746.2002.02674.x. [DOI] [PubMed] [Google Scholar]
- 40.Bullen JW, et al. Protein kinase A-dependent phosphorylation stimulates the transcriptional activity of hypoxia-inducible factor 1. Sci Signal. 2016;9(430):ra56. doi: 10.1126/scisignal.aaf0583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Baloğlu E, Ke A, Abu-Taha IH, Bärtsch P, Mairbäurl H. In vitro hypoxia impairs β2-adrenergic receptor signaling in primary rat alveolar epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2009;296(3):L500–L509. doi: 10.1152/ajplung.90390.2008. [DOI] [PubMed] [Google Scholar]
- 42.Wisler JW, et al. A unique mechanism of β-blocker action: carvedilol stimulates β-arrestin signaling. Proc Natl Acad Sci U S A. 2007;104(42):16657–16662. doi: 10.1073/pnas.0707936104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shenoy SK, et al. β-Arrestin1 mediates metastatic growth of breast cancer cells by facilitating HIF-1-dependent VEGF expression. Oncogene. 2012;31(3):282–292. doi: 10.1038/onc.2011.238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kim JW, Gao P, Liu YC, Semenza GL, Dang CV. Hypoxia-inducible factor 1 and dysregulated c-Myc cooperatively induce vascular endothelial growth factor and metabolic switches hexokinase 2 and pyruvate dehydrogenase kinase 1. Mol Cell Biol. 2007;27(21):7381–7393. doi: 10.1128/MCB.00440-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yeo EJ, Cho YS, Kim MS, Park JW. Contribution of HIF-1α or HIF-2α to erythropoietin expression: in vivo evidence based on chromatin immunoprecipitation. Ann Hematol. 2008;87(1):11–17. doi: 10.1007/s00277-007-0359-6. [DOI] [PubMed] [Google Scholar]
- 46.Shorr RG, Lefkowitz RJ, Caron MG. Purification of the β-adrenergic receptor. Identification of the hormone binding subunit. J Biol Chem. 1981;256(11):5820–5826. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.