

Abstract
Glucose-dependent insulinotropic polypeptide (GIP) is an incretin hormone involved in nutrient homeostasis. GIP receptor (GIPR) is constitutively internalized and returned to the plasma membrane, atypical behavior for a G protein-coupled receptor (GPCR). GIP promotes GIPR downregulation from the plasma membrane by inhibiting recycling without affecting internalization. This transient desensitization is achieved by altered intracellular trafficking of activated GIPR. GIP stimulation induces a switch in GIPR recycling from a rapid endosomal to a slow TGN pathway. GPCR kinases and β-arrestin2 are required for this switch in recycling. A coding sequence variant of GIPR, which has been associated with metabolic alterations, has altered post-activation trafficking characterized by enhanced downregulation and prolonged desensitization. Downregulation of the variant requires β-arrestin2 targeting to the TGN but is independent of GPCR kinases. The single amino acid substitution in the variant biases the receptor to promote GIP stimulated β-arrestin2 recruitment without receptor phosphorylation, thereby enhancing downregulation.
Graphical Abstract
INTRODUCTION
G protein-coupled receptors (GPCRs) are the largest family of signaling receptors. Signal transduction is initiated downstream of ligand binding via activation of trimeric G proteins (Lefkowitz and Shenoy, 2005). Post-activation trafficking of GPCRs contributes significantly to the biological effects of GPCRs (Hanyaloglu and von Zastrow, 2008). In the majority of cases GPCR activation leads to enhanced internalization and redistribution of receptors from the plasma membrane (PM) to the interior of cells (Claing et al., 2002). The resultant reduced PM receptor density desensitizes cells to further ligand stimulation. The length of this refractory period, which varies among receptors, is important in sculpting the cellular response. Some GPCRs are internalized and delivered to lysosomes for degradation, in which case re-sensitization to further ligand stimulation requires synthesis of new receptors. Other GPCRs are not degraded but rather are recycled back to the PM. The intracellular dwell time varies among recycled GPCRs, although typically the time to re-sensitization is shorter than for those GPCRs that require new synthesis.
GPCR kinases (GRK) and β-arrestin proteins are important in the post-activation behavior of GPCRs (Claing et al., 2002; Smith and Rajagopal, 2016). GRKs phosphorylate agonist activated GPCR. β-Arrestins, which bind to phosphorylated GPCRs, impact GPCR behavior in a number of ways (Lohse and Hoffmann, 2014). First, β-arrestin binding blocks trimeric G protein re-binding to the GPCR, inhibiting further G protein activation by the GPCR. Second, β-arrestins serve as adaptors that link GPCRs to membrane protein trafficking machinery, thereby influencing post-activation trafficking of the GPCR. The best-described example is β-arrestin-mediated targeting to the clathrin-coated pits to stimulate internalization and induce desensitization (Goodman et al., 1996). However, β-arrestins can also influence the intracellular trafficking of GPCRs (Shenoy et al., 2009). The duration of β-arrestin interaction with the GPCR also affects intracellular trafficking and recycling of GPCRs and GPCRs can be divided into two classes based on their interaction with β-arrestins. (Lohse and Hoffmann, 2014). Typically, GPCRs with prolonged interactions with β-arrestins undergo sustained downregulation. De-phosphorylation of GPCRs reverses β-arrestin binding, contributing to re-sensitization and GPCR signaling homeostasis (Krueger et al., 1997). Finally, the β-arrestins link GPCRs to signaling cascades other than those mediated by trimeric G proteins, including but not limited to ERK, JNK and AKT signaling pathways, thereby expanding the signaling repertoire downstream of GPCRs (Lefkowitz et al., 2006).
Glucose-dependent insulinotropic polypeptide (GIP) and Glucagon-like polypeptide-1 (GLP-1) are secreted by endocrine cells of the gut in response to nutrients in chyme (Dupre et al., 1973). These peptide hormones, known as the incretins, have prominent roles in the regulation of whole body metabolism, largely through enhancement of glucose-stimulated insulin secretion by pancreatic β-cells, although they also have effects on other cells, including adipocytes, osteoblasts and neurons (McIntosh et al., 2009; Tseng and Zhang, 2000). Both incretins signal through GPCRs. The GIP receptor (GIPR) is coupled to trimeric Gαs signal transduction. Trafficking of GIPR does not conform to the typical GPCR behavior (Mohammad et al., 2014). In adipocytes, a cell type that natively expresses GIPR, the receptor is constitutively internalized and recycled to the PM independent of GIP stimulation. GIP stimulation causes a downregulation of PM GIPR by inducing a slowing of receptor recycling without affecting internalization. Thus, unlike the canonical GPCR behavior, GIP-induced downregulation of GIPR is not achieved through stimulation of internalization but rather by the slowing of recycling (Mohammad et al., 2014).
A naturally occurring coding sequence variant of the human GIPR has been associated in genome wide association studies with obesity, cardiovascular disease and an increased risk of bone fractures (Nitz et al., 2007; Sauber et al., 2010; Saxena et al., 2010). In the variant, a glutamine replaces a glutamate within the 6th transmembrane domain (position 354, GIPR-Gln354, rs1800437) of the GIPR. The Gln354 substitution alters post-activation trafficking of the variant rather than ligand binding or cAMP production. The Gln354 substitution has no effect on receptor trafficking in unstimulated adipocytes. However, upon GIP stimulation, there is enhanced receptor desensitization. In addition the refractory period of the variant receptor is four hours as compared to one hour in the wild type (Mohammad et al., 2014). These data suggest that the altered post-activation trafficking of the GIPR-Gln354 variant underlies the link of this variant to alterations in human metabolism, emphasizing the importance of post-activation trafficking for the biology of the GIPR. GIPR trafficking is not understood in detail at a molecular level nor is it known why the Gln354 substitution affects GIPR trafficking.
RESULTS
β-Arrestin2 is responsible for GIP-induced downregulation of GIPR
GIPR trafficking does not conform to the canonical GPCR mechanism of downregulation based on ligand-stimulated internalization. To determine how this specialization is reflected in the protein machinery that regulates GIPR trafficking, we investigated the roles of proteins that regulate trafficking of other GPCRs. β-Arrestins, mediate GPCR internalization by targeting activated receptors to clathrin-coated pits or other internalization mechanisms (Lefkowitz et al., 2006). To characterize the role of β-arrestins in GIPR trafficking, we investigated the impact of siRNA-mediated knockdown of the β-arrestins on the distribution of GIPR between the PM and intracellular compartments of basal and GIP-stimulated adipocytes. We used quantitative fluorescence microscopy and a previously characterized and validated GIPR reporter, HA-GIPR-GFP (HA epitope fused to the extracellular amino terminus of GIPR and GFP fused to the cytoplasmic C terminus) to study GIPR trafficking (Mohammad et al., 2014). GIP binds, activates and induces downregulation of HA-GIPR-GFP similarly to native GIPR, documenting the HA and GFP modifications do not alter the function of GIPR. HA-GIPR-GFP in the PM is quantified by anti-HA indirect immunofluorescence of fixed non-permeabilized adipocytes, and GFP fluorescence is quantified as a measure of total HA-GIPR-GFP expressed per cell. The ratio of anti-HA to GFP fluorescence is a quantitative measurement of the fraction of HA-GIPR-GFP in PM of cells (Lefkowitz et al., 2006).
In unstimulated adipocytes GIPR is nearly equally distributed between the interior and PM of cells, maintained by constitutive internalization and recycling (Mohammad et al., 2014). Intracellular GIPR is localized to a peri-nuclear compartment and to vesicles (puncta) distributed throughout the cytosol (Fig 1A). The stimulation of GIPR with GIP, leads to a reduction of the GIPR from surface. GPCR kinases (GRKs) and β-arrestins mediate downregulation of most GPCRs (Lefkowitz et al., 2006). We first determined the requirement for β-arrestins in GIPR trafficking. Using siRNA for transient knockdown in adipocytes, we achieved an 85% depletion of nonvisual β-arrestin1 and a 65% depletion of nonvisual β-arrestin2, as determined by rt-PCR 24 hrs following introduction of the siRNAs (Supplementary information, Fig S1 and SuppTable 1). Knockdown of β-arrestin1 or 2 did not grossly alter the overall intracellular localization of GIPR in basal adipocytes (Fig 1A).
Fig. 1. βA2 is required for GIP-stimulated GIPR sequestration and slowed recycling.

(A) Fixed non-permeabilized adipocytes expressing HA-GIPR-GFP, with or without β-arrestin1 (βA1) or β-arrestin2 (βA2) knockdown (KD). Surface GIPR was stained with Cy3 using anti HA-epitope antibody. Epi fluorescence images. Scale bar: 50μm
(B) Quantitation of GIPR plasma membrane to total cell expression (PM-to-Total) distribution in basal and GIP-stimulated (100 nM, 60 min) cells. Adipocytes were electroporated with HA-GIPR-GFP and with no siRNA (WT), βA1, βA2 or βA1+2 siRNA. PM-to-total (Cy3/GFP) were determined as described in materials and methods. Data from individual experiments are normalized to the control cells in basal conditions. Data are averages of 9 independent experiments ± SD., P ≤ 0.05.
(C) GIPR internalization in WT adipocytes was measured as described in materials and methods. The slopes, which are the rate constant of internalization, are plotted in panel E. Data are averages ± SD of 9 independent experiments.
(D) GIPR internalization experiment in βA2 KD adipocytes. Data are averages ± SD of 9 independent experiments. Each βA2 KD experiment was accompanied by an experiment in WT adipocytes (shown in (C)).
(E) Internalization rate constants (Ki) for GIPR internalization in WT or βA2 KD adipocytes. The Ki were calculated as slopes of straight lines from (C) and (D). Data are averages of 9 independent experiments ± SD.
(F) GIPR Exocytosis was measured as described in materials and methods. Cell associated Cy3 normalized to GFP was plotted against time. Data are average ± SD of 9 independent experiments.
(G) GIPR exocytosis experiment in βA2 KD adipocytes. Data are averages ± SD of 9 independent experiments. Each βA2 KD experiment was accompanied by an experiment in WT adipocytes (shown in (F).
(H) Exocytic rate constants (Ke) for GIPR in WT or βA2 KD cells calculated from (F) and (G). The data were fit to a single-phase exponential rise equation. Data are averages of 9 independent experiments ± SD., p<0.05.
See also Fig S1.
GIP stimulation induces a 30% decrease of GIPR in the PM (Mohammad et al., 2014). Depletion of β-arrestin2 but not the depletion of β-arrestin1 abrogated GIP-stimulated receptor downregulation (Fig 1B). Knockdown of neither of the β-arrestins affected the distribution of GIPR between the PM and the interior of unstimulated adipocytes. Simultaneous knockdown of both β-arrestins did not have an affect beyond knockdown of β-arrestin2 alone (Fig 1B). GIPR downregulation was restored to β-arrestin2 knockdown adipocytes by re-expression of β-arrestin2, confirming the effect of the knockdown was due to a depletion of β-arrestin2 (Fig 1B). These data establish that β-arrestin2 is required for GIP-induced receptor downregulation. These data also suggest that neither β-arrestin isoform has a role in GIPR trafficking in unstimulated adipocytes because the distribution of GIPR between the PM and intracellular compartments in basal adipocytes, which is maintained by constitutive internalization and recycling, was unaffected by depletion of β-arrestin isoforms individually or together.
β-Arrestin2 is responsible for GIP-induced slowing of GIPR recycling
GIPR downregulation is achieved by GIP-induced slowing of exocytosis with no effect on endocytosis (Mohammad et al., 2014). We measured GIPR endocytosis to determine if the effect of β-arrestin2 depletion on downregulation was indirect due to an effect on internalization. The HA-GIPR-GFP internalization rate constant was measured by quantifying the uptake of anti-HA antibody divided by the amount of HA-GIPR-GFP on the PM, an assay adapted from studies of trafficking of the Glut4 glucose transporter (Blot and McGraw, 2006). A plot of the GIPR internal-to-PM ratio yields a straight line whose slope is the internalization rate constant. Consistent with our previous studies, GIP activation of GIPR had no effect on the internalization rate constant (Fig 1C & E). β-Arrestin2 depletion did not affect the GIPR internalization in basal or GIP-stimulated adipocytes, establishing that the constitutive internalization of the GIPR is independent of β-arrestin2 (Fig 1D & E). These data together with the result that β-arrestin1 knockdown did not alter the fraction of GIPR on the PM (Fig 1B), support a model in which GIPR internalization is independent of either β-arrestin, an unexpected finding considering the role of β-arrestins in internalization of other GPCRs.
The amount of GIPR in the PM is dynamically maintained by internalization and recycling. Since β-arrestin2 depletion abrogates GIPR downregulation without affecting internalization, we hypothesized a role for β-arrestin2 in regulation of GIPR recycling. The HA-GIPR-GFP recycling rate constant was measured using an assay adapted from studies of Glut4 trafficking (Karylowski et al., 2004). In agreement with our previous work (Mohammad et al., 2014), GIP induced an approximate 2.5 fold inhibition of the GIPR recycling rate constant, a change that underlies GIPR downregulation (Fig 1F & H). β-Arrestin2 knockdown abolished GIP-induced inhibition of recycling without affecting GIPR recycling in unstimulated adipocytes (Fig 1G & H). Thus, GIPR is downregulated by a mechanism involving β-arrestin2-dependent inhibition of recycling, independent of a role for β-arrestin2 in regulation of GIPR endocytosis.
GRK 2 and 5 are required for GIP-induced downregulation of GIPR
Activated GPCRs are phosphorylated by GRKs, stimulating the recruitment of β-arrestins to the receptors and thereby regulating β-arrestin control of GPCR trafficking (Claing et al., 2002). We next undertook studies to determine whether the role of β-arrestin2 in GIPR trafficking is dependent on GRKs. We transiently silenced the three GRK isoforms expressed in adipocytes (Fig S1 and SuppTable 2). Individual knockdowns of GRK 2, 5 or 6 did not alter the PM to intracellular distribution of GIPR in unstimulated adipocytes nor affect GIP-induced GIPR downregulation (Fig 2A). To test for functional redundancy among these GRKs, we silenced them in pairs. Double knockdown of GRK 2 and 5 abolished GIPR downregulation, whereas double knockdowns of GRK 2 and 6, or GRK 5 and 6 did not (Fig 2A). Simultaneous knockdown of all three GRKs did not have an effect beyond the double GRK 2 & 5 knockdown (Fig 2A). These data support a model in which GRK 2 or 5 are functionally redundant in GIP-mediated GIPR downregulation.
Fig. 2. GRKs 2 or 5 are required for GIP-stimulated GIPR sequestration and slowed recycling.

(A) Quantitation of GIPR PM-to-Total distribution in basal and GIP-stimulated WT (no siRNA), and GRK KD adipocytes. Data are averages of 5 independent experiments ± SD., p≤0.05.
(B) GIPR Exocytosis experiment in WT adipocytes. Data are averages ± SD of 5 independent experiments.
(C) GIPR exocytosis in GRK2+5 double KD adipocytes. Data are average ± SD of 5 independent experiments. Each GRK 2+5KD experiment was accompanied by an experiment in WT adipocytes (shown in panel B).
(D) Exocytic rate constants (Ke) for GIPR in WT or GRK2+5 KD cells calculated from (B) and (C). The data were fit to a single phase exponential rise equation. Data are averages of 5 independent experiments ± SD., p<0.05.
(E) GIPR internalization in WT adipocytes. The internalization rate constants are plotted in panel G. Data are averages ± SD of 7 independent experiments.
(F) GIPR internalization in GRK2+5 double KD adipocytes. Data are averages ± SD of 7 independent experiments. Each GRK2+5 double KD was accompanied by an experiment in WT adipocytes (shown in panel E).
(G) Internalization rate constants (Ki) for GIPR internalization in WT or βA2 GRK2+5 double KD. The Ki were calculated as slopes of straight lines from (E) and (F).
See also Fig S1 and S2.
We hypothesized that if GRKs 2 or 5 are required for β-arrestin2 regulation of GIPR trafficking, then silencing these proteins should phenocopy β-arrestin2 knockdown, abrogating GIP inhibition of GIPR recycling without affecting GIPR internalization. In support of our hypothesis, double knockdown of GRKs 2 and 5 abrogated GIP-induced slowing of GIPR recycling without affecting recycling in unstimulated adipocytes, whereas silencing of GRKs 2 and 5 did not affect GIPR internalization in either GIP-stimulated or unstimulated adipocytes (Fig 2B to G). Thus, GRK 2 or 5 and β-arrestin2 are required for GIP-induced GIPR inhibition of recycling and therefore they are molecular components of the machinery responsible for GIPR downregulation. Despite the roles of GRKs and β-arrestin2 in promoting internalization of other GPCRs, the internalization of GIPR is independent of GRKs and β-arrestins.
β-Arrestin2 binding to GIPR requires GIP stimulation and GRK 2 or 5
GRK phosphorylation of GPCRs is involved in the recruitment of β-arrestins to the receptors. Our functional data support a model in which GRK phosphorylation of GIPR promotes β-arrestin2 recruitment to GIPR and subsequent slowing of GIPR recycling back to the PM. To further test this model, we probed for β-arrestin2 binding to GIPR by co-immunoprecipitation. GIP-stimulation promoted β-arrestin2 binding to GIPR consistent with β-arrestin2 having a role in GIPR trafficking (Fig 3A). In addition, simultaneous silencing of GRKs 2 and 5 blocked β-arrestin2 recruitment to GIPR (Fig 3B). These data establish that despite GRK/β-arrestin2 regulating recycling rather than internalization of GIPR, β-arrestin2 recruitment to activated GIPR is dependent on GRK. β-Arrestin1 did not co-immunoprecipitate with GIPR in GIP stimulated or unstimulated adipocytes, consistent with the functional data that β-arrestin1 does not have a role in GIPR trafficking (Fig S2).
Fig. 3. βA2 binding to GIPR is GIP- and GRK2/5-dependent.

(A) Immunoblot for co-immunoprecipitated HA-GIPR-GFP blotted for HA-GIPR-GFP and βA2. IP was done with or without GIP stimulation. (Right) Quantification of co immunoprecipitation experiments like that shown in the left panel. βA2 in each lane was normalized to its input and to the GIPR in the IP. Data are averages ± SD of 3 independent experiments., P<0.05.
(B) Immunoblot for co-IP of HA-GIPR-GFP in GRK 2+5 KD cells. Blot was done for HA-GIPR-GFP and βA2. (Right) Quantification of co immunoprecipitation experiments like that shown in the left panel. βA2 in each lane was normalized to its input and to the GIPR in the IP.
GIP-induced slowing of GIPR recycling by β-arrestin2-dependent targeting to the TGN
The GIP-induced slowing of GIPR could be achieved by altering the recycling itinerary of GIPR or by specifically slowing the movement of GIPR without altering the compartments transited during recycling, as would be the case if β-arrestin2 binding to GIPR directly slowed exit from a compartment. The two main recycling pathways are return to the PM from the endosomal recycling compartment (ERC) and recycling through the TGN (Bard and Malhotra, 2006; Johannes and Popoff, 2008; Maxfield and McGraw, 2004). These two pathways have distinct cargos. The transferrin receptor (TR) is an example of a client of the ERC pathway and TGN46 is an example of a client of the TGN recycling pathway (Banting and Ponnambalam, 1997; Maxfield and McGraw, 2004). The TGN and ERC are poorly resolved by conventional light microscopy in adipocytes, making it impossible to distinguish which of these pathways is utilized in GIPR recycling in adipocytes using conventional microcopy approaches (Blot and McGraw, 2008; Puertollano et al., 2003). We turned to a detergent-free, immunoadsorption method to probe the effect of GIP on the localization of the GIPR. This method has been well established in studies of GLUT4 glucose transporter trafficking in adipocytes (Bruno et al., 2016; Kandror and Pilch, 2006). Briefly, cells are broken in the absence of detergent to preserve membranes with their embedded proteins. An antibody against cytoplasmic domain of a protein is used to immunoadsorb membranes specifically enriched in that protein. Co-adsorbing proteins, determined by Western blotting, are in the same membranes as the target protein. We used an anti-TGN-46 antibody to immunoadsorb the TGN (TGN-46-containing membranes) (Fig 4A and B). Although TGN-46 is concentrated in the TGN, it continually cycles between the TGN and the PM. We used an amount of anti-TGN-46 antibody that adsorbed approximately 70% of TGN-46 as a way to bias the adsorption to those membranes in which TGN-46 is concentrated. Both Syntaxin 6 (Sx-6) and mannose-6 phosphate receptor (M6PR), two proteins enriched in the TGN, co-adsorbed to about the same extent as TGN-46, whereas the cis-Golgi matrix protein, GM-130, did not adsorb with the TGN-46 (Fig 4A & B). These data confirm the specificity of the immunoadsorption. Little GIPR was co-adsorbed with TGN-46 in basal adipocytes, whereas a significant amount of GIPR co-adsorbed in GIP-stimulated cells. The co-adsorption profiles of Syntaxin 6, M6PR and GM-130 were unaffected by GIP stimulation. These data demonstrate increased localization of activated GIPR in the TGN.
Fig. 4. GIPR is localized to TR-containing endosomes in basal conditions and to TGN46-containing membranes upon GIP stimulation.

(A) Immunoblot of TGN-46 immunoadsorption from basal and GIP stimulated adipocytes. TGN-46 membranes were immunoisolated using anti-TGN-46 antibody in the absence of detergent. The Input (In), unbound flow-through (FL) and elution (Bd) were run on SDS-PAGE and blotted for TGN markers Syntaxin 6 and mannose-6-phosphate receptor (M-6-P cation independent) and cis-golgi marker GM-130 and for GIPR.
(B) Quantification of (A). Relative enrichment of proteins in elution were calculated by normalizing to the flowthrough.
(C) Immunoblot of GIPR immunoadsorption from basal and GIP stimulated WT adipocytes. GIPR-GFP membranes were pulled down in absence of detergent. The elution (bound) was run on SDS-PAGE together with input and unbound (FL) and blotted for GIPR, TR and TGN-46.
(D) Quantification of (C). Relative amounts of TR or TGN in the elution were calculated by normalizing to input and GIPR in the IP.
(E) Quantification of GIP induced GIPR downregulation in syntaxin-6 (Sx-6) knockdown adipocytes. Two different siRNAs were used. Both knockdowns show a decrease in GIPR downregulation showing that it depends on Sx-6. Data are averages ± SD of 3 independent experiments., P<0.05.
(F) Exocytosis rate constants for TR and TGN vesicles compared with GIPR in basal and GIP stimulated condition. GIPR ke values are plotted from data in Fig. 1.
(G) Schematic representation of GIPR recycling from the ERC (TR-containing endosomes) or TGN compartments with associated rate constants. ki, internalization rate constant, ‘kERC’, rate from the ERC; ‘kTGN’, rate from TGN; and ‘ksort’, GIPR sorting rate from ERC to the TGN. Additional connectivities between these compartments did not significantly improve the performance of this model.
(H) Calculation of GIPR sorting rate constant (Ksort) by using equation 1 (supplementary methods) and the model in Fig. 4G. Data from Fig. 1F was used. Inset shows the value of ‘r’ calculated under basal or upon GIP stimulation.
(I) GIPR as a fraction of total in different compartments under basal or GIP stimulated conditions. The values were calculated from (F).
(J) And (K) Immunoblot of GIPR immunoadsorption from basal and GIP stimulated βA2 KD adipocytes.
As a complementary approach to investigate GIPR localization, we immunoadsorbed GIPR membranes and Western blotted for endosomes with TR and the TGN with TGN-46. In basal and GIP-stimulated adipocytes, both TR and TGN46 were immunoadsorbed with the GIPR, demonstrating GIPR is in both TR-containing endosomes and in the TGN (Fig 4C). There were, however, significant quantitative differences in the amounts of TR and TGN46 co-adsorbed between basal and GIP-stimulated adipocytes. GIP-stimulation caused a reduction of TR and a concomitant increase in TGN46 immuno-adsorbed with GIPR (Fig 4D). These data demonstrate a redistribution of GIPR from membranes enriched in TR to those enriched in TGN-46 following GIP-stimulation, establishing an altered intracellular GIPR trafficking itinerary in stimulated adipocytes correlates with the slowing of recycling.
Targeting to the TGN from endosomes could be through one of the several retrograde pathways. One of the major pathways depends on sytaxin-6 (Sx-6). Transient knockdown of Sx-6 blunted the GIP stimulated GIPR downregulation (Fig 4E and Fig S3). These data support the conclusion that GIP stimulates GIPR targeting to the TGN. These data also suggest that GIPR targeting to the TGN is via Sx-6 dependent retrograde transport.
To explore the functional consequences of the redistribution, specifically whether the change in GIPR recycling itinerary accounts for the slow recycling following GIP stimulation, we measured the recycling rate constants of the ERC and TGN pathways in adipocytes. We used a Furin-Tac chimera for analyses of the TGN pathway. Furin is a TGN resident transmembrane protease that cycles between the TGN and the PM, and the Furin-Tac chimera has been used to quantify trafficking from the TGN (Voorhees et al., 1995). The recycling rate constant of the ERC pathway (TR recycling) was about 3 times faster than the TGN pathway (Furin-Tac recycling) (Fig 4F). GIP stimulation did not affect the recycling rate constants of either the TR or Furin-Tac. The GIP-induced redistribution of GIPR from the more rapid TR recycling pathway to the slower TGN pathway can account for the GIP-induced slowing of GIPR recycling.
We developed a simple, 2-pool kinetic model based on GIPR recycling from the ERC or from the TGN, and modeled changes in the rate of GIPR trafficking from ERC to the TGN (Fig 4G). The rate constants of this 2-pool model are: ‘Ki’ (internalization from the PM), kERC (recycling rate from the ERC to PM, TR recycling pathway), kTGN (recycling rate from the TGN to PM, TGN-36 recycling pathway) and ksort (pathway from the ERC to TGN) (materials and methods). A fit of the data from Fig 1F yielded a value of ksort (GIPR ERC to TGN rate) in the basal state of near 0 (with an upper limit value of 0.02 per min, Fig S4) that increased to a value of approximately 0.22 min per min in GIP-stimulated adipocytes, revealing a pronounced stimulation of GIPR transit from the ERC to the TGN in GIPstimulated adipocytes (Fig 4H).
The model predicts nearly 60% of GIPR on the PM of basal adipocytes, which is reduced to 45% upon GIPR stimulation (Fig 4I), which is in good agreement with the approximate 30% GIP-induced downregulation of GIPR experimentally determined (e.g., Fig 1B). In fact, using the values estimated in this study, the maximum drop in surface GIPR is predicted to be ~30% (equation 11, supplementary materials and methods). In good agreement with the observed downregulation of PM GIPR measured, indicating that GIP induces the maximum downregulation achievable by the switch in recycling pathways. The kinetic model also predicts nearly all of the intracellular GIPR in basal adipocytes is in the ERC, whereas upon GIP stimulation about 60% of GIPR is in the TGN (Fig 4I). The large redistribution of GIPR to the TGN predicted by modeling is in agreement with the experimentally measured increased co-localization of GIPR and TGN46 in stimulated adipocytes (Fig 4A and D).
We hypothesize that the GIP-induced slowing of GIPR recycling requires β-arrestin2 recruitment to GIPR and subsequent rerouting of GIPR from the endosomal to TGN recycling pathway. This model predicts silencing of β-arrestin2, which abrogates GIP-induced slowing of recycling, will also inhibit the rerouting of GIPR from endosomes to the TGN. This is indeed the case, GIPR was not rerouted from TR-containing endosomes to the TGN in β-arrestin2 knockdown adipocytes, establishing a role for β-arrestin2 in GIP-induced targeting of GIPR to the TGN (Fig 4J & K).
The carboxyl cytoplasmic domain of GIPR is required for GIP-induced downregulation
The requirement for GRK 2 or 5 in β-arrestin2-dependent downregulation of GIPR suggests a role for receptor phosphorylation. A homology model based on the glucagon receptor (also a secretin GPCR) predicts the cytoplasmic domain of GIPR extending from residues 400–466. Several serines in the predicted C-terminal domain are conserved among different species, suggesting potential functional relevance of these conserved serines (Fig 5A). To determine the role of the cytoplasmic domain in GIPR trafficking, we characterized the behavior of a HA-GIPR-GFP in which the 23 carboxyl-terminal amino acids were deleted (GIPRΔ444–460). This deletion construct was distributed between peri-nuclear compartment and the PM, similar to the full length GIPR (Fig 5B). Furthermore, this deletion did not quantitatively affect the distribution of GIPR between the PM and intracellular compartments in unstimulated adipocytes, demonstrating that the constitutive basal internalization and recycling of GIPR is not dependent on the deleted sequences (Fig 5C). GIPRΔ444–460 was not downregulated by GIP stimulation, establishing a role for the carboxyl-terminal 23 amino acids in downregulation (Fig 5C). GIP stimulation of GIPRΔ444–460 activated adenylate cyclase to the same degree as WT GIPR, demonstrating the truncation does not abrogate GIP binding or activation of the receptor (Fig 5D).
Fig. 5. GIPR intracellular domain is required for GIP induced GIPR sequestration.

(A) Amino acid sequence of GIPR predicted cytoplasmic domain (residues 400 to 466). The C-terminal deletion is marked by the red bar. Serines mutated to alanines are noted by arrows. Serines conserved in human, mouse, rat and bovine GIPR are shown in green. The sequence also contains two threonines as potential phosphorylation sites. However, these threonines are not conserved in mammals.
(B) Images of GIPRΔ444-466-GFP construct in basal adipocytes. Plasma membrane GIPR was labeled by indirect immunofluorescence with anti-HA/Cy3 secondary antibody of fixed cells. Scale bar: 50μm.
(C) Quantification of downregulation of GIPRΔ444-466-GFP constructs upon GIP stimulation. The data are the PM-to-Total of GIP-stimulated normalized to the basal PM-to-Total., P<0.05.
(D) GIP-stimulated cAMP production in adipocytes expressing GIPR or GIPRΔ444-466. The endogenous GIPR was knocked down by siRNA such that cAMP production was downstream of the ectopically expressed GIPRs.
(E) GIP induced downregulation of the GIPR serine mutants. For each construct, the downregulation has been plotted as PM-to-Total normalized to their basal. The unstimulated level (basal) is shown with a dashed line.
Five potential serine phosphorylation sites are disrupted by the deletion, the 4 serines within the deleted sequences and serine at position 443 at the junction of the deletion (Fig 5A). Simultaneous mutation of these 5 serines to alanines (GIPR-5A) did not affect GIP-stimulated receptor downregulation, establishing that the deletion of the last 23 amino acids of GIPR had an effect beyond the loss of these potential phosphorylation sites (Fig 5E). Downregulation was impacted when the conserved serine at position 435 was mutated to alanine in the context of the GIPR-5A mutant (GIPR-6A), although mutation of serine 435 to alanine by itself did not affect downregulation (Fig 5E). These data are consistent with GIPR phosphorylation having a role in receptor downregulation while also demonstrating redundancy (or complexity) in the phosphorylation of GIPR required for downregulation.
GIPR-Gln354 variant is downregulated by redistribution to the TGN recycling pathway
We have previously shown differences in the post-activation trafficking of a naturally occurring human variant of the GIPR, GIPR-Gln354, in which glutamine is substituted for glutamate at residue 354 (Mohammad et al., 2014). GIP-stimulated downregulation of the variant is more pronounced than that of GIPR-Glu354 (50% versus 30%, respectively) and the time to restore pre-stimulated levels of GIPR following removal of GIPR is 4 times as long for the GIPR-Gln354 variant. These differences are due to an enhanced GIP-induced slowing of the recycling of the GIPR-Gln354 variant compared to the slowing of recycling of GIPR-Glu354 (Mohammad et al., 2014).
GIP-stimulated downregulation of the GIPR-Gln354 variant corresponds to a switch in recycling pathways, demonstrated by increased co immunoadsorption with TGN46 and a corresponding decrease in co immunoadsorption with TR upon GIP stimulation (Fig 6A and B). Thus, despite the enhanced downregulation of the GIPR-Gln354 variant, it is downregulated by the same mechanism as GIPR-Glu354, which involves GIP-stimulated targeting to the TGN recycling pathway. Consequently, the enhanced downregulation and prolonged intracellular sequestration of the variant are not explained by differences in the intracellular itineraries of GIPR and the GIPR-Gln354 variant.
Fig. 6. Redistribution of the GIPR-Gln354 variant to the TGN is dependent on GIP and βA2 and independent of GRKs.

(A) HA-GIPR-Gln354-GFP Immunoadsorption from WT adipocytes and blotted for GIPR, TR and TGN46.
(B) Quantification of data from experiments like that shown in panel A. TR and TGN46 in elution were calculated as normalized in total (input) and GIPR in the elution. Data are average ± SD from 3 independent experiments.
(C) Effect βA1 or βA2 KD on PM-to-Total distribution of GIPR-Gln354 variant. Data are normalized to their own basal values. Data are averages ± SD of 3 independent experiments., P<0.05.
(D) Effect of GRK2+5+6 triple KD on PM-to-Total distribution of GIPR-Gln354 variant. Data are averages ± SD of 3 independent experiments., P<0.05.
(E) PM-to-Total distribution of GIPR-Gln354 variant and GIPR-354Gln-Δ444-466 deletion mutant. Data are averages ± SD of 3 independent experiments., P<0.05.
(F) Co-immunoprecipitation of βA2 with GIPR-Gln354.
(G) Quantification of data from experiments like that shown in panel F. Data are average ± SD from 3 independent experiments.
(H) Relative βA2 binding to GIPR and GIPR-354Gln. GIPR and GIPR-354Gln Co-IP were blotted for GIPR and βA2 on the same blot.
(I) Quantification of data from experiments like that shown in panel H. Data are average ± SD from 3 independent experiments.
(J) GIPR-354Gln-GFP:βA2 binding in GRK2+5 double KD cells.
(K) Quantification of data from experiments like that shown in panel H. Data are average ± SD from 3 independent experiments.
GIPR-Gln354 variant downregulation is dependent on β-arrestin2 yet independent of GRKs
As is the case for the GIPR-Glu354, β-arrestin2 was required for the GIP-induced downregulation of the GIPR-Gln354 variant, and knockdown of β-arrestin1 did not affect GIP-stimulated or unstimulated GIPR-Gln354 variant trafficking (Fig 6C). However, unlike GIPR-Glu354, downregulation of GIPR-Gln354 variant was not dependent on GRKs, as simultaneous knockdown of the 3 isoforms expressed in adipocytes did not affect GIP-stimulated downregulation (Fig 6D). In agreement with GRK-independent downregulation of the GIPR-Gln354 variant, deletion of the 23 carboxyl-terminal amino acids did not affect downregulation (Fig 6E). Thus, GIP binding is sufficient to lead to the downregulation of the GIPR-Gln354 variant independent of GRK phosphorylation of the receptor.
β-Arrestin2 bound to the GIPR-Gln354 variant, as expected based on the effect of β-arrestin2 depletion on downregulation (Fig 6F). However, significant β-arrestin2 also co immunoprecipitated with the variant in unstimulated adipocytes, demonstrating β-arrestin2 binding to the variant in the absence of GIP stimulation (Fig 6F and G). To directly establish that in unstimulated adipocytes there was enhanced β-arrestin2 binding to the GIPR-Gln354 variant, relative to GIPR-Glu354, we directly compared co-immunoprecipitations on the same gel (Fig 6H and I). There was a ~3 fold increase in β-arrestin2 co-immunoprecipitated with the variant relative to GIPR. However, basal β-arrestin2 binding did not target the GIPR-Gln354 variant to the TGN in unstimulated adipocytes (Fig 6B).
Knockdown of GRKs 2 & 5, which did not block GIP-induced downregulation of the GIPR-Gln354 variant (Fig 6C), did not reduce GIP-stimulated β-arrestin2 binding to the variant (Fig 6J and K). These data establish GIP-dependent and phosphorylation-independent recruitment of β-arrestin2 to the Gln354 variant. Thus, a difference between the predominant form of GIPR and the GIPR-Gln354 variant is that binding of β-arrestin2 to the variant is not controlled by phosphorylation of the receptor.
DISCUSSION
We have previously shown non-typical trafficking behavior of GIPR (Mohammad et al., 2014). Namely, GIPR is constitutively internalized and recycled back to the PM in both unstimulated and GIP-stimulated adipocytes, cells that natively express GIPR. GIP promotes downregulation of GIPR from the PM by slowing GIPR recycling with not effect on internalization. The trafficking of GIPR has been previously studied ectopically expressed in a human embryonic kidney cell line (HEK cells), a cell type that does not natively express GIPR (Ismail et al., 2015; Tseng and Zhang, 2000; Wheeler et al., 1999). In HEK cells, GIP stimulation resulted in a rapid redistribution of GIPR from the PM to lysosomes (Ismail et al., 2015). The differences in those results and ours likely reflect the cell types used for study.
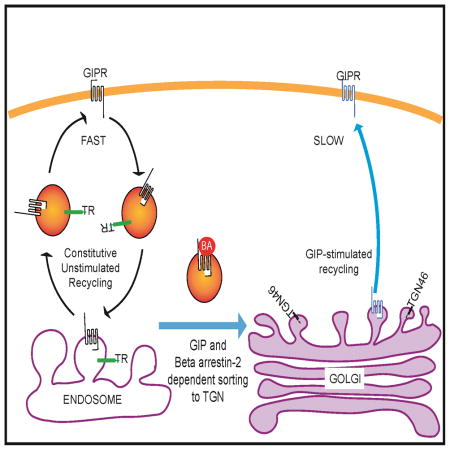
Here we have demonstrated that GIPR downregulation is achieved by a mechanism involving a pronounced GIP-induced redirection of GIPR from the rapid ERC recycling pathway to the slower TGN recycling pathway (Fig 7). This change in recycling kinetics causes a reduction in the steady state PM levels of GIPR. The amount of GIPR on the PM is controlled by GIP modulation of which of the two constitutive recycling pathways is used, without GIP affecting the kinetics of those pathways. The perinuclear endosomal and TGN compartments in the fat cells are poorly resolved by conventional light microscopy. Due to this limitation we have been unable to use microscopy to co-localize GIPR in these compartments. Instead we have used established biochemical method of immunoadsorption of the TGN or GIPR compartments to show that the GIPR indeed traffics to the TGN compartment upon GIP stimulation. Kinetic modeling revealed that the large GIP-induced redistribution of GIPR to the TGN pathway is sufficient to account for the measured 30% reduction on PM GIPR.
Fig. 7. Schematic of the GIPR trafficking pathway.

(A) GIPR is recycled constitutively by the fast TR recycling pathway (black arrows). Upon GIP stimulation, the GIPR is sorted to the slower TGN pathway (blue arrows). This redistribution of recycling pathways results in a dynamic reduction of GIPR from the plasma membrane. The redistribution of GIPR from TR to TGN pathway is regulated by β-arrestin2 (βA2) binding, which in turn is recruited in response to ligand binding and GRK mediated phosphorylation.
(B) Downregulation of the GIPR-354Gln variant is by a similar dynamic sequestration mechanism, requiring GIP stimulation and βA2 targeting of the variant from the TR to the TGN recycling pathway. However, βA2 binding to the variant is independent of GRK phosphorylation.
Other GPCRs traffic through the TGN and therefore the TGN may be a common site of GPCR sequestration. For example, activated CC chemokine receptor 5 (CCR5), somatostatin 2A receptor and β1-adrenergic receptor are all targeted, after ligand activation of internalization, to the TGN (Cheng and Filardo, 2012; Csaba et al., 2007; Escola et al., 2010). Delivery of GPCRs to the TGN may facilitate the transport of internalized GPCRs to different final destinations. For example, CCR5 and somatostatin 2A receptors, like the GIPR, are returned from the TGN to the PM, whereas the β1-adrenergic receptor is targeted from the TGN for degradation.
Knockdown of β-arrestin2 abrogates targeting of GIPR to the TGN and consequently inhibits downregulation of the receptor (Fig 1). Binding of β-arrestin2 to the predominant isoform of the human GIPR, Glu354, is dependent on GRK 2 or 5, consistent with β-arrestin2 recruitment requiring phosphorylation of GIPR (Fig 3). This redundancy in GRKs is not unexpected because GRKs are promiscuous with respect to substrate specificity (Gurevich et al., 2012; Violin et al., 2006; Zidar et al., 2009). Consistent with a role for phosphorylation, deletion or mutation of a group of serines in the carboxyl cytoplasmic domain of the GIPR blocked GIP-stimulated downregulation. Binding of β-arrestins to activated GPCRs requires more than one phosphorylation site within the receptor (Tobin, 2008). The requirement for multiple phosphorylation sites coupled with the promiscuity of GRKs in phosphorylation site specificity provides an explanation as to why mutation of multiple serines in the cytoplasmic domain of GIPR is required to inhibit downregulation (Fig 5).
β-Arrestins have a role in intracellular trafficking of other GPCRs (Kang et al., 2014; Magalhaes et al., 2012), including β-arrestin2-dependent sequestration of β1-adrenergic receptor in the TGN (Cheng and Filardo, 2012). The molecular mechanisms underlying β-arrestins’ regulation of intracellular trafficking have not been fully described. β-Arrestin2 binds to a number of proteins involved in the control of membrane protein trafficking, including clathrin, clathrin adaptor complex AP-2, and ARF6 (Kang et al., 2014). Although future studies are required to define the molecular mechanism of GIPR targeting to the TGN, the most likely scenario is that β-arrestin2 serves as an adaptor to link GIPR to machinery that mediate trafficking from ERC to the TGN, with GIPR in unstimulated cells, when β-arrestin2 is not bound, returned to the PM by the ERC recycling pathway.
The Gln354 variant is associated with altered blood glucose and insulin following glucose ingestion (Nitz et al., 2007), an increased body mass index, impaired glucose homeostasis in obese children (Sauber et al., 2010) and reduced bone mineralization density (Torekov et al., 2014). GIP-stimulated downregulation of the naturally occurring human GIPR-Gln354 variant involves β-arrestin2-dependent targeting to the TGN (Fig 6). Unexpectedly, targeting of GIPR-Gln354 variant to the TGN and its downregulation are independent of GRKs (Fig 6B). In agreement with downregulation of the GIPR-Gln354 variant being independent of receptor phosphorylation, deletion of the cluster of serines in the carboxyl cytoplasmic domain of the variant did not affect downregulation, whereas this deletion in GIPR-Glu354 inhibited downregulation (Figs 5C & 6C). These data suggest a model in which the Gln354 substitution alters the conformation of GIPR in a way that allows for GIP-induced, β-arrestin2-dependent targeting to the TGN independent of GRK phosphorylation of the receptor (Fig 7).
In unstimulated adipocytes, β-arrestin2 binding to GIPR-Gln354 is greater than binding to GIPR-Glu354, (Fig 6I). However, β-arrestin2 binding to GIPR-Gln354 in the absence of GIP is not sufficient to target the receptor to the TGN (Fig 6A). It has recently been shown that different GPCRs impose distinct conformations on bound β-arrestin2 (Lee et al., 2016; Thomsen et al., 2016). These differences in conformations correlate with differences in β-arrestin2-dependent signal transduction and or the stability of β-arrestin2 binding to the GPCR. The GPCR-specific conformation of β-arrestin2 may account for how different GPCRs use β-arrestin2 as a common effector for distinct outputs. Not only can different GPCRs have different effects on β-arrestin2, but structurally distinct ligands binding to the same GPCR can also induce different β-arrestin2 conformations (Lee et al., 2016). Our functional data suggest different conformational states of β-arrestin2 bound to GIPR-Gln354 depending on whether or not GIP is also bound to the receptor. By increasing β-arrestin2 association with GIPR in the absence of GRK phosphorylation, the Gln354 substitution might prolong the β-arrestin2 association. This is in principle similar to observations that switching carboxyl domains of GPCRs can lead to enhanced β-arrestin binding and altered GPCR trafficking (Thomsen et al., 2016). β-Arrestin2 binding to GIPR-Glu354 (the predominant isoform) is phosphorylation dependent and it will be reversed by dephosphorylation, reversing the effects of β-arrestin2 on GIPR behavior. Following GIP removal, GIPR-Glu354 repopulates the PM within 60 min, whereas it takes 4 hrs for the Gln354 variant to be restored to pre-stimulation levels (Mohammad et al., 2014). A consequence of β-arrestin2 binding to the variant independent of phosphorylation is that de-phosphorylation is not a means to accelerate the reversal of the β-arrestin2 effect on GIPR trafficking, thereby providing a mechanism for the enhanced downregulation and prolonged desensitization of the variant GIPR (Mohammad et al., 2014). Consistent with this proposal that the Gln354 substitution has an effect on GIPR trafficking beyond targeting to the TGN, kinetic modeling reveals a 30% maximum downregulation by a switch in recycling mechanism, whereas the variant is downregulated by 50% (Mohammad et al., 2014).
There are at least two retrograde pathways to the TGN, one from the endosomal recycling compartment and the other from late endosomes (Bard and Malhotra, 2006; Johannes and Popoff, 2008; Maxfield and McGraw, 2004). The dependence of GIPR downregulation on Sx-6 (Fig 4E) suggests that GIPR is sorted to the TGN by a sytaxin-6 dependent retrograde pathway from endosomal recycling compartment.
Glu354 of the GIPR, a class ‘B’ GPCR, is a structural counterpart to a Trp that is an element of an “activation micro-switch” in class ‘A’ GPCRs (Cordomi et al., 2015). The micro-switch in class A receptors links transmembrane domains (TM) 6, 7 and 2 (Cordomi et al., 2015; Xue et al., 2015). A change in the conformation of this micro-switch is important in activation of class A GPCRs (Cordomi et al., 2015; Franco et al., 2016). In GIPR, Glu354 (TM 6), Ser381 (TM 7) and Arg183 (TM 2) are structural correlates of the micro-switch, suggesting this region is important in activation of the GIPR (Cordomi et al., 2015). The naturally occurring Gln354 variant change in the micro-switch biases the GIPR to a conformation with enhanced β-arrestin2 binding and altered post-activation behavior of the receptor without activating the receptor. The altered behavior of the variant is conceptually similar to effects of biased agonists in which structurally distinct ligands binding to the same GPCR can, through different structural changes, bias signaling output. In the case of the GIPR-Gln354, a substitution in a region predicted to be important for conformational changes linked to activation, bias the GIPR structure towards binding of β-arrestin2 independent of GRK-dependent phosphorylation. Different substitutions at position 354 have different effects of the conformation of GIPR, emphasizing the importance of this site in GIPR behavior. The natural Gln354 substitution affects the conformation of GIPR in a way that results in recruitment of β-arrestin2 without constitutively activating the receptor (Mohammad et al., 2014), whereas site-directed mutagenesis to an alanine at 354 results in a constitutively active receptor (Cordomi et al., 2015).
Neither β-arrestin1 or 2, or any of the three GRKs expressed in cultured adipocytes were required for internalization of GIPR in basal or GIP-stimulated adipocytes. Although we only achieved a 65% knockdown of β-arrestin2 and therefore residual β-arrestin2 could mediate the internalization, we think this is highly unlikely since knockdown of GRK 2 and 5 results in a near complete reduction of β-arrestin2 binding to GIPR without an effect on internalization (Figs 2 & 3). The most parsimonious interpretation of the data is that GIPR is internalized by a constitutive, non-specialized endocytic mechanism in both unstimulated and GIP-stimulated adipocytes. The internalization rate constant of GIPR was unchanged by GIP stimulation; consistent with the same internalization mechanism being used in both unstimulated and stimulated adipocytes. The biological significance of GRK and β-arrestin independent internalization is not clear at this time.
MATERIALS AND METHODS
Cell culture, transfection, and electroporation
3T3-L1 fibroblasts were used after differentiation into adipocytes, and electroporated as described previously (Mohammad et al., 2014).
Quantitation of GIPR downregulation
HA-GIPR-GFP was transiently expressed in 3T3-L1 adipocytes by electroporation. The following day, the cells were serum starved for two hours prior to the GIP treatment. Cells were then either treated with 100 nM GIP for one hour (stimulated) or incubated without GIP (basal) for one hour. The cells were immediately cooled on ice and fixed with 3.7% formaldehyde. The surface GIPR was stained with anti-HA antibody followed by a Cy3 labeled secondary antibody. The cells were imaged for GFP and Cy3 staining. GFP was used to identify cells expressing the GIPR. The GFP and Cy3 fluorescence was measured for cells expressing GFP (HA-GIPR-GFP) being unbiased to Cy3. Surface associated GIPR was measured as an average of the Cy3:GFP ratio (surface staining normalized to total GIPR expression) for each cell. A minimum of 30 cells was counted. GIPR downregulation was quantified as a decrease in Cy3:GFP upon GIP treatment. Statistical significance was measured for GFP expression and Cy3 staining in cells within each dish (making sure of insignificant variation in expression and staining and identifying outliers). Statistical significance was also measured for all cells among dishes to ensure GIPR downregulation, and between experiments.
GIPR exocytosis
Cells transiently transfected with HA-GIPR-GFP used. Incubated with or without 100 nM GIP for 60 min. Cells were then incubated with anti HA antibody for times ranging from 5 min to 60 min. 100 nM GIP was included for GIP treatment group. Cells were cooled immediately, fixed and stained with Cy3 secondary after permeabilization. Cell associated anti-HA antibody was quantified as Cy3 fluorescence. Cy3 normalized to GFP (Cy3:GFP) was plotted against time. The data were fit to a single exponential rise to derive an apparent exocytic rate constant.
GIPR internalization
Cells electroporated with HA-GIPR-GFP were used. Incubated with or without 100 nM GIP for 60 min. Cells were then incubated in anti-HA antibody with or without GIP for different times from 2 min to 15 min., at the end of each time point, the cells were immediately cooled and fixed. The surface bound antibody was stained with Cy5 secondary in non-permeabilized cells. The internalized antibody was stained with Cy3, after permeabilization. The internalized anti-HA antibody was quantified at each time point as Cy3 fluorescence. Cy3:Cy5 was plotted against time, fitted on the equation for straight line. The slope of the line was calculated as the apparent rate constant of internalization.
Stable expression of HA-GIPR-GFP in adipocytes
HA-GIPR-GFP stable cells were generated by using the ViraPower lentiviral expression system (Life Technologies).
HA-GIPR-GFP co-immunoprecipitation
The method was adapted from (Bruno et al., 2016) for the immunoadsorption of GLUT4 membranes.
HA-GIPR-GFP Immunoadsorption
Cells stably expressing HA-GIPR-GFP were differentiated into adipocytes, treated with or without GIP and lysed in buffer containing 20 mM HEPES, 1 mM EDTA, 250 mM sucrose, and protease inhibitors and no detergent to prevent membranes from solubilizing. The lysate was cleared by centrifugation at 700xg and incubated with anti-GFP microbeads. The lysate containing the beads was loaded on the μMACS magnetic column. HA-GIPR-GFP membranes were eluted after washing the column and elution in 1X lammeli buffer. The lysate (input) flow through (unbound) and elution (bound) were run in SDS-PAGE and blotted for HA-GIPR-GFP, TGN46 or TR.
Data acquisition and processing
Fluorescent images were collected on a DMIRB inverted microscope (Leica Microsystems, Deerfield, IL), using a 20X objective. Fluorescence quantifications were done using MetaMorph image processing software (Molecular Devices, Sunnyvale, CA), as described previously (Blot and McGraw, 2008; Bruno et al., 2016; Mohammad et al., 2014; Sadacca et al., 2013).
Kinetic model derivation and description
The kinetic model has been described in supplementary methods.
Statistical analysis
Statistical significance was calculated by Student’s t-test.
Supplementary Material
Acknowledgments
This work was supported by DK096925 (TEM). We thank members of the McGraw lab for helpful discussions.
Footnotes
Author Contributions.
N.A., Planned and conducted experiments, interpreted data, prepared figures and edited manuscript.
M.B., Planned and conducted experiments, interpreted data, prepared figures and commented on manuscript.
D.S., Conducted experiments, interpreted data, prepared figures.
J.D., Performed kinetic modeling, interpreted data, prepared figures and edited manuscript.
T.E.M., Conceived project, planned experiments, interpreted data, prepared figures, wrote manuscript
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Banting G, Ponnambalam S. TGN38 and its orthologues: roles in post-TGN vesicle formation and maintenance of TGN morphology. Biochimica et biophysica acta. 1997;1355:209–217. doi: 10.1016/s0167-4889(96)00146-2. [DOI] [PubMed] [Google Scholar]
- Bard F, Malhotra V. The formation of TGN-to-plasma-membrane transport carriers. Annu Rev Cell Dev Biol. 2006;22:439–455. doi: 10.1146/annurev.cellbio.21.012704.133126. [DOI] [PubMed] [Google Scholar]
- Blot V, McGraw TE. GLUT4 is internalized by a cholesterol-dependent nystatin-sensitive mechanism inhibited by insulin. The EMBO journal. 2006;25:5648–5658. doi: 10.1038/sj.emboj.7601462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blot V, McGraw TE. Molecular mechanisms controlling GLUT4 intracellular retention. Mol Biol Cell. 2008;19:3477–3487. doi: 10.1091/mbc.E08-03-0236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruno J, Brumfield A, Chaudhary N, Iaea D, McGraw TE. SEC16A is a RAB10 effector required for insulin-stimulated GLUT4 trafficking in adipocytes. J Cell Biol. 2016;214:61–76. doi: 10.1083/jcb.201509052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng SB, Filardo EJ. Trans-Golgi Network (TGN) as a regulatory node for beta1-adrenergic receptor (beta1AR) down-modulation and recycling. The Journal of biological chemistry. 2012;287:14178–14191. doi: 10.1074/jbc.M111.323782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claing A, Laporte SA, Caron MG, Lefkowitz RJ. Endocytosis of G protein-coupled receptors: roles of G protein-coupled receptor kinases and beta-arrestin proteins. Progress in neurobiology. 2002;66:61–79. doi: 10.1016/s0301-0082(01)00023-5. [DOI] [PubMed] [Google Scholar]
- Cordomi A, Navarro G, Aymerich MS, Franco R. Structures for G-Protein-Coupled Receptor Tetramers in Complex with G Proteins. Trends in biochemical sciences. 2015;40:548–551. doi: 10.1016/j.tibs.2015.07.007. [DOI] [PubMed] [Google Scholar]
- Csaba Z, Lelouvier B, Viollet C, El Ghouzzi V, Toyama K, Videau C, Bernard V, Dournaud P. Activated somatostatin type 2 receptors traffic in vivo in central neurons from dendrites to the trans Golgi before recycling. Traffic. 2007;8:820–834. doi: 10.1111/j.1600-0854.2007.00580.x. [DOI] [PubMed] [Google Scholar]
- Dupre J, Ross SA, Watson D, Brown JC. Stimulation of insulin secretion by gastric inhibitory polypeptide in man. The Journal of clinical endocrinology and metabolism. 1973;37:826–828. doi: 10.1210/jcem-37-5-826. [DOI] [PubMed] [Google Scholar]
- Escola JM, Kuenzi G, Gaertner H, Foti M, Hartley O. CC chemokine receptor 5 (CCR5) desensitization: cycling receptors accumulate in the trans-Golgi network. The Journal of biological chemistry. 2010;285:41772–41780. doi: 10.1074/jbc.M110.153460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco R, Martinez-Pinilla E, Lanciego JL, Navarro G. Basic Pharmacological and Structural Evidence for Class A G-Protein-Coupled Receptor Heteromerization. Frontiers in pharmacology. 2016;7:76. doi: 10.3389/fphar.2016.00076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman OB, Jr, Krupnick JG, Santini F, Gurevich VV, Penn RB, Gagnon AW, Keen JH, Benovic JL. Beta-arrestin acts as a clathrin adaptor in endocytosis of the beta2-adrenergic receptor. Nature. 1996;383:447–450. doi: 10.1038/383447a0. [DOI] [PubMed] [Google Scholar]
- Gurevich EV, Tesmer JJ, Mushegian A, Gurevich VV. G protein-coupled receptor kinases: more than just kinases and not only for GPCRs. Pharmacology & therapeutics. 2012;133:40–69. doi: 10.1016/j.pharmthera.2011.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanyaloglu AC, von Zastrow M. Regulation of GPCRs by endocytic membrane trafficking and its potential implications. Annual review of pharmacology and toxicology. 2008;48:537–568. doi: 10.1146/annurev.pharmtox.48.113006.094830. [DOI] [PubMed] [Google Scholar]
- Ismail S, Dubois-Vedrenne I, Laval M, Tikhonova IG, D’Angelo R, Sanchez C, Clerc P, Gherardi MJ, Gigoux V, Magnan R, et al. Internalization and desensitization of the human glucose-dependent-insulinotropic receptor is affected by N-terminal acetylation of the agonist. Molecular and cellular endocrinology. 2015;414:202–215. doi: 10.1016/j.mce.2015.07.001. [DOI] [PubMed] [Google Scholar]
- Johannes L, Popoff V. Tracing the retrograde route in protein trafficking. Cell. 2008;135:1175–1187. doi: 10.1016/j.cell.2008.12.009. [DOI] [PubMed] [Google Scholar]
- Kandror KV, Pilch PF. Isolation of GLUT4 storage vesicles. Curr Protoc Cell Biol. 2006;Chapter 3(Unit 3):20. doi: 10.1002/0471143030.cb0320s30. [DOI] [PubMed] [Google Scholar]
- Kang DS, Tian X, Benovic JL. Role of beta-arrestins and arrestin domain-containing proteins in G protein-coupled receptor trafficking. Current opinion in cell biology. 2014;27:63–71. doi: 10.1016/j.ceb.2013.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karylowski O, Zeigerer A, Cohen A, McGraw TE. GLUT4 is retained by an intracellular cycle of vesicle formation and fusion with endosomes. Molecular biology of the cell. 2004;15:870–882. doi: 10.1091/mbc.E03-07-0517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger KM, Daaka Y, Pitcher JA, Lefkowitz RJ. The role of sequestration in G protein-coupled receptor resensitization. Regulation of beta2-adrenergic receptor dephosphorylation by vesicular acidification. The Journal of biological chemistry. 1997;272:5–8. doi: 10.1074/jbc.272.1.5. [DOI] [PubMed] [Google Scholar]
- Lee MH, Appleton KM, Strungs EG, Kwon JY, Morinelli TA, Peterson YK, Laporte SA, Luttrell LM. The conformational signature of beta-arrestin2 predicts its trafficking and signalling functions. Nature. 2016;531:665–668. doi: 10.1038/nature17154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefkowitz RJ, Rajagopal K, Whalen EJ. New roles for beta-arrestins in cell signaling: not just for seven-transmembrane receptors. Molecular cell. 2006;24:643–652. doi: 10.1016/j.molcel.2006.11.007. [DOI] [PubMed] [Google Scholar]
- Lefkowitz RJ, Shenoy SK. Transduction of receptor signals by beta-arrestins. Science. 2005;308:512–517. doi: 10.1126/science.1109237. [DOI] [PubMed] [Google Scholar]
- Lohse MJ, Hoffmann C. Arrestin interactions with G protein-coupled receptors. Handb Exp Pharmacol. 2014;219:15–56. doi: 10.1007/978-3-642-41199-1_2. [DOI] [PubMed] [Google Scholar]
- Magalhaes AC, Dunn H, Ferguson SS. Regulation of GPCR activity, trafficking and localization by GPCR-interacting proteins. British journal of pharmacology. 2012;165:1717–1736. doi: 10.1111/j.1476-5381.2011.01552.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maxfield FR, McGraw TE. Endocytic recycling. Nat Rev Mol Cell Biol. 2004;5:121–132. doi: 10.1038/nrm1315. [DOI] [PubMed] [Google Scholar]
- McIntosh CH, Widenmaier S, Kim SJ. Glucose-dependent insulinotropic polypeptide (Gastric Inhibitory Polypeptide; GIP) Vitamins and hormones. 2009;80:409–471. doi: 10.1016/S0083-6729(08)00615-8. [DOI] [PubMed] [Google Scholar]
- Mohammad S, Patel RT, Bruno J, Panhwar MS, Wen J, McGraw TE. A naturally occurring GIP receptor variant undergoes enhanced agonist-induced desensitization, which impairs GIP control of adipose insulin sensitivity. Mol Cell Biol. 2014;34:3618–3629. doi: 10.1128/MCB.00256-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitz I, Fisher E, Weikert C, Burwinkel B, Li Y, Mohlig M, Boeing H, Schreiber S, Schrezenmeir J, Doring F. Association analyses of GIP and GIPR polymorphisms with traits of the metabolic syndrome. Molecular nutrition & food research. 2007;51:1046–1052. doi: 10.1002/mnfr.200700048. [DOI] [PubMed] [Google Scholar]
- Puertollano R, van der Wel NN, Greene LE, Eisenberg E, Peters PJ, Bonifacino JS. Morphology and dynamics of clathrin/GGA1-coated carriers budding from the trans-Golgi network. Mol Biol Cell. 2003;14:1545–1557. doi: 10.1091/mbc.02-07-0109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadacca LA, Bruno J, Wen J, Xiong W, McGraw TE. Specialized sorting of GLUT4 and its recruitment to the cell surface are independently regulated by distinct Rabs. Molecular biology of the cell. 2013;24:2544–2557. doi: 10.1091/mbc.E13-02-0103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauber J, Grothe J, Behm M, Scherag A, Grallert H, Illig T, Hinney A, Hebebrand J, Wiegand S, Gruters A, et al. Association of variants in gastric inhibitory polypeptide receptor gene with impaired glucose homeostasis in obese children and adolescents from Berlin. European journal of endocrinology / European Federation of Endocrine Societies. 2010;163:259–264. doi: 10.1530/EJE-10-0444. [DOI] [PubMed] [Google Scholar]
- Saxena R, Hivert MF, Langenberg C, Tanaka T, Pankow JS, Vollenweider P, Lyssenko V, Bouatia-Naji N, Dupuis J, Jackson AU, et al. Genetic variation in GIPR influences the glucose and insulin responses to an oral glucose challenge. Nature genetics. 2010;42:142–148. doi: 10.1038/ng.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shenoy SK, Modi AS, Shukla AK, Xiao K, Berthouze M, Ahn S, Wilkinson KD, Miller WE, Lefkowitz RJ. Beta-arrestin-dependent signaling and trafficking of 7-transmembrane receptors is reciprocally regulated by the deubiquitinase USP33 and the E3 ligase Mdm2. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:6650–6655. doi: 10.1073/pnas.0901083106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JS, Rajagopal S. The beta-Arrestins: Multifunctional Regulators of G Protein-coupled Receptors. The Journal of biological chemistry. 2016;291:8969–8977. doi: 10.1074/jbc.R115.713313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomsen AR, Plouffe B, Cahill TJ, 3rd, Shukla AK, Tarrasch JT, Dosey AM, Kahsai AW, Strachan RT, Pani B, Mahoney JP, et al. GPCR-G Protein-beta-Arrestin Super-Complex Mediates Sustained G Protein Signaling. Cell. 2016;166:907–919. doi: 10.1016/j.cell.2016.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobin AB. G-protein-coupled receptor phosphorylation: where, when and by whom. British journal of pharmacology. 2008;153(Suppl 1):S167–176. doi: 10.1038/sj.bjp.0707662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torekov SS, Harslof T, Rejnmark L, Eiken P, Jensen JB, Herman AP, Hansen T, Pedersen O, Holst JJ, Langdahl BL. A functional amino acid substitution in the glucose-dependent insulinotropic polypeptide receptor (GIPR) gene is associated with lower bone mineral density and increased fracture risk. The Journal of clinical endocrinology and metabolism. 2014;99:E729–733. doi: 10.1210/jc.2013-3766. [DOI] [PubMed] [Google Scholar]
- Tseng CC, Zhang XY. Role of G protein-coupled receptor kinases in glucose-dependent insulinotropic polypeptide receptor signaling. Endocrinology. 2000;141:947–952. doi: 10.1210/endo.141.3.7365. [DOI] [PubMed] [Google Scholar]
- Violin JD, Ren XR, Lefkowitz RJ. G-protein-coupled receptor kinase specificity for beta-arrestin recruitment to the beta2-adrenergic receptor revealed by fluorescence resonance energy transfer. The Journal of biological chemistry. 2006;281:20577–20588. doi: 10.1074/jbc.M513605200. [DOI] [PubMed] [Google Scholar]
- Voorhees P, Deignan E, van Donselaar E, Humphrey J, Marks MS, Peters PJ, Bonifacino JS. An acidic sequence within the cytoplasmic domain of furin functions as a determinant of trans-Golgi network localization and internalization from the cell surface. The EMBO journal. 1995;14:4961–4975. doi: 10.1002/j.1460-2075.1995.tb00179.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler MB, Gelling RW, Hinke SA, Tu B, Pederson RA, Lynn F, Ehses J, McIntosh CH. Characterization of the carboxyl-terminal domain of the rat glucose-dependent insulinotropic polypeptide (GIP) receptor. A role for serines 426 and 427 in regulating the rate of internalization. J Biol Chem. 1999;274:24593–24601. doi: 10.1074/jbc.274.35.24593. [DOI] [PubMed] [Google Scholar]
- Xue L, Rovira X, Scholler P, Zhao H, Liu J, Pin JP, Rondard P. Major ligand-induced rearrangement of the heptahelical domain interface in a GPCR dimer. Nature chemical biology. 2015;11:134–140. doi: 10.1038/nchembio.1711. [DOI] [PubMed] [Google Scholar]
- Zidar DA, Violin JD, Whalen EJ, Lefkowitz RJ. Selective engagement of G protein coupled receptor kinases (GRKs) encodes distinct functions of biased ligands. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:9649–9654. doi: 10.1073/pnas.0904361106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.