

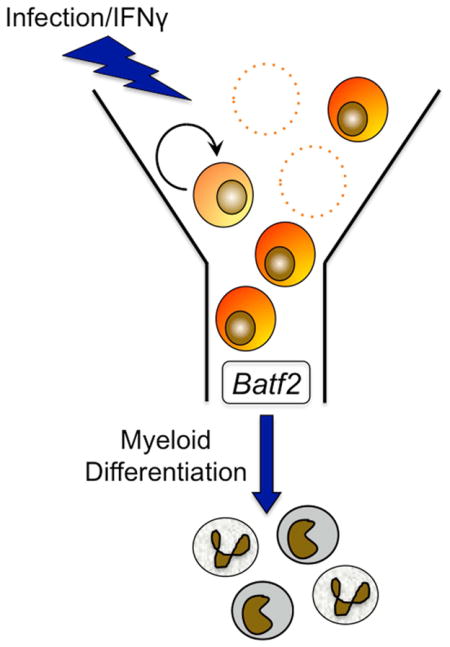
Summary
Chronic infections affect a third of the world’s population and can cause bone marrow suppression, a severe condition that increases mortality from infection. To uncover the basis for infection-associated bone marrow suppression, we conducted repeated infection of WT mice with Mycobacterium avium. After 4–6 months, mice became pancytopenic. Their hematopoietic stem and progenitor cells (HSPCs) were severely depleted and displayed interferon gamma (IFNγ) signaling-dependent defects in self-renewal. There was no evidence of increased HSPC mobilization or apoptosis. However, consistent with known effects of IFNγ, transcriptome analysis pointed towards increased myeloid differentiation of HSPCs and revealed the transcription factor Batf2 as a potential mediator of IFNγ-induced HSPC differentiation. Gain and loss of function studies uncovered a role for Batf2 in myeloid differentiation in both murine and human systems. We thus demonstrate that chronic infection can deplete HSPCs and identify BATF2 as a mediator of infection-induced HSPC terminal differentiation.
Keywords: Hematopoietic stem cell, chronic infection, pancytopenia, bone marrow failure, interferon gamma, terminal differentiation
Graphical Abstract
Introduction
Chronic infections including tuberculosis (2 billion infected worldwide; CDC), hepatitis C virus (180 million; WHO), and HIV (34 million; NIH) are estimated to affect over a third of the world’s population. These diseases are associated with significant health implications including bone marrow suppression and an increased risk for cancer (Ramos-Casals et al., 2003; Scadden et al., 1989). Pancytopenia, a suppression of blood counts across multiple lineages, can affect as many as 12% of people with disseminated or miliary tuberculosis, and increases risk of death from the infection (Achi et al., 2013). Collectively, these observations suggest that chronic infections may significantly affect the function of hematopoietic stem cells (HSCs), the progenitor cells of all blood cells. Thus, understanding the long-term effects of inflammation on HSC number and function is a matter of key clinical importance.
Production of blood cells by the bone marrow is a highly dynamic process. An estimated 1011–1012 blood cells are produced by hematopoietic progenitors in the bone marrow on a daily basis, and this number increases during infection (Takizawa et al., 2012). Primitive hematopoietic stem and progenitor cells (HSPCs) are the precursors of all cells of the peripheral blood (PB) and are responsible for maintaining healthy blood production. HSPCs include not only long-term HSCs, which have self-renewal capacity, but also multipotent progenitors (MPPs), which do not. Recent work suggests that HSPCs can generate restricted subsets of terminally differentiated progeny, bypassing the stepwise progression through common myeloid progenitor (CMP) and common lymphoid progenitor (CLP) stages (Notta et al., 2016). Thus, directed differentiation of blood cells may be driven by signals that act at the earliest stages of the hematopoietic hierarchy.
In fact, HSCs are highly responsive to the inflammatory conditions that exist during a typical infection (Takizawa et al., 2012); and a variety of signals can affect HSC function during infection. HSCs express pathogen pattern recognition receptors such as Toll-like receptor 4. Bacterial products sensed by these receptors alter HSC quiescence and function (Balmer et al., 2014; Nagai et al., 2006). Inflammatory signals propagated by the immune system can also affect HSC function, and interferons (IFN) are particularly powerful regulators of HSCs. For example, IFNα signaling can promote HSC division (Essers et al., 2009; Pietras et al., 2014). We previously showed that IFNγ stimulates cell division of HSCs in a murine model of Mycobacterium avium infection, leading to a defect in repopulation capacity (Baldridge et al., 2010). Furthermore, short-term IFNγ-mediated HSC division appears to accelerate differentiation during infection (Matatall et al., 2014); however, both the long-term impact and the mechanism of these processes remains unknown.
Here we capitalize on our murine model of M. avium infection to evaluate the impact of chronic infection on the HSC pool. M. avium typically produces a chronic (~10 weeks) systemic IFNγ-mediated immune response in mice similar to what can be seen in patients with tuberculosis (Flórido et al., 2005). We show for the first time that chronic infection drives exhaustion of the HSC compartment, with depletion of both PB counts and HSC self-renewal capacity. We use this model to evaluate the mechanisms of HSC loss and identify a new potential mediator of stress-induced myeloid specification. Our study thus provides direct evidence for how infections and persistent inflammation affect the HSC population and elicit diseases associated with HSC loss.
Results
Chronically infected mice develop pancytopenia
To characterize the effects of chronic infection on bone marrow function, we conducted repeated monthly infections of mice with M. avium. PB of infected animals showed a progressive decline in all cell types, reaching statistical significance by 1–2 months of infection (Figure 1A–C). Several mice appeared nearly moribund after 6 months. After 4 months, the hematopoietic effects of infection were irreversible (Figure 1D).
Figure 1. Chronically infected mice develop pancytopenia and severe HSC loss.

Mice were infected with M. avium every 4 weeks for 1 to 6 months. Bone marrow and PB were assessed 4 weeks after the final injection. (A) White blood cell (WBC), (B) Red blood cell (RBC), and (C) Platelet counts decline with chronic infection. (D) WBC counts do not recover following cessation of infections in 4-month infected mice. (E–F) The number of HSCs (KL CD150+ CD48− CD34−) after repeated M. avium infections. (E) % of live WBM cells. (F) Absolute number per bone. (G) Total engraftment of PB, shown as % CD45.2 cells of total blood, 16 weeks after transplant. 2x105 WBM cells from naïve or infected animals (CD45.2) were mixed with 2x105 rescue marrow (CD45.1) and transplanted into lethally irradiated mice. (H) Progenitor populations in the bone marrow of naïve and 6 month infected mice, shown as absolute number of cells per bone. Data are presented as mean ± SEM; * p<0.05, ** p<0.01, *** p<0.001. Data are representative of 2 (A–C, F–G), 3 (E), or 4 (D) independent experiments; n=3–5 per group. See also Figure S1 and Table S1.
Chronic infection depletes HSCs
We characterized hematopoietic progenitors in the bone marrow of chronically infected animals. The number of phenotypically defined long-term HSCs (LK CD150+ CD48− CD34−) declined during the course of infection; by 4 months of infection only 5.7% of the starting number of HSCs remained (Figures 1E&F and S1B&C). This decline in stem cell number outstripped the rate of weight loss (Figure S1A), suggesting that stem cell loss was not due to nutritional deficit. The stem cell marker Sca1 was intentionally excluded because it can be non-specifically activated during infection (Baldridge et al., 2011). In addition, we found that the number of myeloid-biased HSCs was more depressed than the lymphoid-biased HSCs (Figure S1D) (Matatall et al., 2014). This decrease was also manifest by a decline in the absolute number of myeloid cells derived from transplanted marrow (Figure S1E). There was a consistent rebound in the number of HSCs as a percentage of WBM at two months post-infection across 4 repetitions of the chronic infection experiment (Figure 1E), suggesting that the bone marrow can initially adapt to inflammatory responses but that such compensatory processes are eventually overwhelmed.
To assess the number of functionally defined HSCs in chronically infected animals, we transplanted 2x105 WBM cells from infected animals with 2x105 rescue marrow into lethally irradiated naïve recipient animals. As shown in Figure 1G, WBM engraftment declined after chronic infection, mirroring the decline in phenotypically defined HSCs. To determine whether reduced engraftment was due to transmitted infection, we cultured WBM cells in methylcellulose, which does not support the growth of mycobacteria. WBM of chronically infected animals generated significantly fewer total cells after 9 days of incubation compared to control WBM, suggesting that loss of HSPCs was not due to direct infection of the cells (Figure S1F). Collectively these data indicate that HSCs are depleted during chronic infection.
We quantified the presence of committed lymphoid and myeloid progenitors in the marrow during chronic infection. After 6 months of infection, the number of CLPs was steady, but the number of myeloid progenitors including CMPs, granulocyte-monocyte progenitors (GMPs), and megakaryocyte-erythrocyte progenitors (MEPs) was reduced (Figure 1G), indicating myeloid progenitors are more easily exhausted during chronic M. avium infection than lymphoid progenitors.
HSCs from chronically infected animals show a self-renewal defect upon secondary transplant
To ascertain whether cell-autonomous defects occur in HSC function upon chronic infection, we sorted LT-HSCs (SPLSK CD150+) from naïve or infected animals and transplanted 300 cells along with rescue marrow into lethally irradiated recipients. As shown in Figure 2A, sorted LT-HSCs were equally capable of reconstituting the marrow of recipient animals at 16 weeks post-transplant, regardless of infection. Lineage distribution of cells derived from transplanted cells was not affected by chronic infection (Figure 2B). These findings indicate that while the total number of LT-HSCs was decreased in chronically infected animals, their ability to reconstitute long-term hematopoiesis upon primary transplantation was not impaired.
Figure 2. HSCs from chronically infected animals have a self-renewal defect.

(A–B) 300 sorted CD45.2 HSCs (SPLSK CD150+) from naïve or M. avium-infected mice were transplanted with 2x105 CD45.1 rescue marrow into lethally irradiated mice. Donor mice were infected with M. avium 3, 4, 5, or 6 times (once every 4 weeks); the 4 and 5 dose mice were pooled before transplant. PB was assessed at 16 weeks post transplant. (A) Total engraftment is shown as % of CD45.2 cells in total blood. (B) Lineage distribution of T cells, B cells and myeloid cells; shown as % of total CD45.2 cells. (C) Secondary transplant of 250 sorted CD45.2 HSCs (SP LSK CD150+) from previously transplanted animals. Donor HSCs were co-transplanted with 2x105 CD45.1 rescue marrow into lethally irradiated mice. PB was assessed 16 weeks post transplant and engraftment is shown as % of CD45.2 cells in blood. Data are presented as mean ± SEM; * p<0.05. Except for secondary transplant done once with n=5 per group, data are representative of 2 independent experiments with n=5 per group.
To evaluate the self-renewal capacity of HSCs from infected animals, we conducted secondary transplant. Secondary engraftment of sorted HSCs from chronically infected mice was significantly diminished compared to HSCs from naïve animals, and HSCs from animals that had been infected for the longest time were most severely affected (Figure 2C). Thus, secondary transplants revealed a self-renewal defect in HSCs from chronically infected mice, indicating that HSC exhaustion can occur following persistent infectious stimulation.
Loss of HSCs precedes marrow fibrosis
Most medical textbooks ascribe pancytopenia associated with chronic infections such as tuberculosis to marrow fibrosis (Fitzgerald and Haas, 2005). However, a causal relationship between myelofibrosis and bone marrow suppression during infection has not been firmly established (Viallard et al., 2002). Using trichrome staining, we found that patches of marrow fibrosis became evident after only 1 month of infection, but remained limited to small areas of the marrow through 6 months of infection (Figure S2A). Overall, marrow of infected mice demonstrated progressively reduced cellularity (Figure S2B), but neither the loss in cellularity nor the degree of fibrosis was sufficient to account for the ~95% loss in HSCs by 4 months of infection. H&E staining showed a relative increase of granulocytes and monocytes which was confirmed by flow cytometry (Figures 3A&B and S2C&S3). Meanwhile, the absolute number of lymphoid cells in the bone marrow declined, with reductions in B and T cells (Figure 3C and S2D), and all classes of B cell precursors and immature T cells (Figure S2E&F). Altogether, these findings suggest that the rate of HSC loss outpaces the rate of marrow fibrosis and that inflammatory changes, including a relative increase in neutrophils and monocytes, can be seen during chronic infection.
Figure 3. Myeloid cells infiltrate bone marrow during chronic infection.

(A) H&E stained sections of bone marrow from naïve and chronically infected animals. (B–C) Flow cytometry of WBM cells from naïve, 1 month or 4 month infected animals. Absolute number of cells per bone is shown. Data in (B–C) are presented as mean ± SEM; * p<0.05, ** p<0.01, *** p<0.001, **** p<0.0001. Data are representative of 2 independent experiments with n=4–7 per group. See also Figure S2&S3.
Impaired HSC engraftment during M. avium infection is IFNγ-dependent
IFNγ is a key immune mediator during mycobacterial infections and we previously showed that IFNγ alone can induce HSC division and differentiation (Baldridge et al., 2010). Here, we show that IFNγ levels remained high in the serum of infected animals even after 6 months of infection (Figure 4A). We also demonstrate that IFNγ is highly expressed by both T and NK cells in the bone marrow of chronically infected animals (S4A).
Figure 4. Impaired HSC engraftment after M. avium infection is IFNγ-dependent and does not involve HSC mobilization.

(A) Mice were infected with M. avium every 4 weeks for 1 to 6 months. IFNγ levels in serum of chronically infected mice determined by ELISA. One experiment with n=2–5 per group. (B) Engraftment of 2x105 WBM cells from WT or Ifngr1−/− mice that were naïve or infected for one month with M. avium. One experiment with n=9–10 per group. (C) Engraftment of WT or Ifngr1−/− mosaic transplants at 2 weeks pre- and 12 weeks post-infection. Mice were transplanted with either 2x105 WT or 3x105 Ifngr1−/− bone marrow with 2x105 rescue marrow. Transplant recipients were infected with M. avium 6 weeks post transplant. Data represent 4 independent experiments with n=7–14 per group. (D) The number of HSCs in the spleen was assessed in mice that had been treated with G-CSF and/or IFNγ. Shown as % Lin-cKit+CD150+CD48– cells of total leukocytes. Data represent 2 independent experiments with n=2–3 per group. Data are presented as mean ± SEM; * p<0.05, ** p<0.01, *** p<0.001, n.s. not significant. See also Figure S4.
To investigate the role of IFNγ in impaired HSC function during infection, we infected WT or Ifngr1−/− mice with M. avium and then transplanted their marrow into WT recipients. We found that infection reduced the engraftment of HSCs from WT animals. The trend was reversed in infected Ifngr1−/− mice, with WBM from these mice engrafting at least as well as uninfected WT cells (Figure 4B). These findings suggest that IFNγ-dependent effects impair the capacity of WT marrow to engraft following infection. To confirm these findings, we created mosaic animals by transplanting either WT or Ifngr1−/− marrow into lethally irradiated WT recipients. Six weeks after transplant, we infected test animals with M. avium, whereas control animals were uninfected. Engraftment was significantly curtailed in recipients of infected WT marrow. In contrast, infection did not affect engraftment in Ifngr1−/− marrow recipients (Figure 4C). Collectively, these findings suggest that functional deficits in HSCs during M. avium infection are attributable to IFNγ exposure via a cell-autonomous mechanism.
Mathematical modeling of HSC loss during chronic infection
The decline in total HSC number during chronic infection implies an increase in the rate of HSC loss. To estimate the magnitude of HSC loss during infection, we created a mathematical model of hematopoiesis using differential equations based on steady state parameters: total number of HSCs, division rate, rate of self-renewal versus loss, and time (c.f: Estimation of HSC loss, in Methods). We assumed that HSCs can be lost through death, differentiation, or displacement. Thus, we used an aggregate variable d to reflect the overall proportion of HSCs lost per division event, where d=0.5 indicates complete self-renewal and d=1 indicates complete loss (no progeny are HSCs). We applied our data from chronic infection to this model and concluded that the d must increase from d=0.5 at the steady state to a median value of 0.788 during chronic infection to account for the dramatic loss of HSCs observed (Figure S1G). In other words, whereas at steady state each HSC division produces one new HSC and one cell that is “lost” through differentiation, displacement, or death, during infection the percentage of HSC loss goes up by 57%. As might be expected, we observed a very large inter-individual heterogeneity of the response, as reflected in the wide 95% confidence interval (0.729–0.954) for d (Figure S1H and Table S1).
Infection does not induce HSC mobilization
We sought to understand the mechanism by which HSCs were lost during chronic infection. We previously reported that LSK CD150+ CD48− cells are increased in the spleens of M. avium-infected animals, suggesting that displacement from the bone marrow niche is a potential mechanism for HSC loss (Baldridge et al., 2010). However, a more recent report indicated that HSCs are not mobilized from bone marrow during chronic E. chafeensis infection (McCabe et al., 2015). We therefore performed mobilization studies using IFNγ and granulocyte colony-stimulating factor (G-CSF). While G-CSF increased the number of HSCs (LK CD150+ CD48− CD34− Flk2−) in the spleen as expected, IFNγ treatment did not. Co-administration of IFNγ with G-CSF did not further boost the number of HSCs detectible in the spleen (Figure 4D). These results were confirmed upon transplantation of splenocytes from animals after G-CSF or IFNγ treatment (Figure S4B), suggesting that IFNγ does not induce HSC mobilization from the bone marrow.
HSCs from chronically infected animals are not actively apoptotic
We next considered cell death as a potential mechanism of HSC loss during chronic infection. We previously reported that HSCs are induced to divide during infection (Baldridge et al., 2010), and a recent report linked increased cell division with accumulation of DNA damage and attrition of HSCs by apoptosis (Walter et al., 2015). Furthermore, cell death can be triggered in HSCs upon secondary stress after IFNα stimulation(Pietras et al., 2014). Therefore, we investigated whether persistent infection could impose increased replication stress on HSCs, leading to increased apoptosis. We confirmed that cell cycle activity, determined by Ki67 staining, was high in HSCs even after months of infection (Figure 5A). Next, we measured reactive oxygen species (ROS) in the HSCs of infected animals. Compared to HSCs from naïve animals, we saw no increase in ROS in LK CD150+ CD48− HSCs (Figure 5B). However, we did note a slight increase in DNA damage in HSCs from chronically infected mice as measured by pKap1 staining and γH2AX staining (Figure 5C and 5D), but these increases were minor compared to the degree of DNA damage measured in the positive controls.
Figure 5. HSCs from chronically infected animals have increased stress but no increase in rate of apoptosis.

(A) Ki67+ HSCs shown as % of total HSCs (LK CD150+ CD48− CD34−) in naïve, 1 month or 3 month M. avium-infected mice. Data represent 2 independent experiments, with n=6–10 per group. (B) ROS levels in HSCs of naïve and 4 week M. avium infected mice shown as a mean fluorescent intensity (MFI) of CellRox staining. TBHP was the positive control. Data represent 2 independent experiments, n=6–8 per group. (C) DNA damage was assessed using phospho-Kap1 (pKap1) staining of HSCs from naïve or M. avium-infected mice. UV-treated cells were used as positive control. Data are representative of 2 independent studies, n=3–5 per group. (D) % of HSCs with γH2AX staining. Numbers above the bars refer to n for each condition. 1Gy irradiation was used as a positive control. Data are representative of 2 independent experiments. (E–F) Caspase 3/7 activity in 400 HSCs after 12hrs of in vitro culture. (E) HSCs from 1 month or 3 month infected mice. (F) HSCs from control or 24hr IFNγ-treated mice. (G) Caspase 3/7 activity immediately after isolation of 400 HSCs from naïve or M. avium-infected mice. Results for E–G are representative of 2–5 independent experiments, n=3 per group. Data are presented as mean ± SEM; * p<0.05, ** p<0.01, *** p<0.001, n.s. not significant. See also Figure S5.
To test for activation of apoptosis upon secondary stress, we incubated HSCs for 12-hours ex vivo and then measured caspase activation. Following this secondary stress, apoptosis was increased in HSCs from mice that had been infected for 1 or 3 months compared to controls (Figure 5E). To ascertain whether stress-induced apoptosis is related to IFNγ-stimulation, we treated mice with recombinant IFNγ for 24 hours and then incubated HSCs from these mice for 12-hours ex vivo. The IFNγ-treated cells showed elevated caspase activation compared to HSCs from control mice (Figure 5F). We also compared the effect of M. avium infection on stress-induced apoptosis in WT versus Ifngr1−/− mice. In contrast to WT, HSCs from infected Ifngr1−/− mice did not show stress-induced apoptosis, again highlighting that this effect is IFNγ-dependent (Figure S5A).
As a secondary measure of cell survival, we sorted HSCs singly into 96-well plates containing complete stem cell media. The number of colonies formed under these stressful ex vivo conditions was diminished for cells derived from mice that had been infected for 1 or 4 months or from animals treated with IFNγ, consistent with a lower threshold for apoptosis in these cells (Figure S5B). These findings indicate that chronic M. avium infection or transient IFNγ exposure can make HSCs more susceptible to apoptosis upon secondary stress.
As HSCs from infected or IFNγ-treated animals had an increased propensity for apoptosis following ex vivo secondary stress, we investigated whether the rate of apoptosis was increased during infection in vivo. We isolated HSCs from control or infected mice and immediately measured caspase activation (Figure 5G and S5C). Caspase activation was not noted in HSCs from naïve or infected mice immediately after cell sorting and was far from the level that would have been required by mathematical models to account for the loss in HSC number. Thus, even though IFNγ stimulation can lower the threshold for apoptosis in HSCs, an increased rate of apoptosis was not detectible among HSCs during in vivo infection. Therefore, we conclude that apoptosis alone cannot account for significant loss of HSCs observed during chronic infection in vivo (Figure S1G).
Transcriptional profiling of HSCs from chronically infected animals reveals increased differentiation
To further characterize potential mechanisms of HSC loss during chronic infection, we sorted HSCs from naïve mice and mice infected for 1 month with M. avium and conducted whole transcriptome analysis by RNA sequencing (Figure 6A and Table S2). HSCs from infected animals demonstrated increased expression of Stat1, Cxcl9, and the interferon responsive GTPase Irgm1, consistent with an ongoing TH1-type inflammatory response (Figure 6B) (King et al., 2011). Gene ontology analysis of the RNA-Seq data revealed a strong upregulation of genes associated with antigen processing and presentation and immune responses including major histocompatibility complex II genes, suggesting a shift toward gene expression signatures of myeloid cells such as macrophages and dendritic cells (Figure 6C). Genes related to apoptosis and cell death were strikingly absent, further supporting the lack of apoptosis noted in the studies described above. Upon gene set enrichment analysis (GSEA), the only major category significantly enriched was antigen processing and presentation (Figure 6D), pointing towards myeloid differentiation as the major transcriptional change occurring in HSCs during infection.
Figure 6. Transcriptional profiling of HSCs from chronically infected animals reveals increased differentiation.

(A) Heatmap of differentially expressed genes (FDR < 0.1) from RNA-Seq data of naïve compared to M. avium infected HSCs. (B) Fold change in the normalized read counts of Stat1, Irgm1, Cxcl9, and Batf2 between M. avium infected and naïve HSCs. (C) Gene ontology (GO) analysis of RNA-Seq data from HSCs isolated from naïve and 4 week M. avium infected mice. Analyses were performed for differentially expressed genes with an FDR < 0.1. Log10 FDR adjusted p-values are shown on the x axis. (D) Gene set enrichment analysis (GSEA) was performed on RNA-Seq data from naïve and 4 week M. avium infected mice. Antigen processing and presentation pathways are highly enriched (p< 0.001, Normalized Enrichment Score (NES) = 2.05) in the M. avium induced up-regulated genes.
To understand the extent to which IFNγ promotes myeloid differentiation of HSPCs, we isolated CD34+ cells from human Buffy coat samples and treated them with IFNγ in vitro. Within three days, the number of CD34+CD38− stem cells decreased by 7-fold compared to untreated control cells. Meanwhile, there was a dramatic increase in cells with the phenotype of differentiated myeloid cells, particularly CD15+ myelomonocytic cells and CD66b+ granulocytes, consistent with a prior report (Figure S6A&B) (Yang et al., 2005). These data show that IFNγ can induce a major shift towards differentiation in HSCs on a scale that would be sufficient to deplete the HSC population.
Overexpression of Batf2 drives myeloid differentiation of HSPCs
We hypothesized that terminal differentiation may be the major mechanism of HSC loss during chronic infection. To identify molecular drivers of this process, we focused on the 4 transcription factors upregulated during infection. Aside from expected increases in expression of Stat1, NFkappaB, and Ciita, Batf2, an IFNγ-responsive transcription factor, was significantly upregulated in HSCs during M. avium infection (Table S2 and Figure 6B). This transcription factor caught our attention because it has been previously reported to participate in dendritic cell development during stress (Tussiwand et al., 2012). The Batf2 family member Batf acts as a differentiation checkpoint that limits HSC self-renewal in response to DNA damage while promoting lymphoid differentiation (Santos et al., 2014; Wang et al., 2012). We assayed transcript levels from HSCs by qPCR and found that Batf2 was upregulated 8-fold in HSCs during M. avium infection (Figure 7A). Furthermore, we found that Batf2 was not induced when M. avium infection was conducted in Ifngr1−/− mice, suggesting that IFNγ signaling is required for its upregulation. In order to test the relevance of this gene in the human system, we measured BATF2 expression in primary human CD34+ cells. We found that BATF2 expression was detectible in human CD34+ progenitors and was significantly increased upon addition of IFNγ to the culture media (Figure 7B).
Figure 7. Batf2 promotes IFNγ-dependent myeloid differentiation in murine and human cells.

(A) Batf2 RNA expression in HSCs (KL CD150+ CD48− CD34−) of WT or Ifngr1−/− mice 4 weeks after M. avium infection; shown as fold change over WT naïve. Data are representative of 2 independent experiments both performed in triplicate. (B) BATF2 expression measured from umbilical cord blood-derived human CD34+ cells in the presence or absence of rhIFNγ for 3 days. Data are representative of 2 independent experiments using different cord blood samples, performed in triplicate. (C–E) Sca1-enriched cells were transduced with retroviruses overexpressing GFP alone or GFP-Batf2. 2000 CD45.2 Sca1+ transduced cells were transplanted with 2x105 CD45.1 rescue WBM cells. Data represent 5 independent experiments with n=13–14 per group. (C) Lineage distribution of murine PB 8 weeks after transplant. (D) Engraftment of GFP+ CD45.2 cells at 4 and 8 weeks post-transplant. (E) Change in engraftment of GFP+ CD45.2+ cells between 4 and 8 weeks post-transplant. (F) Representative DNA gel of BATF2 exon 1 region amplified from human cord blood-derived CD34+ cells after gene editing with Cas9-only control or Cas9 plus 4 BATF2 sgRNAs. (G) BATF2 RNA expression in human cord blood-derived CD34+ cells after gene editing with Cas9-only control or Cas9 plus sgRNAs. Data are representative of 2 independent experiments performed in triplicate. (H) Percent of cells that express the granulocyte marker CD66b+ in the absence or presence of rhIFNγ x 3 days. Data are representative of 3 independent experiments each in triplicate. Data are presented as mean ± SEM; * p<0.05, ** p<0.01, *** p<0.001, **** p<0.0001. See also Figure S7 and Table S3.
To test the role of Batf2 in hematopoietic progenitors, we overexpressed Batf2 using a retroviral vector in Sca1+ murine progenitors. Overexpression was verified by quantitative PCR (Figure S7A). Hematopoietic progenitors overexpressing Batf2 demonstrated a trend toward increased early myeloid differentiation 8 weeks after transplantation into lethally irradiated WT recipients (Figure 7C). Since Batf family members are known to act in cooperation with cofactors such as Irf1 (Roy et al., 2015), the observation that myeloid differentiation was even modestly increased without additional expression of cofactors was striking. In contrast to control cells overexpressing GFP alone, the overall engraftment of Batf2-overexpressing clones declined between 4 and 8 weeks post-transplant, showing that engraftment capacity is impaired (Figure 7D–E). These findings suggest that Batf2-dependent myeloid differentiation may be a mechanism for HSPC loss during chronic infection.
BATF2 loss of function impairs myeloid differentiation of human HSPCs
To test if Batf2 is required for myeloid differentiation of HSPCs, we used CRISPR-Cas9 gene editing to knock out BATF2 in human CD34+ cord blood-derived progenitors (Gundry et al., 2016; Ran et al., 2013). Four separate single guide RNAs were designed to target the first exon of BATF2; spacing between PAM sequences of these guides were out of frame with one another. Genotyping by PCR revealed that the efficiency of deletion was high, with ~50% of template DNA showing a large 139 bp deletion, representing just one of the 16 potential combinations by which the 4 guides could disrupt the gene (Figure 7F and S7B). Gene expression was no longer detectible by real-time qPCR in the gene-edited cells, indicating a high efficiency of deletion (Figure 7G). We next conducted a differentiation assay of CD34+ progenitors in vitro. Briefly, gene edited CD34+ cells were incubated in complete media in the presence or absence of IFNγ for 3 days, and the number of HSCs (CD34+CD38−), progenitors (CD34+CD38+), and differentiated myeloid cells (CD15+ or CD66b+) were counted. The final cell counts per well were not statistically different between IFNγ-treated and untreated groups. While HSCs were diminished in both Cas9-only control and KO cells (Figure S7C), BATF2 KO CD34+ cells differentiated less efficiently into myeloid progeny. Specifically, the IFNγ-mediated induction of CD66b+ granulocytes was completely abrogated in KO cells (Figure 7H), while the percentage of CD15+ myelomonocytic cells decreased and their absolute number trended down (Figure S7C–D). Meanwhile the number of CD34+CD38+ progenitors in BATF2 KO samples increased, consistent with a differentiation block (Figure S7C–D). These studies identify BATF2 as a novel mediator of IFNγ-dependent terminal differentiation of human hematopoietic progenitors.
Discussion
Here we demonstrated that chronic infection with M. avium leads to pancytopenia, depletion of bone marrow HSCs, and impaired HSC self-renewal. While HSCs from infected animals are exposed to increased replication stress and are more prone to apoptosis upon secondary stress, we found no evidence of increased HSC apoptosis during in vivo infection. Furthermore, there was no evidence of HSC mobilization out of the bone marrow during infection. Instead, RNA profiling data pointed towards differentiation as the most significant transcriptional change. We identified Batf2 as a potential mediator of HSPC differentiation and used gain and loss of function studies to show that Batf2 impairs HSPC persistence and is required for normal IFNγ-dependent myeloid differentiation in human progenitors. These studies suggest that increased terminal differentiation is the major route of HSC loss during chronic infection and provide a direct mechanism by which chronic infection causes HSC depletion.
Bone marrow suppression and pancytopenia are known complications of chronic infectious diseases including mycobacterial infections(Achi et al., 2013). Since mycobacterial infections frequently co-occur with myelofibrosis, many have assumed that pancytopenia is due to physical disruption of the bone marrow (Viallard et al., 2002). However, our data indicate that inflammatory stimulation of HSCs is a more significant contributor to pancytopenia than myelofibrosis. In another study, recruitment of bone marrow macrophages by IFNγ was also found to contribute to HSC dysfunction and loss (McCabe et al., 2015).
Our findings are consistent with several studies using murine knockout models to demonstrate that dysregulated IFN signaling depletes hematopoietic progenitors, including stem cells (Essers et al., 2009; King et al., 2011; Lin et al., 2014; Sato et al., 2009). These studies using genetically modified mice suggest that artificially elevated IFN signaling can damage self-renewal processes and deplete hematopoietic progenitor populations over time. Our study is the first to demonstrate that this occurs in the context of physiologic infection in WT mice.
Although IFNγ did not promote HSC mobilization, our results do not rule out the possibility that HSCs are displaced from cellular niches with the bone marrow architecture as has been shown for IFNα (Kunisaki et al., 2013), nor do they rule out the possibility that HSCs in extramedullary sites could expand through self-renewal events. Prior studies have demonstrated increased apoptosis of HSPCs in aplastic anemia patients with high IFNγ levels through both in vitro and in vivo assays in human cells (Zeng et al., 2006). Our studies suggest that inflammation and IFNγ stimulation may affect HSPCs on a continuum; while we did not detect increased apoptosis during infection, we did find that the threshold for apoptosis was lowered. People with aplastic anemia may have multiple stressors, for example microenvironmental changes in the bone marrow niche in addition to increased IFNγ levels, that together lead to apoptosis.
We identified Batf2 as a potential mediator of IFNγ-dependent terminal HSPC differentiation. Our demonstration that BATF2 has a key role in IFNγ-dependent myeloid differentiation may contribute to the development of future applications to protect stem cells in the context of persistent inflammation. However, Batf2 is likely not the only mediator of IFNγ-dependent myeloid differentiation and our RNASeq data may reveal other key regulators. In addition, although we focused on IFNγ as a powerful mediator of HSC loss during infection, it is almost certainly not the only one. HSCs lacking IFNγ-receptors can also respond to this signal indirectly via the release of IL6 from the microenvironment (Schürch et al., 2014). Chronic LPS exposure was shown to injure HSC function (Esplin et al., 2011). A recent study reported that IL1 proinflammatory signaling accelerates myeloid differentiation of HSPCs through activation of the transcription factor Pu.1 (Pietras et al., 2016). Other infections with less IFNγ-dominant inflammatory signatures may cause distinct hematological effects.
Our demonstration that HSCs are significantly depleted during chronic infection may have significant implications in the field of hematologic malignancies. Infections are epidemiologically linked to acute myelogenous leukemia (Kristinsson et al., 2011), and dysregulation of inflammation is an important contributor to aplastic anemia, which can evolve to cancer (Gañán-Gómez et al., 2015; Young and Maciejewski, 1997). Given recent reports of clonal hematopoiesis as a risk factor for malignancy (Yoshizato et al., 2015), attrition in HSC numbers due to persistent inflammation could provide a pathophysiologic basis for these links. Further work to delineate the effects of inflammation on HSCs is likely to yield important mechanistic insight into other diseases of HSC malfunction, including transient bone marrow suppression, aplastic anemia, myelodysplastic syndrome, and leukemia.
Experimental Procedures
Mice
Wild-type C57Bl/6 (CD45.2) and C57Bl/6.SJL (CD45.1) mice 6–12 weeks of age were used. C57Bl/6 Ifngr1−/− (Stock #3288) mice were obtained from Jackson Laboratories (Bar Harbor, ME [14]). All mice were maintained at an AALAC-accredited, specific-pathogen-free animal facility at Baylor College of Medicine. Genotypes were confirmed by PCR.
Microbial infections
Mice were infected monthly with 2x106 colony-forming units of Mycobacterium avium IV as described (Feng et al., 2008). M. avium was detected by growth on Middlebrook agar and by PCR (Park et al., 2000).
Bone marrow transplantation
Non-competitive bone marrow transplants were performed by IV injection of CD45.2 donor WBM cells from infected mice or naïve controls into lethally irradiated CD45.1 WT recipients. For competitive transplants, CD45.2 donor WBM cells from naïve or infected mice were mixed with wild-type CD45.1 competitor WBM cells prior to injection into CD45.1 recipients. For secondary transplants, sorted CD45.2 donor HSCs were isolated from primary recipients and transplanted with CD45.1 rescue WBM into naïve CD45.1 irradiated recipients.
RNA purification and RNA-Seq
Approximately 70,000 HSCs (SPLSKCD150+) were sorted into Trizol from the pools of naïve or infected mice. RNA was isolated with the RNeasy Micro column (Qiagen, Valencia, CA). Illumina HiSeq was used for sequencing with a paired-end sequencing length of 100bp. FDR < 0.05 was considered statistically significant. Gene ontology and pathway analyses were performed for differentially expressed genes with FDR < 0.1.
Flow cytometry
PB was analyzed with a Hemavet 950. SP staining was performed with Hoechst 33342 (Sigma) as previously described (Goodell et al., 1996). Myeloid-biased and lymphoid-biased HSCs were identified as previously described (Challen et al., 2010). A full list of all staining schemes and antibodies is provided in Table S4&S5.
Colony forming assays
Colony forming assays were performed by plating WBM cells in methylcellulose media and counting colonies after 9 days (Stem Cell Technologies).
Single-cell assays
Single HSCs (KSL CD150+ CD48− CD34−) were sorted into 96 well plates containing StemSpan media and colonies were counted after 10 days.
Cell proliferation and DNA damage
Proliferation and DNA damage of HSCs was determined using flow cytometry of cells co-stained with Ki67 and pKap1. γH2AX staining was performed on sorted HSCs from naïve or 4-month M. avium infected mice as previously described (Walter et al, 2015).
Reactive oxygen species
Levels of ROS within HSCs from control or infected mice were determined by flow cytometry analysis of cells stained with Molecular Probes’ CellRox Green Flow Cytometry Assay Kit (Life Technologies) according to the manufacturer’s protocol.
Apoptosis assays
Apoptosis was determined using the Caspase-Glo 3/7 Assay (Promega). HSCs were sorted directly into 96 well plates containing StemSpan media and Caspase Glo reagent was either immediately added to the wells or was added after 12 hr incubation at 37°C.
Batf2 overexpression and BATF2 deletion
Batf2 was overexpressed in Sca1+ cells using a retrovirus as previously described (Ergen et al., 2012). Sca1+ CD45.2 cells were transplanted into lethally irradiated CD45.1 recipient mice along with rescue marrow, and engraftment of the GFP+ CD45.2 cells was tracked every 4 weeks. BATF2 deletion was conducted as described(Gundry et al., 2016). For each experiment, CD34+ cells were isolated from an unrelated frozen human umbilical cord blood or Buffy coat sample. Four single guide RNAs (Table S3) were mixed with recombinant Cas9 protein and transduced into CD34+ cells by electroporation. Gene deletion was confirmed by PCR and qPCR. Differentiation of human cells was measured by incubation of CD34+ cells with IFNγ for 3 days. HSPCs and differentiated myeloid cells were characterized by flow cytometry.
Mobilization assays
Mice were treated with G-CSF for 6 days and/or rmIFNγ for 24 hours prior to sacrifice. HSCs were detected in the spleen by flow cytometry. For transplants, splenocytes were mixed with CD45.1 rescue WBM cells and transplanted into lethally irradiated CD45.1 recipients.
Estimation of HSC loss
The basis for understanding the dynamics of HSC loss is the differential equation of the form N & (t) = −λN (t) + 2(1 − d)λN (t) where N(t) is the HSC count at time t, λ is the division rate of the HSC, and d is the fraction of HSC progeny that are not HSC (i.e. that constitutes HSC “loss”). The equation admits explicit solution N (t) = N (0) exp[(1 − 2d)λt], where the initial condition N(0) is equal to the pre-infection steady-state HSC count. Logarithm of the HSC count is a linear function of ln[N(0)] and d, and therefore these two quantities can be estimated using standard linear regression, if the value of λ is assumed (we set λ = 0.05 d−1)(Wilson et al., 2008). Given that (1) each measurement corresponds to a different mouse (represented by the asterisks in Figure S1G), and (2) HSC counts show wide interindividual variability, we estimated the variability in any time point with at least two replicates by fitting a gamma distribution. Then we fit regression lines to the quantiles of orders 2.5%, 5%, 25%, 50%, 75%, 95%, and 97.5% of the gamma distributions. These lines (Figure S1G) provide corresponding quantile values of N(0) and d (Table S1), yielding the 95% confidence interval for d.
Statistics
Mean values ± SEM are shown. Student’s t-test or 2-way ANOVA were used for comparisons (GraphPad Prism Version 5.0).
See supplemental methods for more details.
Supplementary Material
Acknowledgments
We thank C. Kadmon, A. Rosen and Y. Zheng for technical assistance; M. Goodell and D. Nakada for useful discussions regarding this work; C. Gillespie for reviewing the manuscript; and the Texas Advanced Computing Center (TACC) at The University of Texas at Austin for proving HPC resources for the data analysis. This project was supported by the Cytometry and Cell Sorting Core (NIAID P30AI036211, NCI P30CA125123, NCRR S10RR024574) and the Integrated Microscopy Core (HD007495, DK56338, CA125123) at Baylor College of Medicine with additional funding from the Dan L. Duncan Cancer Center and the John S. Dunn Gulf Coast Consortium for Chemical Genetics. This work was supported by grants from the NIDDK DK060445 (KAM), the NHLBI HL128173-02 (MK, SC) and K08HL098898 (KYK), and the Department of Defense IDEA award in bone marrow failure research (10505346), the Caroline Wiess Law Foundation for Molecular Medicine, the Aplastic Anemia and MDS International Foundation Liviya Anderson Award, and a March of Dimes Basil O’Connor Starter Scholar Award (KYK).
Footnotes
Author Contributions
Conceptualization, K.Y.K.; Investigation, K.A.M., M.J., and K.Y.K.; Formal Analysis, S.C., F.C., D.S., Q.M., and M.K.; Writing, K.A.M. and K.Y.K.; Supervision, K.Y.K.
Accession Numbers
RNA-Seq data have been deposited in the Gene Expression Omnibus (GEO) under accession number GSE89364.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Achi HV, Ahui BJ, Anon JC, Kouassi BA, Dje-Bi H, Kininlman H. Pancytopenia: a severe complication of miliary tuberculosis. Rev Mal Respir. 2013;30:33–37. doi: 10.1016/j.rmr.2012.08.008. [DOI] [PubMed] [Google Scholar]
- Baldridge MT, King KY, Boles NC, Weksberg DC, Goodell MA. Quiescent haematopoietic stem cells are activated by IFN-γ in response to chronic infection. Nature. 2010;465:793–797. doi: 10.1038/nature09135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldridge MT, King KY, Goodell MA. Inflammatory signals regulate hematopoietic stem cells. Trends Immunol. 2011;32:57–65. doi: 10.1016/j.it.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balmer ML, Schürch CM, Saito Y, Geuking MB, Li H, Cuenca M, Kovtonyuk LV, McCoy KD, Hapfelmeier S, Ochsenbein AF, et al. Microbiota-derived compounds drive steady-state granulopoiesis via MyD88/TICAM signaling. J Immunol. 2014;193:5273–5283. doi: 10.4049/jimmunol.1400762. [DOI] [PubMed] [Google Scholar]
- Challen GA, Boles NC, Chambers SM, Goodell MA. Distinct Hematopoietic Stem Cell Subtypes Are Differentially Regulated by TGF-β1. Stem Cell. 2010;6:265–278. doi: 10.1016/j.stem.2010.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ergen AV, Boles NC, Goodell MA. Rantes/Ccl5 influences hematopoietic stem cell subtypes and causes myeloid skewing. Blood. 2012;119:2500–2509. doi: 10.1182/blood-2011-11-391730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esplin BL, Shimazu T, Welner RS, Garrett KP, Nie L, Zhang Q, Humphrey MB, Yang Q, Borghesi LA, Kincade PW. Chronic exposure to a TLR ligand injures hematopoietic stem cells. J Immunol. 2011;186:5367–5375. doi: 10.4049/jimmunol.1003438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Essers MAG, Offner S, Blanco-Bose WE, Waibler Z, Kalinke U, Duchosal MA, Trumpp A. IFNalpha activates dormant haematopoietic stem cells in vivo. Nature. 2009;458:904–908. doi: 10.1038/nature07815. [DOI] [PubMed] [Google Scholar]
- Feng CG, Weksberg DC, Taylor GA, Sher A, Goodell MA. The p47 GTPase Lrg-47 (Irgm1) links host defense and hematopoietic stem cell proliferation. Cell Stem Cell. 2008;2:83–89. doi: 10.1016/j.stem.2007.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzgerald D, Haas DW. Mycobacterium tuberculosis. In: Mandell GL, Bennett JE, Dolin R, editors. Principles and Practice of Infectious Diseases. Philadelphia, PA: Elsevier; 2005. pp. 2852–2886. [Google Scholar]
- Flórido M, Pearl JE, Solache A, Borges M, Haynes L, Cooper AM, Appelberg R. Gamma interferon-induced T-cell loss in virulent Mycobacterium avium infection. Infection and Immunity. 2005;73:3577–3586. doi: 10.1128/IAI.73.6.3577-3586.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodell MA, Brose K, Paradis G, Conner AS, Mulligan RC. Isolation and functional properties of murine hematopoietic stem cells that are replicating in vivo. J Exp Med. 1996;183:1797–1806. doi: 10.1084/jem.183.4.1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gundry MC, Brunetti L, Lin A, Mayle AE, Kitano A, Wagner D, Hsu JI, Hoegenauer KA, Rooney CM, Goodell MA, et al. Highly Efficient Genome Editing of Murine and Human Hematopoietic Progenitor Cells by CRISPR/Cas9. CellReports. 2016;17:1453–1461. doi: 10.1016/j.celrep.2016.09.092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King KY, Baldridge MT, Weksberg DC, Chambers SM, Lukov GL, Wu S, Boles NC, Jung SY, Qin J, Liu D, et al. Irgm1 protects hematopoietic stem cells by negative regulation of IFN signaling. Blood. 2011;118:1525–1533. doi: 10.1182/blood-2011-01-328682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kristinsson SY, Björkholm M, Hultcrantz M, Derolf ÅR, Landgren O, Goldin LR. Chronic immune stimulation might act as a trigger for the development of acute myeloid leukemia or myelodysplastic syndromes. Journal of Clinical Oncology. 2011;29:2897–2903. doi: 10.1200/JCO.2011.34.8540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunisaki Y, Bruns I, Scheiermann C, Ahmed J, Pinho S, Zhang D, Mizoguchi T, Wei Q, Lucas D, Ito K, et al. Arteriolar niches maintain haematopoietic stem cell quiescence. Nature. 2013;502:637–643. doi: 10.1038/nature12612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin FC, Karwan M, Saleh B, Hodge DL, Chan T, Boelte KC, Keller JR, Young HA. IFN-γ causes aplastic anemia by altering hematopoietic stem/progenitor cell composition and disrupting lineage differentiation. Blood. 2014;124:3699–3708. doi: 10.1182/blood-2014-01-549527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matatall KA, Shen CC, Challen GA, King KY. Type II interferon promotes differentiation of myeloid-biased hematopoietic stem cells. Stem Cells. 2014;32:3023–3030. doi: 10.1002/stem.1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCabe A, Zhang Y, Thai V, Jones M, Jordan MB, MacNamara KC. Macrophage-Lineage Cells Negatively Regulate the Hematopoietic Stem Cell Pool in Response to Interferon Gamma at Steady State and During Infection. Stem Cells. 2015;33:2294–2305. doi: 10.1002/stem.2040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagai Y, Garrett KP, Ohta S, Bahrun U, Kouro T, Akira S, Takatsu K, Kincade PW. Toll-like receptors on hematopoietic progenitor cells stimulate innate immune system replenishment. Immunity. 2006;24:801–812. doi: 10.1016/j.immuni.2006.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Notta F, Zandi S, Takayama N, Dobson S, Gan OI, Wilson G, Kaufmann KB, McLeod J, Laurenti E, Dunant CF, et al. Distinct routes of lineage development reshape the human blood hierarchy across ontogeny. Science. 2016;351:aab2116. doi: 10.1126/science.aab2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park H, Jang H, Kim C, Chung B, Chang CL, Park SK, Song S. Detection and identification of mycobacteria by amplification of the internal transcribed spacer regions with genus- and species-specific PCR primers. J Clin Microbiol. 2000;38:4080–4085. doi: 10.1128/jcm.38.11.4080-4085.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietras EM, Mirantes-Barbeito C, Fong S, Loeffler D, Kovtonyuk LV, Zhang S, Lakshminarasimhan R, Chin CP, Techner JM, Will B, et al. Chronic interleukin-1 exposure drives haematopoietic stem cells towards precocious myeloid differentiation at the expense of self-renewal. Nat Cell Biol. 2016;18:607–618. doi: 10.1038/ncb3346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos-Casals M, García-Carrasco M, López-Medrano F, Trejo O, Forns X, López-Guillermo A, Muñoz C, Ingelmo M, Font J. Severe autoimmune cytopenias in treatment-naive hepatitis C virus infection: clinical description of 35 cases. Medicine (Baltimore) 2003;82:87–96. doi: 10.1097/00005792-200303000-00003. [DOI] [PubMed] [Google Scholar]
- Ran F, Hsu P, Wright J, Agarwala V, Scott D, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat Protoc. 2013;8:2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy S, Guler R, Parihar SP, Schmeier S, Kaczkowski B, Nishimura H, Shin JW, Negishi Y, Ozturk M, Hurdayal R, et al. Batf2/Irf1 induces inflammatory responses in classically activated macrophages, lipopolysaccharides, and mycobacterial infection. J Immunol. 2015;194:6035–6044. doi: 10.4049/jimmunol.1402521. [DOI] [PubMed] [Google Scholar]
- Santos MA, Faryabi RB, Ergen AV, Day AM, Malhowski A, Canela A, Onozawa M, Lee J, Callen E, Gutierrez-Martinez P, et al. DNA-damage-induced differentiation of leukaemic cells as an anti-cancer barrier. Nature. 2014;514:107–111. doi: 10.1038/nature13483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato T, Onai N, Yoshihara H, Arai F, Suda T, Ohteki T. Interferon regulatory factor-2 protects quiescent hematopoietic stem cells from type I interferon-dependent exhaustion. Nat Med. 2009;15:696–700. doi: 10.1038/nm.1973. [DOI] [PubMed] [Google Scholar]
- Scadden DT, Zon LI, Groopman JE. Pathophysiology and management of HIV-associated hematologic disorders. Blood. 1989;74:1455–1463. [PubMed] [Google Scholar]
- Takizawa H, Boettcher S, Manz MG. Demand-adapted regulation of early hematopoiesis in infection and inflammation. Blood. 2012;119:2991–3002. doi: 10.1182/blood-2011-12-380113. [DOI] [PubMed] [Google Scholar]
- Tussiwand R, Lee W, Murphy TL, Mashayekhi M, Wumesh KC, Albring JC, Satpathy AT, Rotondo JA, Edelson BT, Kretzer NM, et al. Compensatory dendritic cell development mediated by BATF-IRF interactions. Nature. 2012;490:502–507. doi: 10.1038/nature11531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viallard JF, Parrens M, Boiron JM, Texier J, Mercie P, Pellegrin JL. Reversible myelofibrosis induced by tuberculosis. Clin Infect Dis. 2002;34:1641–1643. doi: 10.1086/340524. [DOI] [PubMed] [Google Scholar]
- Walter D, Lier A, Geiselhart A, Thalheimer FB, Huntscha S, Sobotta MC, Moehrle B, Brocks D, Bayindir I, Kaschutnig P, et al. Exit from dormancy provokes DNA-damage-induced attrition in haematopoietic stem cells. Nature. 2015;520:549–552. doi: 10.1038/nature14131. [DOI] [PubMed] [Google Scholar]
- Wang J, Sun Q, Morita Y, Jiang H, Gross A, Lechel A, Hildner K, Guachalla LM, Gompf A, Hartmann D, et al. A differentiation checkpoint limits hematopoietic stem cell self-renewal in response to DNA damage. Cell. 2012;148:1001–1014. doi: 10.1016/j.cell.2012.01.040. [DOI] [PubMed] [Google Scholar]
- Wilson A, Laurenti E, Oser G, Wath RCvd, Blanco-Bose W, Jaworski M, Offner S, Dunant CF, Eshkind L, Bockamp E, et al. Hematopoietic Stem Cells Reversibly Switch from Dormancy to Self-Renewal during Homeostasis and Repair. Cell. 2008;135:1118–1129. doi: 10.1016/j.cell.2008.10.048. [DOI] [PubMed] [Google Scholar]
- Yang L, Dybedal I, Bryder D, Nilsson L, Sitnicka E, Sasaki Y, Jacobsen SEW. IFN-gamma negatively modulates self-renewal of repopulating human hemopoietic stem cells. J Immunol. 2005;174:752–757. doi: 10.4049/jimmunol.174.2.752. [DOI] [PubMed] [Google Scholar]
- Yoshizato T, Dumitriu B, Hosokawa K, Makishima H, Yoshida K, Townsley D, Sato-Otsubo A, Sato Y, Liu D, Suzuki H, et al. Somatic Mutations and Clonal Hematopoiesis in Aplastic Anemia. N Engl J Med. 2015;373:35–47. doi: 10.1056/NEJMoa1414799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng W, Miyazato A, Chen G, Kajigaya S, Young NS, Maciejewski JP. Interferon-gamma-induced gene expression in CD34 cells: identification of pathologic cytokine-specific signature profiles. Blood. 2006;107:167–175. doi: 10.1182/blood-2005-05-1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.