

ABSTRACT
Conversion of native cellular prion protein (PrPc) from an α-helical structure to a toxic and infectious β-sheet structure (PrPSc) is a critical step in the development of prion disease. There are some indications that the formation of PrPSc is preceded by a β-sheet rich PrP (PrPβ) form which is non-infectious, but is an intermediate in the formation of infectious PrPSc. Furthermore the presence of lipid cofactors is thought to be critical in the formation of both intermediate-PrPβ and lethal, infectious PrPSc. We previously discovered that the endotoxin, lipopolysaccharide (LPS), interacts with recombinant PrPc and induces rapid conformational change to a β-sheet rich structure. This LPS induced PrPβ structure exhibits PrPSc-like features including proteinase K (PK) resistance and the capacity to form large oligomers and rod-like fibrils. LPS is a large, complex molecule with lipid, polysaccharide, 2-keto-3-deoxyoctonate (Kdo) and glucosamine components. To learn more about which LPS chemical constituents are critical for binding PrPc and inducing β-sheet conversion we systematically investigated which chemical components of LPS either bind or induce PrP conversion to PrPβ. We analyzed this PrP conversion using resolution enhanced native acidic gel electrophoresis (RENAGE), tryptophan fluorescence, circular dichroism, electron microscopy and PK resistance. Our results indicate that a minimal version of LPS (called detoxified and partially de-acylated LPS or dLPS) containing a portion of the polysaccharide and a portion of the lipid component is sufficient for PrP conversion. Lipid components, alone, and saccharide components, alone, are insufficient for conversion.
KEYWORDS: β oligomer, cofactor, conversion, fibril, lipid, lipid A, lipopolysaccharide, prion protein, protein misfolding
INTRODUCTION
Prions are infectious particles composed of misfolded prion proteins (PrPSc). They are known to cause diseases such as scrapie in sheep, bovine spongiform encephalopathy (BSE) in cattle, chronic wasting disease (CWD) in cervids, as well as Kuru, Creutzfeld Jacob Disease (CJD) and Fatal Familial Insomnia (FFI) in humans.1 Prion diseases are uniformly fatal neurodegenerative diseases. Prion infectivity and prion neurotoxicity appears to be due to the progressive conversion the native, non-toxic, α-helix-rich cellular prion protein (PrPc) into an insoluble, toxic, β-sheet rich isoform (PrPSc).2 PrPc is a highly conserved glycosylphosphatidylinositol (GPI) anchored, membrane-bound glycoprotein, that is particularly abundant in the neuronal cells of vertebrates. Although the GPI anchor and glycosylation modulate the strain and species transmissibility properties of PrPSc, they are not required for generating fatal and infectious prion disease.3 The role of PrPSc as the sole prion infectious agent is supported by the ability to generate infectious prions de novo from recombinant (rec) PrPc using POPG (1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-(1′-rac-glycerol)), RNA and protein misfolding cyclic amplification (PMCA).4
The role of cofactors and denaturants in the conversion of α-helical PrPc to β-sheet PrP (PrPβ) is well known. In these methods, the conversion of recPrPc to PrPβ is performed through the addition of denaturants and cofactors such as detergents5, urea6, copper ions7, acid6, RNA8,9, DNA10,11, glycosaminoglycans12, lipids13 and lipopolysaccharides14. However, conformational change to a β-sheet rich structure is not sufficient to create infectious prions (PrPSc). Another critical component of prion disease is the occurrence of template directed replication or propagation of the infectious PrPSc isoform.2,15 Generation of de novo and cell free infectious PrPSc, has been accomplished by first forming a PrPβ intermediate from mammalian PrPc and recPrPc using lipid and RNA induced conversion and then applying PMCA to both select for and amplify the infectious PrPSc isoforms.4,16 Recent work has revealed that some of the structural changes present in PrPβ formed from recPrPc and POPG-induced conversion persist in the final PrPSc conformer.17,18 Furthermore, POPG exposes a region in PrP that is critical for binding the primary PrPSc epitope18 (which is shown to be the N-terminal polybasic region residues 23–33).19 This suggests that the formation of PrPβ is an intermediate in template directed replication of infectious PrPSc. In addition, it has been shown that POPG induces β-sheet conversion of PrP through a transient interaction, which means that the PrPβ isoform is stable after removal of POPG.18 However the continued presence of cofactors (POPG and RNA or phosphatidylethanolamine alone) are needed during PMCA cycles to form and maintain PrPSc.20
We believe that there are other cofactors that will also cause formation of PrPβ. Previously we discovered that lipopolysaccharide (LPS) induces conversion of recPrP to large oligomers and fibrils, rich in β-sheet structure.14 LPS is a major component of the outer membrane of Gram-negative bacteria and is found in or on almost any natural and man-made objects that have been in contact with bacteria.21 LPS is toxic and is known to induce a strong immune response in mammals. The structure of LPS is shown in Fig. 1 and consists of an O antigen polysaccharide repeat, an outer core polysaccharide, an inner core Kdo2 disaccharide and a lipid component called lipid A (Fig. 1). Lipid A consists of a glucosamine disaccharide with ester linked and amide linked fatty acid chains.21
Figure 1.

(A) General structure of LPS redrawn from Magalhaes et al. and Ruiz et al.47,48 with the structure of lipid A, the core oligo-saccharide and the O-Antigen components labeled. Abbreviations represent 2-keto-3-deoxyoctonate (Kdo), L-glycerol-D-manno-heptose (Hep), galactose (Gal), glucose (Glc), N-acetylgalactosamine (Nga) and N-acetylglucosamine (NGc). (B) Structure of Kdo2-lipid A provided by the supplier (Avanti Polar lipids). The inset shows the structure of Kdo. (C) Structure of dLPS according to Muller-Loennies et al.49 The inset shows the structure of Gln (glucosamine).
LPS-induced conversion of prion proteins is unique compared to the addition other denaturants and cofactors, in that the conversion of recPrP occurs at sub-stoichiometric ratios of LPS to recPrP. Furthermore LPS has a unique property of both mediating conversion to PrPβ as well as being a well-known cause of inflammation. LPS induces innate immune responses through toll-like receptors (TLR). This LPS-induced activation of TLR initiates an innate adaptive response that decreases prion propagation.22 However, moderate repeated doses of LPS have been shown to cause systemic inflammation, which accelerates features of neurodegenerative disease in combination with existing disease pathology.23 Furthermore, there is a strong link between a highly pro-inflammatory response in microglia and prion disease progression.24 Intriguingly treatment of the mucosal side of colon tissue with LPS caused up-regulation of genes related to TLR and the inflammasome.25 Treatment of the mucosal side of colon tissue with recPrP and LPS up-regulated genes related to TLR, inflammatory response, attraction of dendritic cells and the JNK-apoptosis pathway.
Given the molecular complexity of LPS and the data from other studies showing that simpler molecules could induce PrP conversion and propagation,4,16 we decided to systematically investigate which portions of LPS are necessary and sufficient to induce PrP conversion. The LPS components that we tested were partially deacylated and detoxified LPS (dLPS), Kdo2-lipid A, lipid A, 2-keto-3-deoxyoctonate (Kdo) and glucosamine (Fig. 1). Other than intact LPS, we found that only dLPS strongly binds recPrPc and also induces conversion to soluble β-sheet rich, large oligomers. The large oligomers generated by both LPS and dLPS have PrPSc features of being β-sheet rich, and partially PK resistant. The concentration dependence of PrP conversion seen from serial dilution of a dLPS-PrP mixture was similar to that seen for LPS-induced PrP conversion. We characterized LPS and dLPS induced PrP conversion of full-length recMoPrP23–231 using resolution enhanced native acidic gel electrophoresis (RENAGE), tryptophan fluorescence, circular dichroism, electron microscopy and PK resistance. The large oligomers formed by dLPS have a PK resistance core above 11 kDa with bands at 18/17 kDa, similar to the large oligomers formed by LPS-induced PrP conversion. This work adds to the understanding of the role of lipids in β-sheet conversion of PrP. Our results are consistent with a mechanism of LPS-induced PrP conversion that requires both a polysaccharide functionality and lipid micelles to provide a scaffold for protein binding and rapid β-sheet formation.
RESULTS
Conversion Capability of LPS Components
LPS is known to induce the conversion of recPrP to fibril-like species and large oligomers.14 A specially designed gel electrophoresis technique called RENAGE allows one to routinely separate monomeric PrP from oligomers, large oligomers and small fibrils.26,27 Small oligomers (8 to 12-mers) are well dispersed in the separating gel, oligomers ∼16 to 24-mers are found near the top of the separating gel, and larger oligomers (megamers) or fibrils migrate just into the top of the stacking gel. RENAGE cannot distinguish oligomers larger than ∼30-mers from fibrils. We previously used this technique to show that LPS induces the formation of large soluble recPrP aggregates or fibrils.14 RENAGE is also well suited to screening for other compounds that will induce PrP oligomerization. Here we used RENAGE to systematically test whether smaller components or fragments of LPS could also cause conversion of native PrP to large oligomers (megamers) or fibrils. In particular, we tested whether partially deacylated and detoxified LPS (dLPS), Kdo2-lipid A, lipid A and 2-keto-3-deoxyoctonate (Kdo) cause recPrP oligomerization using RENAGE. Specifically, 0.5 mg/mL (19.3 µM) of helix-rich recMoPrP23–231 dissolved in 20 mM sodium acetate, pH 5.5 was incubated at 37°C overnight with each of the 4 LPS chemical components. The resulting RENAGE gels are shown in Fig. 2. Only recMoPrP23–231 incubated with dLPS at a weight ratio of 1:0.5 (PrP:dLPS) (estimated to be a molar ratio of 1:1.3 (PrP:dLPS) caused the formation of a large oligomer protein band that migrated a few millimeters into the RENAGE stacking gel. This is similar to what was seen for intact-LPS induced PrP conversion (Fig. 2A). Also the mixture of dLPS with recPrP remained homogeneous, with no visible precipitate. RENAGE gels of truncated forms of the prion protein (recMoPrP90–231) incubated with each of these 4 LPS compounds showed the same result (result not shown).
Figure 2.
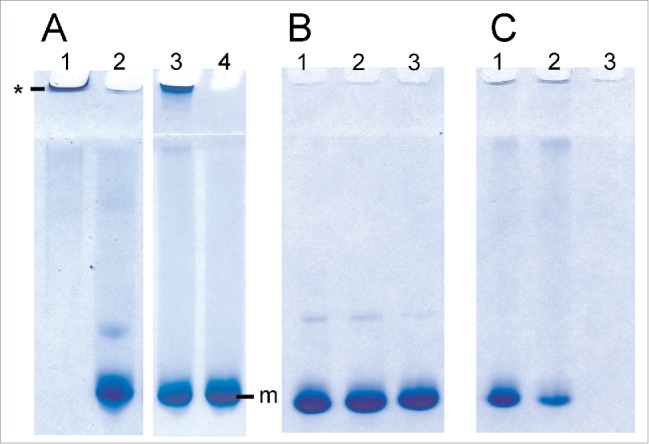
RENAGE gels showing that dLPS is the minimum component of LPS that causes PrP conversion to large oligomers or fibrils. RecMoPrP23–231 at 0.5 mg/mL (19.3 µM) was incubated in 20 mM NaCH3CO2 at pH 5.5 with molar ratios of (A) 1:1.3 PrP:LPS (lane 1), 1:4 lipid A (lane 2), 1:1.3 dLPS (lane 3) and 1:50 PrP:Kdo (lane 4). Panel (B) is recMoPrP23–231 incubated with lipid A at molar ratios of 1:2 (lane 1), 1:4 (lane 2), and 1:8 (lane 3) PrP:lipid A. Panel (C)is recMoPrP23–231 incubated with Kdo2-lipid A at molar ratios of 1:2 (lane 1), 1:4 (lane 2), and 1:8 (lane 3) PrP: Kdo2-lipid A. Both LPS and dLPS-induce PrP conversion to large oligomers (labeled by *). Kdo2-lipid A induced formation of a visible precipitate. PrP in the presence of the other components (Kdo and Lipid A) remained monomeric (labeled by m).
Surprisingly Lipid A did not induce formation of soluble recPrP large oligomers or fibrils, similar to LPS. Initial we solubilized lipid A in 50% methanol:water, which generated a cloudy suspension. When this 50% methanol suspension of lipid A was mixed with recPrP, a protein aggregate was seen in the top of the RENAGE well (Fig. 2A, lane 2). After 10 d of incubating recMoPrP23–231 with this lipid A suspension, a visible precipitate was present. However, it was thought that the poorly solubilized lipid A could have caused the non-specific protein precipitation of PrP. Therefore we solubilized lipid A powder in 0.2% trimethylamine (1 mg/mL stock solution), which fully solubilized lipid A. When lipid A in 0.2% trimethylamine was added to recMoPrP23–231 at molar ratios of 1:2, 1:4, and 1:8 (PrP:lipid A) and incubated overnight at 37°C, no precipitate was found and PrP remained monomeric (Fig. 2B).
Kdo incubated with recMoPrP23–231 was tested at molar ratios of recMoPrP23–231 to Kdo of 1:4 and 1:50 and our results showed that PrP remained monomeric. This indicates that the Kdo component has essentially no effect on the prion protein. Kdo2-lipid A contains the Kdo disaccharide and lipid A. As a result Kdo2-lipid A represents a minimalist form of lipopolysaccharide, containing both oligosaccharide and lipid. However, our results showed that Kdo2-lipid A did not induce conversion to LPS-like soluble, large PrP oligomers. Instead Kdo2-lipid A caused visible protein precipitation, despite being fully solubilized prior to being added to the PrP samples. Precipitate was visible immediately after adding Kdo2-lipid A to recMoPrP23–231. In addition a disappearance of the PrP monomer band is seen in the RENAGE gels at ratios of recMoPrP23–231 to Kdo2-lipid A of 1:2, 1:4, and 1:8 (mol:mol) (Fig. 2C). We cannot conclude whether a specific or a non-specific interaction of Kdo2-lipid A with recMoPrP23–231 caused a precipitate to form. However we can conclude that Kdo2-lipid A does not induce the type of PrP oligomerization which is induced by LPS and dLPS.
Ligand Interaction of LPS Components
LPS binds strongly to recPrP, as was demonstrated using NMR spectroscopy by Saleem et al.14 To facilitate further binding analyses with different LPS components we decided to develop a fluorescent assay to investigate binding. Full length recMoPrP23–231 has 8 tryptophan residues. Truncated recMoPrP90–231 contains 2 tryptophan residues (W98 and W144). To simplify the fluorescence spectra and determine the presence of ligand binding, we employed the truncated PrP construct (MoPrP90–231). We first confirmed the strong binding of LPS to recPrP using tryptophan fluorescence. Emission fluorescence spectra were collected for 15 µM recMoPrP90–231 (corresponding to 0.28 mg/mL) in 20 mM sodium acetate pH 5.5 with added concentrations of 0.05, 0.15, 0.3 and 0.6 mg/mL LPS (Fig. 3A). These LPS concentrations were estimated to be 5, 15, 30 and 60 µM LPS, respectively, from an average LPS molecular weight of 10 kDa. Binding of LPS to recMoPrP90–231 is evident from the fluorescence enhancement and concurrent emission maximum blue shift (inset of Fig. 3A). The background fluorescence from 60 µM LPS alone was negligible (Fig. S1).
Figure 3.
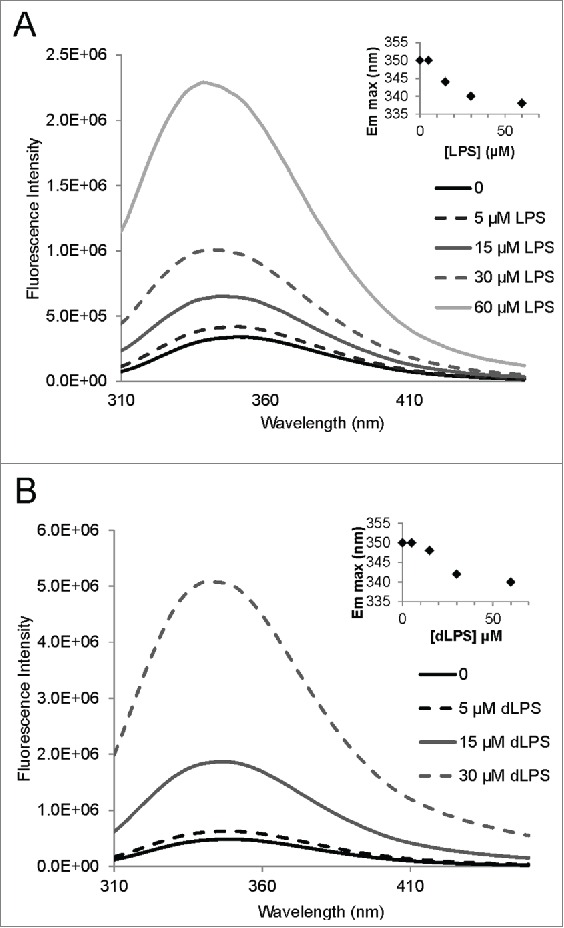
Tryptophan fluorescence of recMoPrP90–231 with LPS and dLPS showing strong binding of the compounds to the PrP protein. Emission spectra were acquired for a 15 µM (0.28 mg/mL) solution of recMoPrP90–231 with 0.05, 0.15, 0.3 and 0.6 mg/mL LPS (A) and dLPS (B). These concentrations of LPS and dLPS correspond to 5, 15, 30 and 60 µM, respectively, based on an estimated molecular weight of 10 kDa for both compounds. The insets on both panels show the emission maximum shift with increasing LPS and dLPS concentration. Note: the 60uM dLPS+MoPrP90–231 is not shown in B.
Tryptophan fluorescence enhancement and an emission maximum blue shift were also observed when 15 µM recMoPrP90–231 was mixed with 0.05, 0.15, 0.3 and 0.6 mg/mL of dLPS (5, 15, 30 and 60 µM, respectively). This result indicates that dLPS strongly binds to PrP and induces a similar conformational change in PrP (Fig. 3B). Intriguingly the emission wavelength blue shift was slightly less for dLPS (340nm) compared to intact LPS (338 nm). This suggests that LPS induces slight different structural conformation changes in PrP than dLPS.
Binding of glucosamine (GlcN), Kdo, lipid A and Kdo2-lipid A to recMoPrP90–231 was also tested by tryptophan fluorescence. Neither glucosamine, Kdo nor lipid A had any effect on tryptophan fluorescence of recPrP, indicating that they either do not bind in the same manner as LPS does or they do not bind at all (results not shown). Although tryptophan fluorescence could miss binding events that do not impact the Trp residues in PrP, it is certain that glucosamine, Kdo or lipid A do not bind to PrP in a similar fashion as LPS or dLPS. For binding to be measured by tryptophan fluorescence Kdo would need to bind near the tryptophan such that the carboxylate (Kdo) group quenched fluorescence or induced a conformational change that result in quenching or change in the immediate environment of the tryptophan.28 In the case of LPS and dLPS binding to PrP, the fluorescence enhancement and blue shifting is indicative of a change in the immediate environment of the tryptophan residues. This fluorescent enhancement is consistent with conformational change, oligomerization, and/or solvation in a lipid environment, upon recMoPrP interacting with LPS and dLPS micelles. If lipid A bound to recPrP we could expect that the interaction with the hydrophobic interface would induce changes in tryptophan fluorescence. The effect of Kdo2-lipid A on recMoPrP90–231 tryptophan fluorescence was also tested, even though Kdo2-lipid A caused protein precipitation. Protein precipitation removes PrP from solution thereby the mixture of PrP and Kdo2-lipid A is not at equilibrium conditions and ligand binding cannot be ascertained.29,30 The formation of protein precipitation could be exacerbated or caused by non-specific interactions with impurities. We were interested to see if fluorescence quenching or fluorescence enhancement occurred at molar ratios of 1:1 or 1:0.33 PrP:Kdo2-lipid A since minimal loss of monomer occurred at a 1:2 ratio as seen by RENAGE (Fig. 2C). Our results showed a decrease in fluorescence emission with 1:0.33 and 1:1 of PrP to Kdo2-lipid A. However, this corresponded directly to the decrease in absorbance at 280 nm, which indicates that protein was precipitating out of solution.
Concentration Dependence of LPS and dLPS Induced PrP Conversion
We investigated the minimal concentration of LPS or dLPS required for PrP conversion as well as tested for potential effects of the propagation of PrP conversion, by serial dilution of converted PrP large oligomers with fresh recPrP. Conversion of recPrP to large oligomers (megamers) occurs within minutes. This is based on the time it takes to prepare, load and initiate a RENAGE gel run (see Fig. 2A*). Incubation of recMoPrP90–231 or recMoPrP23–231 with ratios of PrP to LPS or PrP to dLPS, at or above a weight ratio of 1:1 cause instant and complete conversion to these large oligomers. The required concentration of LPS and dLPS needed to induce PrP conversion of recMoPrP23–231 was followed by 2-fold serial dilution starting with a weight ratio of 1:4 or 1:1 recMoPrP23–231 to lipid. This PrP conversion was conducted in a 20 mM MES buffer at pH 6.5 and incubated at 37°C for 4 d before running the RENAGE gel. Both LPS and dLPS induce 100% conversion of recMoPrP23–231 at a weight ratio of 1:1 (Fig. 4, lanes 7 and 14) and above (results not shown). The PrP conversion efficiency was measured from the ratio of the Coomassie stained bands of the large oligomer (and 16 to 24-mers, if any) versus the total intensity of all bands in that gel lane. Conversion at a ratio of 1:0.5 occurs with 25% efficiency for LPS-induced conversion and 49% for dLPS-induced conversion. The conversion efficiency was similar whether the PrPβ used for 2-fold serial dilution, was incubated for 4 d at 37°C (as shown) or if it was used immediately after mixing LPS/dLPS with PrP (results not shown). The presence of a large oligomer band, at the top of the RENAGE stacking gel, is seen at all dilutions down to weight ratios of 1:0.016 of protein to LPS or dLPS (0.008 mg/mL LPS/dLPS). However the intensity of the 1:0.016 band matches with dilution of the 1:0.032 large oligomer band by half. This result is consistent with results from Saleem et al.,14 who found that LPS-induced PrP conversion does not occur below the LPS critical micelle concentration (CMC) of 0.014 mg/mL31 and that propagation of conversion does not occur. Similarly dLPS-induced PrP conversion has similar concentration dependence with dLPS, where PrP conversion does not occur below the CMC and PrPβ is stable below this dLPS concentration.
Figure 4.
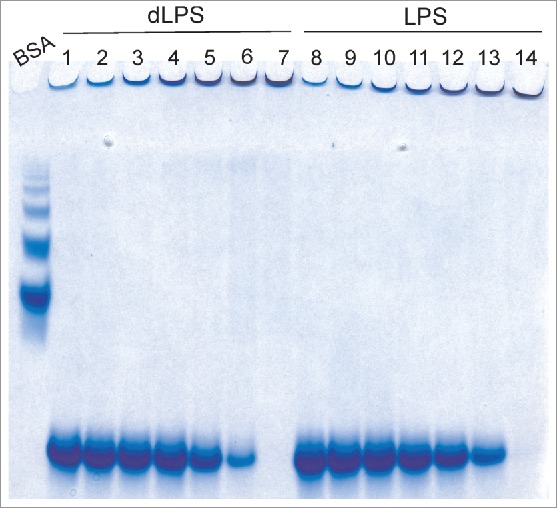
RENAGE gels showing the effects of serial dilution of LPS and dLPS-induced PrPβ conversion with fresh recMoPrP23–231. (A) Samples are from the serial dilution of PrPβ formed from 1:1 (g:g) PrP to LPS (0.5 mg/mL) incubated in 20 mM MES, pH 6.5 at 37°C for 4 d (lane 7 and 14), and diluted with fresh recMoPrP23–231 in buffer. The resulting ratios of PrP to dLPS (lanes 1–6) or PrP to LPS (lanes 8–13) (and the LPS/dLPS concentration) are 1:0.016 (0.008 mg/mL, lane 1 and 8), 1:0.032 (0.016 mg/mL, lane 2 and 9), 1:0.063 (0.031 mg/mL, lane 3 and 10), 1:0.125 (0.063 mg/mL, lane 4 and 11), 1:0.25 (0.125 mg/mL lane 5 and 12), and 1:0.5 (0.25 mg/mL, lane 6 and 13). The size of the oligomers is compared to a BSA ladder.
Intriguingly, the RENAGE gel results show the presence of faint, smaller oligomers (< 25-mers), formed by LPS and dLPS induced conversion of recMoPrP23–231, in addition to large oligomers (Fig. 4). The presence of these small oligomers is more pronounced in dLPS-converted PrP. The size of this small oligomer band from dLPS-induced PrP conversion corresponds to a series of oligomers that are 12-mers (or larger), and was determined using the bovine serum albumin (BSA) ladder, as previously described.26 The presence of these small oligomers is more pronounced in LPS mediated conversion of recombinant Syrian hamster (Sh) PrP90–232 (Fig. 5A). The presence of small oligomers is found when converting 1 mg/mL recShPrP90–232 with 0.09 mg/mL, 0.17 mg/mL and 0.75 mg/mL LPS. These small oligomers range from 9-mers to 24-mers, with the 9-mer, 10-mer and 11-mer clearly separated on the RENAGE gel. The size of these LPS-induced recShPrP90–232 oligomers was determined using shaking-induced PrP oligomers as a standard.27 The formation of approximately 20-mer oligomers is also seen in the LPS-induced conversion of recMoPrP90–231 and recombinant cervid recCePrP94–233, but no 9, 10 or 11-mer bands are visible for these constructs (Fig. 5B). Specific, but as yet undetermined, properties of recShPrP90–232 increase the amount of 9-mer to 24-mers and allow for a banding pattern to be observed under the concentrations loaded.
Figure 5.
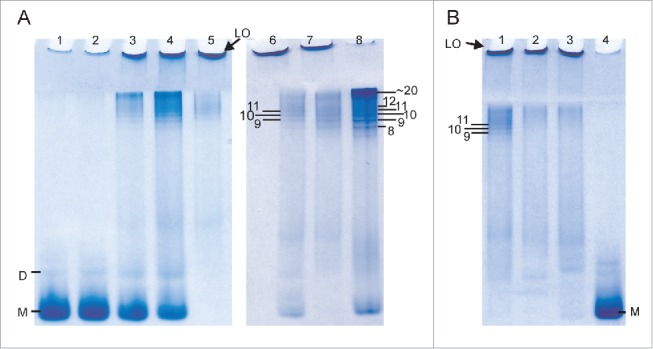
LPS-induced conversion of truncated recPrP generates large oligomers (LO) and small oligomers (9-mer to 20-mers). (A) Concentration dependent conversion of 1 mg/mL recShPrP90–232 in 15 mM NaPO4, 3.3 mM NaCH3CO2 at pH 6.5 with 0.005 mg/mL LPS (lane 1), 0.01 mg/mL LPS (lane 2), 0.09 mg/mL (lane 3), 0.17 mg/mL (lane 4) and 0.75 mg/mL LPS (lane 5). The formation of small oligomers sized 9-mer to 20-mer of 0.5 mg/mL recShPrP90–232 in 150 mM NaCl (unbuffered, pH 5.5) with 0.5 mg/mL LPS (lane 6) was sized by the known size of oligomers generated by shaking at 350 rpm with (lane 7) and without (lane 8) LPS. (B) LPS converts and generates different amounts of small oligomers with 1 mg/mL recShPrP90–232 (lane 1), recMoPrP90–231 (lane 2), and recCePrP94–233 (lane 3) with 0.75 mg/mL LPS in buffer (15 mM NaPO4, 3.3 mM NaCH3CO2 pH 6.5). Lane 4 is monomeric recShPrP90–232. Monomer is labeled with M and dimer is labeled as D.
The results in Figure 5 are also used to determine the concentration dependence of LPS-induced conversion of a truncated PrP construct. We monitored the conversion of recShPrP90–232 using RENAGE at 0.005, 0.01, 0.09, 0.17 and 0.75 mg/mL LPS (Fig. 5A). In this experiment we added LPS at the indicated concentrations to a solution of 1 mg/mL of recShPrP90–232. The recShPrP90–232 construct was used so that we could better compare our results with those from Saleem et al.14 Below the CMC of LPS (0.005 mg/mL) only a faint large oligomer band is seen on RENAGE. This is consistent with conversion not occurring below the CMC. The percentage ratio of monomer and oligomers (small (9 to ∼24-mers) and large (>24-mers)) was plotted against the LPS concentration (Fig. 6). This shows that LPS converts recShPrP90–232 with an exponential trend. Over 50% of the conversion occured when the LPS concentration was above 0.17 mg/mL, corresponding to a weight ratio of 1:0.17 (PrP:LPS; Fig. 6).
Figure 6.
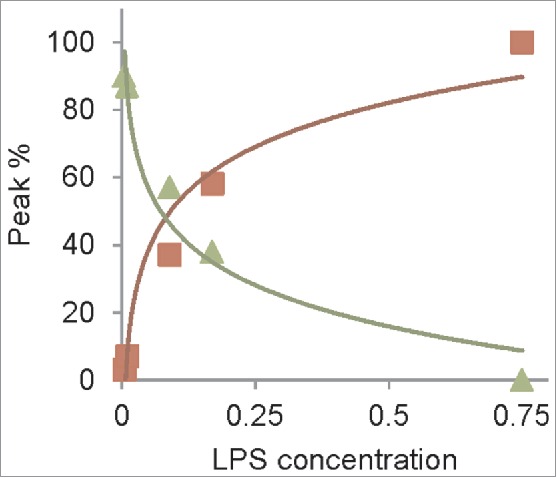
Concentration dependence of recShPrP90–232 with LPS follows exponential trend. Plot of peak area of monomer (triangles) and oligomers (small (9–20-mers) plus large (squares)) vs. LPS concentration (squares), from RENAGE gel in Fig. 5A (lanes 1 to 5).
PrPSc Properties of PrP Oligomers Induced by LPS and dLPS
Our previous work showed that LPS-induced conversion of α-helical recShPrP90–232 and recMoPrP90–231 led to a β-rich oligomeric form with 32% β-sheet.14 Here we collected CD spectra of 0.5 mg/mL recMoPrP23–231 alone and with a weight ratio of 1:1 PrP to LPS or 1:1 PrP to dLPS (Fig. 7). The LPS or dLPS were added 30 to 45 minutes before acquiring the CD spectra. Analysis of the secondary structure content of the resulting CD spectra was performed using BeStSel.32 Our results show that the structure of native recMoPrP23–231 consists of 35.4% α-helix, 7.3% β-sheet, 12.3% turn and 45% disordered – as expected. In contrast, when LPS was added to recMoPrP23–231 the structure consists of 33.2% β-sheet, 13.9% α-helix, 10.8% turn and 42.1 % disordered. Similarly when dLPS was added to recMoPrP23–231 the structure contains 34.1% β-sheet, 14% α-helix, 11.6% turn and 40.3% disordered secondary structure. The LPS/dLPS conversion of recMoPrP23–231 yields a β-sheet rich structure similar to PrPSc, which contains 43% β-sheet.33,34 Both LPS and dLPS induce the formation of large oligomers with similar β-rich structures.
Figure 7.
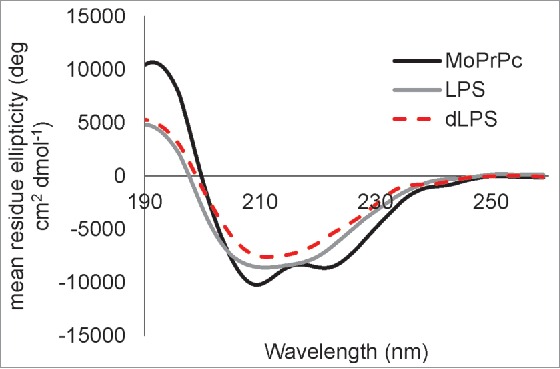
Both LPS and dLPS induce rapid conversion of α-helical recMoPrP23–231 to a β-sheet rich structure. CD spectra are shown for recMoPrP23–231(black line), recMoPrP23–231 plus 1:1 (g:g) LPS (gray line),and recMoPrP23–231 plus 1:1 (g:g) dLPS (red dashed line). The recMoPrP23–231 concentration is at 0.5 mg/mL in water and the LPS or dLPS was added 30 to 45 min before acquiring the spectra. The secondary structure content calculated from BeStSel of recMoPrP23–231 is 35.4% α-helix, 7.3% β-sheet, 12.3% turn and 45% other/disordered; with LPS is 33.2% β-sheet, 13.9% α-helix, 10.8% turn and 42.1 % other/disordered; and with dLPS is 34.1% β-sheet, 14% α-helix, 11.6% turn and 40.3 % other.
The large PrP oligomers that were formed by LPS and dLPS were analyzed by negative stain transmission electron microscopy (TEM). To prepare the samples 0.5 mg/mL of recMoPrP23–231 in 20 mM MES, pH 6.5 was incubated with LPS or dLPS for 4 d at 37°C. Negative stain TEM using uranyl acetate showed that large oligomers or spheroids were formed in both cases (Fig. 8A, B). Less commonly, rod-like fibrils were seen in the electron micrographs of LPS converted PrPβ samples. In comparison, negative stain TEM of recMoPrP23–231 incubated with Kdo2-lipid A, using the same conditions, showed clumps, consistent with amorphous aggregate (Fig. 8C). Electron micrographs of LPS and dLPS alone, contained no oligomer structures. Instead the electron micrograph of LPS alone, contained supramicellular structures and those of dLPS were largely blank, with no defined structures and sometimes spindle-like. (Fig. S2). The sizes of the large PrP oligomers formed in both LPS and dLPS were a range of 17 to 50 nm in width, and they appear to loosely associate into larger aggregates. The formation of large proteinaceous PrP oligomers is also seen with infectious recPrPSc generated using recPrP with POPG, RNA and PMCA.35 Furthermore, the PrP oligomers formed from LPS-induced and dLPS-induced PrP conversion were free of micelles (Fig. 8A, B). This suggests that PrP binds to LPS or dLPS, and undergoes conformational change and conversion to large oligomers.
Figure 8.

Negative stain TEM of recMoPrP23–231 incubated with LPS (A) or dLPS (B) for 4 d or for 20 hours with Kdo2-lipid A (c). The micrographs show the formation of large oligomers or spheroids. The size of the large PrP oligomers formed with LPS or dLPS was 17–40nm. An amorphous aggregate was observed when the precipitate formed with Kdo2-lipid A and PrP was examined. The scale bar = 100 nm.
Proteinase K resistance is one of the hallmarks of PrP fibrils and infectious prions (PrPSc).36 As a result, we tested the β-sheet oligomers formed by LPS and dLPS for evidence of PK resistance (Fig. 9). RecMoPrP23–231 at 0.5 mg/mL in 20 mM MES pH 6.5 was incubated with 0.5 mg/mL LPS or 0.5 mg/mL dLPS for 4 d at 37°C. The resulting PrP oligomer samples were then incubated with PK at weight ratios of 1:50, 1:200 or 1:400 of PK to PrP. They were then separated on a 12% acrylamide SDS-PAGE gel and visualized with Colloidal Coomassie blue staining. PK digestion of native, monomeric recMoPrP23–231 produces PK resistant fragments below 11 kDa (Fig. 9A). In contrast, LPS converted PrP oligomers have PK-resistant fragments at bands corresponding to 17/18 kDa and 13/14 kDa, at a weight ratio of 1:400 PK to PrP (Fig. 9B). PrP oligomers generated with dLPS have the same sized PK-resistant fragments as those formed by intact LPS induced PrP conversion (Fig. 9C). PK resistance between LPS and dLPS differed only slightly. The LPS-induced PrP conversion generated PK resistant fragments with molecular weight bands of 13/14 kDa at a 1:200 ratio, but bands of this size were not seen at this ratio of PK to PrP in dLPS converted PrPβ. However in other experiments we have seen resistance at a PK to PrP ratio of 1:200 with recMoPrP23–231 converted with dLPS (results not shown). The PrP large oligomers that are formed in LPS or dLPS have less PK resistance than found in deglycosylated PrPSc.37 However, we were interested in the 17/18 kDa PK-resistant fragment, present in both LPS-induced and dLPS-induced PrP conversion. This 17/18 kDa band was undetected with PK-digested native monomer. This reflects a similar conformational change present in LPS or dLPS converted PrPβ. We also tested for PK resistance immediately after adding LPS or dLPS to recMoPrP23–231. In these samples we also found 17/18 kDa and 13/14 kDa bands at 1:400 PK to PrP, but fewer of these large PK-fragments were seen after immediate conversion than were found after 4 d incubation (results not shown). Instead, more intense bands were seen below 11 kDa after PK digestion. This result is consistent with the immediate conversion of PrPc to PrPβ due to LPS and dLPS, but longer incubation times generate enhanced PK resistance.
Figure 9.
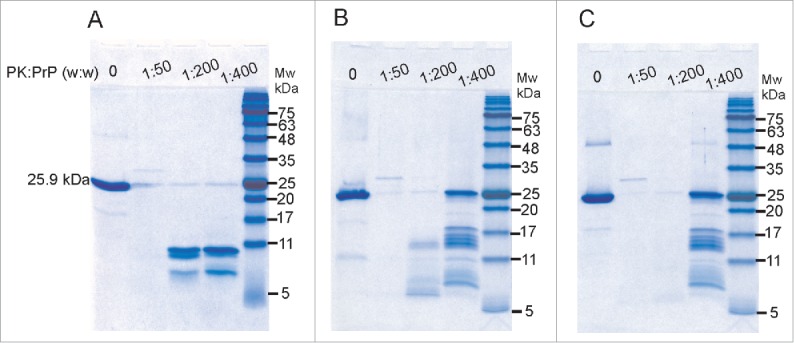
LPS and dLPS-converted recPrP forms a PK resistant core. SDS-PAGE of PK treated samples of recMoPrP23–231 (A), recMoPrP23–231 with 1:1 (g:g) LPS (B) and recMoPrP23–231 with 1:1 (g:g) dLPS (C). The samples were incubated at 37°C for 4 d before PK digestion.
DISCUSSION
We previously demonstrated LPS-induced conversion of recPrPc to PrPβ.14 As LPS contains multiple lipid and saccharide components, we examined whether a chemical constituent of LPS will induced PrP conversion. Our results show that both the polysaccharide and lipid component of LPS are required to induce PrP conversion to PrPβ. This conversion is not possible with either lipid A or Kdo2-lipid A or any of the smaller chemical components of LPS such as Kdo or glucosamine. Only a modestly de-acylated form of LPS, called dLPS, is able to generate PrPβ in a similar fashion to intact LPS. Our results also indicate that the intact structure of lipid A is not required, but that the scaffolding afforded by the micellar nature of LPS and dLPS is required for conversion.
The strong binding of LPS to recPrP, as originally recorded by solution NMR14, is also demonstrated here using tryptophan fluorescence. Binding of LPS or dLPS to PrP actually increased, not quenched, tryptophan fluorescence. The use of tryptophan fluorescence also allowed us to detect similar interactions with PrP and to monitor recPrP binding to dLPS. However, our tryptophan fluorescence results showed that Kdo, glucosamine, and Lipid A, did not bind to recPrP. Because a precipitate formed when Kdo2-lipid A was added to recMoPrP23–231 and recMoPrP90–231, we could not conclude if Kdo2-lipid A interacts specifically with PrP, and thus induces the formation of PrP aggregates. Alternatively the precipitate/aggregates may be caused by poorly dissolved Kdo2-lipid A or impurities non-specifically interacting with PrP. However, we can conclude that Kdo2-lipid A does not induce the type of PrP oligomerization which is induced by LPS and dLPS. The strong binding of both LPS and dLPS to recPrP is strengthened by the β-sheet conformational change (as determined by CD analysis). The conformational change (to a beta-rich PrPβ) and the concomitant oligomerization induced by LPS is expected to cause both a loss of NMR signal, as seen previously14, as well as the fluorescence enhancement and blue shift seen in this study. Our results are consistent with a transient interaction of a conversion-inducing cofactor binding to PrPc and promoting rapid formation of PrPβ. Moreover, our data also shows that PrPβ, once formed, is stable without the presence of LPS of dLPS micelles. This PrPβ form is stable and can be seen on RENAGE gels (Fig. 4) after the LPS or dLPS is diluted beyond the CMC. TEM images also show the formation of large protein oligomers with no detectable LPS micelles (Fig. 8). These results agree with those reported for POPG-induced PrP conversion, where the interaction of PrP with POPG is not required to maintain PrPβ and the interaction is transient.18
The PrPβ oligomers that we generated with LPS and dLPS-induced conversion of full-length recMoPrP23–231 have similarly high β-sheet content as Saleem et al.14 observed with LPS-converted truncated Syrian hamster PrP (recShPrP). In particular, Saleem et al. showed that recShPrP90–232 converted to PrPβ oligomers with a weight ratio as low as 1:0.75 of LPS to PrP and generated a PrPβ form with up to 32% β-sheet.14 Similarly, CD of the PrPβ oligomers we generated via LPS and dLPS-induced conversion of full length recMoPrP23–231 produced large oligomers with 33% and 34% β-sheet content, respectively. We can use the concentration dependence of LPS-induced conversion presented here (using RENAGE – Fig. 5A and Fig. 6) to compare to the conformational change measured by CD in Saleem et al.14 Both RENAGE and CD analysis of LPS converted recShPrP90–231, studied by Saleem et al., show conversion at sub-stoichiometric ratios of PrP to LPS. RENAGE of converted recShPrP90–232 show that a ratio as low as 1:0.09 (PrP to LPS) there is 37% conversion to PrPβ small and large oligomers (Fig. 5A and Fig. 6). CD analysis of this 1:0.09 sample contained 20% β-sheet. Therefore, both our studies and those of Saleem et al. indicated that conversion occurs when the LPS concentration is above the CMC.
Both LPS and dLPS are able to induce recPrPc to form large PrPβ oligomers with PK resistant fragments larger than those in native recPrPc. Oligomeric isoforms of PrPβ are also formed from other PrP conversion methods.6,38 Oligomerization has shown to be critical for conversion potency in the propagation of native PrPc to PrPSc in PMCA and QuIC assays.39 Furthermore, oligomers are linked to a form of relatively less protease resistant prions that peak at the transition from asymptomatic to symptomatic prion disease.40 Despite oligomers usually displaying less protease resistance than fibrils,41,42 the PrPβ formed by LPS and dLPS contain a 17/18 kDa PK resistant fragment at a weight ratio of 1:400 PK to PrP. The presence of 17/18 kDa PK resistance bands is intriguing because a 17 kDa PK resistant band is found in deglycosylated PrPSc.37 In addition, when infectious recPrPSc is generated via PMCA and POPG with RNA, a 17 kDa PK resistance band is prominent.4 Therefore, the presence of a 17 kDa PK resistance fragments in our study suggests that the large β-sheet rich PrP oligomers generated by both LPS and dLPS share similar structural features with infectious prions. Notably, infectious prions generated from recombinant PrP also contained large oligomers that were 26 nm in diameter.35 The size of PrPβ oligomers formed with LPS and dLPS is a heterogeneous mix of oligomers 17–50 nm in diameter.
Overall this study was designed to provide additional biophysical insight into the mechanism and generality of the LPS-induced PrP conversion phenomenon first observed by Saleem et al.14 Our data shows that LPS converts many forms of PrP (full length and truncated), from many species of PrP (mouse, Syrian hamster, cervid) and under a wide range of LPS to PrP concentrations. We have also shown that a minimal version of LPS (called detoxified and partially de-acylated LPS or dLPS) containing a portion of the polysaccharide and a portion of the lipid component is sufficient for PrP conversion. Lipid components, alone, and polysaccharide components, alone, are insufficient for conversion. Because PrP conversion by LPS or dLPS occurred only at concentrations at or above the CMC, this suggests that transiently formed LPS micelles catalyze the binding and conversion of PrPc to PrPβ. Characterization of the PrPβ products formed by LPS and dLPS via fluorescence, EM, CD, PK digestion and RENAGE demonstrated that these oligomers exhibit many of the physical properties of infectious PrPSc particles.
MATERIALS AND METHODS
Materials
For the experiments described here, recombinant mouse PrP full length 23–231 (recMoPrP23–231), truncated 90–231 (recMoPrP90–231) as well as recombinant Syrian hamster truncated 90–232 (recShPrP90–232) and recombinant cervid truncated 94–233 (recCePrP94–233) prion proteins were used. These constructs contain a His6x purification tag and were expressed and purified as previously described.27,43 Other supplies were purchased from various vendors. These included: LPS E. coli O111:B4 from Sigma-Aldrich (L3024), dLPS E.coli O111:B4 from Sigma-Aldrich (L3023), Lipid A (monophosphorylated from Salmaonella minnesota R595) from Avanti Polar Lipids (699200P), Kdo2-Lipid A (diphosphorylated) from Avanti Polar Lipids (699500P), 2-keto-3-deoxyoctonate (Kdo) from Sigma-Aldrich (K2755), glucosamine HCl from Sigma-Aldrich (G4875) and proteinase K from Promega (V3021). dLPS (L3023) from Sigma-Aldrich is partially delipidated by alkaline hydrolysis, which will preferentially remove the ester-linked fatty acids and thus leave the N-linked fatty acids intact.44
Stocks of the LPS components were prepared as follows. For intact LPS solutions the powder was suspended in water to give a solution with a concentration of 5 mg/mL and then vortexed for 1 min, heated at 70°C for 1 min and sonicated in water bath sonicator for 1 min. For the dLPS stock solutions the powder was suspended in water to give dLPS solution with a concentration of 5 mg/mL and the solution vortexed for 5 min. To prepare 1 mg/mL of lipid A the powder was suspended in water and frozen. Then to an aliquot 0.2% trimethylamine was added and then heated at 65–70°C for 20 sec, vortexed for 1 min, heated at 65–70°C for 20 sec and sonicated for 5 minutes. This heating, vortexing, heating and sonication process was repeated to acquire a clear solution. A 1 mg/mL stock of Kdo2-lipid A was prepared by dissolving the powder in 20 mM sodium phosphate pH 7.4, sonicating 5 min, heating at 70°C and vortexing to obtain a clear solution.
Resolution Enhanced Native Acidic Gel Electrophoresis (RENAGE)
The loss of monomeric PrP and conversion to small and large PrP oligomers/fibrils was visualized using a specially developed technique called RENAGE.26 Gels were prepared using a 8% acrylamide pH 4.3 running gel and a 3% acrylamide pH 5.2 stacking gel as previously described.26 The running buffer consisted of 0.35 M β-alanine and 0.14 M acetate at pH 4.3. Samples were mixed with 5X dissolving buffer (37% glycerol, 128 mM acetate-KOH, pH 5.2 and 0.01% crystal violet (Sigma-Aldrich Canada)). Gels were pre-run at a current of 30 mA per gel at normal polarity (anions move down the gel) for 17 to 20 minutes and then 5 μg of the different PrP samples were loaded in the dissolving buffer. The gels were then run at 30 mA with reverse polarity (cations move down the gel) for ∼85 minutes. Gels were stained with colloidal Coomassie blue for at least 4 hrs and then destained in water.45 The size of the recMoPrP23–231 oligomers was compared to a ladder of bovine serum albumin (BSA) oligomers that form when lyophilized BSA (Sigma-Aldrich, L7906) is dissolved in water.
The area of protein bands was determined by converting the gel lanes to a band-intensity chromatogram using ImageJ (http://rsbweb.nih.gov/ij/index.html). Chromatograms were then plotted using the Origin software package (OriginLab Corp., version 9) and the peaks manually marked and integrated, using Origin's “peak analyzer” module. Percentages of monomer, small oligomers and large oligomers were then calculated from the area of each peak compared to the total integrated area.
Tryptophan Fluorescence
The ability of LPS and its various components (dLPS, Kdo2-lipid A, lipid A, Kdo and glucosamine) to bind to PrP was measured by the quenching or enhancement of tryptophan fluorescence of recMoPrP90–231. Samples were prepared with a concentration of 15 µM recMoPrP90–231 in 20 mM sodium acetate at pH 5.5 with 5, 15, 30 and 60 µM of the LPS or LPS components including: Kdo, lipid A, Kdo2-lipid A, and dLPS, in a total volume of 0.1 mL. The LPS or LPS components were added 30 min to 1 hr before collecting the fluorescence spectra. Emission spectra were acquired on a QuantaMaster 400 spectrofluorimeter (Photon Technology International Inc., London, ON, Canada) with an excitation wavelength of 295 nm and emission wavelengths collected from 310 to 450 nm. The excitation slits covered a 2 nm window and the emission slits covered a 5 nm window. The step size was 2 nm with an integration time of 1 sec for each point. The absorbance at 280 nm of the samples was measured after acquiring the spectra. This was used as a measure of the amount of PrP that precipitated during the experiment, and where there was no precipitate to correct for effect of LPS or dLPS absorbance. The quartz cuvette path length was 3 mm. We corrected for the inner filter effect caused by increase in absorbance using the following equation.46
where Fobs is the measured fluorescence intensity; Fcorr is the calculated corrected fluorescence intensity; Aex is the change in excitation absorbance caused by ligand; and dex is the 3 mm path length.
Circular Dichroism
The secondary structure of recMoPrP23–231 treated with LPS or dLPS was determined using CD. Samples were prepared with 0.5 mg/mL PrP in water with a weight ratio of 1:1 LPS or dLPS added 30 min before collecting the spectra. The LPS stock at 5 mg/mL was reclarified by vortexing for 1 min followed by 2 cycles of heating at 70°C and sonicating for 1 min in a water bath sonicator. CD spectra were acquired on a Jasco J-810 circular dichroism spectropolarimeter using a 0.1 mm quartz cell with samples dissolved in 20 mM sodium acetate, pH 5.5. Spectra were recorded as the average of 3 or 4 scans from 190 to 260 nm. All spectra were acquired with a scan rate of 20 nm/min and smoothed with a Savitzky–Golay window of 13 points. The secondary structure content was determined using BeStSel.32
Negative Stain Electron Microscopy
PrP samples converted with LPS or dLPS were applied to freshly UV irradiated 300 mesh copper grids with a support film of Formvar with carbon (Ted Pella Inc., Cat# 01753-F, Redding, CA, USA). The PrP samples were diluted to 2 μM in water and then 10 μL of the PrP solution was spotted onto Parafilm. The grid was placed on top of the droplet for 30 seconds. Grids were washed once with 10 µL double-deionized water and stained twice with 10 µL 4% uranyl acetate, for 30 seconds each. Residual solution was wicked away using filter paper. Micrographs were acquired on a Philips/FEI (Morgagni) transmission election microscope at 80 kV.
Proteinase K Digestion
The susceptibility/resistance to proteinase K (PK) digestion of the LPS and dLPS-converted PrP samples was determined using a method adapted from Atarashi et al.37. Samples were generated by incubating 0.5 mg/mL recMoPrP23–231 in 20 mM MES pH 6.5 with a weight ratio of 1:1 LPS or dLPS at 37°C for 4 d. Alternatively samples were analyzed immediately after adding LPS or dLPS. For digestion, 12 µL of PrP samples at a 0.5 mg/mL concentration were mixed with 2.4 µL of 1 M Tris buffer at pH 8 and 8.1 µL milli-Q water. Then proteinase K (Promega, Madison, WI, USA) was added at ratios of 1:400, 1:200 and 1:50, PK to PrP (weight-to-weight). The final concentrations were 0.25 mg/mL PrP and 100 mM Tris at pH 8. The sample was digested for 45 minutes at 37°C. After digestion, 2.4 µL of 1 mM PMSF was added (resulting in a 0.1 mM PMSF solution) and the samples were placed on ice until boiling. Next 16.5 µL Laemmli sample buffer (2% SDS, 5% glycerol, 2 mM DTT, 50 mM Tris, pH 6.8, 0.01% bromophenol blue) was added followed by 10 µL of 10 M urea (resulting in a final concentration of 2 M urea). These samples were boiled at 95°C for 5 min and 3 µg protein samples were loaded onto a 12% SDS-PAGE gel with a Tris-Glycine buffer system. The gel was run at 180 V for 45 minutes and visualized with colloidal Coomassie blue.
ABBREVIATIONS
- BSE
bovine spongiform encephalopathy
- CWD
chronic wasting disease
- CJD
Creutzfeldt Jakob disease
- FFI
fatal familial insomnia
- PrPc
cellular prion protein
- PrPSc
infectious particle of misfolded prion protein
- PrP
prion protein
- PrPβ
β-sheet converted prion protein
- recPrP
recombinant prion protein
- TLR
toll-like receptor
- PMCA
Protein Misfolding Cyclic Amplification
- EM
electron microscopy
- CD
circular dichroism
- TEM
transmission electron microscopy
- PK
proteinase K
- RENAGE
resolution enhanced native acidic gel electrophoresis
- recMoPrP23–231
recombinant full-length mouse prion protein residues 23–231
- LPS
lipopolysaccharide
- dLPS
detoxified and partially deacylated LPS
- Kdo
2-keto-3-deoxyoctonate
- POPG
1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-(1′-rac-glycerol)
- PMSF
phenylmethanesulfonyl fluoride
- DTT
dithiothreitol
- CMC
critical micelle concentration
- NMR
nuclear magnetic resonance
Supplementary Material
DISCLOSURE OF POTENTIAL CONFLICTS OF INTEREST
There are no potential conflicts of interest or financial interests to disclose.
ACKNOWLEDGMENTS
EM data was acquired with the help of Arlene Oatway in the Biological Sciences Microscopy Unit. We also thank Gareth Lambkin for assistance and guidance using the SpectroMax i3X spectrofluorimeter.
FUNDING
This work was supported by an Alberta Prion Research Institute (APRI) grant to DSW.
REFERENCES
- [1].Collinge J, Clarke AR. A general model of prion strains and their pathogenicity. Science 2007; 318:930-6; PMID:17991853; http://dx.doi.org/ 10.1126/science.1138718 [DOI] [PubMed] [Google Scholar]
- [2].Caughey B, Baron GS, Chesebro B, Jeffrey M. Getting a Grip on Prions: Oligomers, Amyloids, and Pathological Membrane Interactions. Annu Rev Biochem 2009; 78:177-204; PMID:19231987; http://dx.doi.org/ 10.1146/annurev.biochem.78.082907.145410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Race B, Phillips K, Meade-White K, Striebel J, Chesebro B. Increased Infectivity of Anchorless Mouse Scrapie Prions in Transgenic Mice Overexpressing Human Prion Protein. J Virol 2015; 89:6022-32; PMID:25810548; http://dx.doi.org/ 10.1128/JVI.00362-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Wang F, Wang XH, Yuan CG, Ma JY. Generating a Prion with Bacterially Expressed Recombinant Prion Protein. Science 2010; 327:1132-5; PMID:20110469; http://dx.doi.org/ 10.1126/science.1183748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Stohr J, Weinmann N, Wille H, Kaimann T, Nagel-Steger L, Birkmann E, Panza G, Prusiner SB, Eigen M, Riesner D. Mechanisms of prion protein assembly into amyloid. Proc Natl Acad Sci USA 2008; 105:2409-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Baskakov IV, Legname G, Baldwin MA, Prusiner SB, Cohen FE. Pathway complexity of prion protein assembly into amyloid. J Biol Chem 2002; 277:21140-8; PMID:11912192; http://dx.doi.org/ 10.1074/jbc.M111402200 [DOI] [PubMed] [Google Scholar]
- [7].Wong E, Thackray AM, Bujdoso R. Copper induces increased β-sheet content in the scrapie-susceptible ovine prion protein Prp(VRQ) compared with the resistant allelic variant Prp(ARR). Biochem J 2004; 380:273-82; PMID:14969585; http://dx.doi.org/ 10.1042/bj20031767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Deleault NR, Lucassen RW, Supattapone S. RNA molecules stimulate prion protein conversion. Nature 2003; 425:717-20; PMID:14562104; http://dx.doi.org/ 10.1038/nature01979 [DOI] [PubMed] [Google Scholar]
- [9].Gomes MPB, Millen TA, Ferreira PS, Silva NLCE, Vieira TCRG, Almeida MS, Silva JL, Cordeiro Y. Prion protein complexed to N2a cellular RNAs through its N-terminal domain forms aggregates and is toxic to murine neuroblastoma cells. J Biol Chem 2008; 283:19616-25; PMID:18456654; http://dx.doi.org/ 10.1074/jbc.M802102200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Nandi PK, Leclerc E, Nicole JC, Takahashi M. DNA-induced partial unfolding of prion protein leads to its polymerisation to amyloid. J Mol Biol 2002; 322:153-61; PMID:12215421; http://dx.doi.org/ 10.1016/S0022-2836(02)00750-7 [DOI] [PubMed] [Google Scholar]
- [11].Cordeiro Y, Machado F, Juliano L, Juliano MA, Brentani RR, Foguel D, Silva JL. DNA converts cellular prion protein into the β-sheet conformation and inhibits prion peptide aggregation. J Biol Chem 2001; 276:49400-9; PMID:11604397; http://dx.doi.org/ 10.1074/jbc.M106707200 [DOI] [PubMed] [Google Scholar]
- [12].Ellett LJ, Coleman BM, Shambrook MC, Johanssen VA, Collins SJ, Masters CL, Hill AF, Lawson VA. Glycosaminoglycan sulfation determines the biochemical properties of prion protein aggregates. Glycobiology 2015; 25:745-55; PMID:25701659; http://dx.doi.org/ 10.1093/glycob/cwv014 [DOI] [PubMed] [Google Scholar]
- [13].Kazlauskaite J, Sanghera N, Sylvester I, Venien-Bryan C, Pinheiro TJT. Structural changes of the prion protein in lipid membranes leading to aggregation and fibrillization. Biochemistry-Us 2003; 42:3295-304; http://dx.doi.org/ 10.1021/bi026872q [DOI] [PubMed] [Google Scholar]
- [14].Saleem F, Bjorndahl TC, Ladner CL, Perez-Pineiro R, Ametaj BN, Wishart DS. Lipopolysaccharide induced conversion of recombinant prion protein. Prion 2014; 8:221-32; http://dx.doi.org/ 10.4161/pri.28939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Jucker M, Walker LC. Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature 2013; 501:45-51; PMID:24005412; http://dx.doi.org/ 10.1038/nature12481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Deleault NR, Harris BT, Rees JR, Supattapone S. Formation of native prions from minimal components in vitro. Proc Natl Acad Sci USA 2007; 104:9741-6; PMID:17535913; http://dx.doi.org/ 10.1073/pnas.0702662104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Miller MB, Wang DW, Wang F, Noble GP, Ma J, Woods VL Jr, Li S, Supattapone S. Cofactor Molecules Induce Structural Transformation during Infectious Prion Formation. Structure 2013; 21:2061-8; PMID:24120764; http://dx.doi.org/ 10.1016/j.str.2013.08.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Zurawel AA, Walsh DJ, Fortier SM, Chidawanyika T, Sengupta S, Zilm K, Supattapone S. Prion Nucleation Site Unmasked by Transient Interaction with Phospholipid Cofactor. Biochemistry-Us 2014; 53:68-76; http://dx.doi.org/ 10.1021/bi4014825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Solforosi L, Bellon A, Schaller M, Cruite JT, Abalos GC, Williamson RA. Toward molecular dissection of PrPC-PrPSc interactions. J Biol Chem 2007; 282:7465-71; PMID:17218310; http://dx.doi.org/ 10.1074/jbc.M610051200 [DOI] [PubMed] [Google Scholar]
- [20].Deleault NR, Walsh DJ, Piro JR, Wang F, Wang X, Ma J, Rees JR, Supattapone S. Cofactor molecules maintain infectious conformation and restrict strain properties in purified prions. Proc Natl Acad Sci USA 2012; 109:8546-51; PMID:22586108; http://dx.doi.org/ 10.1073/pnas.1204498109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Kell DB, Pretorius E. On the translocation of bacteria and their lipopolysaccharides between blood and peripheral locations in chronic, inflammatory diseases: the central roles of LPS and LPS-induced cell death. Integr Biol-Uk 2015; 7:1339-77; http://dx.doi.org/ 10.1039/C5IB00158G [DOI] [PubMed] [Google Scholar]
- [22].Kang S-G, Kim C, Cortez LM, Garza MC, Yang J, Wille H, Sim VL, Westaway D, McKenzie D, Aiken J. Toll-like Receptor-mediated Immune Response Inhibits Prion Propagation. Glia 2016; 64:937-51; PMID:26880394 [DOI] [PubMed] [Google Scholar]
- [23].Cunningham C. Microglia and neurodegeneration: The role of systemic inflammation. Glia 2013; 61:71-90; PMID:22674585; http://dx.doi.org/ 10.1002/glia.22350 [DOI] [PubMed] [Google Scholar]
- [24].Vincenti JE, Murphy L, Grabert K, McColl BW, Cancellotti E, Freeman TC, Manson JC. Defining the microglia response during the time course of chronic neurodegeneration. J Virol 2016; 90:3003-17; http://dx.doi.org/ 10.1128/JVI.02613-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Dervishi E, Lam TH, Dunn SM, Zwierzchowski G, Saleem F, Wishart DS, Ametaj BN. Recombinant mouse prion protein alone or in combination with lipopolysaccharide alters expression of innate immunity genes in the colon of mice. Prion 2015; 9:59-73; PMID:25695140; http://dx.doi.org/ 10.1080/19336896.2015.1019694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Ladner CL, Wishart DS. Resolution-enhanced native acidic gel electrophoresis: A method for resolving, sizing, and quantifying prion protein oligomers. Anal Biochem 2012; 426:54-62; PMID:22490465; http://dx.doi.org/ 10.1016/j.ab.2012.04.005 [DOI] [PubMed] [Google Scholar]
- [27].Ladner-Keay CL, Griffith BJ, Wishart DS. Shaking alone induces de novo conversion of recombinant prion proteins to β-sheet rich oligomers and fibrils. Plos One 2014; 9:e98753; PMID:24892647; http://dx.doi.org/ 10.1371/journal.pone.0098753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Lakowicz JR. Principles of Fluorescence Spectroscopy. New York: Springer, 2006 [Google Scholar]
- [29].Hulme EC, Trevethick MA. Ligand binding assays at equilibrium: validation and interpretation. Brit J Pharmacol 2010; 161:1219-37; http://dx.doi.org/ 10.1111/j.1476-5381.2009.00604.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Pollard TD. A guide to simple and informative binding assays. Mol Biol Cell 2010; 21:4061-7; PMID:21115850; http://dx.doi.org/ 10.1091/mbc.E10-08-0683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Santos NC, Silva AC, Castanho MARB, Martins-Silva J, Saldanha C. Evaluation of lipopolysaccharide aggregation by light scattering spectroscopy. Chembiochem 2003; 4:96-100; PMID:12512082; http://dx.doi.org/ 10.1002/cbic.200390020 [DOI] [PubMed] [Google Scholar]
- [32].Micsonai A, Wien F, Kernya L, Lee YH, Goto Y, Refregiers M, Kardos J. Accurate secondary structure prediction and fold recognition for circular dichroism spectroscopy. Proc Natl Acad Sci USA 2015; 112:E3095-103; PMID:26038575; http://dx.doi.org/ 10.1073/pnas.1500851112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Caughey BW, Dong A, Bhat KS, Ernst D, Hayes SF, Caughey WS. Secondary structure-analysis of the scrapie-associated protein PRP 27-30 in water by infrared-spectroscopy. Biochemistry-Us 1991; 30:7672-80; http://dx.doi.org/ 10.1021/bi00245a003 [DOI] [PubMed] [Google Scholar]
- [34].Pan KM, Baldwin M, Nguyen J, Gasset M, Serban A, Groth D, Mehlhorn I, Huang ZW, Fletterick RJ, Cohen FE, et al.. Conversion of α-helices into β-sheets features in the formation of the scrapie prion proteins. Proc Natl Acad Sci USA 1993; 90:10962-6; PMID:7902575; http://dx.doi.org/ 10.1073/pnas.90.23.10962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Piro JR, Wang F, Walsh DJ, Rees JR, Ma JY, Supattapone S. Seeding specificity and ultrastructural characteristics of infectious recombinant prions. Biochemistry-Us 2011; 50:7111-6; http://dx.doi.org/ 10.1021/bi200786p [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Silva CJ, Vazquez-Fernandez E, Onisko B, Requena JR. Proteinase K and the structure of PrPSc: The good, the bad and the ugly. Virus Res 2015; 207:120-6; PMID:25816779; http://dx.doi.org/ 10.1016/j.virusres.2015.03.008 [DOI] [PubMed] [Google Scholar]
- [37].Atarashi R, Moore RA, Sim VL, Hughson AG, Dorward DW, Onwubiko HA, Priola SA, Caughey B. Ultrasensitive detection of scrapie prion protein using seeded conversion of recombinant prion protein. Nat Methods 2007; 4:645-50; PMID:17643109; http://dx.doi.org/ 10.1038/nmeth1066 [DOI] [PubMed] [Google Scholar]
- [38].Yang C, Lo WL, Kuo YH, Sang JC, Lee CY, Chiang YW, Chen RPY. Revealing structural changes of prion protein during conversion from α-helical monomer to β-oligomers by means of ESR and nanochannel encapsulation. Acs Chem Biol 2015; 10:493-501; PMID:25375095; http://dx.doi.org/ 10.1021/cb500765e [DOI] [PubMed] [Google Scholar]
- [39].Kim C, Haldiman T, Surewicz K, Cohen Y, Chen W, Blevins J, Sy MS, Cohen M, Kong Q, Telling GC, et al.. Small protease sensitive oligomers of PrPSc in distinct human prions determine conversion rate of PrPC. Plos Pathog 2012; 8:e1002835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Mays CE, van der Merwe J, Kim C, Haldiman T, McKenzie D, Safar JG, Westaway D. Prion infectivity plateaus and conversion to symptomatic disease originate from falling precursor levels and increased levels of oligomeric PrPSc species. J Virol 2015; 89:12418-26; PMID:26423957; http://dx.doi.org/ 10.1128/JVI.02142-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Simoneau S, Rezaei H, Sales N, Kaiser-Schulz G, Lefebvre-Roque M, Vidal C, Fournier JG, Comte J, Wopfner F, Grosclaude J, et al.. In vitro and in vivo neurotoxicity of prion protein oligomers. Plos Path 2007; 3:1175-86; http://dx.doi.org/ 10.1371/journal.ppat.0030125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Bocharova OV, Breydo L, Parfenov AS, Salnikov VV, Baskakov IV. In vitro conversion of full-length mammalian prion protein produces amyloid form with physical properties of PrPSc. J Mol Biol 2005; 346:645-59; PMID:15670611; http://dx.doi.org/ 10.1016/j.jmb.2004.11.068 [DOI] [PubMed] [Google Scholar]
- [43].Bjorndahl TC, Zhou GP, Liu XH, Perez-Pineiro R, Semenchenko V, Saleem F, Acharya S, Bujold A, Sobsey CA, Wishart DS. Detailed biophysical characterization of the acid-induced PrP(c) to PrP(β) conversion process. Biochemistry-Us 2011; 50:1162-73; http://dx.doi.org/ 10.1021/bi101435c [DOI] [PubMed] [Google Scholar]
- [44].Ding HF, Nakoneczna I, Hsu HS. Protective Immunity Induced in Mice by Detoxified Salmonella Lipopolysaccharide. J Med Microbiol 1990; 31:95-102; PMID:2406449; http://dx.doi.org/ 10.1099/00222615-31-2-95 [DOI] [PubMed] [Google Scholar]
- [45].Neuhoff V, Stamm R, Eibl H. Clear background and highly sensitive protein staining with coomassie blue dyes in polyacrylamide Gels - a systematic analysis. Electrophoresis 1985; 6:427-48; http://dx.doi.org/ 10.1002/elps.1150060905 [DOI] [Google Scholar]
- [46].van de Weert M, Stella L. Fluorescence quenching and ligand binding: A critical discussion of a popular methodology. J Mol Struct 2011; 998:144-50; http://dx.doi.org/ 10.1016/j.molstruc.2011.05.023 [DOI] [Google Scholar]
- [47].Magalhaes PO, Lopes AM, Mazzola PG, Rangel-Yagui C, Penna TCV, Pessoa A. Methods of endotoxin removal from biological preparations: a review. J Pharm Pharm Sci 2007; 10:388-404; PMID:17727802 [PubMed] [Google Scholar]
- [48].Ruiz N, Kahne D, Silhavy TJ. TIMELINE Transport of lipopolysaccharide across the cell envelope: the long road of discovery. Nat Rev Microbiol 2009; 7:677-83; PMID:19633680; http://dx.doi.org/ 10.1038/nrmicro2184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Muller-Loennies S, Lindner B, Brade H. Structural analysis of oligosaccharides from lipopolysaccharide (LPS) of Escherichia coli K12 strain W3100 reveals a link between inner and outer core LPS biosynthesis. J Biol Chem 2003; 278:34090-101; PMID:12819207; http://dx.doi.org/ 10.1074/jbc.M303985200 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.