

Abstract
Pancreatic dysfunction during diabetes is linked to the induction of endoplasmic reticulum (ER) stress on pancreatic β (beta)-cells. Our laboratory recently discovered that p21 protects from diabetes by modifying the outcome of ER stress response. In the present study we explored the antidiabetic acticvity of ciclopirox (CPX), an iron chelator and recently described activator of p21 expression. The effects of CPX in beta cell survival and function were assessed in cultured islets in vitro as well as in diabetic mice in vivo. The consequences of CPX in high glucose-induced insulin released and reactive oxygen species (ROS) production were also evaluated. Islet survival assays confirmed the significance of p21 in the regulation of glucotoxicity and suggested that CPX, counteracts glucotoxicity in a manner that depends on p21. In vivo, administration of CPX in wt diabetic mice restored glucose homeostasis. In wt cultured islets, CPX suppressed the expression of ER stress markers BiP, GRP94 and CHOP and reduced the levels of ROS during culture at high glucose. This reduction of ER stress may be associated with the ability of CPX to inhibit insulin release. Iron citrate stimulated insulin release, which was inhibited by CPX that functions as an iron chelator. It is conceivable that inhibition of insulin production constrains ER stress in islets promoting their survival and thus protecting from diabetes in vivo. This UPR-antagonizing activity of CPX suggests application for the management not only of diabetes, but other conditions related to ER stress.
Keywords: Unfolded protein response, glucotoxicity, glucose, chaperone, beta cells
Introduction
Endoplasmic reticulum (ER) stress is intrinsic to the pathogenesis of diabetes by mechanisms that involve the progressive loss of β cell mass due to the inability of β cells to cope with the increased demands for insulin production and secretion [1,21]. Thus, alleviation of the pro-apoptotic activity of ER stress during diabetes emerges as a strategy of choice during diabetes development. We have reported that deregulation of p21 activity occurs during ER stress and is associated with the cytotoxic effects of the unfolded protein response (UPR) [14–16]. In the context of diabetes we have found that stimulation of p53/p21 expression by nutlin-3a, a pharmacological activator of p53 stability [34], is beneficial for diabetes in mice [14,18]. Similar results, on the beneficial activity of nutlin-3a in diabetes, have also been reported by others [30] while the specific suppression of p21 during ER stress and its prosurvival activity during DNA damage-induced apoptosis have also been suggested [19].
The fact that besides p21 stimulation, p53 stabilization by nutlin-3a also elicits pro-apoptotic effects that may likely compromise its beneficial activities for diabetes management suggests that bypassing this death-inducing function of p53 may bear significant anti-diabetic value. Consistently with this notion, p21-deficient diabetic mice not only were not responsive to nutlin-3a but were rather more sensitive to diet-induced diabetes than their wild type counterparts [4]. This is in line with the notion that in the absence of p21 the pro-apoptotic effects of p53 persist, reducing islets’ survival and function during treatment of diabetic mice with nutlin-3a.
In the present study, we explored how Ciclopirox (CPX), that stimulates p21 expression [12] affects glucose homeostasis in diabetic mice and insulin release in cultured pancreatic islets. CPX (IUPAC name: 6-cyclohexyl-1-hydroxy-4-methylpyridin-2(1H)-one; Trade name: Loprox) is an iron chelator that besides the stimulation of p21 expression can also cause accumulation of HIF1 transcription factor in the nucleus [35]. HIF1 is the master regulator of oxygen homeostasis that stimulates angiogenesis at conditions of limited oxygen supply and CPX was shown to stimulate HIF-1-dependent gene expression [11,35]. Importantly, CPX was also shown to possess antitumor activity by mechanisms that are likely p53-independent, further justifying its use as an activator of p21 expression that operates independently of p53 stabilization [17,36]. Our results suggest that CPX possesses antidiabetic activity that is associated to its ability to modulate the intensity of ER stress.
Materials and Methods
Animals and Islets
C57Bl/6 mice used in these studies were originally purchased from the Jackson Laboratory (Bar Harbor, ME, USA) and maintained in our facilities. Animal care and experiments were carried out in accordance with the guidelines of the Animal Facilities by the Athens University Medical School Ethics Committee in agreement with the European Union (approval no. 7924 /12/12/2014).
Islet isolation and Viability Assessment
Pancreatic islets from wild type, p21-deficiency, p53-deficiency and CHOP-deficiency were manually isolated from female C57Bl/6 mice by the collagenase digestion method (Sigma–Aldrich;) [10] -. After isolation, the islets were incubated at 37°C with 95% air and 5% CO2 in RPMI 1640 medium (GIBCO, cat. no. 11879) containing 5.5 mM glucose, supplemented with 10% FBS and 100 units/mL streptomycin/ampicilin. After culture, mouse islets were treated with dithizone (DTZ) DTZ (Merck; Whitehouse Station, NJ; http://www.merck.com) for their specificity, by DTZ staining. The stock solution was prepared with 50 mg of DTZ in 5 ml of dimethyl sulfoxide (DMSO) and stored briefly at −20°C. In vitro DTZ staining was performed by adding 10 μl of the stock solution to 1 ml Krebs-Ringer bicarbonate buffer (pH 7.4) with HEPES (10 mM) (KRBH) and incubated at 37°C for 10–15 min. After the dishes were thrice rinsed with HBSS, clusters stained crimson red were examined with a stereomicroscope. After examination, the dishes were refilled with DMEM containing 10% FBS. After 5 hours, all traces of stain had disappeared. All the islets stained with DTZ were stained by crimson red. After 5 hours, all traces of stain had disappeared. Islets were then incubated with extracellular Krebs–Ringer (KR) solution (Sigma–Aldrich) for 30 min at 37°C prior to the experiment. Islet cell viability was assessed in intact islets by using the CellTiter 96 AQueous One Solution Cell Proliferation Assay from Promega (Madison, USA). The assay is based on the reduction of a tetrazolium compound [3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS), inner salt by metabolically active cells into a colored product and recently has been used successfully by other groups for assessment of islets’ viability [29]. Mouse islets of each of the four genotypes above, of the first survival experiment were seeded onto a 96-well plate at 50 islets per well in 100 μL of RPMI 1640 medium (GIBCO, cat. no. 11879) in 5.5, 17, 25 mM glucose or containing media for 24h and 48h. For the second survival experiment islet cells of each of the four genotypes above were incubated onto a 96-well plate at 50 islets per well in 100 μL of RPMI 1640 medium following with increasing concentration amounts of normal (5.5mM) and high (17mM) and very high (25mM) glucose in the culture media for 24h, then they were let to recover for 48h in normal glucose (5mM) -containing media. After the incubation period, 10μl of the MTS was added to each well. The microplate was incubated in a humidified atmosphere for 4 h. Then absordance is recorded at a wavelength of 490nm with a 96-well plate using a Varioskan Flash micro-plate reader (Thermo-Scientific). The quantity of formazan produced is proportional to the number of living cells in culture. Each experiment was done in triplicate.
Western blotting
The mouse islets were lysed in RIPA buffer [50 mM Tris, pH 7.2; 150 mM NaCl; 1% sodium deoxycholate; 0.1% SDS; 1% Triton-X 100; 10 mM NaF; 1 mM Na3VO4; protease inhibitor cocktail (1:1000, Sigma, St. Louis, MO)] and were subjected to immunoblot analysis by standard methods. Proteins were resolved on a 10% SDS-PAGE and transferred to a nitrocellulose membrane (Bio-Rad, Hercules, CA). Membranes were probed overnight with GADD153 (F-168), sc-575 from Santa Cruz Biotechnology (1:50); p21 (F-5), sc-6246 from Santa Cruz Biotechnology (1:200); actin, clone C4 MAB1501 from Millipore (Billerica, MA, USA) (1:2000); BiP antibody (cat. no. 3183) from Cell Signaling (Danvers, MA, USA) (1:1000); GRP94 antibody (H-212), sc-11402 from Santa Cruz Biotechnology (1:500); insulin rabbit polyclonal H-86, sc-9168 from Santa Cruz Biotechnology (1:500) antibodies followed by incubation with horseradish peroxidase-conjugated (HRP) anti-mouse (1:10000), goat anti-rabbit IgG (1:10000, Santa Cruz biotechnology Inc., Santa Cruz, CA, USA) for 1 hour. After incubation with enhanced chemiluminescence reagents (Santa Cruz biotechnology Inc.), membranes were exposed to X-Ray film (Fuji, Fischer Scientific) and then were analyzed using an Image J software for Windows.
In vivo analysis of anti-diabetic effect of CPX
Mice were randomly grouped. The first group of C57Bl/6 mice 4–5 weeks old, were fed with a control chow diet containing 10% of kcal as fat (low-fat diet) Open Source Diets (New Brunswick, NJ, USA) D12450B and drinking water with no added sucrose (control). The second group of C57Bl/6 mice 4–5 weeks old were fed with a diet containing 40% kcal as a high-fat diet (HFD; Open Source Diets (New Brunswick, NJ, USA) D12451, with free access to (10%) sweetened water (sucrose). Mice with a glucose concentration after 6h fasting exceeding 180mg/dL were considered diabetic, after 2 consecutive glucose measurements. The blood glucose levels were measured using an Accu-Check blood glucose monitor (Roche, Indianapolis, IN, USA) using blood samples collected from the tail vein. All glucose readings were performed after 6h fasting. Diabetic mice were treated daily by oral gavages (in this procedure a bulb tipped gastric gavage needle is attached to a syringe and used to deliver the compound into the stomach. After the needle is passed to the correct length, the compound may be injected) for 20 days with CPX (25 mg/kg) prepared in a solution (4% ethanol, 5.2% Tween 80, and 5.2%PEG 400) or vehicle control. The mice were then sacrificed and the pancreata were preserved in 10% buffered formalin for later histological examination.
Glucose Level Determination and Intraperitoneal Insulin tolerance test
Mice were fasted for 6 h. Glucose levels of mice were measured by an Accu-Chek Active meter with blood sample obtained from the tail. For glucose tolerance mice were injected i.p. with 2 g glucose/kg and blood glucose was checked at different time points as graphically indicated (at either 0, 30, 60, 90, 120, 160, 220 min). For insulin resistance test, mice were injected intraperitoneally at a dose of 0.75 units/kg with human insulin (0.75 U/kg body weight; Sigma) at different time points as indicated (at either 0, 30, 60, 90, 120, 160 min), as for glucose tolerance after insulin injection and blood glucose was measured as described above.
Islet number measurements
Pancreatic tissue samples were fixed with 10% buffered formalin, processed by standard methods for paraffin-embedding, sectioned into 4-μm tissue sections, mounted on polyl-lysine (Sigma) -coated slides and dried overnight at 37°C (7). Sections were then dewaxed in xylene and then rehydrated. Finally, sections were stained with hematoxylin and eosin. Islet numbers were assessed in serial sections of pancreata and expressed as the product of islet numbers and pancreatic weight.
Determination of insulin secretion
Mouse islet cells were seeded onto a 96-well plate at 50 islets per well in 100 μL of RPMI 1640 medium (GIBCO, cat. no. 11879) containing 5.5 mM glucose. Islets were resuspended in extracellular Krebs–Ringer (KR) solution (Sigma–Aldrich) for 30 min at 37°C. After washing with the above buffer, the islets were further incubated with Krebs-Ringer bicarbonate buffer (KRBH) supplemented with 5.5 or 17 or 25 mM glucose for 30 min and 60 min alone or in combination with CPX (20μΜ). Released insulin in the supernatant was measured by the Ultra Sensitive Mouse Insulin ELISA kit (Crystal Chem Inc.) in accordance with the manufacturer’s guidelines.
Measurement of cellular ROS production
ROS measurements were measured by using the DCFDA Cellular ROS Detection Assay Kit (Abcam/Mitosciences) in a 96-well dark microplate in accordance with the manufacturer’s guidelines. Briefly, 30 islets in 100μl RPMI culture medium 1640 containing 5.5 mM glucose were seeded into each well. On the day of the experiment the islets were incubated with KR buffer containing 0.01% (w/v) bovine serum albumin (BSA), 16 mM HEPES, and 2.8 mM glucose for 30 min at 37 °C. Then the islets were incubate with Krebs-Ringer bicarbonate buffer (KRBH) supplemented with with either 5.5 mM glucose or 25 mM glucose alone or in combination with CPX (20μΜ) treatment for 24h and iron citrate alone with increasing concentrations (20–200mg/l) or in combination with CPX treatment for 24h. The islets were washed with PBS and stained with 20 μM DCFH-DA in the dark for 30 min at 37 °C. After incubation, DCFH-DA was removed, and the islets were washed with 1 × buffer solution. Samples were analyzed using a Varioskan Flash micro-plate reader (Thermo-Scientific) with excitation wavelength at 485nm and an emission wavelength at 535nm. Tert-Butyl hydroperoxide (TBHP) was used as positive control. The results shown reflect the average of 2 independent experiments, each of which was performed in triplicates.
Measurement of serum ferritin levels
Animals were anesthetized using injectable (ketamine/xylazyne). Blood samples were collected in EDTA-coated Eppendorf tubes, centrifuged at 1,500 rpm (453×g) for 10min. The supernatant (serum) was carefully transferred into 0.5 ml Eppendorf tubes and stored at −80°C for further analysis. Serum Ferritin levels were determined using the Ferritin (FTL) ELISA Kit (ab157713, Abcam, Cambridge, MA) in accordance with the manufacturer’s guidelines. The results shown reflect the average of 2 independent experiments, each of which was performed in triplicates.
Statistics
Statistical analysis was performed by the student’s T test. Differences were considered significant when P< 0.05. All values are expressed as average +/− SD.
Results
Islets’ survival during glucotoxicity in relation to p53, p21 and CHOP status
Initially we explored how p21-deficency, p53-deficiency and CHOP-deficiency affect the sensitivity of pancreatic islets to glucotoxicity. As shown in Figure 1 culture of wt pancreatic islets at increasing amounts of glucose induced toxicity by a time-dependent mechanism. Consistently with the pro-apoptotic role of CHOP during ER stress, CHOP deficiency conferred resistance since only culture at high glucose for 48h significantly inhibited islet growth (Fig. 1a). In agreement with the protective role of p21 and p53 during the UPR, ablation of any of these cell cycle regulators sensitized islets to the cytotoxic effects of high glucose (Fig. 1a). A trend, albeit not confirmed statistically, for more potent prosurvival activity conferred by p21 than p53, especially for 17mM, was unveiled because for the latter it may be masked by its concomitant pro-apoptotic activity. The contribution of each of p21, p53 and CHOP in determining glucotoxicity is shown in Figure 1b which illustrates the relative survival of islets at 17mM and 25mM glucose in comparison with their survival at normal glucose.
Figure 1. Islet survival in relation to genotype at increasing glucose concentrations.
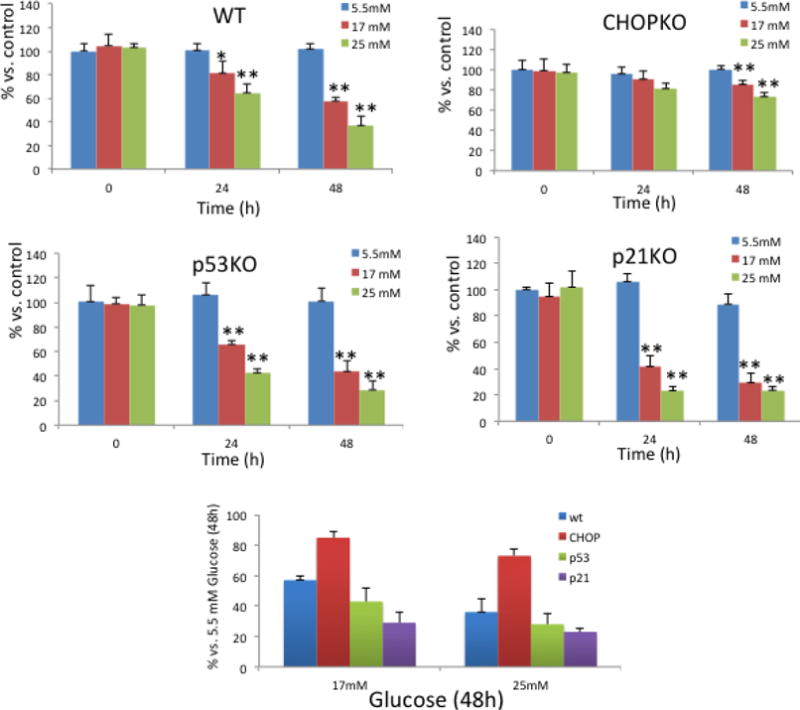
a. Survival of cultured pancreatic islets of each genotype at increasing amounts of glucose for 24h and 48h as indicated in the figure. *, P<0.05; **, P<0.005 Student’s T test. b. Relative survival of islets of each genotype in high glucose for 48h expressed as % vs. survival at normal glucose. All values are expressed as average +/− SD.
CPX promotes islet survival under glucotoxic conditions
In view of the potent sensitixing activity of p21 ablation against glucotoxicity we evaluated how pharmacological stimulation of p21 expression promotes islets’ survival. To that end we utilized CPX, a previously described stimulator of p21 expression. Specifically, islets from wt, p21-deficient, p53-deficient and CHOP-deficient mice were exposed to CPX at 20μΜ for 24h under increasing glucose amounts in the culture media, then they were let to recover for 48h in normal glucose-containing media and their survival was assessed (Fig. 2a). The 48h recovery in normal media was deemed necessary because CPX, by inducing p21 expression would inflict cell cycle arrest in islets masking the effects of glucotoxicity. Indeed, CPX treatment of wt islets under normal glucose significantly inhibited islet survival. As shown in fig. 2b, despite this cytostatic effect in wt islets under normal glucose, CPX protected wt but not p21-deficient animals from glucotoxicity suggesting that intact p21 expression is required for the pro-survival activity of CPX. A similar trend was also found for p53, the major regulator of p21 expression. Considering though that the p21-stimulatory activity of CPX in the islets is also p53-independent, as described earlier for cancer cells [39], this observation suggests that p53 deregulation may alleviate the pro-survival activity of CPX by alternative mechanisms. Thus, p21/p53 expression is required for CPX to produce its protective effects at glucotoxic conditions. Consistently with its pro-apoptotic role during ER stress CHOP ablation conferred resistance of islets to glucotoxicity (Fig. 2b).
Figure 2. Assessment of pancreatic islet survival in the presence of CPX.
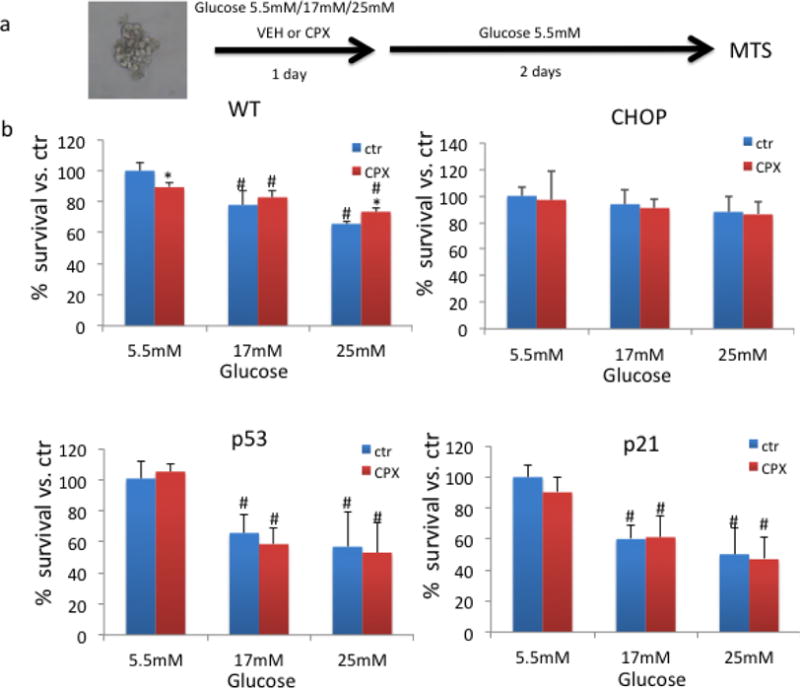
a. Experimental design of how islet survival was assessed in the presence of CPX at 20μΜ or vehicle (VEH) for 24h under different increasing glucose amounts in the culture media, then they were let to recover for 48h in normal glucose-containing media. A pool of several islets has been used as described in the methods. b. Survival of islets of different genotypes in relation to glucose concentration in the presence or absence of CPX. *, P<0.05 vs. untreated with CPX. #, P<0.05 vs. islets cultured in normal glucose, Student’s T test. All values are expressed as average +/− SD.
Ciclopirox produces antidiabetic activity in wt mice
Subsequently, we evaluated the effects of CPX in glucose homeostasis in wt diabetic mice. First asked if CPX treatment induces p21 levels in the pancreas. To that end mice received orally CPX at 0–25 mg/kg daily for 5 days and the levels of p21 were assessed. CPX administration from as low as 5mg/kg effectively stimulated the expression of p21 in the pancreas of wild type mice (Fig. 3a). Subsequently, we explored the consequences of daily CPX at 25mg/kg administration in glucose homeostasis of wt mice that became diabetic by receiving high fat diet (HFD) combined with sucrose at 10% in their drinking water. This experimental treatment started about 50 days after initiation of the diabetes-inducing diet, when fasting glucose levels were about 185 mg/dl. We used the higher dosage despite the efficient p21 stimulation at lower dosages on the basis of earlier results on the beneficial CPX administration at 25mg/ml [39]. As shown in Figure 3b CPX at 25mg/kg daily effectively improved glucose homeostasis causing reduction of blood glucose levels. Noteworthy, CPX administration in a diabetic mouse with glucose levels at about 180 mg/dl was also effecting in stimulating p21, albeit at higher concentrations (Fig. 3a). Glucose tolerance (Fig. 3c) and insulin sensitivity (Fig. 3d) tests suggested that CPX did not affect dramatically the kinetics of plasma glucose in the diabetic mice implying that its effects were not primarily in glucose metabolism. Ferritin levels were elevated in the sera of the diabetic mice and decreased significantly (P<0.05) following CPX treatment (Fig. 3e). The effects of CPX in glucose homeostasis were limited to the diabetic mice because glucose levels in wt non-diabetic mice were not affected by CPX administration (Fig. 4). Histological analysis of pancreata from mice treated with CPX suggested that total islet numbers increased significantly only in the diabetic mice, but not in the non-diabetic mice (Fig. 5). It is noted that areas in the exocrine pancreas with cells exhibiting intense eosinophilic granular cytoplasm were detected in the CPX-treated groups which is consistent with increased activity.
Figure 3. CPX restores glucose homeostasis in diabetic mice.
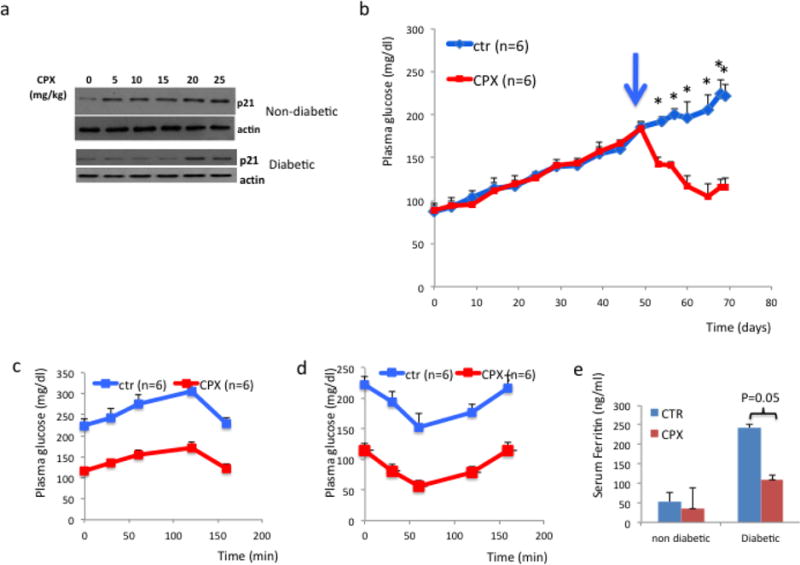
a. p21 expression in the pancreas of wild type mice non-diabetic (upper panel) or diabetic with glucose at 180 mg/dl (lower panel) that received CPX. Mice received orally CPX at 0–25 mg/kg daily for 5 days and the levels of p21 were assessed. Actin levels were used as positive control.
b. Fasting glucose levels of wild type mice fed with HFD and sucrose after CPX at 25mg/kg daily administration. Administration of CPX was initiated about 50 days after initiation of the diabetes-inducing diet when glucose levels exceeded 180 mg/dl (arrows) and continued daily thereafter. c. Glucose tolerance and insulin sensitivity (d) of diabetic mice that have been treated with saline or CPX. For this experiment mice with similar levels of fasting glucose were selected between experimental groups. e. Ferritin levels in the sera of non-diabetic and diabetic mice that received CPX. All values are expressed as average +/− SD.
Figure 4. Fasting blood glucose levels of non-diabetic wt mice non affected by CPX.
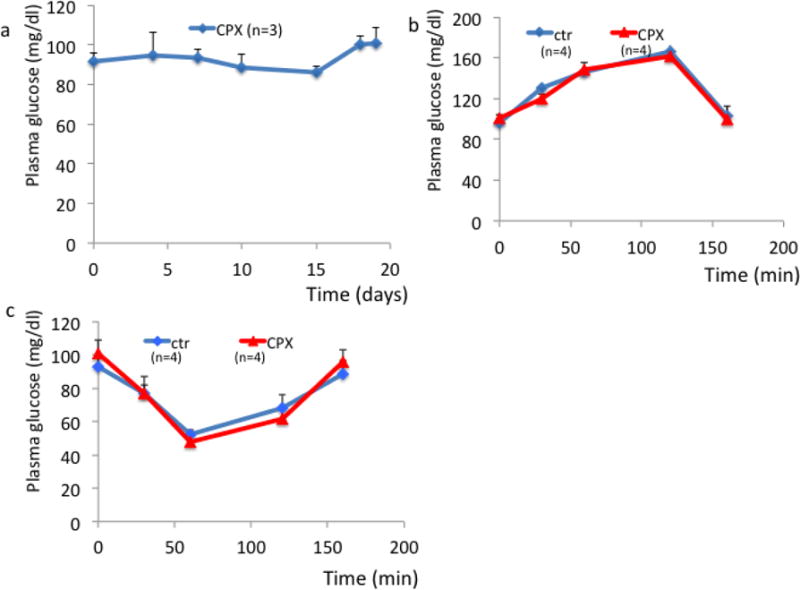
Fasting glucose for non-diabetic wt mice treated with CPX (a) glucose tolerance (b) and insulin sensitivity (c) of wt mice receiving CPX. All values are expressed as average +/− SD.
Figure 5. Islet cell morphology after Ciclopirox treatment in HFD-induced diabetic mice and control mice.
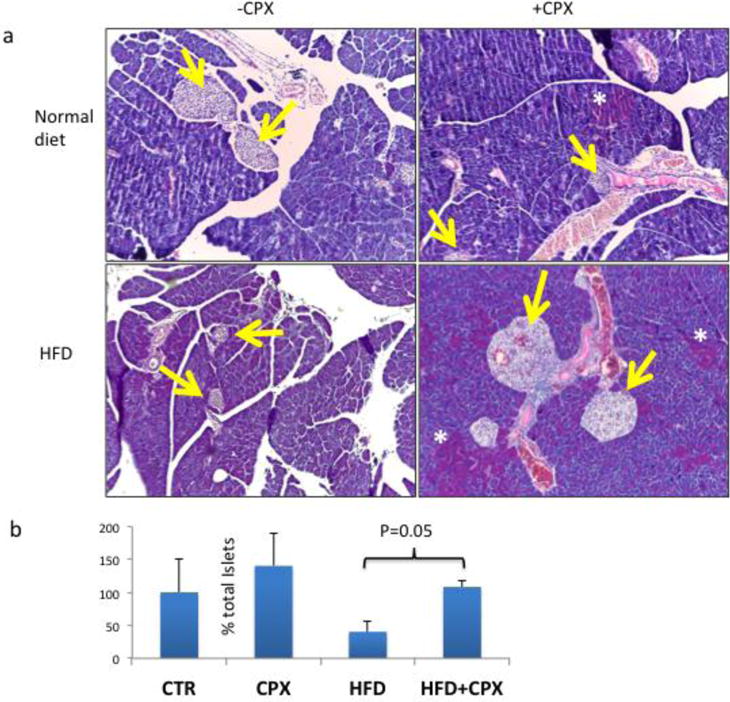
a. Pancreatic islet morphology (H&E staining) of wt non-diabetic (upper panel) or diabetic (lower panel) mice receiving CPX or saline. Yellow arrows show islets. White asterisks indicate areas in the exocrine panceas with cells exhibiting intense eosinophilic granular cytoplasm. b. Quantification of total islets expressed as the % vs. controls of the product between islet numbers in cross-sections and the weight of each pancreas. All values are expressed as average +/− SD.
Effects of CPX in insulin release and glucose-induced ER stress in islets
In order to understand better how CPX operates in β cells, we tested if it affects the efficiency by which insulin is produced following glucose stimulation. As shown in Figure 6a CPX suppressed the release of insulin. In view of the antidiabetic activity of CPX in vivo, the inhibition of insulin release appears contradictory. Thus, we hypothesized that reduced insulin secretion may be linked to reduced ER stress and blunted UPR, which in turn could explain in the long run the increased survival and functionality of the animals that received CPX. To test this hypothesis, we exposed islets to CPX (20μΜ) and high glucose (25mΜ) for 24h and tested the levels of chaperones BiP, GRP94 and CHOP that reflect the intensity of the UPR. As shown in Figure 6b CPX alleviated the levels of BiP, GRP94 and CHOP in the wt islets suggesting that the execution of the UPR was compromised. In line with these observations and consistently with the reduced insulin levels in the media (Fig. 6a) the levels of insulin in the β islets were also suppressed (Fig. 6b). In line with these findings is also the inhibitory activity of CPX in insulin release triggered by KCl that operates by promoting membrane depolarization (Fig. 6a).
Figure 6. CPX decreases insulin release and attenuates the consequences of glucose-induced ER stress in islets.
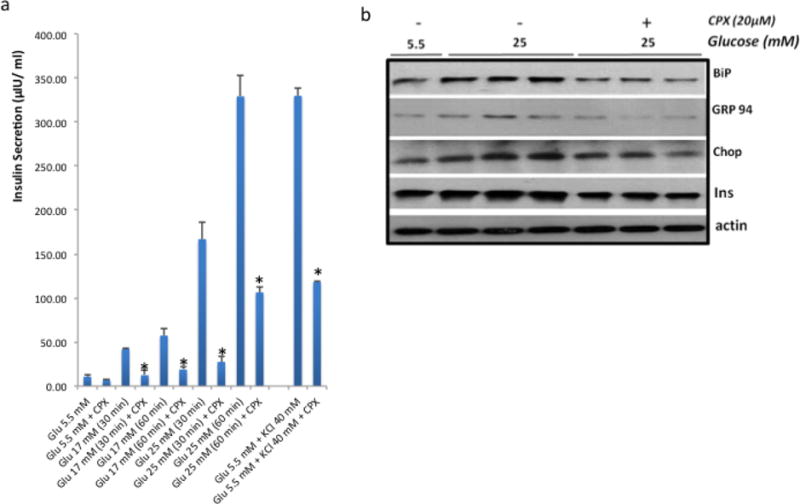
a. Insulin release in wt islets following glucose stimulation at 17mM or 25mM, or 40 mM KCl and treated with CPX at 20μΜ for 30min and 60min respectively. *, P<0.01 vs. untreated. b. Expression of insulin, CHOP, GRP94 and BiP in wt islets treated with high glucose alone or in combination with CPX (in KRB). Actin levels were assessed as loading control. High glucose experiment was performed in triplicates. All values are expressed as average +/− SD.
Effects of CPX in insulin release, ER stress and ROS production induced by iron citrate
While modulation of insulin release by CPX constitutes a challenging mechanism by which CPX may affect ER stress, how CPX regulates insulin expression remains obscure. Being an iron chelator, it is conceivable that CPX modulates insulin production by adjusting the levels of bioavailable iron in the islets. To test this hypothesis, we exposed islets to increasing amounts of iron citrate for 24h, alone or combined with CPX for 24h, and recorded the levels of chaperones BiP, GRP94 and CHOP, as well as of insulin. As shown in Fig. 6a increasing amounts of iron citrate triggered insulin production and induced the expression of ER stress-markers BiP, GRP94 and CHOP. Concomitant exposure though of islets with CPX compromised the effects of iron citrate on insulin production and ER stress induction (Fig. 7a). In addition to ER stress, CPX was also a potent inhibitor of ROS that were produced by the culture of islets to iron citrate and glucose at high or moderate concentrations (Fig. 7b).
Figure 7. Iron citrate elevates ER-stress markers, whereas Ciclopirox restores ER-stress homeostasis.
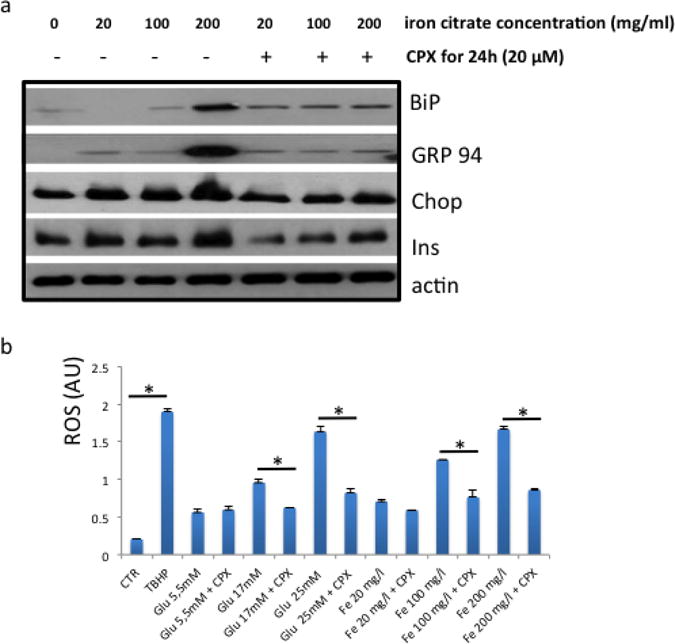
a. The effects of iron citrate and ciclopirox at the indicated concentrations in insulin, BiP, GRP94 and CHOP levels in islets cultured at 5.5mM glucose. Actin levels were assessed as loading control. The high glucose experiment was performed in triplicates. b. Effect of CPX at 20μM (in KRB) for 24h in ROS production (in arbitrary units, AU) that was triggered by high (25mM) or moderate (17mM) glucose (Glu) or iron citrate (Fe) (in 5.5mM glucose). TBHP was used as positive control for ROS production. *. P<0.05, Student’s t-test. All values are expressed as average +/− SD.
Discussion
Promotion of β cell survival emerges as an attractive target for the long–term management of diabetic patients. Recent results of our laboratory suggested that p21 protects mice from diet and STZ-induced diabetes by mechanisms involving a shift of the UPR from its pro-apoptotic to its adaptive function [14–16]. In line with these findings, genetic activation of p21 in β cells of transgenic mice protected them from STZ-induced pancreatic dysfunction [37] while pharmacological, p53-dependent activation of p21 by nutlin, produced anti-diabetic activity [15,30]. In the present study, we confirmed the protective role of p21 in islets against glucotoxicity (Fig. 1) and evaluated the antidiabetic activity of CPX, a recently described activator of p21 expression [12]. Administration of CPX, an iron chelator that stimulates p21 by p53 independent mechanisms [39], was strongly anti-diabetic in mice at which diabetes was induced by HFD and sucrose-sweetened water (Fig. 3). At least in part this was due to the protection of islets from glucotoxicity (Fig. 2). The ability of CPX to inhibit the production of high or moderate glucose-stimulated ROS production [38] may account for its protective effects in islets (Fig. 7b).
The fact though that only in diabetic, but not in non-diabetic controls serum glucose levels decreased by the CPX prompted us to explore if the antidiabetic activity of CPX is also related to metabolic effects associated with the stress-induced by glucotoxicity. Indeed, assessment of insulin release in islets treated with CPX showed strong inhibition of insulin secretion triggered by high glucose (Fig. 6a). Considering the antidiabetic action of CPX in mice this inhibition of insulin secretion was rather unexpected. It is conceivable that CPX, besides its effects in islets may also affect peripheral tissues in terms of insulin sensitivity in insulin-dependent organs, by modulating levels and activity of GLUT transporters, insulin receptor and signaling pathways and gluconeogenesis. An additional possibility could be that by inhibiting the production of insulin the β cells are less prone to develop ER stress which may compromise pancreatic function (Fig. 7). To that end an intriguing possibility is that CPX, by inhibiting insulin release reduces ER stress in pancreatic islets which in turn improves long-term survival and function. Consistently with this notion, adequate regulation of glucose homeostasis may be attained by submaximal levels of insulin secretion, which however would not exhaust islets that could survive and function normally for more prolonged time periods. This concept raises the possibility that besides modulating the outcome of UPR (efficiency of ER stress-associated death) CPX also regulates the efficiency of UPR induction. This notion was also implied before [14] by our earlier observations that nutlin-3a, another stimulator of p21 expression, alleviates ER stress that is induced by high glucose. In addition, we have also reported that p53 ablation intensifies the UPR in mice inducing certain ER stress – associated pathologies in hepatocytes [4]. Indeed, assessment of the levels of the ER stress markers BiP, GRP94 and CHOP, in islets cultured in high glucose-containing media, showed that CPX reduced the intensity of the UPR (Fig. 6). Whether this effect of CPX is limited to islets under glucotoxic conditions or CPX operates, directly or indirectly, as a chaperone that assists protein folding thus decreasing the risk of UPR remains to be determined. It is noted though that CPX blocked the release of insulin that was triggered by KCl. The latter induces insulin release, by causing membrane depolarization through closing KATP channels, causing elevation of cytosolic free Ca2+ concentration [7, 20,23,24]. Thus CPX, at least in part, may interfere with Ca2+ homeostasis and membrane polarization, which in turn implies a wider role of CPX in the regulation of UPR and islets’ function, beyond regulation of glucotoxicity.
A moderate suppression in the levels of intracellular insulin was also recorded however, it was less pronounced as compared to that recorded for the insulin released in the media (Fig. 5). The fact that CPX only modestly suppressed insulin expression in the cells, but potently inhibited its release suggests that this stimulator of p21 expression interferes mostly with insulin secretion than production. Considering that insulin accumulation was not recorded during CPX treatment and also that ER stress was not induced, but rather suppressed, implies that CPX operates upstream of insulin secretion, reducing the islets’ sensitivity to glucose. To that end, in mice, during diet-induced diabetes, β cells retained their functionality and glucose homeostasis was attained despite that insulin production was compromised. It is noted that the rapid response to CPX treatment in vivo may imply additional levels at which CPX interferes with glucose homeostasis, beyond the promotion of islets’ survival during glucose-induced stress of the ER.
How CPX regulates insulin secretion remains vague. In view of its function as iron chelator, and considering the relationship between iron and diabetes, CPX may reduce the cells’ iron load, thus reducing the risk for diabetes onset [9,32,33]. Studies in both humans and experimental animals show association between the levels of ferritin and diabetes onset [31,32]. In the present study increased levels of ferritin were found in the sera of the diabetic mice while CPX treatment significantly reduced it (Fig 3e). Noteworthy, iron chelation with deferoxamine or deferasirox was also shown to be beneficial for diabetes management by a mechanism that depends on HIF-1a [3]. In this study, exposure of islets to iron citrate caused some induction in the expression of insulin, confirming and extending earlier observations on the stimulatory activity of iron to insulin [5]. This effect was dose-dependent and was associated with the induction of ER stress associated markers, especially at the higher doses of iron citrate (Fig. 6a). Importantly, CPX that binds and titrates out iron alleviated the consequences of iron citrate in insulin production and reduced the intensity of ER stress (Fig. 7a). In addition to ER stress, CPX also alleviated the production of ROS that was stimulated by iron citrate-in addition to high glucose – (Fig. 7b) further complicating the mechanism(s) contributing to the beneficial activity of CPX in islets’ function and survival. Indeed, the ability of CPX to scavenge directly hydroxyl radicals has been demonstrated [28]. Noteworthy, CPX alleviated ROS production produced by excessive amounts of glucose, at 17mM and 25mM, and iron citrate further confirming its function as a modulator of stress responses during stress. This ROS scavenging activity of CPX may also be relevant to the recorded inhibition of insulin secretion since previously described results suggest the stimulatory role of ROS in insulin production [22,27]. In addition, the beneficial effects of anti-oxidants in islets’ function has also been demonstrated [6,13].
It is noted that while our present and earlier findings are consistent with the prosurvival role of p21 during ER stress in β cells, a recent study demonstrated in INS-1-derived 832/13 cells and rat islets exogenous expression of p21 induces apoptosis [8]. It is conceivable that during prolonged cell cycle arrest induced especially by the forced expression p21, cells and islets undergo apoptosis without the latter reflecting a direct causative effect of p21 activation. Indeed, a series of studies in diverge cellular systems demonstrated that p21 is pro-survival following different forms of stress including ER stress [19] and is capable of activating various mitogenic ques such as those trigerred by CDK8 [2,25,26,36]. Finally, our earlier studies showed that p21’s undergoes stimulation followed by suppression during intense/prolonged ER stress [14,16]. The overall role of p21 in β cells is rather complex and the actual fate of cells represents the collective outcome of the combined intensity of the pro-apoptotic (ER stress) and the prosurvival signals (p21).
Although no evidence of toxicity was detected in the CPX-treated groups such as loss of body weight, signs of cachexia or gross morphologicl alterations during necropsy, it is formally possible that at least partially the effects of CPX in vivo were due to kidney toxicity and increased excretion of glucose in the urine. However, the in vitro results on pancreatic islets strongly support that at least partially these effects are due to the promotion of islets’ survival during glucotoxicity.
Collectively, our findings suggest that CPX is beneficial for diabetes. It is likely that this is attained by converging mechanisms related both to the modulation of the p21-dependent sensitivity of β cells to ER stress-related apoptosis and by the regulation of the induction of UPR as such, through inhibition of insulin release that constitutes a major stimulator of ER stress in β cells. These activities involve mechanisms that appear to be both β cell-specific, such as regulation of insulin production, and mechanisms that may operate in additional cell types as well such as the alleviation of ROS production (Fig. 8). Thus, the beneficial activity of CPX may be not limited only to the management of diabetes, but also to other ER stress – associated diseases.
Figure 8.
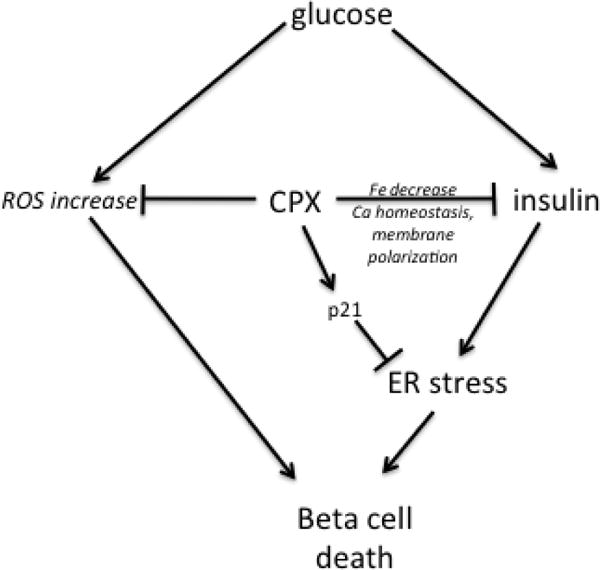
Diagrammatic depiction of the presumed role of CPX in regulating islets’ survival by modulating p21 expression, insulin release and ER stress induction.
Acknowledgments
This study was supported by grant RAG051976A from NIH/NIA
Footnotes
Disclosure statement: The authors have nothing to declare
References
- 1.Back SH, Kaufman RJ. Endoplasmic reticulum stress and type 2 diabetes. Annual Review of Biochemistry. 2012;81:767–93. doi: 10.1146/annurev-biochem-072909-095555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Blagosklonny MV. Are p27 and p21 cytoplasmic oncoproteins? Cell Cycle. 2002;1:391–3. doi: 10.4161/cc.1.6.262. [DOI] [PubMed] [Google Scholar]
- 3.Cheng K, Ho K, Stokes R, Scott C, Lau SM, Hawthorne WJ, et al. Hypoxia-inducible factor-1alpha regulates beta cell function in mouse and human islets. J Clin Invest. 2010;120:2171–2183. doi: 10.1172/JCI35846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dioufa N, Chatzistamou I, Farmaki E, Papavassiliou AG, Kiaris H. P53 Antagonizes the Unfolded Protein Response and Inhibits Ground Glass Hepatocyte Development During Endoplasmic Reticulum Stress. Experimental Biology and Medicine (Maywood, N.J) 2012;237:1173–80. doi: 10.1258/ebm.2012.012140. [DOI] [PubMed] [Google Scholar]
- 5.Erion KA, Ferrante T, Corkey B, Deeney J. Iron stimulates insulin secretion in clonal pancreatic β-cells and dissociated rat islets. FASEB J. 2013;27 1010.13. [Google Scholar]
- 6.Han J, Song B2, Kim J, Kodali VK, Pottekat A, Wang M3, Hassler J, Wang S, Pennathur S, Back SH, Katze MG, Kaufman RJ. Antioxidants Complement the Requirement for Protein Chaperone Function to Maintain β-Cell Function and Glucose Homeostasis. Diabetes. 2015;62:2892–904. doi: 10.2337/db14-1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Henquin JC, Mourad NI, Nenquin M. Disruption and stabilization of β-cell actin microfilaments differently influence insulin secretion triggered by intracellular Ca2+ mobilization or store-operated Ca2+ entry. FEBS Lett. 2012;586:89–95. doi: 10.1016/j.febslet.2011.11.030. [DOI] [PubMed] [Google Scholar]
- 8.Hernandez AM1, Colvin ES, Chen YC, Geiss SL, Eller LE, Fueger PT. Upregulation of p21 activates the intrinsic apoptotic pathway in β-cells. Am J Physiol Endocrinol Metab. 2013;304:E1281–90. doi: 10.1152/ajpendo.00663.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huang J, Jones D, Luo B, Sanderson M, Soto J, Abel ED, et al. Iron overload and diabetes risk: a shift from glucose to Fatty Acid oxidation and increased hepatic glucose production in a mouse model of hereditary hemochromatosis. Diabetes. 2011;60:80–7. doi: 10.2337/db10-0593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li DS, Yuan YH, Tu HJ, Liang QL, Dai LJ. A protocol for islet isolation from mouse pancreas. Nature Protocols. 2009;4:1649–1652. doi: 10.1038/nprot.2009.150. [DOI] [PubMed] [Google Scholar]
- 11.Linden T, Katschinski DM, Eckhardt K, Scheid A, Pagel H, Wenger RH. The antimycotic ciclopirox olamine induces HIF-1alpha stability, VEGF expression, and angiogenesis. The FASEB Journal : Official Publication of the Federation of American Societies for Experimental Biology. 2003;17:761–3. doi: 10.1096/fj.02-0586fje. [DOI] [PubMed] [Google Scholar]
- 12.Ma TC, Langley B, Ko B, Wei N, Gazaryan IG, Zareen N, Yamashiro DJ, Willis DE, Ratan RR. A screen for inducers of p21waf1/cip1 identifies HIF prolyl hydroxylase inhibitors as neuroprotective agents with antitumor properties. Neurobiol Dis. 2013;49:13–21. doi: 10.1016/j.nbd.2012.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Malhotra JD, Miao H, Zhang K, Wolfson A, Pennathur S, Pipe SW, Kaufman RJ. Antioxidants reduce endoplasmic reticulum stress and improve protein secretion. Proc Natl Acad Sci U S A. 2008;105:18525–30. doi: 10.1073/pnas.0809677105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mihailidou C, Chatzistamou I, Papavassiliou A, Kiaris H. Improvement of chemotherapeutic drug efficacy by endoplasmic reticulum stress. Endocrine-Related Cancer. 2015;22(2):229–38. doi: 10.1530/ERC-15-0019. [DOI] [PubMed] [Google Scholar]
- 15.Mihailidou C, Chatzistamou I, Papavassiliou AG, Kiaris H. Regulation of P21 during diabetes-associated stress of the endoplasmic reticulum. Endocr Relat Cancer. 2015;22:217–28. doi: 10.1530/ERC-15-0018. [DOI] [PubMed] [Google Scholar]
- 16.Mihailidou C, Papazian I, Papavassiliou AG, Kiaris H. CHOP-dependent regulation of p21/waf1 during ER stress. Cell Physiol Biochem. 2010;25:761–766. doi: 10.1159/000315096. [DOI] [PubMed] [Google Scholar]
- 17.Minden MD, Hogge DE, Weir SJ, Kasper J, Webster DA, Patton L, et al. Oral ciclopirox olamine displays biological activity in a phase I study in patients with advanced hematologic malignancies. Am J Hematol. 2014;89:363–368. doi: 10.1002/ajh.23640. [DOI] [PubMed] [Google Scholar]
- 18.Mkrtchian S. Targeting unfolded protein response in cancer and diabetes. Endocr Relat Cancer. 2015;22(3):C1–4. doi: 10.1530/ERC-15-0106. [DOI] [PubMed] [Google Scholar]
- 19.Mlynarczyk C, Fåhraeus R. Endoplasmic reticulum stress sensitizes cells to DNA damage-induced apoptosis through p53-dependent suppression of p21(CDKN1A.) Nature Communications. 2014;5:5067. doi: 10.1038/ncomms6067. [DOI] [PubMed] [Google Scholar]
- 20.Olofsson CS, Göpel SO, Barg S, Galvanovskis J, Ma X, Salehi A, Rorsman P, Eliasson L. Fast insulin secretion reflects exocytosis of docked granules in mouse pancreatic B-cells. Pflügers Arch. 2002;444:43–51. doi: 10.1007/s00424-002-0781-5. [DOI] [PubMed] [Google Scholar]
- 21.Papa FR. Endoplasmic reticulum stress, pancreatic β-cell degeneration, and diabetes. Cold Spring Harbor Perspectives in Medicine. 2012;2:1–17. doi: 10.1101/cshperspect.a007666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pi J, Bai Y, Zhang Q, Wong V, Floering LM, Daniel K, et al. Reactive oxygen species as a signal in glucose-stimulated insulin secretion. Diabetes. 2007;56:1783–91. doi: 10.2337/db06-1601. [DOI] [PubMed] [Google Scholar]
- 23.Pizarro-Delgado J, Braun M, Hernández-Fisac I, Martin-del-Río R, Tamarit-Rodriguez J. Glucose promotion of GABA metabolism contributes to the stimulation of insulin secretion in β-cells. Biochem J. 2010;431:381–389. doi: 10.1042/BJ20100714. (2010) [DOI] [PubMed] [Google Scholar]
- 24.Pizarro-Delgado J, Deeney JT, Martín-del-Río R, Corkey BE, Tamarit-Rodriguez J. KCl-Permeabilized Pancreatic Islets: An Experimental Model to Explore the Messenger Role of ATP in the Mechanism of Insulin Secretion. PLoS ONE. 2015;10:e0140096. doi: 10.1371/journal.pone.0140096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Porter DC, Farmaki E, Altilia S, Schools GP, West DK, Chen M, Chang BD, Puzyrev AT, Lim CU, Rokow-Kittell R, Friedhoff LT, Papavassiliou AG, Kalurupalle S, Hurteau G, Shi J, Baran PS, Gyorffy B, Wentland MP, Broude EV, Kiaris H, Roninson IB. Cyclin-dependent kinase 8 mediates chemotherapy-induced tumor-promoting paracrine activities. Proc Natl Acad Sci U S A. 2012;109:13799–804. doi: 10.1073/pnas.1206906109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Roninson IB. Oncogenic functions of tumour suppressor p21(Waf1/Cip1/Sdi1): association with cell senescence and tumour-promoting activities of stromal fibroblasts. Cancer Lett. 2002;179:1–14. doi: 10.1016/s0304-3835(01)00847-3. [DOI] [PubMed] [Google Scholar]
- 27.Saadeh M, Ferrante TC, Kane A, Shirihai O, Corkey BE, Deeney JT. Reactive oxygen species stimulate insulin secretion in rat pancreatic islets: studies using mono-oleoyl-glycerol. PloS One. 2012;7:e30200. doi: 10.1371/journal.pone.0030200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sato E, Kohno M, Nakashima T, Niwano Y. Ciclopirox olamine directly scavenges hydroxyl radical. Int J Dermatol. 2008;47:15–18. doi: 10.1111/j.1365-4632.2007.03359.x. [DOI] [PubMed] [Google Scholar]
- 29.Schubert U, Schmid J, Lehmann S, et al. Transplantation of pancreatic islets to adrenal gland is promoted by agonists of growth-hormone-releasing hormone. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:2288–2293. doi: 10.1073/pnas.1221505110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Secchiero P, Toffoli B, Melloni E, Agnoletto C, Monasta L, Zauli G. The MDM2 inhibitor Nutlin-3 attenuates streptozotocin-induced diabetes mellitus and increases serum level of IL-12p40. Acta Diabetol. 2013;50:899–906. doi: 10.1007/s00592-013-0476-8. [DOI] [PubMed] [Google Scholar]
- 31.Simcox JA, McClain DA. Iron and Diabetes Risk. Cell metabolism. 2013;17:329–341. doi: 10.1016/j.cmet.2013.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Swaminathan S, Fonseca V, Alam M, Shah SV. The role of iron in diabetes and its complications. Diabetes Care. 2007;30:1926–33. doi: 10.2337/dc06-2625. [DOI] [PubMed] [Google Scholar]
- 33.Simcox JA, McClain DA. Iron and diabetes risk. Cell Metab. 2013 Mar 5;17(3):329–41. doi: 10.1016/j.cmet.2013.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science (New York, N.Y.) 2004;303:844–848. doi: 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]
- 35.Wanner RM, Spielmann P, Stroka DM, Camenisch G, Camenisch I, Scheid A, et al. Epolones induce erythropoietin expression via hypoxia-inducible factor-1 alpha activation. Blood. 2000;96:1558–1565. [PubMed] [Google Scholar]
- 36.Weiss RH. p21Waf1/Cip1 as a therapeutic target in breast and other cancers. Cancer Cell. 2003;4:425–9. doi: 10.1016/s1535-6108(03)00308-8. [DOI] [PubMed] [Google Scholar]
- 37.Yang J, Zhang W, Jiang W, Sun X, Han Y, Ding M, et al. P21cip-overexpression in the mouse beta cells leads to the improved recovery from streptozotocin-induced diabetes. PloS One. 2009;4:e8344. doi: 10.1371/journal.pone.0008344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yu T, Jhun BS, Yoon Y. High-glucose stimulation increases reactive oxygen species production through the calcium and mitogen-activated protein kinase-mediated activation of mitochondrial fission. Antioxidants & Redox Signaling. 2011;14:425–437. doi: 10.1089/ars.2010.3284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhou H, Shen T, Luo Y, Liu L, Chen W, Xu B, et al. The antitumor activity of the fungicide ciclopirox. International Journal of Cancer. Journal International Du Cancer. 2010;127:2467–77. doi: 10.1002/ijc.25255. [DOI] [PMC free article] [PubMed] [Google Scholar]