

Abstract
Mitochondrial dysfunction is a hallmark of ageing, and mitochondrial maintenance may lead to increased healthspan. Emerging evidence suggests a crucial role for signalling from the nucleus to mitochondria (NM signalling) in regulating mitochondrial function and ageing. An important initiator of NM signalling is nuclear DNA damage, which accumulates with age and may contribute to the development of age-associated diseases. DNA damage-dependent NM signalling constitutes a network that includes nuclear sirtuins and controls genomic stability and mitochondrial integrity. Pharmacological modulation of NM signalling is a promising novel approach for the prevention and treatment of age-associated diseases.
Mitochondria are present in the cytoplasm of almost all mammalian cells. They provide energy by generating ATP and are crucial for many signalling pathways. Mitochondria constantly adjust their morphology and function in response to changes in the microenvironment, such as exposure to endogenous or exogenous stress1,2. Healthy mitochondria are maintained by multiple processes, including fusion and fission, DNA repair and mitophagy (clearance of damaged mitochondria)2–6. Impairment of any of these processes can lead to mitochondrial dysfunction and contribute to mitochondrial diseases, neurodegenerative disorders, cancer, diabetes, heart disease, immunodeficiency and ageing1,7–10. For example, impairment of mitophagy is implicated in Parkinson disease, Alzheimer disease and pathological ageing11–13. Although the aetiology of mitochondrial diseases has largely been attributed to defects in mitochondrial proteins, mutations in non-mitochondrial proteins or impairment of nucleus-to-mitochondria signalling (NM signalling) can also promote mitochondrial dysfunction. Thus, a better understanding of the molecular signalling mechanisms that influence mitochondrial functions is important in relation to human health.
Owing to its central function in energy metabolism, a healthy mitochondrial pool is crucial for organismal health. Indeed, some data suggest that damaged mitochondria accumulate with age in organisms spanning the evolutionary spectrum, from unicellular organisms to humans10,12,14–16. However, age-associated changes in gross mitochondrial function, such as oxidative phosphorylation, have not been consistently detected17,18. Thus, age- associated mitochondrial changes may not be the primary drivers, but rather a downstream consequence, of nuclear signalling pathways that drive the ageing process17,19,20.
Recent evidence suggests that compromised NM signalling is a key component of mammalian ageing. DNA damage is well known to affect DNA replication, transcription and many signalling pathways, but how this damage invokes signalling to mitochondria is a new area of investigation. Several DNA repair-deficient, premature ageing diseases, such as Cockayne syndrome, xeroderma pigmentosum group A (XPA) and ataxia telangiectasia, involve mitochondrial alterations consisting of increased mitochondrial membrane potential and increased oxygen consumption rates — traits that are indicative of adaptive responses3,12,21–23. In this Review, we focus on NM signalling pathways that are activated in response to genotoxic stimuli, especially DNA damage, and their roles in the maintenance of mitochondrial integrity and ageing (FIG. 1). We also discuss emerging findings suggesting that intervention in NM signalling can alleviate age-associated physiological changes. Importantly, NM signalling is closely coupled with signalling from mitochondria to the nucleus, including the mitochondrial unfolded protein response (UPRmt). Recent findings suggest that the nuclear DNA damage response regulates protein homeostasis in mitochondria through activation of the UPRmt (for more details about the molecular pathways involved, see REFS 16, 24–27).
Figure 1. An overview of DNA damage-induced nucleus-to-mitochondria signalling and ageing.

Nuclear DNA damage can cause mitochondrial dysfunction. DNA damage leads to the activation of a number of proteins, resulting in widespread downstream changes in various cellular pathways. Among these pathways, signalling from the nucleus to mitochondria (NM signalling) is less studied than many others, but it may be very important in the ageing process and in age-associated diseases, and interventions in this process may slow ageing. A number of factors contribute to NM signalling, such as upstream DNA damage sensors including poly(ADP-ribose) polymerase 1 (PARP1), ataxia telangiectasia mutated (ATM) and the transcription factor p53. The NAD-dependent protein deacetylase sirtuin 1 (SIRT1) and the transcription regulator AMP-activated protein kinase (AMPK) then propagate the NM signal through post-translational modifications (deacetylation or phosphorylation, respectively) of DNA histones, peroxisome proliferator-activated receptor-γ co-activator 1α (PGC1α) and other proteins. The combined effect of the activation of the DNA damage response is downstream changes in the transcriptome, epigenome and metabolome, and in cellular bioenergetics. These contribute to ageing and the development of age-associated diseases such as neurodegeneration and cancer.
Nuclear DNA repair and mitochondrial health
Progressive damage to nuclear DNA and mitochondrial DNA (mtDNA) is considered to be a prominent contributing factor to the ageing process and may underlie several age-associated diseases4,20,28,29. Cells have multiple DNA repair pathways to rectify the myriad types of DNA lesions that occur and to maintain DNA integrity in the nucleus and in mitochondria (BOX 1). Key nuclear DNA repair pathways include DNA double- strand break repair (DSBR), base excision repair (BER) and nucleotide excision repair (NER). Multiple mitochondria-associated adaptive processes are regulated by nuclear transcription regulators, such as peroxi-some proliferator-activated receptor-γ co-activator 1α (PGC1α), hypoxia inducible factor 1α (HIF1α), the fork-head box O (FOXO) proteins and some sirtuin family members. Sirtuins are a group of NAD+-dependent deacetylases that can alter the activities of diverse proteins (BOX 2). Coordination between nuclear DNA repair proteins and nuclear sirtuins (SIRT1, SIRT6 and SIRT7) reveals a facet of how nuclear DNA repair proteins may regulate mitochondrial homeostasis (FIG. 2).
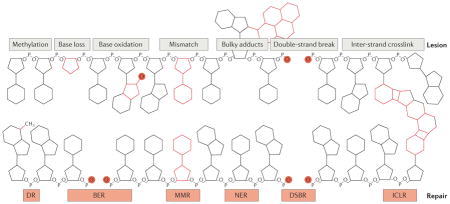
Box 1. Nuclear and mitochondrial DNA damage repair.
Nuclear DNA damage is regarded as a major culprit in cancer, neurodegeneration, mitochondrial dysfunction and many age-related diseases, and unrepaired DNA damage can result in pathological levels of nucleus-to-mitochondria signalling (NM signalling), which may exacerbate disease progression. As DNA is constantly assaulted by both endogenous (for example, reactive oxygen species and hydrolysis) and exogenous (such as ultraviolet and ionizing radiation) stresses, cells have evolved several DNA repair pathways to maintain the integrity of DNA3,4,141–144. Major DNA repair pathways in the nucleus include direct reversal (DR) of the lesion (usually methylation), base excision repair (BER), mismatch repair (MMR), nucleotide excision repair (NER; comprising global genome NER and transcription-coupled NER), double-strand break repair (DSBR, which includes non-homologous end-joining (NHEJ) and homologous recombination (HR)) and inter-strand crosslink repair (ICLR), among others (see the figure). DNA repair pathways in mitochondria are less well understood; the available data suggest that BER is the major DNA repair pathway, whereas NER does not exist in mitochondria.
DR, MMR and DSBR may exist in mitochondria, although their enzymatic constituents remain to be elucidated3,145,146. Intriguingly, a number of predominantly nuclear DNA repair proteins are also present in mitochondria. Many of the proteins involved in BER are present in both compartments, and for many other DNA metabolic proteins, it is unclear whether they are normally present in the mitochondria or imported under conditions of stress. Some DNA repair proteins, such as RECQL4, petite integration frequency 1 (PIF1), DNA replication helicase/nuclease 2 (DNA2) and Suppressor of Var1 3-like protein 1 (SUPV3L1) are localized at both the nucleus and mitochondria and may maintain mitochondrial DNA stability and/or mitochondrial function, in addition to promoting nuclear DNA integrity147,148. For example, RECQL4 is one of the five human RecQ helicases, and it is mutated in Rothmund–Thomson syndrome. RECQL4 primarily localizes to the nucleus and participates in nuclear DNA repair. A portion of RECQL4 also localizes to the mitochondria and regulates the translocation of p53 to mitochondria, as well as maintaining mitochondrial DNA integrity147,148.
Box 2. Functions of sirtuins in ageing and age-associated diseases.
Sirtuins are NAD+-dependent deacetylase and deacylase enzymes that control metabolism and ageing. Sirtuins modify proteins, and, in the process, they consume NAD+ and generate nicotinamide (NAM) and ADP-ribose (ADPR; see the figure). The Saccharomyces cerevisiae SIR2 gene (silent information regulator 2; encoding the protein Sir2p) was the first sirtuin to be discovered; it affects replicative lifespan in yeast through histone deacetylation and chromatin silencing at ribosomal DNA repeats and sub-telomeric sequences33,149,150. The SIR2 orthologue in Caenorhabditis elegans is sir-2.1. Overexpression of sir-2.1 extended lifespan in some studies but not others16,151–153. There are seven mammalian sirtuins, SIRT1 to SIRT7. SIRT1, which is a nuclear protein, is defective in many age-associated diseases and regulates diverse ageing-related processes, including mitochondrial homeostasis and genomic stability12,33. More than 50 proteins are deacetylated by SIRT1, including some that are involved in energy metabolism (such as peroxisome proliferator-activated receptor-α (PPARα) and PPARγ), mitochondrial homeostasis (peroxisome proliferator-activated receptor-γ co-activator 1α (PGC1α), hypoxia inducible factor 1α (HIF1α), autophagy-related protein 7 (ATG7) and ATG8), and some that are involved in DNA repair (such as ataxia telangiectasia mutated (ATM), xeroderma pigmentosum group A-complementing protein (XPA), Werner syndrome ATP-dependent helicase (WRN), KU70 and others)12,33,47,48,154. SIRT2 is primarily cytoplasmic but is observed in the nucleus during mitosis, where it regulates the mitotic checkpoint. SIRT3, SIRT4 and SIRT5 are mitochondrial enzymes, with SIRT3 being responsible for the majority of deacetylation of mitochondrial metabolic enzymes155. The nuclear SIRT6 is linked to telomere maintenance and genome stabilization. In mice, SIRT6 loss leads to genomic instability and premature ageing phenotypes, whereas overexpression of SIRT6 extends mouse lifespan54,55,60,62. SIRT7, which is also a nuclear protein, promotes epigenetic stabilization of cancer-associated gene expression programmes and regulates mitochondrial homeostasis through the regulation of nuclear-encoded mitochondrial genes and the mitochondrial unfolded protein response (UPRmt)25,68. Mammalian sirtuins affect cancer biology through tumour suppressive and oncogenic effects33. Thus, the sirtuins participate in multiple cellular pathways, and future work should further interrogate the crosstalk between sirtuins and ageing.
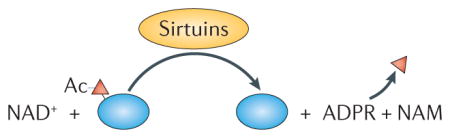
Figure 2. PARP1–NAD+–SIRT1-mediated nuclear DNA damage to mitochondria signalling.

Activation of poly(ADP-ribose) polymerase 1 (PARP1) upon DNA damage facilitates DNA repair but leads to loss of NAD+ and acetyl-CoA, the latter being an important molecule in cellular metabolism. This results in inhibition of sirtuins such as SIRT1, owing to competition for NAD+. Loss of sirtuin activity leads to an increase in mitochondrial reactive oxygen species (O2−), owing to decreased activation of downstream stress response factors such as AMP-activated protein kinase (AMPK), forkhead box O proteins (FOXOs) and peroxisome proliferator-activated receptor-γ co-activator 1α (PGC1α). Collectively, these enzymes regulate a large number of genes that are involved in coping with oxidative stress, such as those encoding uncoupling proteins (UCPs) like UCP2 and superoxide dismutases (SODs). In addition, SIRT1, as well as SIRT6, has been shown to positively regulate several DNA repair pathways, such as homologous recombination (HR), non-homologous end-joining (NHEJ), base excision repair (BER) and nucleotide excision repair (NER). Specifically, SIRT1 has been shown to deacetylate repair factors such as xeroderma pigmentosum group A-complementing protein (XPA, which is involved in NER), KU70 (involved in NHEJ), ataxia telangiectasia mutated (ATM; involved in HR and NHEJ) and thymine DNA glycosylase (TDG; involved in BER). Activation of PARP1 and subsequently less-robust DNA repair, as well as increased oxidative stress, may represent a vicious cycle that could contribute to the ageing process. Thus, PARP1 is pivotal to the initiation of different DNA repair pathways, and it interacts with SIRT1 to execute cellular signalling from the nucleus to mitochondria.
The PARP1 and NAD+–SIRT1–PGC1α axis
A prominent pathway linking nuclear DNA damage to mitochondrial homeostasis is the NAD+–SIRT1–PGC1α axis, in which NAD+ has a pivotal role. NAD+ is a rate-limiting metabolite for poly(ADP-ribose) polymerase 1 (PARP1) and SIRT1 activities, making the cellular content of NAD+ an important index for cell fate and organismal health (see below). Intracellular NAD+ levels are usually approximately 0.2–0.5 mM, depending on cell type and condition27. There is a subcellular difference in NAD+ content between the nucleus and mitochondria, as the nuclear NAD+ content is ~0.07 mM — much lower than in mitochondria, where it is 0.25 mM or greater27,30. The cellular NAD+ level is tightly regulated by three biosynthetic pathways, the Preiss–Handler pathway, de novo biosynthesis and the salvage pathway27,31. There is an age-dependent decrease in NAD+, and NAD+ is also reduced in several pathologies, suggesting a contributory role for NAD+ depletion in ageing and age-related diseases. The regulation of NAD+ metabolism and its importance in health and ageing are discussed in two excellent recent reviews27,31.
Mammalian SIRT1 is thought to be involved in epigenetic regulation, mitochondrial maintenance and the ageing process. SIRT1, which is a prominent nuclear sirtuin, regulates various cellular pathways and may delay the progression of ageing through deacetylation of several substrates, including PGC1α, which is a master regulator of mitochondrial biogenesis32. The deacetylation activity of SIRT1 and other sirtuins requires consumption of NAD+, which is a cofactor also involved in DNA damage repair12,33–35. When DNA breaks occur, the DNA break sensor PARP1 detects break sites and initiates DNA repair signalling through generation of PAR, a process called PARylation, which consumes NAD+ (REF. 36) (FIG. 2). Interestingly, PARP1 and SIRT1 have comparable Km values for NAD+ (50–97 μM and 94–96 μM, respectively)27. PARylation promotes the rapid recruitment of PAR-binding DNA repair proteins to locate and efficiently repair the DNA damage, as well as promoting the decondensation of chromatin around the damage site. Aged and DNA repair-deficient laboratory animal models exhibited persistent hyperactivation of PARP1, lower SIRT1 activity and NAD+ depletion. PARP1 inhibition or NAD+ supplementation restored NAD+ levels and SIRT1 activity12,16,27. Thus, PARP1 and SIRT1 can compete for NAD+, and increased consumption of NAD+ by one of these proteins may limit the activity of the other (FIGS 2,3a). When cells with a very high energy requirement, such as neurons or cardiac myocytes, are subjected to severe reductions in energy availability — such as during an ischaemic event (stroke or myocardial infarction) — increased consumption of NAD+ by PARP1 (REFS 37,38) and SIRT1 (REF. 39) may contribute to a cellular energy crisis that triggers cell death. The molecular details of the nature of the competition between PARP1 and SIRT1 for NAD+ as a substrate merit further investigation.
Figure 3. DNA damage-induced nucleus-to-mitochondria signalling is linked to metabolic dysfunction and age-associated diseases.

a | Poly(ADP-ribose) polymerase 1 (PARP1) activation leads to alterations in central bioenergetic pathways. A consequence of loss of NAD+ is change in the NAD+/NADH ratio. Notably, PARP1 activation also leads to increased nicotinamide (NAM), a competitive inhibitor of sirtuins such as SIRT1, as well as loss of NAD+, which is a substrate for sirtuins. The decrease in the NAD+/NADH ratio also leads to the conversion of pyruvate to lactate by lactate dehydrogenase, while decreasing the formation of acetyl-CoA by pyruvate dehydrogenase. An isoform of pyruvate dehydrogenase has recently been found in the cell nucleus, and the DNA damage response could thereby regulate the formation of acetyl-CoA locally. Loss of acetyl-CoA decreases histone acetylation (Ac) and changes the epigenome, resulting in chromatin condensation and gene silencing, while also leading to decreased availability of acetyl-CoA for the mitochondria for the generation of ATP. Histone acetylation is also reduced by the activity of sirtuins such as SIRT1 and of other classes of histone Lys deacetylases (HDACs). Ketones, which are a group of metabolites made in the liver, increase acetyl-CoA levels and histone acetylation and could counteract the effects of age-associated DNA damage. b | Ataxia telangiectasia mutated (ATM) regulates multiple metabolic pathways following DNA damage through phosphorylation of p53, liver kinase B1 (LKB1) and AMP-activated kinase (AMPK). AMPK is a central kinase in the adaptive cellular response and phosphorylates a number of key factors. These include SIRT1, which is a known regulator of mitochondrial biogenesis; forkhead box O transcription factors (FOXOs), which are involved in cellular stress responses; glucose transporters (GLUTs), which are involved in glucose transport and glycolysis; UNC51-like kinase 1 (ULK1), which is a regulator of autophagy; peroxisome proliferator-activated receptor-γ co-activator 1α (PGC1α), which is a master-regulator of mitochondrial biogenesis; sterol regulatory element-binding protein 1 (SREBP1), which is a regulator of fatty acid oxidation; and hypoxia inducible factor 1α (HIF1α), which is a central positive regulator of glycolysis. Collectively the activity of these factors leads to changes in cellular bioenergetics.
Imbalances of the activities of PARP1 and SIRT1 have been associated with ageing and pathological processes. An age-dependent increase in PARylation was observed in various experimental animals (including nematodes and mice)12,16, and PARP activation has been detected in a wide range of human diseases, including chronic heart failure, stroke, Alzheimer disease and Parkinson disease40. Deletion of the Parp1 gene increased NAD+ levels, SIRT1 activity and mitochondrial biogenesis in mice41, and pharmacological inhibition of PARP1 increased mitochondrial biogenesis and extended lifespan in wild-type nematodes16. Compromised balance between the activities of PARP1 and SIRT1 signalling may also contribute to premature ageing syndromes, such as in XPA and Cockayne syndrome. Individuals with XPA are UV-sensitive, with a high risk of skin cancers, and they suffer from neurodegeneration, including progressive cerebral and cerebellar atrophy, neuropathy and sensorineural hearing loss12,42. XPA is caused by mutation of the gene encoding the protein XPA, which participates in NER43. Patients with Cockayne syndrome show photosensitivity (but no cancer susceptibility), with major clinical manifestations including severe early-onset neurodegeneration, hearing loss, muscle weakness and cachectic dwarfism44. This is caused by mutations in genes encoding Cockayne syndrome group A or B (CSA or CSB) proteins, which are also involved in the NER pathway of DNA repair. Recent studies in patient cells and various experiments in animal models suggest that both XPA and Cockayne syndrome have a noticeable phenotype of mitochondrial disease, as evidenced by severe mitochondrial clinical features and dysfunction including altered mitochondrial metabolism and accumulation of damaged mitochondria12,21,35. In patient cells and animal models of XPA and CSB, there is a reduction of SIRT1 activity, owing to persistent activation of PARP, induced by unrepaired DNA damage. Inhibition of PARP1 or supplementation of NAD+ precursors restored SIRT1 activity and improved mitochondrial homeostasis12,21,35. Thus, short-term activation of PARP1 is beneficial for genomic stability, because it promotes DNA repair; however, persistent activation of PARP1 in age-related diseases probably contributes to pathology.
In addition to the NAD+–SIRT1–PGC1α axis, other processes are affected by PARP1/SIRT1 competition. SIRT1 modulates the activities of mitochondria-related transcription factors other than PGC1α, including HIF1α, FOXO1 and FOXO3A, through deacetylation or degradation (for HIF1α)45,46. DAF-16, a homologue of mammalian FOXO3A, is the only FOXO family member in Caenorhabditis elegans. The C. elegans SIR-2.1 (a homologue of mammalian SIRT1) can activate the DAF-16–superoxide dismutase (SOD-3) pathway to protect against reactive oxygen species (ROS)16, which can trigger PARP1 (FIG. 2). Many other pathway steps in NM signalling following DNA damage are being investigated.
SIRT1 participates in nuclear DNA repair
SIRT1 affects multiple DNA repair pathways. It deacetylates and activates some major DSBR proteins, including KU70, nibrin, histone deacetylase 1 (HDAC1) and the RecQ helicase protein Werner (WRN)47–49. In neurons, SIRT1 has a role in DSBR through non-homologous end-joining (NHEJ). Specifically, it participates in the initial sensing and signalling of DSBs through stabilization of ataxia telangiectasia mutated (ATM) at DSB sites and deacetylates HDAC1, which is implicated in another step of NHEJ47. In proliferating cells, SIRT1 participates in homologous recombination and BER through deacetylation of WRN49. Even though ATM is a crucial sensor of DSBs, SIRT1 can promote DSBR in an ATM-independent manner, which may involve WRN and other proteins50. SIRT1 also increases BER activity through deacetylation of key pathway proteins, including apurinic/apyrimidinic endonuclease 1 and thymine DNA glycosylase51,52. SIRT1 also promotes NER through deacetylation of XPA, resulting in enhanced interaction of XPA with other NER proteins53. Thus, in addition to direct regulation of mitochondrial homeostasis through the aforementioned pathways, SIRT1 promotes genomic stability (FIG. 2).
Other sirtuins
Two other nuclear sirtuins, SIRT6 and SIRT7, have important roles in chromatin regulation and genome maintenance and, like SIRT1, have intriguing links to mitochondrial homeostasis. SIRT6 is a chromatin-remodelling factor that maintains genomic stability through various mechanisms and represses gene expression programmes associated with ageing, metabolism and cancer. SIRT6 is implicated in longevity regulation: overexpression of SIRT6 increases lifespan in male transgenic mice, and SIRT6 deficiency leads to shortened mouse lifespan, genomic instability and, in mouse cancer models, to increased tumour growth and aggressiveness54–56. Additionally, ageing-associated phenotypes observed in SIRT6-deficient mice have some features similar to those of Hutchinson–Gilford progeria syndrome (HGPS), which is a rare and fatal human premature ageing disease caused by mutations in the lamin A gene55,57,58. Lamin A may be an endogenous activator of SIRT6 by promoting its DNA repair-mediating activity58. Despite its nuclear localization and functions, SIRT6 influences mitochondrial homeostasis and has important effects on cancer cell metabolism. SIRT6 increases mitochondrial respiration by repressing transcription of key glycolytic enzymes through inhibition of HIF1α; in cancer cells, this prevents tumorigenic glycolytic metabolism (known as the ‘Warburg effect’)56,59.
SIRT6 also regulates genomic stability and promotes DSBR through several mechanisms. It prevents telomere dysfunction and interacts physically or functionally with many DSBR proteins, including PARP1, DNA-dependent protein kinase (DNA-PK), C-terminal-binding protein-interacting protein (CTIP), WRN and the ATP-dependent chromatin remodeller SWI/SNF-related matrix-associated actin-dependent regulator of chromatin A5 (SNF2H)60–64. Thus, SIRT6 is involved in DNA damage recognition64 and affects mitochondrial homeostasis and tumour suppression. In addition to DNA repair, anti-ageing effects of SIRT6 may be associated with its role in repressing long interspersed element 1 (LINE1) retrotransposons, a group of transposable elements that induce genomic instability65. LINE1 transposition increases upon DNA damage or cellular and tissue ageing and is associated with depletion of SIRT6 at LINE1 promoters. Interestingly, retrotransposons can trigger innate immune responses, which may involve mitochondrial stress signalling1, thus suggesting linkage between SIRT6 and mitochondria. This area of research is a ripe topic for future investigations to better understand the role of SIRT6 in DNA damage-induced NM signalling.
SIRT7 is less well characterized than SIRT6, but studies indicate that it is a dynamic nuclear regulator of both chromatin and mitochondrial functions. SIRT7 promotes chromatin-dependent repression of genes involved in the epigenetic maintenance of oncogenic transformation, including cytoplasmic and mito-chondrial ribosomal proteins66. In SIRT7-deficient mouse liver, increased ribosomal protein expression is associated with endoplasmic reticulum (ER) stress and hepatosteatosis67, and, in haematopoietic stem cells, SIRT7 loss leads to increased levels of mitochondrial ribosomal proteins and mitochondrial protein folding stress25. SIRT7 may regulate mitochondrial homeostasis by deacetylating GA-binding protein β1 (GABPβ1), which is a master regulator of nuclear-encoded mitochondrial genes; accordingly, SIRT7-deficient mice exhibit multi-systemic mitochondrial dysfunction68.
In addition to nuclear sirtuins, the mitochondrial SIRT3 and SIRT4 may participate in DNA damage- induced NM signalling. SIRT3, a major mitochondrial deacetylase, prevents both age-related and noise-induced hearing loss through reduction of oxidative mtDNA damage (such as apurinic and apyrimidinic sites and 8-oxo-dGuo) in the cochlea and brain, or through reduction of neurite degeneration caused by noise69,70. The roles of SIRT3 in neuroprotection may be mediated by augmentation of the mitochondrial antioxidant defence system and deacetylation of several proteins, including mitochondrial isocitrate dehydrogenase 2, SOD2 and cyclophilin D69,71. SIRT4 is induced by many genotoxic agents, and it regulates the cellular metabolic response to nuclear DNA damage response pathways through repression of mitochondrial glutamine metabolism. Although the roles of mitochondrial sirtuins in ageing, metabolism and mitochondrial function have been established33,72,73, further work on how these mitochondrial sirtuins link mitochondrial maintenance and genomic stability will improve our understanding of their roles in NM signalling.
Although we discuss a few of the nuclear sirtuins separately, there is evidence for functional interactions among them. The expression of SIRT6 may be dependent on SIRT1 through the formation of a SIRT1–FOXO3A–nuclear respiratory factor 1 (NRF1) transcription complex on the SIRT6 promoter74. SIRT7 and SIRT1 interact physically, and this interaction is important for regulation of the epithelial–mesenchymal transition (EMT)-like process of metastatic progression in cancer cells75. Proteomic studies of SIRT6- or SIRT7-interacting factors uncovered more than 110 common proteins, although it is not known whether these interactions occur in the same or in distinct protein complexes76. Several of these proteins, including nucleophosmin and nucleolin, are associated with ageing-related processes such as DNA repair and cellular senescence, but they can also have an impact on mitochondrial biology76, indicating a possible involvement in DNA damage-induced NM signalling.
Metabolic NM signalling pathways
DNA damage leads to metabolic alterations at the cellular and organismal levels. This is evident in premature ageing disorders such as Werner syndrome, ataxia telangiectasia, Cockayne syndrome and Hutchinson–Gilford progeria, in which weight loss is highly prevalent35,77,78. Notably, weight loss is common in normal human ageing, as well as in neurodegenerative diseases such as Parkinson disease, Huntington disease and amyotrophic lateral sclerosis (ALS)79. Indeed, in cell and animal models of accelerated ageing, weight loss is prominent and may be caused by increased metabolic rates12,35,80. Pharmacological inhibition of the DNA damage sensor PARP1 decreases the metabolic rate of old Csbm/m mice (with a truncation mutation in CSB), supporting the idea that DNA damage-induced hyperactivation of this enzyme contributes to metabolic alterations in accelerated ageing disorders35. In the process of PARylation, PARP1 consumes NAD+, which leads to alterations in the NAD+/NADH ratio, resulting in increased lactate production35,81. This phenomenon is observed in the brain during normal and accelerated ageing82 (FIG. 3a). Lactate is produced by lactate dehydrogenase from pyruvate. Pyruvate dehydrogenase decarboxylates pyruvate, thereby creating the central bioenergetic molecule acetyl-CoA. This process occurs in mitochondria; however, recent findings suggest that the pyruvate dehydrogenase complex is also present in the nucleus, where acetyl-CoA is utilized by histone acetyltransferases to control the epigenome83. Intriguingly, DNA damage- induced PARP1 activation can lead to decreases in the NAD+/NADH ratio, decreased formation of acetyl-CoA, and thus loss of histone acetylation, perhaps contributing to the global transcriptional silencing that is known to occur after DNA damage84,85. Accordingly, age-associated accumulation of DNA damage and persistent activation of PARP1 could contribute to age- associated alterations in the epigenome. Importantly, cellular acetyl-CoA levels can be increased through simple dietary interventions such as fasting or ingestion of a ketogenic diet. Indeed, these two diets increase lifespan and/or healthy life expectancy (healthspan) across species86.
Alterations in food intake regulate ageing across numerous species. As is the case with DNA damage signalling, fasting also leads to the activation of downstream factors. One conserved and key cellular energy sensor is AMP-activated protein kinase (AMPK), which facilitates cell survival through the orchestrated activation of multiple pathways that maintain cellular energy homeostasis. AMPK is activated by increases in ADP and AMP levels during periods of either low energy availability or increased energy demand45. In addition, AMPK is activated by ROS and elevated cytoplasmic calcium levels through Ca2+/calmodulin-dependent protein kinase kinase-β (CaMKKβ). AMPK is also activated by ATM, which is a master regulator of the DNA damage response (as described below), and by p53. ATM can either directly phosphorylate and activate AMPK or activate the upstream AMPK activator liver kinase B1 (LKB1)87 (FIG. 3b).
Additionally, p53 stabilization has been reported to induce transcriptional activation of sestrin 1 and sestrin 2, resulting in AMPK activation88. AMPK activation leads to increased glucose and fatty acid oxidation through activation of glucose transporters (GLUTs), HIF1α and sterol regulatory element- binding proteins (SREBPs), as well as activation of mitochondrial maintenance signalling through phosphorylation and activation of the promitophagic factor UNC51-like kinase 1 (ULK1)89. In addition, AMPK phosphorylates and activates the mTORC1 inhibitor tuberous sclerosis complex 2 (TSC2), thereby further activating autophagy and inhibiting cell growth. AMPK also positively regulates PGC1α activity, which stimulates mitochondrial biogenesis, and FOXO3A activity, which leads to activation of stress response and pro- survival pathways45. Thus, it is clear that AMPK has a central role in regulating mitochondrial function both directly and indirectly (FIG. 3b). Importantly, AMPK represents a pharmacological target, with specific activators having potential beneficial effects in various age-related diseases.
Mitophagy–apoptosis crosstalk
DNA damage activates certain pivotal proteins that lead to amplification and propagation of a signal, ultimately resulting in cellular outcomes such as mitophagy and apoptosis. Several mediators seem to be important for the choice between autophagy and mitophagy or apoptosis. Importantly, low levels of DNA damage stress may stimulate mitophagy and antagonize apoptosis, whereas high levels of stress inhibit mitophagy and stimulate apoptosis.
Mitochondria not only have a central role in metabolic pathways, they also regulate cell fate through crosstalk between mitophagy and apoptosis. Macroautophagy (autophagy) and apoptosis are intimately interconnected to determine whether cells survive or die (FIG. 4). DNA damage constitutes a strong intrinsic apoptotic signal. Apoptosis can occur by several different mechanisms, including caspase 8-dependent apoptosis, mitochondria- mediated caspase 9- and caspase 3-dependent apoptosis, and caspase-independent necrosis or autophagic cell death90 (FIG. 4a). The B cell lymphoma 2 (BCL-2) family of proteins are closely involved in the regulation of apoptosis and consist of both pro-survival members, including BCL-2, BCL-XL, BCL-W, A1A and MCL2; and pro-apoptotic members such as BAX, BAK, BIM, BID (BCL-2 homologous 3-interacting domain death agonist) and PUMA91. Apoptosis is initiated when the DNA lesion burden exceeds a specific threshold, leading to the initiation of intrinsic and/or extrinsic apoptotic pathways.
Figure 4. DNA damage signalling in the regulation of mitophagy and apoptosis.

a | Apoptosis can be initiated through intrinsic or extrinsic pathways. The intrinsic pathways can be initiated by genotoxic stress, leading to the activation of p53 and transcriptional upregulation of pro-apoptotic factors such as BAD, BAX and BID. If the stress is sustained or high enough, BAD and BAX accumulation results in the permeabilization of the outer mitochondrial membrane and release of cytochrome C (CytC) that can associate with apoptosis-activating factor 1 (APAF1) and activate caspase 9 (CASP9). CASP9 activation leads to CASP3 activation and initiation of apoptosis. The extrinsic pathway can be initiated through activation of the FAS receptor. This leads to FAS-associated death domain protein (FADD)-mediated activation of CASP8, resulting in CASP3 activation. Notably, there is considerable crosstalk between intrinsic and extrinsic pathways. b | Selective mitophagy is regulated through factors such as PTEN-induced putative kinase 1 (PINK1) and the E3-ubiquitin ligase parkin. Initially, mitochondrial damage causes loss of the mitochondrial membrane potential (Δψ). This leads to the retention of PINK1 at the outer mitochondrial membrane and phosphorylation of outer membrane proteins. Concomitantly, parkin is activated, leading to ubiquitylation of outer membrane proteins. PINK1 phosphorylates ubiquitin that recruits adaptor proteins such as optineurin (OPT), nuclear dot protein 52 (NDP52) and p62, leading to recruitment of the UNC51-like kinase 1 (ULK1) complex (consisting of proteins such as autophagy-related protein 101 (ATG101), ATG13 and FIP200) and to the formation of a double lipid membrane-bound vesicle, the autophagosome. The adaptor proteins (OPT, p62 and NDP52) interact with the autophagosome through light chain 3 (LC3), a small protein that coats the autophagosome. The entire mitochondrion will eventually become engulfed in the autophagosome, which will fuse with a lysosome leading to degradation of the mitochondrion. c | Ataxia telangiectasia mutated (ATM) is activated by breaks in DNA and possibly also by oxidative stress (O2−). At low levels of DNA damage stress, ATM activation leads to phosphorylation, ubiquitylation and activation of the NEMO JNK (NF-κB essential modulator Jun N-terminal kinase) pathway that stimulates mitophagy. ATM also phosphorylates the pro-apoptotic factor BID to inhibit apoptotic signalling. At high levels of stress, ATM phosphorylates and activates p53, which propagates pro-apoptotic signals. d | p53 has been well characterized in the response to genotoxic stimuli, in which it transcriptionally activates pro-apoptotic proteins such as BAX and p21 while simultaneously inhibiting the ULK1-containing autophagy-initiating complex. In addition, p53 activation may lead to decreased expression of parkin and decreased activation of parkin PINK1-mediated mitophagy. At lower levels of stress, p53 can stimulate mitophagy through the activation of DNA damage-regulated autophagy modulator protein 1 (DRAM1), which stimulates p62-mediated mitophagy. e | Sirtuin 1 (SIRT1) regulates both mitophagy and apoptosis. At low levels of DNA damage, nuclear SIRT1 can be activated to facilitate DNA repair. After lethal levels of nuclear DNA damage, SIRT1 is inhibited by the DNA damage response, leading to p53 acetylation and cell death. Loss of SIRT1 activity decreases the stimulation of mitophagy through peroxisome proliferator-activated receptor-γco-activator 1α (PGC1α)- and AMP-activated kinase (AMPK)-dependent pathways. SIRT1 regulates mitochondrial biogenesis and mitophagy through deacetylation of PGC1α, and it also interacts with AMPK to regulate mitophagy through mutual activation: AMPK activates SIRT1 by increasing the ratio of NAD+/NADH, and it is activated by SIRT1 through deacetylation of liver kinase B1 (LKB1), an AMPK activator. Furthermore, AMPK phosphorylates and activates the mTORC1 inhibitor protein tuberous sclerosis complex 2 (TSC2), thereby further activating autophagy. AMPK also positively regulates PGC1α activity, which stimulates mitochondrial biogenesis and activates forkhead box protein O 3A, leading to activation of stress response and pro-survival pathways. NAM, nicotinamide.
Autophagy (auto, self; phagy, eating of) generally functions as a pro-survival mechanism, and defective autophagy is linked with neurodegenerative diseases and ageing. Autophagy is a process whereby cells sequester damaged or unused cytoplasmic substrates within a double-membrane vesicle — the autophagosome — that is then fused with lysosomes, leading to the degradation of the engulfed substrates92. Mitophagy is the selective degradation of mitochondria by autophagy. In mammalian cells, there are several mitophagy pathways, such as NIX (also known as BNIP3L)-regulated programmed mitophagy, which is found in blood cells; and selective mitophagy, which can be regulated in a PTEN-induced putative kinase 1 (PINK1)-dependent or PINK1-independent manner3,93–95. Genes encoding PINK1 and the ubiquitin ligase parkin are often mutated in autosomal recessive parkinsonism5. In mitophagy, full-length PINK1 located at the outer mitochondrial membrane phosphorylates ubiquitin to activate parkin, which generates ubiquitin chains to recruit downstream autophagy receptors and signals robust mitophagy95 (FIG. 4b). In normal, steady-state conditions, the mitochondrial membrane potential drives full-length PINK1 through the translocase of the inner membrane (TIM) complex, where mitochondrial processing peptidase (MPP) and presenilin-associated rhomboid-like protein (PARL) cleave the mitochondrial targeting sequence and transmembrane domain of PINK1. The cytoplasmic part of PINK1 is further degraded by the ubiquitin proteasome5,12,96. Thus, mitophagy targets damaged and depolarized mitochondria, as mitochondria with higher mitochondrial membrane potentials are spared from PINK1-dependent mitophagy.
It is increasingly clear that mitophagy plays an important part in the maintenance of neuronal health, as well as in the ageing process itself11. Current knowledge of the relationship between autophagy and apoptosis suggests that autophagy precedes apoptosis in many instances, and that autophagy and apoptosis are often antagonistic22,90,92 (FIG. 4). In this section, the interplay between classic apoptotic mediators and mitophagy, and the role of NM signalling in the regulation of this crosstalk, will be illustrated in three major pathways.
ATM-mediated mitophagy–apoptosis crosstalk
The protein kinase ATM is a major activator of the DNA damage response. It has a key role in the repair of DSBs, and it is closely linked to health through its roles in the maintenance of genomic stability and mitochondrial homeostasis (FIG. 4c). ATM triggers multiple cellular events to promote cell survival at low levels of DSBs; conversely, it drives cellular apoptosis after extensive DSB damage23,97. ATM promotes survival by activating repair of DSBs, upregulating pro-survival genes and inducing autophagy and mitophagy. In response to DNA damage, cells recruit DNA repair proteins, including ATM, to DSBs to initiate DNA repair. This process also involves ubiquitylation of the nuclear factor-κB (NF-κB) essential modifier (NEMO) protein, which translocates from the nucleus to the cytoplasm, where it activates the Jun N-terminal kinase (JNK)97,98. JNK modulates the balance between apoptosis and autophagy and mitophagy through the phosphorylation of specific BCL-2 protein family members, such as BCL-2 and BIM. This releases the pro-autophagy protein Beclin 1 from BCL-2 or BIM complexes, and autophagy is initiated99. When DSBs are extensive, JNK instead promotes apoptosis through at least two independent pathways: the inactivation of the anti-apoptotic BCL-2 and the activation of the FAS-associated death domain protein (FADD)–caspase 8 pro-apoptotic pathway90,97. Thus, in response to DSBs, ATM uses NEMO and JNK to regulate autophagy/ mitophagy–apoptosis crosstalk (FIG. 4c). The ATM–BID axis is an additional regulator of mitochondrial function following DNA damage. ATM may inhibit ROS production by phosphorylating BID and preventing its translocation to mitochondria and apoptosis100 (FIG. 4c).
A role for ATM in mitochondrial homeostasis and longevity is supported by the phenotypes of ataxia telangiectasia patients and Atm−/− laboratory animal models. Patients with ataxia telangiectasia show neurodegeneration, cancer predisposition and sterility23. Cells from these patients have mitochondrial dysfunction and alterations in mtDNA copy number, higher mitochondrial levels of ROS, elevated cellular respiratory capacity and an accumulation of damaged mitochondria12,80,101. The importance of mitochondrial dysfunction in the aetiology or progression of ataxia telangiectasia is further supported by the notion that ataxia telangiectasia patient cells and Atm−/− mouse models are highly sensitive to oxidants. Conversely, mitochondrial antioxidants protect Atm−/− cells against oxidative stress and mitigate some ataxia telangiectasia phenotypes in Atm−/− mice23. It is likely that defective mitophagy significantly contributes to the mitochondrial dysfunction in ataxia telangiectasia12,80. Using a bio-informatics tool, Mito DB, it was reported that patients with ATM mutations clinically resemble patients with mitochondrial diseases12,102. Further studies into pathways involved in mitochondrial dysfunction in ataxia telangiectasia are warranted.
p53-mediated mitophagy–apoptosis crosstalk
The tumour suppressor p53 is at the hub of numerous signalling pathways, but although the role of p53 in apoptosis is well established, it has only recently been associated with mitophagy (FIG. 4d). Under lethal genotoxic stress, p53 transcriptionally upregulates pro-apoptotic proteins such as p21, RB, BAX, FAS, death receptor 5 (DR5; also known as TNFRSF10B), NOXA, PIG3, p53AIP1 and PUMA, which leads to cellular apoptosis, senescence or growth arrest103,104. Of note, p53 in the cytoplasm may enhance apoptosis through inhibition and destabilization of the autophagy-initiation ULK1–FIP200–autophagy-related protein 13 (ATG13)–ATG101 complex, as well as through inhibition of PINK1–Parkin-mediated mitophagy105. However, in cells under low levels of stress, nuclear p53 can bind the promoter regions of genes that encode pro-autophagy proteins, including DNA damage-regulated autophagy modulator 1 (DRAM1), thereby inducing mitophagy89,106. DRAM1 is a lysosomal membrane protein involved in autophagy induction; when mitochondrial protein synthesis is impaired, mitochondria generate more ROS to activate autophagy and mitophagy through the ROS–p53–DRAM1 pathway107. These examples illustrate how p53-mediated nuclear signalling regulates apoptosis and mitophagy (FIG. 4d).
In recent years, evidence has accumulated to suggest that p53 has crucial roles in ageing through interconnected regulation of apoptosis, senescence and autophagy. Studies from animal models support the view that p53 signalling prolongs lifespan, as evidenced by decreased levels of tumour incidence, senescence and ageing-associated damage in model organisms108–110. In C. elegans, it was reported that daf-2 (encoding an insulin-like receptor) mutants had extended lifespan through inhibition of tumour growth in a DAF-16–p53-dependent manner110. Compared with wild type, mice with moderately high levels of p53 showed high cancer resistance, increased antioxidant activity and delayed ageing109. In view of the role of p53 in the induction of autophagy, it would be of interest to investigate the role of autophagy in p53-mediated longevity. Collectively, the available data suggest that p53 can determine cell fate by influencing mitophagy–apoptosis crosstalk, and that an imbalance of this crosstalk in favour of apoptosis may contribute to degenerative diseases and ageing.
SIRT1-mediated mitophagy–apoptosis crosstalk
In addition to the well-described role of SIRT1 in mitochondrial biogenesis through PGC1α, SIRT1 also regulates the crosstalk between mitophagy and cell death. SIRT1 deacetylates and inactivates p53, allowing cells to escape apoptosis after excessive toxic stress, including DNA damage111,112 (FIG. 4e). It also inhibits apoptosis by activating autophagy and mitophagy. In yeast, Sir2 (silent information regulator 2; the homologue of mammalian SIRT1) protects against ageing, partially through induction of mitophagy by transcriptional upregulation of the mitophagy executor Atg32 (REF. 113). In mammals, SIRT1 can activate autophagy through several pathways, including deacetylation of light chain 3 (LC3; also known as MAP1LC3B), which facilitates its nuclear export and its conjugation on the autophagic membrane through interaction with another cytoplasmic autophagy regulator, ATG7 (REF. 114). SIRT1 can also regulate mitophagy by maintaining PINK1 integrity through regulation of the mitochondrial membrane potential12. In XPA, a high mitochondrial membrane potential destabilizes full-length PINK1, causing defective mitophagy. Activation of the NAD+–SIRT1–PGC1α axis in XPA cells restored mitophagy through upregulation of mitochondrial uncoupling protein 2 (UCP2)12.
SIRT1 also interacts cooperatively with AMPK, which is a central regulator of metabolism and energy homeostasis, to stimulate mitophagy (FIG. 4e). Either physiological (through fasting or exercise) or pharmacological activation of AMPK can activate SIRT1 through increasing the NAD+/NADH ratio12,45,89. Interestingly, AMPK can also be activated by SIRT1 through deacetylation of LKB1, leading to the phosphorylation and activation of AMPK. This may suggest the existence of a positive-feedback loop in the regulation of SIRT1–AMPK signalling115. Thus, SIRT1 may counteract ageing by maintaining mitochondrial integrity and inhibiting apoptosis through tight regulation of several major cell fate-determining proteins. Importantly, hyperactivation of the DNA damage response may inhibit SIRT1 activity through the consumption of NAD+, even though SIRT1 promotes genomic stability through participation in DNA repair, as discussed above.
Collectively, autophagy and mitophagy are key regulators of mitochondrial homeostasis, and defective mitophagy may accelerate the ageing process and the development of age-related neurodegenerative diseases. Enhancement of autophagy and mitophagy may therefore represent a therapeutic strategy for these diseases.
Strategies for improving healthspan
Recent findings demonstrate the possibility of interventions in ageing that will reduce or delay morbidity and increase healthspan116. The NM signalling network is emerging as a promising target for improving healthspan.
Enhancing the NAD+–SIRT1 pathway
The NAD+–SIRT1 pathway is an important target for intervention. SIRT1 activity can be enhanced by increasing NAD+ levels or by the use of SIRT1 activators. One way to increase NAD+ levels is to reduce NAD+ consumption by, for example, using inhibitors of PARPs (such as olaparib and veliparib (FIG. 5a)) or of other NAD+-consuming enzymes such as CD38 (REFS 12,16). As PARP1 is a key protein in DNA repair, and because its inhibition may lead to genomic instability, it is necessary to proceed cautiously with such long-term intervention in humans. As CD38 participates in several cellular functions (such as immunity, signal transduction and calcium signalling), and even though inhibition of CD38 may increase SIRT1 activity, the side-effects of CD38 inhibition are unclear and thus warrant caution when applying this strategy in humans27.
Figure 5. Pharmacological interventions in the DNA damage response that may lead to increases in lifespan and healthspan.

a | Inhibition of poly(ADP-ribose) polymerase 1 (PARP1) by olaparib and other PARP inhibitors has been shown to increase lifespan in model organisms. b | Impaired cellular metabolism can be counteracted by treatment with NAD+ precursors such as nicotinamide riboside (NR), by activation of NAD+-generating enzymes such as nicotinamide phosphoribosyltransferase by P7C3, or by replenishment of acetyl-CoA levels with ketones such as β-hydroxybutyrate (βOHB). c | Signalling factors and targets in the DNA damage response (DDR) may constitute additional targets for interventions. These include activation of sirtuin 1 by compounds such as SRT1720, or AMP-activated kinase (AMPK) activation using the AMP analogue AICAR. d | Mitochondrial function may be augmented by stimulation of autophagy by rapamycin or its newer analogues, or by stimulation of autophagy through alternative pathways using compounds such as spermidine.
NAD+ precursors such as nicotinamide riboside and nicotinamide mononucleotide (NMN) also increase SIRT1 activity and have potential as anti-ageing drugs (FIG. 5b). In C. elegans, NAD+ levels are decreased during ageing, in part, by age-associated induction of NAD+-consuming PARP1. Consistently, supplementation with nicotinamide riboside increased worm lifespan in a SIR2.1-dependent manner12,16. In 22-month-old mice, 7-day supplementation with NMN significantly rescued mitochondrial dysfunction and normalized the mitochondrial parameters to those of young mice46. Nicotinamide riboside was also reported to be beneficial in both C. elegans and mouse models of XPA and Cockayne syndrome, which exhibit impaired NM signalling12,35. Encouraged by these data, several nicotinamide riboside clinical trials are underway117–120. Intriguingly, the P7C3 class of aminopropyl carbazole chemicals, which increase cellular NAD+ levels by enhancing the activity of the NAD+- generating enzyme nicotinamide phosphoribosyl-transferase (NAMPT), show strong neuroprotective activity121. Furthermore, NAMPT levels and SIRT1 activity are increased by treatment with AMPK activators such as 5-aminoimidazole-4-carboxamide-1-β-D-riboside (AICAR)45 (FIG. 5c).
A group of small molecules called SIRT1-activating compounds (STACs) may also have some health benefits. STACs are either natural or synthetic small bio-active compounds, which allosterically activate SIRT1 by binding to the SIRT1-activation domain (amino acids 190–240)122,123. Some STACs, such as the natural polyphenol resveratrol and its synthetic analogues SRT2104 and SRT1720 (FIG. 5c), can extend healthspan and lifespan in mice, and some human studies have shown protective effects of resveratrol and SRT2104 (REFS 123–125). Ongoing laboratory and clinical studies to determine the long-term safety and clinical applicability of STACs are necessary before their potential as anti-ageing agents can be explored. Collectively, the development of NAD+–SIRT1 pathway pharmaceuticals is highly desirable.
Ketones
Ketones (or ketone bodies) are a group of molecules produced through fatty acid oxidation in the liver by the conjugation of two acetyl-CoA molecules. Importantly, ketogenesis is induced by fasting, which is a well-characterized pro-longevity intervention. Ketones enter the blood circulation and are transported into the brain and other organs, where they are used as substrates for energy production. Notably, ketones can rescue features of accelerated ageing in Cockayne syndrome and can attenuate neurodegeneration in models of ALS, Parkinson disease, Huntington disease and Alzheimer disease86,126. Indeed, a recent synthetic ketone ester was developed that ameliorated behavioural deficits and amyloid pathology in a mouse model of Alzheimer disease, thereby providing a new pharmacological approach for tackling age-associated neurodegenerative diseases126. Furthermore, the ketone body D-β-hydroxybutyrate (β-OHB) (FIG. 5b) is a histone deacetylase inhibitor that was shown to extend lifespan in C. elegans, possibly through activating the SIR-2.1, AMPK, DAF-16–FOXO and SKN-1–NRF pathways127. The potential health benefits of β-OHB may be at least partially attributed to its ability to suppress oxidative stress by mediating increased histone acetylation at the FOXO3A and metallothionein 2 (MT2) promoters128. Although still speculative, bio-energetic interventions could thus be a way to decrease age-associated genome instability.
Inducing mitophagy
Inducing autophagy and mitophagy to diminish the load of damaged proteins and mitochondria, and to increase cell survival, may ameliorate age-related diseases accompanied by mitochondrial dysfunction. Common features of Alzheimer disease, Parkinson disease and Huntington disease include mitochondrial dysfunction, impaired autophagy and neuronal death13,92,129. Accumulation of damaged mitochondria may induce neuronal death in Parkinson disease, and stimulation of autophagy can ameliorate disease pathology in laboratory Parkinson disease models13,92. In addition, defective mitophagy has been suggested as a trigger of disease progression, especially neurodegeneration, in some premature ageing diseases including XPA, Cockayne syndrome and ataxia telangi-ectasia12,35,80. In C. elegans and mouse models of XPA and Cockayne syndrome, increased mitophagy through restoration of the NAD+–SIRT1 (SIR2.1) pathway improves healthspan12,35.
Another major strategy to enhance autophagy and mitophagy is to inhibit mTORC1, which is a negative regulator of autophagy (FIG. 5d), using compounds such as rapamycin and associated rapamycin esters. Indeed, rapamycin can increase mouse lifespan and rejuvenate the ageing heart, and it is beneficial when administrated in mouse models of Alzheimer disease, Parkinson disease, Huntington disease and Hutchinson–Gilford progeria; thus, it is being considered as an anti-ageing drug92,130,131. However, potential toxicity of rapamycin and its analogues have been noticed132, raising the need for discovery of new mTORC1-inhibiting compounds with fewer side-effects. New autophagy-inducing compounds that may function independently of mTORC1 have been identified, including spermidine (FIG. 5d) and some US Food and Drug Administration-approved drugs such as L-type Ca2+-channel antagonists and imidazoline receptor agonists133,134. Clearly, the next generation of autophagy stimulators could be promising drugs for various age-associated diseases.
Open questions and future challenges
Impairment of DNA damage-mediated NM signalling can cause mitochondrial dysfunction, which is linked to ageing and various age-associated pathologies. Our understanding of the mechanisms of mitochondrial maintenance has greatly expanded over the decades, implicating both mitochondrial processes and signalling between mitochondria and other cellular compartments, especially the nucleus. Because of their central physiological role, mitochondria continue to be a focus of investigations in a great number of diseases and ageing.
In light of the importance of DNA damage-induced NM signalling in mitochondrial maintenance, genomic stability and longevity, associated pathways are of interest. For instance, modulation of DNA damage-induced NM signalling can activate UPRmt, which is a signalling pathway that contributes to longevity16,24–27. Inhibition of the DNA repair signalling protein PARP1 seems to extend lifespan in C. elegans through activation of UPRmt (REF. 16). There may be inducers of UPRmt other than DNA damage, and other ways to coordinate NM signalling with signalling from mitochondria to the nucleus. A related issue is the linkage between telomere maintenance and NM signalling. Telomere attrition may induce mitochondrial dysfunction through the p53–PGC1α axis135. Some telomeric proteins, including telomerase reverse transcriptase and TRF1-interacting nuclear protein 2 (TIN2) may localize to both the nucleus and mitochondria and thereby link nuclear and mitochondrial events through DNA damage-induced NM signalling136,137.
Additional mechanistic investigations are needed to propose a unified role for PARP1–sirtuin crosstalk in ageing. It seems contradictory that inhibition of PARP1 extends lifespan in both wild-type and disease animal models12,16, but that high PARP1 activity may be associated with longevity138,139. We hope that future studies will uncouple the conceptual link between PARP1-induced PARylation and high levels of DNA repair activity. High levels of PARP1-induced PARylation will facilitate DNA repair when the latter is proficient, but it will over-consume NAD+ without enhancing lesion repair when DNA repair pathways are compromised. PARylation is regulated in a dynamic manner and can also be reversed by poly(ADP-ribose) glycohydrolase138. Thus, PARylation levels do not necessarily correlate with PARP activity. However, the sirtuin family of NAD+-dependent deacetylase and deacylase enzymes affect both intra-mitochondrial and NM signalling mechanisms. Although SIRT1 and SIRT6 both influence longevity, genomic stability and normal mitochondrial function, they may differ in their relationship with PARP1. SIRT6 is an upstream activator of PARP1, whereas SIRT1 and PARP1 can mutually affect the activity of one another in response to a range of stimuli, including genotoxic, metabolic and circadian changes140. In addition, further mechanistic studies of DNA repair proteins that have nuclear and mitochondrial localization (BOX 1) are needed to extend our understanding of DNA damage-induced NM signalling.
Novel strategies to target different NM pathways and mitochondrial maintenance, such as sustaining energy metabolism, upregulating mitophagy and regulating DNA repair, should be explored in depth in experimental models with a view towards intervention in humans. For compounds targeting the NM pathways mentioned here (FIG. 5), some major questions remain. Do these compounds cross the blood–brain barrier? What are their optimal pharmacokinetic conditions? So far, indications are that certain compounds, such as nicotinamide riboside and ketones, may have little or no toxicity in humans, but this requires further exploration. This research area is ripe for development, and new mechanistic insights and intervention strategies will probably soon emerge from ongoing research.
Acknowledgments
The authors acknowledge the valuable work of the many investigators whose published articles they were unable to cite owing to space limitations. They thank Prabhat Khadka and Anne Tseng for critical reading of the manuscript. This research was supported entirely by the Intramural Research Program of the US National Institutes of Health (NIH) National Institute on Ageing (NIA). K.F.C. was supported by the Department of Veterans Affairs (Merit Award), research awards from the Glenn Foundation for Medical Research and the NIH/NIA (R56AG050997).
Glossary
- Cockayne syndrome
A rare accelerated-ageing disease with progressive neurodegeneration, caused by mutations in genes encoding two DNA repair proteins, CSA and CSB
- Xeroderma pigmentosum
A rare autosomal-recessive disorder characterized by severe sun sensitivity and skin cancer, associated with mutation of genes encoding a group of DNA repair proteins, XPA to XPG
- Ataxia telangiectasia
A genomic instability disease with progressive cerebellar neurodegeneration caused by mutation of the ataxia telangiectasia mutated (ATM) gene, encoding the kinase ATM, which is a master regulator of DNA damage processing
- Preiss–Handler pathway
A NAD+ biosynthetic process that consumes dietary nicotinic acid
- Salvage pathway
A NAD+ biosynthetic pathway that uses nicotinamide to generate nicotinamide mononucleotide (NMN), which is then transformed into NAD+
- Reactive oxygen species (ROS)
By-products of cellular metabolism, which at low levels provide health benefits, whereas at high levels they become increasingly noxious, with broad pathological consequences
- Ataxia telangiectasia mutated (ATM)
A 350 kDa Ser/Thr protein kinase that is required for activation of the DNA damage response to double-strand breaks through phosphorylation of >700 downstream DNA repair proteins
- Hepatosteatosis
Also known as hepatic steatosis (fatty liver). A common liver abnormality, in which patients have excessive accumulation of triglycerides (lipid droplets) in the liver
- Apurinic and apyrimidinic sites
(AP sites; also known as abasic sites). Sites of DNA sugar without a base, which is a common DNA lesion and is typically repaired by DNA base excision repair through sugar cleavage by AP endonuclease 1 (APE1)
- 8-oxo-dGuo
(8-oxo-7,8-dihydro-2′-deoxyguanosine). An oxidative DNA lesion, which can be repaired by DNA base excision repair
- NAD+/NADH ratio
NAD exists in cells in both oxidized (NAD+) and reduced (NADH) forms. The ratio of NAD+ and NADH regulates many cellular processes, including energy metabolism and mitochondrial functions
- Ketogenic diet
A diet that is high-fat, high-protein and low-carbohydrate
- Stress response
A cellular response to certain types of stress, such as caloric restriction or increase in reactive oxygen species, in which different stress- counteracting pathways are upregulated
- Autophagosome
A key structure of autophagy, an autophagosome is a spherical, double-membrane vesicle that sequesters cytoplasmic contents for degradation
- Rapamycin
A natural metabolite from the bacterium Streptomyces hygroscopicus, which inhibits mTOR and extends lifespan in species from yeast to fruit flies and mice
Footnotes
Competing interests statement
The authors declare competing interests: see Web version for details.
DATABASES
Mito DB (The mitochondrial disease database): http://www.mitodb.com/
ALL LINKS ARE ACTIVE IN THE ONLINE PDF
References
- 1.West AP, et al. Mitochondrial DNA stress primes the antiviral innate immune response. Nature. 2015;520:553–557. doi: 10.1038/nature14156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wallace DC. Mitochondrial DNA variation in human radiation and disease. Cell. 2015;163:33–38. doi: 10.1016/j.cell.2015.08.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Scheibye-Knudsen M, et al. Protecting the mitochondrial powerhouse. Trends Cell Biol. 2014;25:158–170. doi: 10.1016/j.tcb.2014.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Maynard S, Fang EF, Scheibye-Knudsen M, Croteau DL, Bohr VA. DNA damage, DNA repair, aging, and neurodegeneration. Cold Spring Harb Perspect Med. 2015;5:a025130. doi: 10.1101/cshperspect.a025130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Youle RJ, van der Bliek AM. Mitochondrial fission, fusion, and stress. Science. 2012;337:1062–1065. doi: 10.1126/science.1219855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Randow F, Youle RJ. Self and nonself: how autophagy targets mitochondria and bacteria. Cell Host Microbe. 2014;15:403–411. doi: 10.1016/j.chom.2014.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wallace DC. Mitochondria and cancer. Nat Rev Cancer. 2012;12:685–698. doi: 10.1038/nrc3365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Coskun P, et al. A mitochondrial etiology of Alzheimer and Parkinson disease. Biochim Biophys Acta. 2012;1820:553–564. doi: 10.1016/j.bbagen.2011.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mattson MP. Pathways towards and away from Alzheimer’s disease. Nature. 2004;430:631–639. doi: 10.1038/nature02621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wallace DC. Mitochondrial diseases in man and mouse. Science. 1999;283:1482–1488. doi: 10.1126/science.283.5407.1482. [DOI] [PubMed] [Google Scholar]
- 11.Palikaras K, Lionaki E, Tavernarakis N. Coordination of mitophagy and mitochondrial biogenesis during ageing in C. elegans. Nature. 2015;521:525–528. doi: 10.1038/nature14300. Provides evidence for a pivotal role of mitophagy in healthspan and lifespan in C. elegans. [DOI] [PubMed] [Google Scholar]
- 12.Fang EF, et al. Defective mitophagy in XPA via PARP-1 hyperactivation and NAD+/SIRT1 reduction. Cell. 2014;157:882–896. doi: 10.1016/j.cell.2014.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Menzies FM, Fleming A, Rubinsztein DC. Compromised autophagy and neurodegenerative diseases. Nat Rev Neurosci. 2015;16:345–357. doi: 10.1038/nrn3961. [DOI] [PubMed] [Google Scholar]
- 14.Petersen KF, et al. Mitochondrial dysfunction in the elderly: possible role in insulin resistance. Science. 2003;300:1140–1142. doi: 10.1126/science.1082889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Conley KE, Jubrias SA, Esselman PC. Oxidative capacity and ageing in human muscle. J Physiol. 2000;526:203–210. doi: 10.1111/j.1469-7793.2000.t01-1-00203.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mouchiroud L, et al. The NAD+/sirtuin pathway modulates longevity through activation of mitochondrial UPR and FOXO signaling. Cell. 2013;154:430–441. doi: 10.1016/j.cell.2013.06.016. Shows that the UPRmt contributes to SIR2.1-related longevity in C. elegans. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hey-Mogensen M, et al. A novel method for determining human ex vivo submaximal skeletal muscle mitochondrial function. J Physiol. 2015;593:3991–4010. doi: 10.1113/JP270204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Capel F, et al. Due to reverse electron transfer, mitochondrial H2O2 release increases with age in human vastus lateralis muscle although oxidative capacity is preserved. Mech Ageing Dev. 2005;126:505–511. doi: 10.1016/j.mad.2004.11.001. [DOI] [PubMed] [Google Scholar]
- 19.Maynard S, et al. Relationships between human vitality and mitochondrial respiratory parameters, reactive oxygen species production and dNTP levels in peripheral blood mononuclear cells. Aging. 2013;5:850–864. doi: 10.18632/aging.100618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013;153:1194–1217. doi: 10.1016/j.cell.2013.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Scheibye-Knudsen M, et al. Cockayne syndrome group B protein prevents the accumulation of damaged mitochondria by promoting mitochondrial autophagy. J Exp Med. 2012;209:855–869. doi: 10.1084/jem.20111721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Scheibye-Knudsen M, Fang EF, Croteau DL, Bohr VA. Contribution of defective mitophagy to the neurodegeneration in DNA repair-deficient disorders. Autophagy. 2014;10:1468–1469. doi: 10.4161/auto.29321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shiloh Y, Ziv Y. The ATM protein kinase: regulating the cellular response to genotoxic stress, and more. Nat Rev Mol Cell Biol. 2013;14:197–210. [PubMed] [Google Scholar]
- 24.Houtkooper RH, et al. Mitonuclear protein imbalance as a conserved longevity mechanism. Nature. 2013;497:451–457. doi: 10.1038/nature12188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mohrin M, et al. A mitochondrial UPR-mediated metabolic checkpoint regulates hematopoietic stem cell aging. Science. 2015;347:1374–1377. doi: 10.1126/science.aaa2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pellegrino MW, Nargund AM, Haynes CM. Signaling the mitochondrial unfolded protein response. Biochim Biophys Acta. 2013;1833:410–416. doi: 10.1016/j.bbamcr.2012.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Canto C, Menzies KJ, Auwerx J. NAD+ metabolism and the control of energy homeostasis: a balancing act between mitochondria and the nucleus. Cell Metab. 2015;22:31–53. doi: 10.1016/j.cmet.2015.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Michikawa Y, Mazzucchelli F, Bresolin N, Scarlato G, Attardi G. Aging-dependent large accumulation of point mutations in the human mtDNA control region for replication. Science. 1999;286:774–779. doi: 10.1126/science.286.5440.774. [DOI] [PubMed] [Google Scholar]
- 29.Croteau DL, Popuri V, Opresko PL, Bohr VA. Human RecQ helicases in DNA repair, recombination, and replication. Annu Rev Biochem. 2014;83:519–552. doi: 10.1146/annurev-biochem-060713-035428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang H, et al. Nutrient-sensitive mitochondrial NAD+ levels dictate cell survival. Cell. 2007;130:1095–1107. doi: 10.1016/j.cell.2007.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Verdin E. NAD+ in aging, metabolism, and neurodegeneration. Science. 2015;350:1208–1213. doi: 10.1126/science.aac4854. [DOI] [PubMed] [Google Scholar]
- 32.Rodgers JT, et al. Nutrient control of glucose homeostasis through a complex of PGC-1α and SIRT1. Nature. 2005;434:113–118. doi: 10.1038/nature03354. [DOI] [PubMed] [Google Scholar]
- 33.Chalkiadaki A, Guarente L. The multifaceted functions of sirtuins in cancer. Nat Rev Cancer. 2015;15:608–624. doi: 10.1038/nrc3985. [DOI] [PubMed] [Google Scholar]
- 34.Imai S, Armstrong CM, Kaeberlein M, Guarente L. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature. 2000;403:795–800. doi: 10.1038/35001622. The first evidence linking NAD+ to a sirtuin. [DOI] [PubMed] [Google Scholar]
- 35.Scheibye-Knudsen M, et al. A high fat diet and NAD+ rescue premature aging in Cockayne syndrome. Cell Metab. 2014;20:840–855. doi: 10.1016/j.cmet.2014.10.005.. Together with reference 12, establishes a causative link from DNA damage to mitochondrial dysfunction and premature ageing.
- 36.Gibson BA, Kraus WL. New insights into the molecular and cellular functions of poly(ADP-ribose) and PARPs. Nat Rev Mol Cell Biol. 2012;13:411–424. doi: 10.1038/nrm3376. [DOI] [PubMed] [Google Scholar]
- 37.Eliasson MJ, et al. Poly(ADP-ribose) polymerase gene disruption renders mice resistant to cerebral ischemia. Nat Med. 1997;3:1089–1095. doi: 10.1038/nm1097-1089. [DOI] [PubMed] [Google Scholar]
- 38.Virag L, Szabo C. The therapeutic potential of poly(ADP-ribose) polymerase inhibitors. Pharmacol Rev. 2002;54:375–429. doi: 10.1124/pr.54.3.375. [DOI] [PubMed] [Google Scholar]
- 39.Liu D, Gharavi R, Pitta M, Gleichmann M, Mattson MP. Nicotinamide prevents NAD+ depletion and protects neurons against excitotoxicity and cerebral ischemia: NAD+ consumption by SIRT1 may endanger energetically compromised neurons. Neuromolecular Med. 2009;11:28–42. doi: 10.1007/s12017-009-8058-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pacher P, Szabo C. Role of the peroxynitrite-poly(ADP-ribose) polymerase pathway in human disease. Am J Pathol. 2008;173:2–13. doi: 10.2353/ajpath.2008.080019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bai P, et al. PARP-1 inhibition increases mitochondrial metabolism through SIRT1 activation. Cell Metab. 2011;13:461–468. doi: 10.1016/j.cmet.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.DiGiovanna JJ, Kraemer KH. Shining a light on xeroderma pigmentosum. J Invest Dermatol. 2012;132:785–796. doi: 10.1038/jid.2011.426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cleaver JE. Defective repair replication of DNA in xeroderma pigmentosum. Nature. 1968;218:652–656. doi: 10.1038/218652a0. [DOI] [PubMed] [Google Scholar]
- 44.Lindenbaum Y, et al. Xeroderma pigmentosum/ Cockayne syndrome complex: first neuropathological study and review of eight other cases. Eur J Paediatr Neurol. 2001;5:225–242. doi: 10.1053/ejpn.2001.0523. [DOI] [PubMed] [Google Scholar]
- 45.Canto C, et al. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature. 2009;458:1056–1060. doi: 10.1038/nature07813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gomes AP, et al. Declining NAD+ induces a pseudohypoxic state disrupting nuclear-mitochondrial communication during aging. Cell. 2013;155:1624–1638. doi: 10.1016/j.cell.2013.11.037. Demonstrates a role for NAD+ in the regulation of nuclear–mitochondrial communication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dobbin MM, et al. SIRT1 collaborates with ATM and HDAC1 to maintain genomic stability in neurons. Nat Neurosci. 2013;16:1008–1015. doi: 10.1038/nn.3460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cohen HY, et al. Calorie restriction promotes mammalian cell survival by inducing the SIRT1 deacetylase. Science. 2004;305:390–392. doi: 10.1126/science.1099196. [DOI] [PubMed] [Google Scholar]
- 49.Li K, et al. Regulation of WRN protein cellular localization and enzymatic activities by SIRT1-mediated deacetylation. J Biol Chem. 2008;283:7590–7598. doi: 10.1074/jbc.M709707200. [DOI] [PubMed] [Google Scholar]
- 50.Uhl M, et al. Role of SIRT1 in homologous recombination. DNA Repair. 2010;9:383–393. doi: 10.1016/j.dnarep.2009.12.020. [DOI] [PubMed] [Google Scholar]
- 51.Yamamori T, et al. SIRT1 deacetylates APE1 and regulates cellular base excision repair. Nucleic Acids Res. 2010;38:832–845. doi: 10.1093/nar/gkp1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Madabushi A, Hwang BJ, Jin J, Lu AL. Histone deacetylase SIRT1 modulates and deacetylates DNA base excision repair enzyme thymine DNA glycosylase. Biochem J. 2013;456:89–98. doi: 10.1042/BJ20130670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fan W, Luo J. SIRT1 regulates UV-induced DNA repair through deacetylating XPA. Mol Cell. 2010;39:247–258. doi: 10.1016/j.molcel.2010.07.006. [DOI] [PubMed] [Google Scholar]
- 54.Kanfi Y, et al. The sirtuin SIRT6 regulates lifespan in male mice. Nature. 2012;483:218–221. doi: 10.1038/nature10815. [DOI] [PubMed] [Google Scholar]
- 55.Mostoslavsky R, et al. Genomic instability and aging-like phenotype in the absence of mammalian SIRT6. Cell. 2006;124:315–329. doi: 10.1016/j.cell.2005.11.044. [DOI] [PubMed] [Google Scholar]
- 56.Sebastian C, et al. The histone deacetylase SIRT6 is a tumor suppressor that controls cancer metabolism. Cell. 2012;151:1185–1199. doi: 10.1016/j.cell.2012.10.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liu GH, et al. Recapitulation of premature ageing with iPSCs from Hutchinson–Gilford progeria syndrome. Nature. 2011;472:221–225. doi: 10.1038/nature09879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ghosh S, Liu B, Wang Y, Hao Q, Zhou Z. Lamin A is an endogenous SIRT6 activator and promotes SIRT6-mediated DNA repair. Cell Rep. 2015;13:1396–1406. doi: 10.1016/j.celrep.2015.10.006. [DOI] [PubMed] [Google Scholar]
- 59.Zhong L, et al. The histone deacetylase Sirt6 regulates glucose homeostasis via Hif1α. Cell. 2010;140:280–293. doi: 10.1016/j.cell.2009.12.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kaidi A, Weinert BT, Choudhary C, Jackson SP. Human SIRT6 promotes DNA end resection through CtIP deacetylation. Science. 2010;329:1348–1353. doi: 10.1126/science.1192049. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 61.Mao Z, et al. SIRT6 promotes DNA repair under stress by activating PARP1. Science. 2011;332:1443–1446. doi: 10.1126/science.1202723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Michishita E, et al. SIRT6 is a histone H3 lysine 9 deacetylase that modulates telomeric chromatin. Nature. 2008;452:492–496. doi: 10.1038/nature06736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.McCord RA, et al. SIRT6 stabilizes DNA-dependent protein kinase at chromatin for DNA double-strand break repair. Aging. 2009;1:109–121. doi: 10.18632/aging.100011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Toiber D, et al. SIRT6 recruits SNF2H to DNA break sites, preventing genomic instability through chromatin remodeling. Mol Cell. 2013;51:454–468. doi: 10.1016/j.molcel.2013.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Van Meter M, et al. SIRT6 represses LINE1 retrotransposons by ribosylating KAP1 but this repression fails with stress and age. Nat Commun. 2014;5:5011. doi: 10.1038/ncomms6011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Barber MF, et al. SIRT7 links H3K18 deacetylation to maintenance of oncogenic transformation. Nature. 2012;487:114–118. doi: 10.1038/nature11043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Shin J, et al. SIRT7 represses Myc activity to suppress ER stress and prevent fatty liver disease. Cell Rep. 2013;5:654–665. doi: 10.1016/j.celrep.2013.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ryu D, et al. A SIRT7-dependent acetylation switch of GABPβ1 controls mitochondrial function. Cell Metab. 2014;20:856–869. doi: 10.1016/j.cmet.2014.08.001. Provides evidence that SIRT7 regulates mitochondrial function. [DOI] [PubMed] [Google Scholar]
- 69.Someya S, et al. Sirt3 mediates reduction of oxidative damage and prevention of age-related hearing loss under caloric restriction. Cell. 2010;143:802–812. doi: 10.1016/j.cell.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Brown KD, et al. Activation of SIRT3 by the NAD+ precursor nicotinamide riboside protects from noise-induced hearing loss. Cell Metab. 2014;20:1059–1068. doi: 10.1016/j.cmet.2014.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Cheng A, et al. Mitochondrial SIRT3 mediates adaptive responses of neurons to exercise and metabolic and excitatory challenges. Cell Metab. 2016;23:128–142. doi: 10.1016/j.cmet.2015.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hirschey MD, et al. SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature. 2010;464:121–125. doi: 10.1038/nature08778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jeong SM, et al. SIRT4 has tumor-suppressive activity and regulates the cellular metabolic response to DNA damage by inhibiting mitochondrial glutamine metabolism. Cancer Cell. 2013;23:450–463. doi: 10.1016/j.ccr.2013.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kim HS, et al. Hepatic-specific disruption of SIRT6 in mice results in fatty liver formation due to enhanced glycolysis and triglyceride synthesis. Cell Metab. 2010;12:224–236. doi: 10.1016/j.cmet.2010.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Malik S, et al. SIRT7 inactivation reverses metastatic phenotypes in epithelial and mesenchymal tumors. Sci Rep. 2015;5:9841. doi: 10.1038/srep09841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lee N, et al. Comparative interactomes of SIRT6 and SIRT7: implication of functional links to aging. Proteomics. 2014;14:1610–1622. doi: 10.1002/pmic.201400001. [DOI] [PubMed] [Google Scholar]
- 77.Woods CG, Taylor AM. Ataxia telangiectasia in the British Isles: the clinical and laboratory features of 70 affected individuals. Q J Med. 1992;82:169–179. [PubMed] [Google Scholar]
- 78.Merideth MA, et al. Phenotype and course of Hutchinson-Gilford progeria syndrome. N Engl J Med. 2008;358:592–604. doi: 10.1056/NEJMoa0706898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Dahl AK, et al. Body mass index, change in body mass index, and survival in old and very old persons. J Am Geriatr Soc. 2013;61:512–518. doi: 10.1111/jgs.12158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Valentin-Vega YA, et al. Mitochondrial dysfunction in ataxia-telangiectasia. Blood. 2012;119:1490–1500. doi: 10.1182/blood-2011-08-373639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Williamson DH, Lund P, Krebs HA. The redox state of free nicotinamide-adenine dinucleotide in the cytoplasm and mitochondria of rat liver. Biochem J. 1967;103:514–527. doi: 10.1042/bj1030514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ross JM, et al. High brain lactate is a hallmark of aging and caused by a shift in the lactate dehydrogenase A/B ratio. Proc Natl Acad Sci USA. 2010;107:20087–20092. doi: 10.1073/pnas.1008189107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sutendra G, et al. A nuclear pyruvate dehydrogenase complex is important for the generation of acetyl-CoA and histone acetylation. Cell. 2014;158:84–97. doi: 10.1016/j.cell.2014.04.046. [DOI] [PubMed] [Google Scholar]
- 84.Mone MJ, et al. Local UV-induced DNA damage in cell nuclei results in local transcription inhibition. EMBO Rep. 2001;2:1013–1017. doi: 10.1093/embo-reports/kve224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Shanbhag NM, Rafalska-Metcalf IU, Balane-Bolivar C, Janicki SM, Greenberg RA. ATM-dependent chromatin changes silence transcription in cis to DNA double-strand breaks. Cell. 2010;141:970–981. doi: 10.1016/j.cell.2010.04.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Longo VD, Mattson MP. Fasting: molecular mechanisms and clinical applications. Cell Metab. 2014;19:181–192. doi: 10.1016/j.cmet.2013.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Tripathi DN, et al. Reactive nitrogen species regulate autophagy through ATM-AMPK-TSC2-mediated suppression of mTORC1. Proc Natl Acad Sci USA. 2013;110:E2950–E2957. doi: 10.1073/pnas.1307736110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Budanov AV, Karin M. p53 target genes sestrin1 and sestrin2 connect genotoxic stress and mTOR signaling. Cell. 2008;134:451–460. doi: 10.1016/j.cell.2008.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Egan DF, et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science. 2011;331:456–461. doi: 10.1126/science.1196371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Marino G, Niso-Santano M, Baehrecke EH, Kroemer G. Self-consumption: the interplay of autophagy and apoptosis. Nat Rev Mol Cell Biol. 2014;15:81–94. doi: 10.1038/nrm3735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Youle RJ, Strasser A. The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol. 2008;9:47–59. doi: 10.1038/nrm2308. [DOI] [PubMed] [Google Scholar]
- 92.Rubinsztein DC, Marino G, Kroemer G. Autophagy and aging. Cell. 2011;146:682–695. doi: 10.1016/j.cell.2011.07.030. [DOI] [PubMed] [Google Scholar]
- 93.Allen GF, Toth R, James J, Ganley IG. Loss of iron triggers PINK1/Parkin-independent mitophagy. EMBO Rep. 2013;14:1127–1135. doi: 10.1038/embor.2013.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Chu CT, et al. Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells. Nat Cell Biol. 2013;15:1197–1205. doi: 10.1038/ncb2837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lazarou M, et al. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature. 2015;524:309–314. doi: 10.1038/nature14893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Pickrell AM, Youle RJ. The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron. 2015;85:257–273. doi: 10.1016/j.neuron.2014.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Biton S, Ashkenazi A. NEMO and RIP1 control cell fate in response to extensive DNA damage via TNF-α feedforward signaling. Cell. 2011;145:92–103. doi: 10.1016/j.cell.2011.02.023. [DOI] [PubMed] [Google Scholar]
- 98.Picco V, Pages G. Linking JNK activity to the DNA damage response. Genes Cancer. 2013;4:360–368. doi: 10.1177/1947601913486347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Luo S, et al. Bim inhibits autophagy by recruiting Beclin 1 to microtubules. Mol Cell. 2012;47:359–370. doi: 10.1016/j.molcel.2012.05.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Maryanovich M, et al. The ATM-BID pathway regulates quiescence and survival of haematopoietic stem cells. Nat Cell Biol. 2012;14:535–541. doi: 10.1038/ncb2468. [DOI] [PubMed] [Google Scholar]
- 101.Mercer JR, et al. DNA damage links mitochondrial dysfunction to atherosclerosis and the metabolic syndrome. Circ Res. 2010;107:1021–1031. doi: 10.1161/CIRCRESAHA.110.218966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Scheibye-Knudsen M, Scheibye-Alsing K, Canugovi C, Croteau DL, Bohr VA. A novel diagnostic tool reveals mitochondrial pathology in human diseases and aging. Aging. 2013;5:192–208. doi: 10.18632/aging.100546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Oda K, et al. p53AIP1, a potential mediator of p53-dependent apoptosis, and its regulation by Ser-46-phosphorylated p53. Cell. 2000;102:849–862. doi: 10.1016/s0092-8674(00)00073-8. [DOI] [PubMed] [Google Scholar]
- 104.Murray-Zmijewski F, Slee EA, Lu X. A complex barcode underlies the heterogeneous response of p53 to stress. Nat Rev Mol Cell Biol. 2008;9:702–712. doi: 10.1038/nrm2451. [DOI] [PubMed] [Google Scholar]
- 105.Hoshino A, et al. Cytosolic p53 inhibits Parkin-mediated mitophagy and promotes mitochondrial dysfunction in the mouse heart. Nat Commun. 2013;4:2308. doi: 10.1038/ncomms3308. [DOI] [PubMed] [Google Scholar]
- 106.Crighton D, et al. DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell. 2006;126:121–134. doi: 10.1016/j.cell.2006.05.034. [DOI] [PubMed] [Google Scholar]
- 107.Xie X, Le L, Fan Y, Lv L, Zhang J. Autophagy is induced through the ROS-TP53-DRAM1 pathway in response to mitochondrial protein synthesis inhibition. Autophagy. 2012;8:1071–1084. doi: 10.4161/auto.20250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Poyurovsky MV, Prives C. P53 and aging: a fresh look at an old paradigm. Aging. 2010;2:380–382. doi: 10.18632/aging.100179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Matheu A, et al. Delayed ageing through damage protection by the Arf/p53 pathway. Nature. 2007;448:375–379. doi: 10.1038/nature05949. [DOI] [PubMed] [Google Scholar]
- 110.Pinkston JM, Garigan D, Hansen M, Kenyon C. Mutations that increase the life span of C elegans inhibit tumor growth. Science. 2006;313:971–975. doi: 10.1126/science.1121908. [DOI] [PubMed] [Google Scholar]
- 111.Vaziri H, et al. hSIR2SIRT1 functions as an NAD-dependent p53 deacetylase. Cell. 2001;107:149–159. doi: 10.1016/s0092-8674(01)00527-x. [DOI] [PubMed] [Google Scholar]
- 112.Luo J, et al. Negative control of p53 by Sir2α promotes cell survival under stress. Cell. 2001;107:137–148. doi: 10.1016/s0092-8674(01)00524-4. [DOI] [PubMed] [Google Scholar]
- 113.Sampaio-Marques B, et al. SNCA (α-synuclein)-induced toxicity in yeast cells is dependent on sirtuin 2 (Sir2)-mediated mitophagy. Autophagy. 2012;8:1494–1509. doi: 10.4161/auto.21275. [DOI] [PubMed] [Google Scholar]
- 114.Huang R, et al. Deacetylation of nuclear LC3 drives autophagy initiation under starvation. Mol Cell. 2015;57:456–466. doi: 10.1016/j.molcel.2014.12.013. [DOI] [PubMed] [Google Scholar]
- 115.Price NL, et al. SIRT1 is required for AMPK activation and the beneficial effects of resveratrol on mitochondrial function. Cell Metab. 2012;15:675–690. doi: 10.1016/j.cmet.2012.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Longo VD, et al. Interventions to slow aging in humans: are we ready? Aging Cell. 2015;14:497–510. doi: 10.1111/acel.12338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.US National Library of Medicine. ClinicalTrials.gov. 2014 https://clinicaltrials.gov/ct2/show/NCT02300740.
- 118.US National Library of Medicine. ClinicalTrials.gov. 2014 https://clinicaltrials.gov/ct2/show/NCT02303483.
- 119.US National Library of Medicine. ClinicalTrials.gov. 2014 https://clinicaltrials.gov/ct2/show/NCT02191462.
- 120.US National Library of Medicine. ClinicalTrials.gov. 2016 https://clinicaltrials.gov/ct2/show/NCT02678611.
- 121.Wang G, et al. P7C3 neuroprotective chemicals function by activating the rate-limiting enzyme in NAD salvage. Cell. 2014;158:1324–1334. doi: 10.1016/j.cell.2014.07.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Hubbard BP, et al. Evidence for a common mechanism of SIRT1 regulation by allosteric activators. Science. 2013;339:1216–1219. doi: 10.1126/science.1231097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Sinclair DA, Guarente L. Small-molecule allosteric activators of sirtuins. Annu Rev Pharmacol Toxicol. 2014;54:363–380. doi: 10.1146/annurev-pharmtox-010611-134657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.van der Meer AJ, et al. The selective sirtuin 1 activator SRT2104 reduces endotoxin-induced cytokine release and coagulation activation in humans. Crit Care Med. 2015;43:e199–202. doi: 10.1097/CCM.0000000000000949. [DOI] [PubMed] [Google Scholar]
- 125.Timmers S, et al. Calorie restriction-like effects of 30 days of resveratrol supplementation on energy metabolism and metabolic profile in obese humans. Cell Metab. 2011;14:612–622. doi: 10.1016/j.cmet.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Kashiwaya Y, et al. A ketone ester diet exhibits anxiolytic and cognition-sparing properties, and lessens amyloid and tau pathologies in a mouse model of Alzheimer’s disease. Neurobiol Aging. 2013;34:1530–1539. doi: 10.1016/j.neurobiolaging.2012.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Edwards C, et al. D-β-hydroxybutyrate extends lifespan in C. elegans. Aging. 2014;6:621–644. doi: 10.18632/aging.100683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Shimazu T, et al. Suppression of oxidative stress by β-hydroxybutyrate, an endogenous histone deacetylase inhibitor. Science. 2013;339:211–214. doi: 10.1126/science.1227166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Ravikumar B, et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet. 2004;36:585–595. doi: 10.1038/ng1362. Provides the first evidence that compromised autophagy contributes to the pathology of the neurodegenerative disorder Huntington disease. [DOI] [PubMed] [Google Scholar]
- 130.Leslie M. A putative antiaging drug takes a step from mice to men. Science. 2013;342:789. doi: 10.1126/science.342.6160.789. [DOI] [PubMed] [Google Scholar]
- 131.Dai DF, et al. Altered proteome turnover and remodeling by short-term caloric restriction or rapamycin rejuvenate the aging heart. Aging Cell. 2014;13:529–539. doi: 10.1111/acel.12203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Soefje SA, Karnad A, Brenner AJ. Common toxicities of mammalian target of rapamycin inhibitors. Target Oncol. 2011;6:125–129. doi: 10.1007/s11523-011-0174-9. [DOI] [PubMed] [Google Scholar]
- 133.Eisenberg T, et al. Induction of autophagy by spermidine promotes longevity. Nat Cell Biol. 2009;11:1305–1314. doi: 10.1038/ncb1975. [DOI] [PubMed] [Google Scholar]
- 134.Pavel M, Rubinsztein DC. In: Antitumor Potential and Other Emerging Medicinal Properties of Natural Compounds. Fang EF, Ng TB, editors. Springer; 2013. pp. 227–238. [Google Scholar]
- 135.Sahin E, et al. Telomere dysfunction induces metabolic and mitochondrial compromise. Nature. 2011;470:359–365. doi: 10.1038/nature09787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Saretzki G. Extratelomeric functions of human telomerase: cancer, mitochondria and oxidative stress. Curr Pharm Des. 2014;20:6386–6403. doi: 10.2174/1381612820666140630095606. [DOI] [PubMed] [Google Scholar]
- 137.Chen LY, et al. Mitochondrial localization of telomeric protein TIN2 links telomere regulation to metabolic control. Mol Cell. 2012;47:839–850. doi: 10.1016/j.molcel.2012.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Mangerich A, Burkle A. Pleiotropic cellular functions of PARP1 in longevity and aging: genome maintenance meets inflammation. Oxid Med Cell Longev. 2012;2012:321653. doi: 10.1155/2012/321653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Grube K, Burkle A. Poly(ADP-ribose) polymerase activity in mononuclear leukocytes of 13 mammalian species correlates with species-specific life span. Proc Natl Acad Sci USA. 1992;89:11759–11763. doi: 10.1073/pnas.89.24.11759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Luna A, Aladjem MI, Kohn KW. SIRT1/PARP1 crosstalk: connecting DNA damage and metabolism. Genome Integr. 2013;4:6. doi: 10.1186/2041-9414-4-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Hoeijmakers JH. DNA damage, aging, and cancer. N Engl J Med. 2009;361:1475–1485. doi: 10.1056/NEJMra0804615. [DOI] [PubMed] [Google Scholar]
- 142.Cleaver JE, Lam ET, Revet I. Disorders of nucleotide excision repair: the genetic and molecular basis of heterogeneity. Nat Rev Genet. 2009;10:756–768. doi: 10.1038/nrg2663. [DOI] [PubMed] [Google Scholar]
- 143.Hanawalt PC, Spivak G. Transcription-coupled DNA repair: two decades of progress and surprises. Nat Rev Mol Cell Biol. 2008;9:958–970. doi: 10.1038/nrm2549. [DOI] [PubMed] [Google Scholar]
- 144.Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature. 2009;461:1071–1078. doi: 10.1038/nature08467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Lakshmipathy U, Campbell C. Double strand break rejoining by mammalian mitochondrial extracts. Nucleic Acids Res. 1999;27:1198–1204. doi: 10.1093/nar/27.4.1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Alexeyev M, Shokolenko I, Wilson G, LeDoux S. The maintenance of mitochondrial DNA integrity — critical analysis and update. Cold Spring Harb Perspect Biol. 2013;5:a012641. doi: 10.1101/cshperspect.a012641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Ding L, Liu Y. Borrowing nuclear DNA helicases to protect mitochondrial DNA. Int J Mol Sci. 2015;16:10870–10887. doi: 10.3390/ijms160510870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Croteau DL, et al. RECQL4 localizes to mitochondria and preserves mitochondrial DNA integrity. Aging Cell. 2012;11:456–466. doi: 10.1111/j.1474-9726.2012.00803.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Lin SJ, Defossez PA, Guarente L. Requirement of NAD and SIR2 for life-span extension by calorie restriction in Saccharomyces cerevisiae. Science. 2000;289:2126–2128. doi: 10.1126/science.289.5487.2126. [DOI] [PubMed] [Google Scholar]
- 150.Dang W, et al. Histone H4 lysine 16 acetylation regulates cellular lifespan. Nature. 2009;459:802–807. doi: 10.1038/nature08085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Viswanathan M, Guarente L. Regulation of Caenorhabditis elegans lifespan by sir-2.1 transgenes. Nature. 2011;477:E1–E2. doi: 10.1038/nature10440. [DOI] [PubMed] [Google Scholar]
- 152.Tissenbaum HA, Guarente L. Increased dosage of a sir-2 gene extends lifespan in Caenorhabditis elegans. Nature. 2001;410:227–230. doi: 10.1038/35065638. [DOI] [PubMed] [Google Scholar]
- 153.Burnett C, et al. Absence of effects of Sir2 overexpression on lifespan in C elegans and Drosophila. Nature. 2011;477:482–485. doi: 10.1038/nature10296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Oberdoerffer P, et al. SIRT1 redistribution on chromatin promotes genomic stability but alters gene expression during aging. Cell. 2008;135:907–918. doi: 10.1016/j.cell.2008.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Finley LW, Haigis MC. Metabolic regulation by SIRT3: implications for tumorigenesis. Trends Mol Med. 2012;18:516–523. doi: 10.1016/j.molmed.2012.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]