

Abstract
Recent studies have demonstrated that amides can be used in nickel-catalyzed reactions that lead to cleavage of the amide C–N bond, with formation of a C–C or C–heteroatom bond. However, the general scope of these methodologies has been restricted to amides where the carbonyl is directly attached to an arene or heteroarene. We now report the nickel-catalyzed esterification of amides derived from aliphatic carboxylic acids. The transformation requires only a slight excess of the alcohol nucleophile and is tolerant of heterocycles, substrates with epimerizable stereocenters, and sterically congested coupling partners. Moreover, a series of amide competition experiments establish selectivity principles that will aid future synthetic design. These studies overcome a critical limitation of current Ni-catalyzed amide couplings and are expected to further stimulate the use of amides as synthetic building blocks in C–N bond cleavage processes.
Keywords: nickel, catalysis, cross-coupling, aliphatic amides, esterification
Graphical Abstract
Recent studies on nickel-catalyzed activation of amides have been shown to forge C–C or –heteroatom bonds through cleavage on the amide C–N bond. We report the first nickel-catalyzed activation of amides derived from aliphatic carboxylic acids, thus overcoming the key limitation associated with nickel-catalyzed couplings of amides.
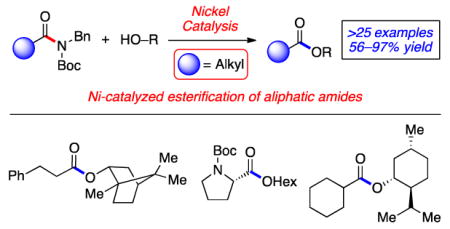
Amides are prevalent functional groups found in synthetic intermediates, natural products, proteins, and various other molecules of importance.[ 1 ] Accordingly, the development of methods to construct amide C–N bonds has been a popular topic of study for many decades.[1] In contrast, the ability to harness amides as synthetic building blocks in C–N bond cleavage reactions has remained underdeveloped.[ 2 ],[ 3 ] The modest synthetic utility of amides can be attributed to the strength of the amide C–N bond, which benefits from well-known resonance stabilization.[4]
With this longstanding challenge in mind, recent efforts have focused on the metal-catalyzed cleavage of amide C–N bonds, with applications in C–C and C–heteroatom bond formation.[ 5 ],[ 6 ],[ 7 ],[ 8 ],[ 9 ],[ 10 ] Breakthroughs in this area include palladium-catalyzed decarbonylative[6c] and non-decarbonylative[5],[6a],[6d],[6e] C–C bond formations using twisted amides or other activated amide substrates. Additionally, we have shown that nickel catalysis is effective to promote the non-decarbonylative cross-coupling of anilides and Ts- or Boc-activated amide derivatives (Figure 1).[7] In turn, esters,[7a] ketones,[7b],[c] or other amides[7d] can be readily prepared under mild reaction conditions and in a predictable manner, using non-precious metal catalysis.[ 11 ] Despite the promise of these methodologies, a notable drawback has been pervasive in nickel-mediated amide cross-coupling chemistry. Specifically, such methodologies have largely required that the amide substrate bear an aromatic (or heteroaromatic) ring attached to the carbonyl.[12] For amide activation methodologies to become generally useful, we sought to overcome this limitation and enable the activation and cross-coupling of amides derived from aliphatic carboxylic acids.
Figure 1.

Nickel-catalyzed activation of benzamides and derivatives (prior studies) and nickel-catalyzed esterification of aliphatic amides (present study); Boc=tert-butyloxycarbonyl, Ts=tosyl, Bn=benzyl.
Herein, we show that amides derived from aliphatic carboxylic acids can indeed undergo nickel-catalyzed cross-coupling, as demonstrated in the conversion of amides to esters (Figure 1, 1 → 2). These studies not only provide a facile means to convert amides to esters, which itself is a challenging transformation,[ 13 ] but should also greatly enable the use of amides as building blocks in chemical synthesis.[2]
Based on prior computational studies, oxidative addition was believed to be the rate-determining step of our proposed catalytic cycle.[14] We hypothesized that the choice of ligand in the conversion of aliphatic amides to esters could have a dramatic effect on the ease of oxidative addition.[7a] Thus, we performed an extensive survey of reaction parameters in collaboration with the Catalysis Group of Boehringer Ingelheim. The screening efforts, which involved the testing of over 100 ligands,[15] were ultimately fruitful and led us to focus on the use of pyridine-type ligands in optimization studies (Table 1). The challenging coupling of cyclohexyl amide 3 with (−)-menthol (4, 1.25 equiv), a sterically hindered nucleophile, was selected for these efforts. Although the use of bipyridine or phenanthroline led to no measureable yield of 5 (entries 1 and 2), the coupling product was obtained in 24% yield when terpyridine was employed (entry 3). Doubling the ligand loading was also attempted (entry 4), but 15 mol% (1:1 Ni to terpyridine) was found to be ideal. Additionally, increasing the equivalents of 4 did not lead to improvement (entry 5). However, it was found that concentration had an effect on the reaction (entries 6 and 7), with higher concentrations giving slightly increased yield of product 5. Finally, we found that increasing the temperature to 100 °C delivered ester 5 in 56% yield (entry 8).[16],[17] As this test transformation allowed for the coupling of two significantly hindered fragments with the remaining mass being only unreacted starting material, we opted to evaluate the scope of our methodology under these reaction conditions.
Table 1.
Optimization of reaction conditions.[a]
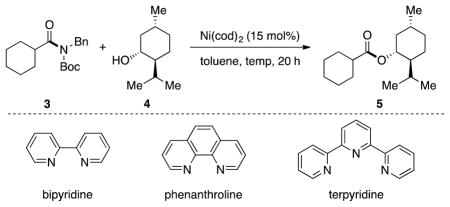
| |||||
|---|---|---|---|---|---|
| Entry | Ligand (loading) | Equivs of 4 | Concentration | Temp. | Yield |
| 1 | bipyridine (15 mol%) | 1.25 equiv | 0.5 M | 80 °C | 0% |
| 2 | phenanthroline (15 mol%) | 1.25 equiv | 0.5 M | 80 °C | 0% |
| 3 | terpyridine (15 mol%) | 1.25 equiv | 0.5 M | 80 °C | 24% |
| 4 | terpyridine (30 mol%) | 1.25 equiv | 0.5 M | 80 °C | 31% |
| 5 | terpyridine (15 mol%) | 2.5 equiv | 0.5 M | 80 °C | 30% |
| 6 | terpyridine (15 mol%) | 1.25 equiv | 0.33 M | 80 °C | 19% |
| 7 | terpyridine (15 mol%) | 1.25 equiv | 1.0 M | 80 °C | 35% |
| 8 | terpyridine (15 mol%) | 1.25 equiv | 1.0 M | 100 °C | 56% |
Yields were determined by 1H NMR analysis using hexamethylbenzene as an internal standard;
Bn=benzyl, Boc=tert-butyloxycarbonyl, cod=bis(1,5-cyclooctadiene)nickel(0).
Having identified suitable reaction conditions, we evaluated the scope of the amide substrate using 1.25 equivalents of 1-hexanol (7) as the alcohol coupling partner (Figure 2). We were delighted to find that lower catalyst loadings of 5 or 10 mol% nickel could be employed in all cases to give synthetically useful yields of products. Non-α-branched substrates were well tolerated, as illustrated by the formation of 9–11.[18] Of note, an aryl chloride was tolerated as shown by the formation of ester 9b.[19] Moreover, the preparation of 11 in 92% yield (using 2.5 equivalents of 7), demonstrates the feasibility of converting two amides to esters in the same pot. Although our focus has been on amides derived from aliphatic substrates, it should be noted that vinyl amides can also be employed, as shown by the synthesis of 12. With regard to α-branched aliphatic substrates, amides derived from cyclopentane and cyclohexane carboxylic acids could be used, as seen by the formation of 13 and 14, respectively. The high-yielding preparation of 15 and 16 further exemplifies the tolerance of our methodology toward α-branching, in addition to olefins and an oxygen-containing heterocycle. It was also shown that a cyclopropane-containing amide and a sterically hindered tertiary amide substrate were well tolerated, as seen by the formation of 17 and 18, respectively.[20] Finally, it should be noted that the esterification can be performed on the benchtop using our laboratory’s recent protocol involving Ni(cod)2–paraffin capsules (see the SI for details).[7e]
Figure 2.

Evaluation of amide substrates. Yields reflect the average of two isolation experiments. Yield was determined by 1H NMR analysis using 1,3,5-trimethoxybenzene as an internal standard [a] Reaction was performed using 2.5 equivalents of 7. [b] Reaction was performed at 80 °C with 2.0 equivalents of 7; Bn=benzyl, Boc=tert-butyloxycarbonyl, cod=bis(1,5-cyclooctadiene)nickel(0).
With regard to the alcohol component, the scope of the methodology was found to be broad (Figure 3). Primary alcohols were well tolerated, including alcohols that bear olefins or ethers, as determined by the formation of 21 and 22, respectively. In addition to oxygen-containing heterocyclic esters (i.e., 23–25), we found that sulfur- and nitrogen-containing heterocycles could be employed to give 26–28. The tolerance of our methodology to furans, thiophenes, unprotected indoles, and pyrrolidines bodes well for future applications in natural product synthesis and medicinal chemistry. Secondary alcohols also underwent coupling as shown by the formation of esters 29–32.[21] The fact that this methodology can be used to couple hindered alcohols, such as borneol to give 32, is especially noteworthy.[22]
Figure 3.

Scope of the alcohol coupling partner. Yields reflect the average of two isolation experiments; Bn=benzyl, Boc=tert-butyloxycarbonyl, cod=bis(1,5-cyclooctadiene)nickel(0).
As a further test of this methodology, we assessed several heterocyclic substrates that bear epimerizable α-stereocenters (Figure 4). We were delighted to find that Ts- and Boc-protected proline-derived substrates coupled well with 1-hexanol (7) to yield esters 33 and 34. Moreover, an indoline and a Cbz-protected piperidine substrate underwent esterification to furnish products 35 and 36, respectively. In all cases, the reactions proceeded without significant loss of stereochemical integrity.[23] The mild nature of this methodology bodes well for future synthetic applications.
Figure 4.

Coupling of substrates bearing epimerizable α-stereocenters. Yields reflect the average of two isolation experiments. [a] Reaction run for 40 h; Bn=benzyl, Boc=tert-butyloxycarbonyl, cod=bis(1,5-cyclooctadiene)nickel(0), Ts=tosyl, Cbz=carboxybenzyl.
Finally, we performed a series of competition experiments to establish selectivity trends (Figure 5). First, we compared substrates 3 and 37 in the coupling with 1-hexanol (7) and found that aliphatic amide 3 reacts exclusively in the presence of benzamide 37 to give ester 14. Ester 38 was not observed. Next, we performed two competition experiments between aliphatic substrates to probe steric and electronic effects. Amide 3 underwent preferential esterification in the presence of the more sterically hindered amide 39. Additionally, a competition experiment between dioxane derivative 40 and cyclohexyl substrate 3 led to selective coupling to furnish ester 16 in 86% yield. Finally, in an alcohol competition experiment between 1-hexanol (7) and (–)-menthol (4) using substrate 3, we exclusively observed the formation of the corresponding n-hexyl ester, 14. These selectivity findings should aid the judicious design of future synthetic applications.
Figure 5.

Competition experiments. Yields were determined by 1H NMR analysis using 1,3,5-trimethoxybenzene as an internal standard; Bn=benzyl, Boc=tert-butyloxycarbonyl, cod = bis(1,5-cyclooctadiene)nickel(0).
We have discovered the first nickel-catalyzed activation of amides derived from aliphatic carboxylic acids, as demonstrated by the esterification of Boc-activated amides. The methodology is tolerant of heterocycles, substrates with epimerizable stereocenters, and sterically congested coupling partners. Finally, competition experiments reveal selectivity trends of this methodology. This study is expected to prompt the use of aliphatic amides in other C–C and C–X bond forming reactions.
Supplementary Material
Acknowledgments
The authors are grateful to the NIH-NIGMS (R01 GM117016 for N.K.G), the Dreyfus Foundation, the UCLA Gold Shield Alumnae, and the University of California, Los Angeles for financial support. We thank Bristol-Myers Squibb (L.H.), the Foote Family (L.H. and E.L.B.), the Majeti–Alapati family (L.H.), and the University of California, Los Angeles Graduate Division (L.H.) for fellowship support. E.L.B. acknowledges a National Science Foundation predoctoral fellowship (DGE-1144087). These studies were also supported by shared instrumentation grants from the NSF (CHE-1048804) and the National Center for Research Resources (S10RR025631).
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- 1.Greenberg A, Breneman CM, Liebman JF, editors. The Amide Linkage: Structural Significance in Chemistry, Biochemistry, and Materials Science. Wiley-Interscience; 2003. [Google Scholar]
- 2.Amides are considered to be ideally suited for use as synthons in chemical synthesis because of their directing group ability and stability to a variety of reaction conditions. For select recent examples involving amide directing groups in transition metal-catalyzed C–H functionalization methodologies, see: Topczewski JJ, Cabrera PJ, Saper NI, Sanford MS. Nature. 2016;531:220–224. doi: 10.1038/nature16957.He J, Jiang H, Takise R, Zhu R-Y, Chen G, Dai H-X, Murali Dhar TG, Shi J, Zhang H, Cheng PTW, Yu J-Q. Angew Chem Int Ed. 2016;55:785–789. doi: 10.1002/anie.201509996.Angew Chem. 2016;128:795–799.Zhang XG, Dai HX, Wasa M, Yu JQ. J Am Chem Soc. 2012;134:11948–11951. doi: 10.1021/ja305259n.Shang R, Ilies L, Nakamura E. J Am Chem Soc. 2015;137:7660–7663. doi: 10.1021/jacs.5b04818.Matsubara T, Ilies L, Nakamura E. Chem Asian J. 2016;11:380–384. doi: 10.1002/asia.201501095.Yokota A, Aihara Y, Chatani N. J Org Chem. 2014;79:11922–11932. doi: 10.1021/jo501697n.Fruchey ER, Monks BM, Cook SP. J Am Chem Soc. 2014;136:13130–13133. doi: 10.1021/ja506823u.Roane J, Daugulis O. Org Lett. 2013;15:5842–5845. doi: 10.1021/ol402904d.Nishino M, Hirano K, Satoh T, Miura M. Angew Chem Int Ed. 2013;52:4457–4461. doi: 10.1002/anie.201300587.Angew Chem. 2013;125:4553–4557.
- 3.For select reviews on amides as directing groups in ortho-metalation, see: Whisler MC, MacNeil S, Snieckus V, Beak P. Angew Chem Int Ed. 2004;43:2206–2225. doi: 10.1002/anie.200300590.Snieckus V. Chem Rev. 1990;90:879–933.Beak P, Snieckus V. Acc Chem Res. 1982;15:306–312.
- 4.Pauling L, Corey RB, Branson HR. Proc Natl Acad Sci USA. 1951;37:205–211. doi: 10.1073/pnas.37.4.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.(a) Li X, Zou G. Chem Commun. 2015;51:5089–5092. doi: 10.1039/c5cc00430f. [DOI] [PubMed] [Google Scholar]; (b) Li X, Zou G. J Organomet Chem. 2015;794:136–145. [Google Scholar]
- 6.(a) Meng G, Szostak M. Org Biomol Chem. 2016;14:5690–5705. doi: 10.1039/c6ob00084c. [DOI] [PubMed] [Google Scholar]; (b) Meng G, Szostak M. Org Lett. 2016;18:796–799. doi: 10.1021/acs.orglett.6b00058. [DOI] [PubMed] [Google Scholar]; (c) Meng G, Szotak M. Angew Chem Int Ed. 2015;54:14518–14522. doi: 10.1002/anie.201507776. [DOI] [PubMed] [Google Scholar]; Angew Chem. 2015;127:14726–14730. [Google Scholar]; (d) Meng G, Szostak M. Org Lett. 2015;17:4364–4367. doi: 10.1021/acs.orglett.5b02209. [DOI] [PubMed] [Google Scholar]; (e) Shi S, Szostak M. Chem Eur J. 2016;22:10420–10424. doi: 10.1002/chem.201602202. [DOI] [PubMed] [Google Scholar]; (f) Liu C, Meng G, Liu Y, Liu R, Lalancette R, Szostak R, Szostak M. Org Lett. 2016;18:4194–4197. doi: 10.1021/acs.orglett.6b01836. [DOI] [PubMed] [Google Scholar]
- 7.(a) Hie L, Fine Nathel NF, Shah T, Baker EL, Hong X, Yang Y-F, Liu P, Houk KN, Garg NK. Nature. 2015;524:79–83. doi: 10.1038/nature14615. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Weires NA, Baker EL, Garg NK. Nat Chem. 2016;8:75–79. doi: 10.1038/nchem.2388. [DOI] [PubMed] [Google Scholar]; (c) Baker EL, Yamano MM, Zhou S, Anthony SM, Garg NK. Nat Commun. 2016;7:11554. doi: 10.1038/ncomms11554. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Simmons BJ, Weires NA, Dander JE, Garg NK. ACS Catal. 2016;6:3176–3179. doi: 10.1021/acscatal.6b00793. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Dander JE, Weires NA, Garg NK. Org Lett. 2016;18:3934–3936. doi: 10.1021/acs.orglett.6b01758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.For the nickel-catalyzed decarbonylative coupling of amides with arylboronic esters, see: Shi S, Meng G, Szostak M. Angew Chem Int Ed. 2016;55:6959–6963. doi: 10.1002/anie.201601914.Angew Chem. 2016;128:7073–7077.
- 9.For the nickel-catalyzed decarbonylative borylation of amides, see: Hu J, Zhao Y, Liu J, Zhang Y, Shi Z. Angew Chem Int Ed. 2016;55:8718–8722. doi: 10.1002/anie.201603068.Angew Chem. 2016;128:8860–8864.
- 10.Yada A, Okajima S, Murakami M. J Am Chem Soc. 2015;137:8708–8711. doi: 10.1021/jacs.5b05308. [DOI] [PubMed] [Google Scholar]
- 11.Nickel catalysis is considered to be attractive because nickel is less expensive, more abundant, and displays a smaller CO2 print, compared to its precious metal counterpart, palladium. For pertinent reviews, see: Rosen BM, Quasdorf KW, Wilson DA, Zhang N, Resmerita AM, Garg NK, Percec V. Chem Rev. 2011;111:1346–1416. doi: 10.1021/cr100259t.Tasker SZ, Standley EA, Jamison TF. Nature. 2014;509:299–309. doi: 10.1038/nature13274.Mesganaw T, Garg NK. Org Process Res Dev. 2013;17:29–39.Ananikov VP. ACS Catal. 2015;5:1964–1971.
- 12.No examples of non-decarbonylative aliphatic amide couplings using nickel catalysis have been reported. In the context of decarbonylative nickel-catalyzed couplings of amides, two examples involving the decarbonylative borylation of activated benzylic substrates have been reported (see ref 9).
- 13.The conversion of amides to esters typically requires harshly acidic or basic reaction conditions. For Keck’s mild, albeit limited, approach using Meerwin’s salt, see: Keck GE, McLaws MD, Wager TT. Tetrahedron. 2000;56:9875–9883.
- 14.The oxidative addition barrier for N-methyl-N-phenylbenzamide + Ni/SIPr was calculated to be 26 kcal/mol. For comparison, the corresponding oxidative addition barrier involving N-methyl-N-phenylacetamide was found to be 31.8 kcal/mol (see reference 7a).
- 15.The use of mono(oxazolines), bis(oxazolines), pyridinebis(oxazolines), N-heterocyclic carbenes, N-heterocyclic bis(carbenes), mono- and bidentate phosphines, and Buchwald-type phosphines led either to no reaction or very low conversions.
- 16.Heating beyond 100 °C led to diminished yields, whereas increased reaction times led to minimal improvement in conversion.
- 17.In the absence of Ni(cod)2, terpyridine, or both, the esterification reaction fails. Thus, we conclude that Ni catalysis is indeed operative. Analogous control experiments were performed for all reactions presented in this manuscript to validate nickel catalysis.
-
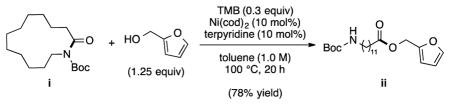
18.A cyclic substrate, macrolactam i, was coupled with furfuryl alcohol to give ester ii in 78% yield (by 1H NMR analysis using 1,3,5-trimethoxybenzene (TMB) as an internal standard).
- 19.In addition to the aryl chloride, couplings in the presence of aryl bromides and aryl nitriles show promise. For robustness screening results see the Supporting Information.
- 20.The coupling of 19a and 7 was performed in the presence of the radical scavengers galvinoxyl, BHT, and TEMPO. In all cases, the formation of the corresponding ester product 9a was observed, suggesting that a radical mechanism is likely not operative.
- 21.Coupling of t-butanol with 19a under the standard reaction conditions did not lead to product formation.
- 22.Coupling of phenol with 19a under the standard reaction conditions led to the recovery of starting material.
- 23.See the Supporting Information for details.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.