

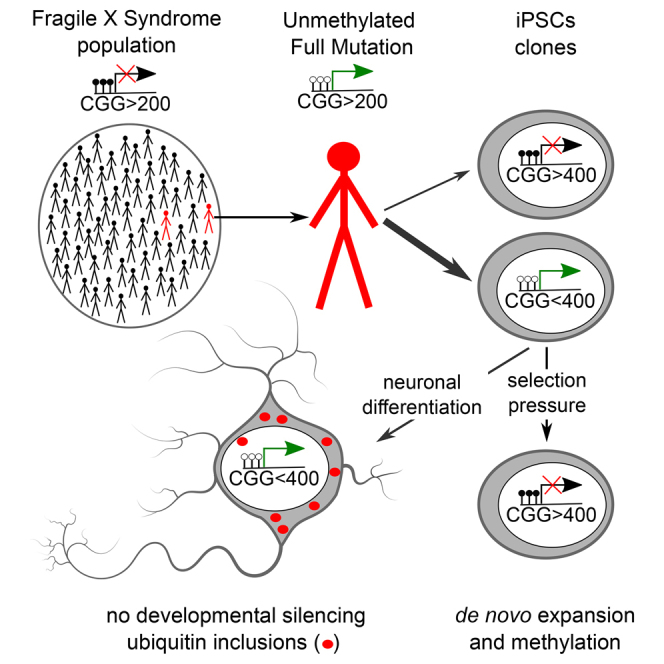
Summary
In fragile X syndrome (FXS), CGG repeat expansion greater than 200 triplets is believed to trigger FMR1 gene silencing and disease etiology. However, FXS siblings have been identified with more than 200 CGGs, termed unmethylated full mutation (UFM) carriers, without gene silencing and disease symptoms. Here, we show that hypomethylation of the FMR1 promoter is maintained in induced pluripotent stem cells (iPSCs) derived from two UFM individuals. However, a subset of iPSC clones with large CGG expansions carries silenced FMR1. Furthermore, we demonstrate de novo silencing upon expansion of the CGG repeat size. FMR1 does not undergo silencing during neuronal differentiation of UFM iPSCs, and expression of large unmethylated CGG repeats has phenotypic consequences resulting in neurodegenerative features. Our data suggest that UFM individuals do not lack the cell-intrinsic ability to silence FMR1 and that inter-individual variability in the CGG repeat size required for silencing exists in the FXS population.
Keywords: fragile X syndrome, unmethylated full mutation, DNA methylation, triplet expansion, fragile X tremor ataxia syndrome, neuron, ubiquitin inclusion, FMR1, CGG repeat, de novo silencing
Graphical Abstract
Highlights
-
•
Unmethylated full mutation (UFM) iPSCs and neurons maintain active FMR1
-
•
UFM iPSCs have the capacity to silence FMR1
-
•
CGG repeat size required for silencing in UFM is higher than 200 described for FXS
-
•
UFM iPSCs derived neurons show signs of neurodegeneration
Rare individuals do not silence FMR1 and do not develop FXS despite carrying the disease causing CGG expansion. Using iPSCs, Fodor, Di Giorgio, and colleagues show that these individuals have not lost the ability to silence FMR1 but the CGG repeat number required for silencing is higher than the one described for FXS patients.
Introduction
FMR1 is an X-linked gene containing an array of CGG repeats located within the 5′ UTR, which normally range from 6 to 55 and may be unstable upon transmission to the next generation (Biancalana et al., 2015).
Repeat numbers from 55 to 200, so-called premutation, result in expression of mRNA with expanded CGG repeats. Individuals carrying the premutation are at risk of developing fragile X tremor ataxia syndrome (FXTAS, OMIM #300623), a late-onset neurodegenerative disease (Hagerman et al., 2001). One of the hallmarks of this disease is ubiquitin-positive inclusion bodies, which have been detected in postmortem brain samples (Greco et al., 2006).
A repeat expansion of more than 200 triplets (full mutation) triggers gene silencing of FMR1, causing fragile X syndrome (FXS, OMIM #300624), the most common inherited form of intellectual disability and autism (Verkerk et al., 1991). Silencing is initiated during early embryonic development and involves establishment of heterochromatin at the FMR1 promoter, including DNA methylation (Sutcliffe et al., 1992). FMR1 encodes the fragile X mental retardation protein (FMRP) and its absence impairs synaptic functions (Willemsen et al., 2011).
Mosaicism in CGG repeat length is often observed in FXS patients, who carry both premutation and full mutation alleles and therefore differ in the proportion of cells with silenced FMR1, which contributes to the clinical spectrum of FXS phenotypes (Rousseau et al., 1994). Mosaicism in the methylation pattern of the expanded CGG repeats has also been described (Hagerman et al., 1994). So-called unmethylated full mutation (UFM) individuals represent an extreme case with all expanded alleles above 200 CGG repeats being unmethylated. These individuals display no signs of intellectual disability (Smeets et al., 1995, Tabolacci et al., 2008, Wohrle et al., 1998) and only a handful of cases have been identified worldwide. Molecular properties of the FMR1 promoter have been studied in lymphoblastoid cell lines and primary fibroblasts derived from UFM individuals (Pietrobono et al., 2005, Tabolacci et al., 2008). Normal or slightly elevated FMR1 transcription, with reduced FMRP level due to translational inefficiency, as well as euchromatic configuration of the FMR1 promoter have been demonstrated in these lines (Pietrobono et al., 2005, Tabolacci et al., 2008). However, it is not clear whether these cells have completely lost the ability to methylate FMR1.
Human embryonic stem cells (ESCs) with more than 200 CGG repeats in the FMR1 locus as well as induced pluripotent stem cells (iPSCs) from FXS patients have been used to study the disease properties at a cellular level (Avitzour et al., 2014, Colak et al., 2014, Eiges et al., 2007, Sheridan et al., 2011, Telias et al., 2013, Urbach et al., 2010). These human ESCs serve as a model for developmental silencing of FMR1. In a fraction of ESC lines FMR1 is already repressed, whereas in some it is still active and becomes silenced during in vitro neuronal differentiation (Avitzour et al., 2014, Colak et al., 2014). In contrast, iPSCs derived from FXS patients do not reactivate FMR1, suggesting that the gene is locked in a silenced state that is resistant to epigenetic reprogramming (Sheridan et al., 2011, Urbach et al., 2010). Therefore, these cells are not used to study the mechanism of FMR1 silencing, but for modeling of neurological phenotypes of FXS (Sheridan et al., 2011, Telias et al., 2013). In a recent study, iPSCs have been also derived from one UFM individual (de Esch et al., 2014). It has been reported that the cells gained silencing of the FMR1 promoter upon reprogramming, hindering the use of these cells for further analyses of the UFM phenotype.
In this study we used somatic reprogramming to dissect the relationship between repeat lengths and silencing status in iPSCs from two unrelated UFM individuals. We found that in the majority of iPSC clones FMR1 remained unmethylated and active. However, in a small proportion of clones which carried more than 400 CGGs FMR1 was silenced, suggesting that the CGG repeat number necessary to induce the silencing is ∼400 in UFM individuals and not ∼200 as described for FXS. Moreover, we demonstrate that upon selective pressure, unmethylated UFM clones gained methylation accompanied by an expansion of the CGG repeats above this higher threshold.
Furthermore, the persistence of the UFM phenotype in iPSC-derived neurons allowed us to investigate whether cells carrying the expanded CGG repeat number and active FMR1 develop a neurodegenerative phenotype. Indeed, we found ubiquitin inclusion bodies in these cells, a phenotypic feature of FXTAS patients. We also observed that in UFM as well as in premutation, iPSC-derived neurons form FMRP inclusions that may contribute to the FXTAS pathology.
Results
Genetic and Epigenetic Characterization of FMR1 Locus in UFM Individuals
Here we describe one previously reported (Pietrobono et al., 2005, Tabolacci et al., 2008) and one new UFM individual, identified within two unrelated FXS families (Figure 1A). Blood samples were obtained from both UFM individuals, from their two healthy brothers N-B1 and N-B2, and the FXS nephew of UFM1, F-N1 (Figure 1A). In addition, we sampled buccal swabs from the additional living members of the families (Figure 1A). For all, we determined CGG repeat length by PCR amplification (Figure 1B). The full expansion status (greater than 200 CGGs) was confirmed for UFM and FXS individuals, as well as the premutation status of their mothers. As expected, in premutation and full mutation females we detected both expanded and wild-type (WT) alleles. The CGG repeat expansion of UFM individuals did not have a discrete length but displayed a continuous spectrum of sizes. This pattern is observed for all reported UFM individuals to date and is attributed to the somatic instability of unmethylated repeats (Biancalana et al., 2015). For UFM1 repeat sizes ranged from 200 to 500 CGGs and for UFM2 premutation bands of 50 and 150 and a smear from 200 to 370 repeats were detected, indicating a mosaic status between a premutation and full mutation (Figure 1B). Southern blot analysis and capillary electrophoresis gave comparable results for UFM1 (Tabolacci et al., 2008) and UFM2, respectively (Figure S1).
Figure 1.

UFM Families
(A) Pedigrees of families with UFM subjects. Oval, female; square, male; crossed, deceased; white, normal; gray, premutation; black, FXS; black on white pattern, UFM; blue, buccal swap sample; red, blood sample.
(B) CGG repeat size in 5′ UTR of FMR1 analyzed by PCR in UFMs and their families' members. Additional analysis of the expansion size by capillary electrophoresis in UFM2 is provided in Figure S1.
(C) Percentage DNA methylation of 22 CpGs of the FMR1 promoter analyzed by bisulfite pyrosequencing in PBMC population. UFM individuals display a minor proportion of methylated alleles. Graph based on data in Table S1.
We compared the DNA methylation status of the FMR1 promoter in the most closely related male individuals with UFM, WT, and FXS phenotypes. We analyzed 22 CpGs within the FMR1 promoter using bisulfite pyrosequencing in peripheral blood mononuclear cells (PBMCs) purified from blood. In cells from both UFMs the mean DNA methylation was 4% and 6% for UFM1 and UFM2, respectively, compared with 0.1% and 0.3% in two WT (N-B1 and N-B2) individuals and 79% in an FXS patient (F-N1) (Figure 1C and Table S1). The low level of methylation in UFM PBMCs suggests the presence of fully methylated alleles and, therefore, a low level of methylation mosaicism in these individuals.
CGG Repeat Sizes and FMR1 Expression States in UFM iPSC Clones
PBMCs are composed of cells with heterogeneous CGG repeat size and the FMR1 promoter methylation status. To dissect the relationship between repeat size, methylation status, and FMR1 expression, we derived iPSCs from PBMCs and performed analyses on multiple distinct clones.
Activated T cells were used for somatic reprogramming by Sendai viruses harboring the SOX2, OCT4, c-MYC, and KLF4 reprogramming factors (Takahashi et al., 2007). A total of 11–12 clones were characterized from each individual. All clones expressed markers of pluripotency (Figure S2A) and were analyzed for the CGG repeat size, the DNA methylation status of the promoter, and the expression level of FMR1 (Figure 2 and Table S1).
Figure 2.

Increased Silencing Threshold in iPSC Clones Derived from UFM Subjects
(A) CGG repeat size in 5′ UTR of FMR1 analyzed by PCR in iPSCs derived from PBMCs from UFM1 and UFM2. Background color of a clone name indicates expression of FMR1 based on data in (B), expressing (blue), not expressing, or expressing below 5% of WT level (red). Circles under the clone names indicate mean percentage of DNA methylation across 22 CpGs of the FMR1 promoter based on data in Table S1. Silent and fully methylated clones are observed in both UFM subjects. Yellow lines indicate the proposed approximate thresholds of CGG repeat numbers triggering FMR1 silencing in a given individual. The grey line corresponds to 200 CGG repeats. See also Figure S2.
(B) Expression of FMR1 mRNA in iPSC clones from PBMCs from UFM1 and UFM2 analyzed by TaqMan assay. WT iPSC clone 86-14 derived from a normal individual is included as a WT reference. Data are presented as a mean of three independent biological replicates. Error bars represent SD.
The majority of clones obtained from both UFM individuals had repeat sizes corresponding to the spectrum of sizes observed in the PBMCs (Figure 2A). Three clones from UFM1 displayed shorter CGG repeat sizes, with either WT (UFM1-1) or premutation (UFM1-2 and UFM1-3) size. This may represent either a contraction event or preexisting lowly abundant alleles in the original PBMC samples.
The majority of UFM clones with repeat sizes above 200 CGGs retained the hypomethylated FMR1 promoter and expressed FMR1 at levels comparable with WT (Figure 2). Most of them showed discrete repeat sizes as expected from the iPSC clonal derivation procedure. All clones were analyzed between passages 5 and 10. A representative UFM clone UFM1-5 was followed until passage 22 and showed no changes in the hypomethylated status of the FMR1 promoter and only slight CGG repeat instability (Figure S2B). We conclude that the UFM phenotype is stable upon somatic reprogramming and is maintained in iPSCs.
Interestingly, 2 of 11 clones from UFM1 (UFM1-10 and UFM1-11) with the highest CGG repeat sizes (>450 CGGs) were hypermethylated and did not express FMR1. In UFM2 we also identified 1 of 12 clones (UFM2-12) that was silenced, hypermethylated, and carried more than 400 CGGs. However, a second silenced clone from UFM2 (UFM2-4) carried less than 200 CGGs. These silenced clones may originate from the 5% methylated alleles in PBMC samples or represent a de novo silencing event during reprogramming.
We conclude that in the UFM individuals analyzed in this study, the silencing threshold lies at 450 repeats in UFM1 and 400 repeats in UFM2 (yellow lines in Figure 2A). However, this threshold is less evident in UFM2 where an outlier clone, UFM2-4, has been identified.
Silencing Threshold of CGG Repeat Length in FXS iPSCs
The above results prompted us to investigate whether the increased silencing threshold is a property exclusive to these two UFM individuals. To this end, we analyzed iPSCs from FXS patient F-N1, a nephew of UFM1 (Figure 3). In the majority of F-N1 clones the FMR1 promoter was fully methylated, an observation in line with the described resistance of FMR1 silencing to the somatic reprogramming (Sheridan et al., 2011, Urbach et al., 2010). However, we also identified two clones with active FMR1 (F-N1-1 and F-N1-2) and repeat sizes above 200 CGG, but shorter than in the silenced clones. The majority of the alleles in these clones were hypomethylated (Figure 3 and Table S1). We conclude that in this FXS patient the silencing threshold lies around 400 CGG (yellow line in Figure 3A). Clones carrying the same size of 420 CGG were active in UFM1 (UFM1-7, UFM1-8, UFM1-9) and silenced in F-N1 (FN1-3).
Figure 3.

UFM-Like Clones and Altered Silencing Threshold in iPSCs Derived from FXS Patients
(A) CGG repeat size in 5′ UTR of FMR1 analyzed by PCR in iPSC clones derived from FXS patients. Background color of a clone name indicates expression of FMR1 based on data in (B), expressing (blue), not expressing or expressing below 5% of WT level (red). Circles under the clone names indicate mean percentage of DNA methylation across 22 CpGs of the FMR1 promoter based on data in Table S1. Active, hypomethylated clones with more than 200 CGGs are observed in both FXS patients. Yellow line indicates the proposed approximate threshold of CGG repeat numbers triggering FMR1 silencing in these individuals. The grey line corresponds to 200 CGG repeats.
(B) Expression of FMR1 mRNA in iPSC clones derived from FXS patients analyzed by TaqMan assay. WT iPSC clone 86-14 derived from a normal individual is included as a WT reference. Data are presented as a mean of three independent biological replicates. Error bars represent SD.
We also derived iPSCs from an FXS individual not related to the UFM families, from which iPSCs with the active unmethylated full mutation allele of FMR1 have been previously described (Avitzour et al., 2014). Similarly to the previous report, one of three analyzed clones was active and fully unmethylated (Figure 3). The repeat size of this clone ranged from 250 to 320 CGGs. A faint premutation band of 80 CGGs was also detected. Nevertheless, 98% of alleles were unmethylated. A smear indicating instability of active expanded alleles was also observed. Consistent with a shift of the repeat threshold in this individual, the two silenced clones FX97-2 and FX97-3 had repeat sizes larger than 400 CGGs. We conclude that the threshold in CGG repeat size resulting in silencing of FMR1 is subject to inter-individual variation not only in UFM individuals but also in a proportion of FXS patients.
De Novo Methylation of FMR1 in UFM iPSCs Is Coupled with an Increase in the CGG Repeat Size
To assess whether UFM cells have intrinsic capacity to de novo silence the FMR1 locus, we evaluated whether an active allele with a given repeat size can spontaneously gain DNA methylation. Under extended cell culture up to passage 22 we did not observe a spontaneous appearance of methylated alleles for a representative clone UFM1-5 (Figure S2B). Therefore, we established a genetic system to select for FMR1 silencing events. Our approach was based on the fact that replicating mammalian cells expressing the herpes simplex virus thymidine kinase (HSV-TK) enzyme become sensitive to the prodrug ganciclovir causing cell lethality, while non-expressing cells survive and proliferate upon treatment. We deployed genome engineering to drive expression of a hygromycin-HSV-TK fusion protein from the endogenous FMR1 promoter (Figure 4A).
Figure 4.

Gain in CGG Repeat Number Is Coupled with FMR1 Silencing in UFM iPSC Clones
(A) Strategy to target a hygromycin resistance-HSV thymidine kinase (HyTK) positive/negative selection cassette into FMR1. In this setup transgene expression is driven by the endogenous FMR1 promoter. See also Figure S3.
(B) Experimental design to select for iPSCs that silence the FMR1 promoter driven HyTK. Continuous hygromycin administration selects for cells that express the transgene making them sensitive to ganciclovir. Four weeks without hygromycin allows appearance of cells that spontaneously downregulate the transgene. These cells can be selected for by ganciclovir treatment. Subsequent DNA analysis identifies clones with FMR1 promoter methylation events.
(C and D) Crystal violet staining of surviving cells after ganciclovir treatment of two knockin iPSC clones (UFM1-5-6 and UFM1-5-7) that were maintained: (C) under hygromycin selection or (D) without hygromycin selection for 4 weeks.
(E) DNA methylation status and number of CGG repeats of FMR1 from knockin iPSC lines before and after hygromycin withdrawal and subsequent ganciclovir selection. Lanes UFM1-5-6 and UFM1-5-7 are the original clones before hygromycin withdrawal. Subclones UFM1-5-7-1 and UFM1-5-7-2 are derived from UFM1-5-7 after 4 weeks of hygromycin withdrawal and subsequent ganciclovir selection. Circles under each lane indicate mean percentage of DNA methylation across 22 CpGs of the FMR1 promoter for a given line based on data in Table S1. Two ganciclovir-resistant clones, UFM1-5-7-1 and UFM1-5-7-2, gained ∼90% methylation and have increased CGG repeats (∼800 CGG) compared with the parental UFM1-5-7 (320 CGG). See also Figure S4.
(F) Crystal violet staining of surviving cells after hygromycin treatment of ganciclovir-resistant subclone UFM1-5-7-1. For cells maintained with ganciclovir, no surviving colonies are observed. Upon ganciclovir withdrawal, spontaneous reactivation of FMR1 promoter is possible and appearance of surviving colonies is observed.
(G) Analysis of two hygromycin-resistant colonies obtained from subclone UFM1-5-7-1 after withdrawal of ganciclovir as indicated in (F). Parental clone UFM1-5-7 is analyzed in the first lane for direct comparison. CGG repeat sizes 320 and 150 indicate contraction events. Black circles under each lane indicate mean percentage of DNA methylation across 22 CpGs of the FMR1 promoter based on data in Table S1. No methylation is observed in the surviving colonies, indicating that contraction below the silencing threshold is associated with loss of DNA methylation.
We inserted a selection cassette coding for a fusion of a P2a peptide-hygromycin-HSV-thymidine kinase (Hyg-TK) protein into exon 4 of the FMR1 gene in iPSC clone UFM1-5 using the CRISPR/Cas9 system (Figures 4A and S3A). This setup allowed for the direct hygromycin selection of correct insertions. After selection of clones we confirmed a single integration event and analyzed their CGG repeat length, FMR1 methylation, and mRNA expression (Figures S3B–S3E). The clones were maintained in hygromycin-supplemented medium to assure the absence of cells with methylated FMR1 promoter. Two clones, UFM1-5-6 and UFM1-5-7, with 250 and 320 CGG repeats, respectively, were selected for further experiments. Unlike the parental line UFM1-5 (Figure S3F), the knockin clones were ganciclovir sensitive at concentrations of 5 and 10 μM after 3 days of prodrug treatment in the absence of hygromycin (Figures 4B, 4C, and S3F). To allow the appearance of spontaneous FMR1 silencing events, we cultured cells without hygromycin for 4 weeks (∼8 passages). Subsequent selection for silenced clones was performed using 10 μM ganciclovir for 3 days. The 4 weeks of hygromycin withdrawal was necessary to observe reproducible appearance of ganciclovir-resistant clones in UFM1-5-7, and ganciclovir selection after shorter periods of hygromycin withdrawal yielded no survivals. For the line UFM1-5-6 we did not observe any stable ganciclovir-resistant clones (Figure 4D).
Most ganciclovir selected subclones did not survive passaging, especially the ones which lost the iPSC morphology. Importantly, we identified two clones with iPSC morphology and stable ganciclovir resistance (Figure 4E). Both subclones UFM1-5-7-1 and UFM1-5-7-2 showed full methylation of the FMR1 promoter and CGG repeat sizes of ∼800 CGGs, indicating that the expansion of the CGG repeat led to the silencing of FMR1. We used clone UFM1-5-7-1 in a follow-up experiment to test the reversibility of the CGG expansion and methylation by applying hygromycin selection. Interestingly, upon withdrawal of ganciclovir we observed the appearance of “revertant” hygromycin-resistant clones which coincided with contraction of CGG repeats and loss of DNA methylation (Figures 4F and 4G).
We conclude that the silencing of FMR1 in UFM iPSCs directly depends on the size of the CGG repeat. Furthermore, UFM cells have not lost the capacity to silence FMR1. Consistently, we did not find any mutation common to the two UFMs that would obviously impair the silencing machinery (Figure S4A; Tables S2 and S3). Neither did we identify a common mutation in the proximal regulatory sequences of FMR1 (Figures S4B–S4D).
FMR1 Silencing Status Is Not Affected by Differentiation of UFM iPSC Clones
The conversion of an active UFM allele to a silenced one required an increase of the CGG repeat length. However, switching from an active to a silenced state, without changes in the repeat size, underlies the process of developmental silencing of FMR1 in FXS embryos and has been modeled by neuronal differentiation of FXS human ESCs (Colak et al., 2014, Eiges et al., 2007, Sutcliffe et al., 1992, Telias et al., 2013). Therefore, we evaluated the stability of FMR1 expression during neuronal differentiation of UFM iPSCs.
The exact time window of FMR1 silencing during in vitro differentiation of FXS human ESCs to cortical neurons has been reported at day 45 (Colak et al., 2014). We applied the same differentiation protocol to iPSCs from two UFM individuals with variable CGG repeat sizes and FMR1 silencing status (Figure 5). We used four iPSC lines with unmethylated expanded repeats, namely UFM1-5 (320 CGG), UFM1-9 (420 CGG), UFM2-5 (200 CGG), and UFM2-9 (260 CGG), as well as iPSC clone UFM1-11 with methylated expanded repeats of 480 CGG. Two WT lines, either with UFM1 background (UFM1-1) or from an unrelated healthy donor 86-14, were used as controls. All lines were efficiently differentiated into class III β-tubulin/MAP2-positive neurons (Figure S5). We analyzed the expression level of FMR1 mRNA at four time points, up to 90 days of neuronal differentiation (Figure 5A). At day 60 we additionally confirmed the presence of FMRP by immunofluorescence (Figure 5B). For all the lines with active FMR1 we observed a gradual increase in the expression during differentiation (Figure 5A), consistent with the described dynamics of FMR1 expression during neuronal differentiation of WT human ESC lines (Telias et al., 2013). We also did not detect the appearance of any methylated FMR1 alleles at day 90 (Figure 5A). No major changes in the size of the CGG repeats were found at this time point (data not shown). Also the methylation status of clone UFM1-9, which had 8% of its alleles methylated in the iPSC state, did not change during differentiation into neurons (7%–10% in three independent differentiations). In addition, line UFM1-11, with the longest CGG repeat size (480), retained full methylation of the FMR1 promoter.
Figure 5.

UFM iPSC Derived Neurons Do Not Undergo Developmental Silencing of FMR1
(A) Expression of FMR1 mRNA during 90 days of neuronal differentiation of iPSC lines with variable repeat length and methylation. Timing of FMR1 silencing in FXS human ESCs as reported by Colak et al. (2014) is indicated and no significant drop of FMR1 mRNA expression is observed before or after this time in any line. Black circles next to each line indicate mean percentage of DNA methylation across 22 CpGs of the FMR1 promoter at day 90 of differentiation. Data points represent mean of three independent differentiations ± SEM. See also Figure S5.
(B) Antibody staining of FMRP, the protein product of FMR1, in iPSC-derived neurons at day 60 of neuronal differentiation. FMRP, red; DAPI, blue. Except for the UFM1-11 clone with fully methylated and silent FMR1, FMRP was detected in all clones. Scale bar, 20 μm.
We conclude that the silencing status of FMR1 is a stable feature of a given repeat size in the UFM background. Furthermore, this result shows that UFM cells are not subjected to developmental silencing induced by neuronal differentiation.
iPSC-Derived Neurons from UFM Subjects Show Phenotypic Properties of FXTAS
Ubiquitin-positive inclusion bodies (IBs) are found in postmortem brain samples of FXTAS patients (Greco et al., 2006) and in mouse models of FXTAS (Wenzel et al., 2010) expressing the FMR1 with repeat sizes between 50 and 200 CGG. Therefore, to analyze the phenotypic consequences of the expression of FMR1 with more than 200 repeats, we analyzed the numbers of ubiquitin-positive IBs in UFM iPSC-derived neurons (Figure 6). To enhance the maturation of neuronal precursors we used a co-culture system, whereby iPSC-derived neuronal progenitors are transiently transfected with a GFP expression vector and injected into organotypic mouse brain slices (OTBS) containing cortex, striatum, and hippocampus (Pecho-Vrieseling et al., 2014). After 6 weeks of co-culture we compared the number of ubiquitin IBs in isogenic iPSC-derived neurons only differing in the repeat size. UFM1-5 (320 CGG) and UFM1-9 (420 CGG), which expressed FMR1, were evaluated for the FXTAS phenotype whereas UFM1-1 (20 CGG), UFM1-3 (150 CGG), and UFM1-11 (480 CGG) with silenced FMR1 served as WT, premutation, and FXS controls, respectively (Figure 2). We found multiple ubiquitin inclusions throughout the cell bodies of GFP-positive human neurons (Figures 6A and 6B). We quantified the number of inclusions in both cytoplasm and nucleus and found significantly higher numbers in the UFM clone (UFM1-5) compared with WT (UFM1-1) (Figures 6A and 6B). This effect was not observed for premutation (UFM1-3) neurons, indicating that expression of mRNA with 150 CGG repeats is not sufficient to trigger the effect. All the quantified cells in clones UFM1-1, UFM1-3, and UFM1-5 expressed FMR1 as judged from FMRP staining. For UFM clone UFM1-9 with 8% methylated alleles we observed that only a proportion of neurons expressed FMRP and found a significantly higher number of inclusions only in these cells. The proportion of neurons that did not express FMRP did not show this effect, similar to neurons from FXS clone UFM1-11 with fully methylated FMR1 (Figures 6A and 6B). Thus, expression of FMR1 with expanded repeat is required for the accumulation of ubiquitin IBs.
Figure 6.

Neurodegenerative Features of UFM iPSC-Derived Neurons
(A) Representative images of staining for ubiquitin and FMRP inside GFP-labeled, isogenic, iPSC-derived neurons with spectrum of CGG repeat sizes corresponding to WT (UFM1-1), classical premutation (UFM1-3), UFM (UFM1-5 and UFM1-9), and FXS (UFM1-11). The iPSC-derived neurons were cultured within murine brain slices for 6 weeks. Ubiquitin (Ubi), red; FMRP, magenta; GFP, green; DAPI, blue. Multiple ubiquitin-positive inclusion bodies (IBs) are detected in UFM lines UFM1-5 and UFM1-9. Punctuated staining of FMRP is observed in UFM1-3, UFM1-5, and UFM1-9. Scale bars, 2 μm.
(B) Number of ubiquitin IBs per cell body of GFP-labeled human neuron. IBs ≥0.5 μm were counted. Black and gray bars represent FMRP-positive and -negative cells, respectively. Increased number of IBs is observed in both UFM clones but only for cells expressing FMRP. n = 15–50 cells per line coming from three independent rounds of brain slice injections (three mice). Data are presented as mean ± SEM; ∗∗∗p < 0.0001, unpaired Student's t test.
(C) Number of FMRP foci per cell body of GFP-labeled human neuron. FMRP foci ≥0.5 μm were counted. For UFM1-9 only cells expressing FMRP are quantified. n = 15–50 cells per line coming from three independent rounds of brain slice injections (three mice). Data are presented as mean ± SEM; ∗∗∗p < 0.0001, unpaired Student's t test.
In addition, we observed a dotted, aggregate-like pattern of FMRP staining in neurons derived from premutation (UFM1-3) and both UFM clones (UFM1-5 and UFM1-9) compared with an even distribution of FMRP in WT (UFM1-1) neurons (Figures 6A and 6C). We quantified the number of these FMRP aggregate-like structures in neuronal cell bodies and found a significant increase in premutation and UFM neurons compared with WT (Figure 6C). As expected, no FMRP staining was observed in FXS (UFM1-11) neurons.
These data together suggest that UFM neurons may develop FXTAS neurodegenerative pathology. Moreover, some pathological phenotypes were only present or more pronounced in UFM when being compared with premutation neurons.
Discussion
Genotype-phenotype correlation is greatly affected by inter-individual genetic differences and external factors. This variability is particularly relevant for the penetrance of genetic variants and may explain the spectrum of clinical phenotypes. In extreme cases, individuals who carry a pathological mutation may lack disease features. Dissecting the correlation between genotype and phenotype in these individuals may provide novel insights into the disease.
FXS is caused by a CGG trinucleotide repeat expansion in the 5′ UTR of FMR1 that leads to its developmental silencing. There is a broadly accepted consensus that 200 CGGs represent the threshold above which the FMR1 promoter becomes methylated and its transcription is turned off (Biancalana et al., 2015, Willemsen et al., 2011). An exception to this rule has been identified in rare UFM individuals who do not silence FMR1 despite the expansion above this threshold (Smeets et al., 1995, Tabolacci et al., 2008, Wohrle et al., 1998). Whether these individuals have lost the capacity to silence FMR1 locus has so far remained elusive.
In this study, we demonstrate that in two unrelated UFM cases FMR1 silencing is not completely impaired. However, we found that the CGG repeat number necessary to trigger silencing is around 400 CGG rather than 200 CGG described for FXS patients. Analysis of iPSC clones with a spectrum of discrete repeat sizes allowed us to dissect the relation of CGG repeats with the silencing status of FMR1, previously only evaluated in primary cells and tissues with complex mosaic patterns of CGG repeat lengths. We show that in the majority of iPSC clones derived from the two UFM subjects, the FMR1 promoter was unmethylated and active (Figure 2). In contrast, clones from FXS subject F-N1 in majority were silenced (Figure 3). However, also in a fraction of UFM clones with more than 400 CGG repeats FMR1 was methylated and not expressed. An exception to this was the methylated clone UFM1-4 with 180 CGG repeats. It is not fully exceptional for a high premutation to be methylated, although very infrequent (Rousseau et al., 1994; G.N., unpublished data). Moreover, we detected ∼5% methylated alleles in blood of both UFMs, indicating that silencing of FMR1 in UFM background is not an artifact of iPSCs but occurs in vivo as well (Figure 1C).
To test whether UFM iPSCs are capable of de novo methylation and silencing of FMR1, we applied a selection pressure paradigm to unmethylated expanded alleles (Figure 4). Indeed, we found very rare events of methylation gain that were associated with a major increase in the number of CGG repeats. This demonstrates that UFM iPSCs are capable of gaining DNA methylation at the FMR1 promoter when the CGG size is higher than their silencing threshold. By using a reporter knocked into the FMR1 gene we monitored the silencing events in the endogenous locus that allowed us to evaluate if both cis and trans components of silencing machinery are in place in UFM cells. This is especially relevant, as the CGG-mediated silencing of FMR1 has not been recapitulated in the transient transfection or randomly integrated reporter assays (Solvsten and Nielsen, 2011). The result of our experiment suggests that UFM individuals possess all the necessary components for the silencing of FMR1. Consistent with this notion, we have not identified any obvious genetic mutation common to both UFM individuals by analyzing the exome sequence of components of the epigenetic machinery and the regulatory sequences of the FMR1 locus, including the sequence of the CGG repeat implicated in silencing initiation (Colak et al., 2014) (Figure S4).
Little is known about why the silencing threshold lies at 200 CGG repeats in FXS. In human ESCs carrying unmethylated CGG expansions a formation of a DNA:RNA hybrid over the CGG repeat is required for silencing initiation (Colak et al., 2014). Recently the direct dependence of FMR1 silencing maintenance on the expanded CGG repeat has been demonstrated by removal of the repeat with CRISPR/Cas9 in FXS iPSCs, which resulted in demethylation and reactivation of the FMR1 (Park et al., 2015). Here, by using a selection strategy we demonstrate that by increasing the length of the repeat an active FMR1 allele becomes silenced and furthermore that it can be reverted to an active state upon a contraction event (Figure 4). Therefore, the dependence of the FMR1 activity on the repeat length is bidirectional. Our selection system provides a tool to further study the dynamics of FMR1 silencing.
The FMR1 gene with more than 200 CGG repeats is silenced during early embryonic development (Sutcliffe et al., 1992). This process has been modeled in human ESCs derived from embryos carrying the FMR1 full mutation. In these cells FMR1 is silenced during in vitro neuronal differentiation (Colak et al., 2014, Eiges et al., 2007, Telias et al., 2013). Unfortunately, it is not possible to obtain human ESCs from UFM individuals. However, a recent study suggests that there is genetic and epigenetic equivalence of human ESCs and iPSCs with matched genetic background (Choi et al., 2015). Therefore, we investigated whether UFM iPSC clones with a spectrum of unmethylated repeat lengths would silence FMR1 in this developmental model (Figure 5). We neither observed a drop in the expression nor an appearance of methylated alleles at all analyzed stages of neuronal differentiation. Additionally, the silenced methylated status of cells with more than 450 CGG repeats did not change. These data suggest that the activity status of an allele with a given repeat size is maintained in iPSC-derived neurons. Furthermore, the expanded unmethylated alleles are resistant to silencing during differentiation. If the lack of methylation in UFM was a result of a maternal effect or stochastic events, FMR1 would have been silenced during in vitro differentiation as observed in human ESCs carrying the FMR1 full mutation (Colak et al., 2014, Eiges et al., 2007, Telias et al., 2013). However, our data suggest that the UFM cells possess intrinsic properties that affect their silencing threshold causing the lack of FMR1 methylation during neuronal differentiation.
Recently, iPSCs were derived from fibroblasts obtained from another unrelated UFM subject (de Esch et al., 2014). The lymphoblastoid cell line from this subject has been previously characterized and showed the same epigenetic profile of the FMR1 promoter-like lymphoblastoid cells from UFM1 (Pietrobono et al., 2005, Tabolacci et al., 2008). However, the authors reported that upon iPSC derivation the FMR1 promoter gained methylation and the FMR1 expression was shut off in all derived clones (de Esch et al., 2014). Inter-individual heterogeneity of the UFM group may be the cause of different results of UFM reprogramming in our study. Alternatively, different sources of the material being fibroblasts and PBMCs in de Esch et al. (2014) in their two respective studies may have led to this discrepancy. However, we reproduced the results of Avitzour et al. (2014) obtaining a UFM-like clone from an independent FXS fibroblast line (GM09497, Figure 3), which suggest that different cell type origins alone would not result in clones with different silencing status. Furthermore, the results from de Esch et al. (2014) are in agreement with our observation that UFM subjects retain the ability to silence FMR1. In addition, the published data may well be interpreted in favor of an altered silencing threshold. The fibroblasts used by de Esch et al. (2014) for reprogramming carried the repeat sizes of 200–230 CGGs. The CGG size disclosed for two silent iPSC clones in de Esch et al. (2014) were 330 and 380 repeats. The difference between the original repeat size of the fibroblasts and the one reported in the iPSC clones with silent FMR1 is in line with our hypothesis of a shifted silencing threshold in UFM individuals.
Our observation of the variability in the silencing threshold is not limited to the UFM group, as we also detected this phenomenon in FXS patients. From a UFM-related FXS subject F-N1 and from an unrelated patient, FX97 (derived from GM09497 fibroblasts), we derived iPSC clones (Figure 3). All clones from both individuals carried FMR1 alleles with more than 200 CGG repeats, most of them being silenced as previously described for FXS iPSCs (Sheridan et al., 2011, Urbach et al., 2010). However, some of the clones were active, hypomethylated, and had lower CGG repeat sizes than the silenced ones, consistent with a shift in the FMR1 silencing threshold in these cells. Interestingly, in a recent report iPSCs were derived from the same fibroblasts GM09497 (Avitzour et al., 2014). Among four clones one was carrying an active FMR1 allele with a fully expanded repeat, while the remaining three were silenced. The exact repeat size of the clones is not reported. Nevertheless, these data show the reproducibility of our results and indicate that the shift in the repeat threshold is an intrinsic property of the cells and not a random phenomenon. Our data support the hypothesis that the number of CGG repeats that triggers the epigenetic silencing of the FMR1 promoter may vary between individuals.
In our model, the persistence of the increased silencing threshold in UFM iPSC-derived neurons prevents the development of the FXS phenotype. However, the expressed expanded CGG repeat is predicted to give rise to an additional phenotype in these neurons. Individuals that carry active FMR1 alleles with 50–200 CGG repeats (permutation) are at risk of developing the late-onset neurodegenerative disease FXTAS, and there have been reports of FXTAS diagnosis in UFM individuals (Basuta et al., 2015, Loesch et al., 2012). Key hallmarks of this disease are relatively large, single per cell, intra-nuclear ubiquitin-positive IBs found in postmortem brain samples from FXTAS patients as well as in mouse models (Greco et al., 2006, Wenzel et al., 2010). At the cellular level FXTAS phenotype has been explored in iPSCs from a premutation individual carrying an expansion of 94 CGG repeats (Liu et al., 2012). However, the authors did not report on the presence of ubiquitin IBs. In the FXTAS brain samples a correlation of the CGG repeat number and the percentage of neurons with IBs has been observed (Greco et al., 2006). Therefore, iPSCs that express FMR1 mRNA with more than 200 CGG repeats may potentially show enhanced or accelerated FXTAS phenotypes. Indeed, we find that neurons carrying more than 200 CGG repeats showed significantly increased numbers of ubiquitin-positive IBs compared with WT neurons (Figure 6B). This effect was not present in cells with silenced FMR1 as judged by FMRP staining, indicating that not the expansion per se but the expression of the expended repeat is necessary to trigger the effect.
We did not observe an increased number of ubiquitin IBs in neurons derived from an isogenic iPSC line with a classical premutation length. It is likely that the expression of mRNA with longer CGG repeat lengths in UFM neurons results in accelerated pathological development compared with premutation neurons. Furthermore, in contrast to a single large intra-nuclear IB observed in FXTAS mouse models or in postmortem brain samples of FXTAS patients, we found multiple IBs in both cytoplasm and nucleus. The iPSC-derived neurons represent an early developmental stage in comparison with an aging brain. Developmental progression of the number and size of the inclusions is reported in FXTAS mice (Wenzel et al., 2010). Therefore, the pattern of ubiquitin inclusions observed here may reflect an early stage of the IB formation at the onset of the disease. Additionally we detected that both premutation and UFM neurons show increased numbers of FMRP aggregate-like structures (Figure 6C). FMRP has been reported to have a tendency to aggregate and spontaneously misfold toward β-rich structures in vitro (Sjekloca et al., 2011). Therefore, aggregation of FMRP may be contributing to the FXTAS pathology. Overall, our data provide evidence for an increased accumulation of ubiquitin and FMRP inclusions in UFM iPSC-derived neurons, which may be signs of an accelerated FXTAS phenotype in the UFM lines compared with the classical premutation.
In summary, our analyses reveal that UFM individuals have not lost the ability to silence FMR1, but the size of the CGG expansion triggering the silencing is higher than the one described in FXS patients (200 CGG). Furthermore, inter-individual variability in the CGG size requited for silencing is present not only in UFM but also in two FXS patients analyzed in this study. We propose a model in which the threshold size together with the proportion of the FMR1 alleles below this threshold delineates UFM and FXS phenotypes. UFM do not silence FMR1 and are spare of FXS pathology; nevertheless, our data suggest that the expression of FMR1 gene with large CGG expansion may increase their risk of developing FXTAS.
Experimental Procedures
Ethics and Sample Collection
The sample collection and all of the Experimental Procedures were approved by the Ethics committees of the Catholic University School of Medicine in Rome and the Ethikkommission Nordwest-und Zentralschweiz. Proper informed consent was obtained from all donors. Blood samples were collected from UFM individuals, their healthy brothers, and FXS nephew of UFM1. Buccal swab samples were obtained from additional family members. Primary skin fibroblasts GM09497 were obtained from Coriell Institute for Medical Research. Experiments involving mice were carried out in accordance with authorization guidelines of the Swiss Federal and Cantonal veterinary offices for care and use of laboratory animals and were approved by the Swiss Cantonal veterinary office and performed according to Novartis animal license number 2063.
FMR1 DNA Methylation, CGG Repeat Length, and Expression
CGG repeat number in the 5′ UTR of FMR1 was analyzed by PCR amplification using an AmplideX FMR1 PCR kit (Asuragen) and agarose gel electrophoresis if not stated differently. DNA methylation of 22 CpGs of the FMR1 promoter was analyzed using bisulfite pyrosequencing by EpigenDx. Expression of FMR1 was quantified using TaqMan assay Hs00924547_m1 (Thermo Fisher). See Supplemental Experimental Procedures for the details.
iPSC Derivation and Neuronal Differentiation
iPSCs were derived from activated T cells isolated from PBMCs using a CytoTune-iPS Reprogramming Kit (Invitrogen). iPSCs were cultured on Matrigel (Corning) in mTeSR medium (Stem Cell Technologies) or Nutristem (Biological Industries). iPSCs were differentiated to neurons using the dual SMAD inhibition protocol (Chambers et al., 2009). Neuronal precursor cells derived from iPSCs were differentiated either on Matrigel or on OTBS using a previously described method (Pecho-Vrieseling et al., 2014). For detailed iPSC derivation and differentiation protocols, see Supplemental Experimental Procedures. The iPSC lines generated in this study are available upon request for research to study FXS and related diseases if all legal and ethical standards are met.
Immunostaining and Image Analysis
Immunostaining was performed using standard procedures explained in detail in Supplemental Experimental Procedures, using the following antibodies: rabbit anti-class III β-tubulin (PRB-435P, BioLegend), chicken anti-MAP2 (ab5392, Abcam), mouse anti-FMRP (Sc-101048, Santa Cruz Biotechnology), and rabbit anti-ubiquitin clone 10H4L21 (701339, Thermo Fisher). Images were acquired with a Zeiss confocal microscope. Ubiquitin and FMRP spots lying within the GFP-positive human neurons were counted manually using visualization with Imaris software as described in Supplemental Experimental Procedures.
Thymidine Kinase Knockin and Positive-Negative Selection
A P2a peptide-hygromycin-HSV thymidine kinase-stop codon-SV40 poly(A)(Hyg-TK) cassette was inserted into exon 4 of FMR1 using CRISPR-mediated homologous recombination. For the details of construct generation, validation, and selection procedure, see Supplemental Experimental Procedures. Knockin lines were maintained in Nutristem medium (Biological Industries) supplemented with 25 μg/mL hygromycin. Positive-negative selection was performed with 10 μM ganciclovir without hygromycin.
Exome Sequencing, FMR1 Promoter, and CGG Sequencing
Details of exome sequencing, Sanger sequencing of FMR1 promoter, and single-molecule real-time sequencing (SMRT) of the CGG repeat tract are described in Supplemental Experimental Procedures.
Statistical Analysis
Statistical analysis in Figure 6 was performed with Prism (Graphpad) software. The unpaired Student's t test was used to determine significant differences between groups. Random samples from each mouse were taken. No mice and data points were excluded from the analysis. OTBSs that showed a severe degeneration of both hemispheres (measured as holes in the cultures) were excluded from the analysis.
Author Contributions
U.B., E.P.-V., M.M., B.G.M., G.R., E.J.O., M. Buehler, P.C., G.N., T.B., F.P.d.G., and B.D.F. designed the experiments; U.B., E.P.-V., A.T., J.K., I.F., S.F., T.D., C.M., M.I., U.N., and E.T. performed the experiments; U.B., M. Beibel, N.K., and B.D.F. performed the NGS data analysis; U.B., F.P.D., and B.D.F. wrote the manuscript with input from T.B., M. Buehler, E.P.-V., G.N., E.T., and G.R.
Acknowledgments
We thank Kathrin Wagner, Daniel Kaiser, and Andreas Katopodis from Novartis Institutes for Biomedical Research (NIBR) for purification of PBMCs and isolation of T cells. We thank Shola Richards, Ieuan Clay, Caroline Gubser Keller, Sarah Brasa, Remi Terranova, and Ivan Galimberti from NIBR for discussions on data analysis and interpretation. We thank Yi Yang from NIBR for Cas9-gRNA vector. We thank Stephen Helliwell for comments on the manuscript. Research in the E.T., P.C., and G.N. laboratories was funded by Telethon grant to E.T. (GGP15257A).
Published: November 10, 2016
Footnotes
Supplemental Information includes Supplemental Experimental Procedures, five figures, and three tables and can be found with this article online at http://dx.doi.org/10.1016/j.stemcr.2016.10.004.
Contributor Information
Francesco Paolo Di Giorgio, Email: francesco.di_giorgio@novartis.com.
Barna D. Fodor, Email: barna.fodor@novartis.com.
Accession Numbers
Exome sequencing data have been deposited at the European Genome-Phenome Archive (EGA, https://ega-archive.org), under accession number EGAS00001001737. The data are accessible to the FXS research community via the controlled access procedure of the EGA.
Supplemental Information
References
- Avitzour M., Mor-Shaked H., Yanovsky-Dagan S., Aharoni S., Altarescu G., Renbaum P., Eldar-Geva T., Schonberger O., Levy-Lahad E., Epsztejn-Litman S. FMR1 epigenetic silencing commonly occurs in undifferentiated fragile X-affected embryonic stem cells. Stem Cell Rep. 2014;3:699–706. doi: 10.1016/j.stemcr.2014.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basuta K., Schneider A., Gane L., Polussa J., Woodruff B., Pretto D., Hagerman R., Tassone F. High functioning male with fragile X syndrome and fragile X-associated tremor/ataxia syndrome. Am. J. Med. Genet. A. 2015;167A:2154–2161. doi: 10.1002/ajmg.a.37125. [DOI] [PubMed] [Google Scholar]
- Biancalana V., Glaeser D., McQuaid S., Steinbach P. EMQN best practice guidelines for the molecular genetic testing and reporting of fragile X syndrome and other fragile X-associated disorders. Eur. J. Hum. Genet. 2015;23:417–425. doi: 10.1038/ejhg.2014.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers S.M., Fasano C.A., Papapetrou E.P., Tomishima M., Sadelain M., Studer L. Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat. Biotechnol. 2009;27:275–280. doi: 10.1038/nbt.1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi J., Lee S., Mallard W., Clement K., Tagliazucchi G.M., Lim H., Choi I.Y., Ferrari F., Tsankov A.M., Pop R. A comparison of genetically matched cell lines reveals the equivalence of human iPSCs and ESCs. Nat. Biotechnol. 2015;33:1173–1181. doi: 10.1038/nbt.3388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colak D., Zaninovic N., Cohen M.S., Rosenwaks Z., Yang W.Y., Gerhardt J., Disney M.D., Jaffrey S.R. Promoter-bound trinucleotide repeat mRNA drives epigenetic silencing in fragile X syndrome. Science. 2014;343:1002–1005. doi: 10.1126/science.1245831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Esch C.E., Ghazvini M., Loos F., Schelling-Kazaryan N., Widagdo W., Munshi S.T., van der Wal E., Douben H., Gunhanlar N., Kushner S.A. Epigenetic characterization of the FMR1 promoter in induced pluripotent stem cells from human fibroblasts carrying an unmethylated full mutation. Stem Cell Rep. 2014;3:548–555. doi: 10.1016/j.stemcr.2014.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eiges R., Urbach A., Malcov M., Frumkin T., Schwartz T., Amit A., Yaron Y., Eden A., Yanuka O., Benvenisty N. Developmental study of fragile X syndrome using human embryonic stem cells derived from preimplantation genetically diagnosed embryos. Cell Stem Cell. 2007;1:568–577. doi: 10.1016/j.stem.2007.09.001. [DOI] [PubMed] [Google Scholar]
- Greco C.M., Berman R.F., Martin R.M., Tassone F., Schwartz P.H., Chang A., Trapp B.D., Iwahashi C., Brunberg J., Grigsby J. Neuropathology of fragile X-associated tremor/ataxia syndrome (FXTAS) Brain. 2006;129:243–255. doi: 10.1093/brain/awh683. [DOI] [PubMed] [Google Scholar]
- Hagerman R.J., Hull C.E., Safanda J.F., Carpenter I., Staley L.W., O'Connor R.A., Seydel C., Mazzocco M.M., Snow K., Thibodeau S.N. High functioning fragile X males: demonstration of an unmethylated fully expanded FMR-1 mutation associated with protein expression. Am. J. Med. Genet. 1994;51:298–308. doi: 10.1002/ajmg.1320510404. [DOI] [PubMed] [Google Scholar]
- Hagerman R.J., Leehey M., Heinrichs W., Tassone F., Wilson R., Hills J., Grigsby J., Gage B., Hagerman P.J. Intention tremor, parkinsonism, and generalized brain atrophy in male carriers of fragile X. Neurology. 2001;57:127–130. doi: 10.1212/wnl.57.1.127. [DOI] [PubMed] [Google Scholar]
- Liu J., Koscielska K.A., Cao Z., Hulsizer S., Grace N., Mitchell G., Nacey C., Githinji J., McGee J., Garcia-Arocena D. Signaling defects in iPSC-derived fragile X premutation neurons. Hum. Mol. Genet. 2012;21:3795–3805. doi: 10.1093/hmg/dds207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loesch D.Z., Sherwell S., Kinsella G., Tassone F., Taylor A., Amor D., Sung S., Evans A. Fragile X-associated tremor/ataxia phenotype in a male carrier of unmethylated full mutation in the FMR1 gene. Clin. Genet. 2012;82:88–92. doi: 10.1111/j.1399-0004.2011.01675.x. [DOI] [PubMed] [Google Scholar]
- Park C.Y., Halevy T., Lee D.R., Sung J.J., Lee J.S., Yanuka O., Benvenisty N., Kim D.W. Reversion of FMR1 methylation and silencing by editing the triplet repeats in fragile X iPSC-derived neurons. Cell Rep. 2015;13:234–241. doi: 10.1016/j.celrep.2015.08.084. [DOI] [PubMed] [Google Scholar]
- Pecho-Vrieseling E., Rieker C., Fuchs S., Bleckmann D., Esposito M.S., Botta P., Goldstein C., Bernhard M., Galimberti I., Muller M. Transneuronal propagation of mutant huntingtin contributes to non-cell autonomous pathology in neurons. Nat. Neurosci. 2014;17:1064–1072. doi: 10.1038/nn.3761. [DOI] [PubMed] [Google Scholar]
- Pietrobono R., Tabolacci E., Zalfa F., Zito I., Terracciano A., Moscato U., Bagni C., Oostra B., Chiurazzi P., Neri G. Molecular dissection of the events leading to inactivation of the FMR1 gene. Hum. Mol. Genet. 2005;14:267–277. doi: 10.1093/hmg/ddi024. [DOI] [PubMed] [Google Scholar]
- Rousseau F., Heitz D., Tarleton J., MacPherson J., Malmgren H., Dahl N., Barnicoat A., Mathew C., Mornet E., Tejada I. A multicenter study on genotype-phenotype correlations in the fragile X syndrome, using direct diagnosis with probe StB12.3: the first 2,253 cases. Am. J. Hum. Genet. 1994;55:225–237. [PMC free article] [PubMed] [Google Scholar]
- Sheridan S.D., Theriault K.M., Reis S.A., Zhou F., Madison J.M., Daheron L., Loring J.F., Haggarty S.J. Epigenetic characterization of the FMR1 gene and aberrant neurodevelopment in human induced pluripotent stem cell models of fragile X syndrome. PLoS One. 2011;6:e26203. doi: 10.1371/journal.pone.0026203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjekloca L., Pauwels K., Pastore A. On the aggregation properties of FMRP—a link with the FXTAS syndrome? FEBS J. 2011;278:1912–1921. doi: 10.1111/j.1742-4658.2011.08108.x. [DOI] [PubMed] [Google Scholar]
- Smeets H.J., Smits A.P., Verheij C.E., Theelen J.P., Willemsen R., van de Burgt I., Hoogeveen A.T., Oosterwijk J.C., Oostra B.A. Normal phenotype in two brothers with a full FMR1 mutation. Hum. Mol. Genet. 1995;4:2103–2108. doi: 10.1093/hmg/4.11.2103. [DOI] [PubMed] [Google Scholar]
- Solvsten C., Nielsen A.L. FMR1 CGG repeat lengths mediate different regulation of reporter gene expression in comparative transient and locus specific integration assays. Gene. 2011;486:15–22. doi: 10.1016/j.gene.2011.06.034. [DOI] [PubMed] [Google Scholar]
- Sutcliffe J.S., Nelson D.L., Zhang F., Pieretti M., Caskey C.T., Saxe D., Warren S.T. DNA methylation represses FMR-1 transcription in fragile X syndrome. Hum. Mol. Genet. 1992;1:397–400. doi: 10.1093/hmg/1.6.397. [DOI] [PubMed] [Google Scholar]
- Tabolacci E., Moscato U., Zalfa F., Bagni C., Chiurazzi P., Neri G. Epigenetic analysis reveals a euchromatic configuration in the FMR1 unmethylated full mutations. Eur. J. Hum. Genet. 2008;16:1487–1498. doi: 10.1038/ejhg.2008.130. [DOI] [PubMed] [Google Scholar]
- Takahashi K., Tanabe K., Ohnuki M., Narita M., Ichisaka T., Tomoda K., Yamanaka S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- Telias M., Segal M., Ben-Yosef D. Neural differentiation of Fragile X human Embryonic Stem Cells reveals abnormal patterns of development despite successful neurogenesis. Dev. Biol. 2013;374:32–45. doi: 10.1016/j.ydbio.2012.11.031. [DOI] [PubMed] [Google Scholar]
- Urbach A., Bar-Nur O., Daley G.Q., Benvenisty N. Differential modeling of fragile X syndrome by human embryonic stem cells and induced pluripotent stem cells. Cell Stem Cell. 2010;6:407–411. doi: 10.1016/j.stem.2010.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verkerk A.J., Pieretti M., Sutcliffe J.S., Fu Y.H., Kuhl D.P., Pizzuti A., Reiner O., Richards S., Victoria M.F., Zhang F.P. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell. 1991;65:905–914. doi: 10.1016/0092-8674(91)90397-h. [DOI] [PubMed] [Google Scholar]
- Wenzel H.J., Hunsaker M.R., Greco C.M., Willemsen R., Berman R.F. Ubiquitin-positive intranuclear inclusions in neuronal and glial cells in a mouse model of the fragile X premutation. Brain Res. 2010;1318:155–166. doi: 10.1016/j.brainres.2009.12.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willemsen R., Levenga J., Oostra B.A. CGG repeat in the FMR1 gene: size matters. Clin. Genet. 2011;80:214–225. doi: 10.1111/j.1399-0004.2011.01723.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wohrle D., Salat U., Glaser D., Mucke J., Meisel-Stosiek M., Schindler D., Vogel W., Steinbach P. Unusual mutations in high functioning fragile X males: apparent instability of expanded unmethylated CGG repeats. J. Med. Genet. 1998;35:103–111. doi: 10.1136/jmg.35.2.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.