

Abstract
Introduction
Fusion of BCR-ABL1 genes causes chronic myeloid leukemia (CML). As a reliable marker of disease burden, it also serves as the target of tyrosine kinase inhibitors (TKIs). New more sensitive molecular diagnostic tools for BCR-ABL1 can contribute to therapeutic decision-making, especially in considering drug discontinuation for patients enjoying prolonged deep molecular response.
Areas covered
Several novel platforms are transforming CML molecular diagnostics to enable faster point-of-care devices, better understanding of clonal diversity and resistance mutations. Here, we review these molecular platforms, knowing implementation in other hematological malignancies will ensue.
Expert commentary
Treatment with TKI in CML is the first example of a highly effective targeted therapy. Monitoring of BCR-ABL1 mRNA is standard in assessing disease burden being highly predictive of outcomes recommended by both ELN and NCCN; however, studies has demonstrated poor adherence to these recommendations. In both clinical practice and assay performance, further optimizing of BCR-ABL1 monitoring can be envisioned including point-of-care methods for increased availability of rapid, standardized testing and increasingly sensitive molecular assays that allow for quantification of MRD and detecting resistance mutations.
Keywords: chronic myeloid leukemia, monitoring, BCR-ABL1 transcript levels, molecular diagnostics, tyrosine kinase inhibitor resistance mutations
1.0 INTRODUCTION
The BCR-ABL1 fusion gene causes chronic myeloid leukemia (CML), and its expressed chimeric mRNA is a marker of disease burden, while its protein product is the therapeutic target of tyrosine kinase inhibitors (TKIs). The advent of TKIs simply revolutionized the therapy of CML, which previously was only curative with allogeneic transplantation.
The survival outcomes in chronic phase CML patients are now similar to those in the general population [1]. CML patients treated with imatinib have excellent response rates, reportedly achieving a complete cytogenetic response (CCyR) at 1 and 5 years in 69% and 87%, respectively [2]. In the IRIS trial, ~90% of patients treated with imatinib were still alive after 6 years of follow-up [3]. In comparison to imatinib, second-generation TKIs (ie, dasatinib and nilotinib) have shown even higher rates of short term response, including improved rates of achieving 3 month early molecular response (EMR), 12 month cytogenetic remissions (CCyR) and major molecular response (MMR). However, thus far a clear overall survival advantage for second generation TKIs has not been clearly demonstrated thus far when compared to imatinib [4-8].
The international scale (IS) was implemented in 2006 with detailed recommendations for standardized RQ-PCR protocols and international validation control materials [9]. When patients achieve a major molecular response (MMR; BCR-ABL1/control gene ratio ≤0.1% on the International Scale [IS]), resistance and progression are unlikely[10]. This is possible through molecular monitoring for BCR-ABL1 mRNA from the peripheral blood and marrow, which is highly sensitive as well as reliable measure of disease burden, and correlates well with outcome. With the sensitive diagnostics modalities, when patients achieve complete molecular remission (CMR), it may possible to further identify patients that can discontinue TKI therapy [11-13]. Current trend in molecular reporting include the use of MR 4 or 4.5 to indicate the deepest log reduction in molecular response based on IS. Initial reports show approximately 40% of patients who sustained either CMR or MR 4.5 (depending on the specific studies’ endpoint) remained in molecular remission after stopping TKI therapy for up to two years [11, 14]. Currently, multiple clinical trials with the goal of TKI discontinuation are underway and will help define the optimal setting for when a patient may attempt treatment-free remission.
Failure in TKI therapy can necessitate switching of TKIs, or eventually moving to transplant or a clinical trial. The NCCN and ELN guidelines [15, 16] have divided failure to reach treatment milestones into primary resistance, toxicity, and resistance/progression (after initial achievement of guidelines). In patients with imatinib resistance, BCR-ABL1 mutations may be detected in approximately half [17, 18]. Assorted point mutations in the ABL tyrosine kinase domain have been described which confers different resistance characteristics by either directly inhibit TKI binding or favor the active conformation of the kinase [19].
In approximately 30-50% of patients in whom frontline imatinib fails, salvage therapy containing second-generation TKIs can produce a CCyR [20-22]. When initial therapy with a second-generation TKI fails, salvage with third-generation TKIs (bosutinib or ponatinib) and even with another second-generation TKI can still produce a major and complete cytogenetic remission but at lower likelihoods [23, 24]. Patients responding to second-line salvage therapy generally relapse and often will show a new mutation. Although treatment with appropriate TKIs can be considered, if cure is the goal, allogeneic transplant remains the best option for highly resistant disease with a proven track record for generating new mutations [25].
The realm of molecular pathology has exploded in the past decade, and the constant push to create “better” technologies (roughly defined as faster, more sensitive, and cheaper) to detect disease has transformed the field. There are three main diagnostic utilities in CML with minor overlapping testing strategies. We will describe both the “tried and true” methods in addition to up and coming technologies that may change CML management practices in the future.
2.0 Molecular Testing for the Diagnosis of CML
The Philadelphia chromosome (Ph) is unique to CML, and indicates a reciprocal translocation, t(9;22) between the BCR and ABL1 genes. Varied breakpoint regions lead to the production of different isoforms of the fusion gene products. In CML, the fusion transcripts are formed when exons 13 or 14 of the BCR gene translocate to exon 2 of the ABL1 gene, notated as e13a2 (b2a2) and e14a2 (b3a2), respectively and results in a 210-kDa fusion protein (commonly referred to as “p210”) [26]. Variant Ph translocations have been described in 5-10% of CML patients [27] and are more often associated with partial deletion of chromosome 9q [28]. In contrast, Ph-positive acute lymphoblastic leukemia typically results from the fusion between exon 1 of BCR gene with exon 2 of ABL1 gene, notated as e1a2, and yields a 190-kDa fusion protein (commonly referred to as the “p190”, and seen in <1% of CML patients) [26, 29]. Depending on the specific molecular laboratory, most have their BCR-ABL1 quantitative RT-PCR (qRT-PCR) assays designed to detect both p210 transcripts (e13a2 and e14a2) in a single reaction. In general, separate primers are needed for detection of the p190 transcript.
Methods of Ph detection are compared in Table I. Conventional cytogenetic karyotyping may detect the Ph but is generally not the diagnostic modality of choice due to its requirement of highly skilled staff, require culturing of marrow cells [30], have the longest turnaround times, and require evaluation of ≥20 metaphases to be useful [16]. Despite these limitations, conventional cytogenetics should still be routinely performed especially at diagnosis to detect additional clonal abnormalities. Fluorescence in situ hybridization (FISH) is more sensitive than karyotyping in the detection of the BCR-ABL fusion and can be performed on dividing and non-dividing cells in marrow, peripheral blood, and tissue [30]. One advantage of FISH is that it can detect some very rare BCR-ABL translocations that are not detectable by the vast majority of commercial and laboratory developed qRT-PCR assays. Labs with specialized multiplex-PCR utilizing hundreds of primers can detect rare BCR-ABL1 translocations in a single assay [31].
Table I.
Relative sensitivities of molecular diagnostic tests for CML
| Method | Target | Sensitivity | Advantages | Disadvantages |
|---|---|---|---|---|
| Conventional Cytogenetics | Chromosomal translocation = t(9;22) | 10%-5% | The gold standard, and detection of other large chromosomal changes. | Require dividing cells and highly skilled technician. Very labor and time intensive. Miss cryptic translocations. |
| Fluorescence in situ hybridization (FISH) | BCR-ABL1 DNA | 0.1%-5% | Can use wider range of specimens (PB, BM, FFPE). Can detect cryptic translocations. | Relatively insensitive compared with qRT-PCR. Requires skilled technicians. Highly specific to targeted region and will miss other chromosomal changes. |
| Quantitative reverse transcription polymerase chain reaction (qRT-PCR) | BCR-ABL1 mRNA | 0.001%-0.01% | Very sensitive. Can use wider range of specimens (PB, BM, FFPE). Can detect cryptic translocations. | Not well standardized across laboratories. More suceptable to contamination or RNA degradation issues. |
CML = chronic myelogenous leukemia, PB = peripheral blood, BM = bone marrow, FFPE = formalin fixed parraffin embedded tissue
2.1 Point of care and automated PCR
The demand for a simpler workflow by clinical laboratories have pushed the advancement of automated PCR molecular testing [32]. Cartridge based designs have reduced the need for high complexity manipulations and dramatically reduced the technologist bench time. These cartridge-based systems have been available since 2007 and showed that detection of BCR-ABL1 by these platforms remain highly sensitive and robust [33]. Novel designs for smaller portable point of care devices that utilize combinations of super tiny extraction chambers, chemistry and microfluidics that allow for PCR (including some with isothermal PCR chemistry), and end point probe hybridization are available for molecular diagnosis of infectious diseases and potential application to rapid diagnosis of CML can be implemented [34].
2.2 Isothermal PCR
The concept of isothermal PCR for molecular testing was initially reported in 1990 [35] for infectious disease testing and many of the current platforms utilizing this technology has been associated with a point of care devices or made for use in resource poor areas [36, 37]. Soon after in 1993 BCR-ABL1 fusion transcript detection by isothermal PCR termed nuclei acid sequence based amplification (NASBA) [38] and subsequently by loop mediated isothermal amplification (LAMP) [39] followed suit. NASBA functions by directly amplification of RNA in a series of initial steps utilizing reverse transcriptase to create a double stranded DNA molecule then utilizing T7 RNA polymerase for continuous amplification of complementary RNA transcripts. LAMP utilizes the Bst polymerase enzyme and specialized primers, a combination allowing for high replication activity and increased strand displacement, to increase the amount of DNA produced, which can be grossly seen by DNA stains (Figure 4). For BCR-ABL1 detection by LAMP and other isothermal PCR assays, an initial step with reverse transcriptase is required [39].
Figure 4.
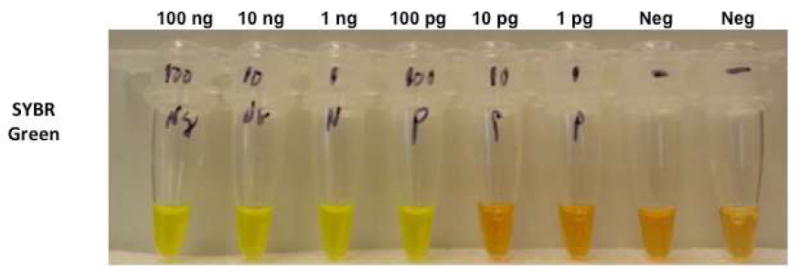
Isothermal PCR reaction with SYBR green.
From left to right, reactions with input of 100ng, 10ng, 1ng, and 100pg K562 RNA demonstrate grossly visible change in color indicating positive fusion signals after one hour at 63°c. The mid right two reactions contained 10pg and 1pg of K562 RNA did not show an obvious grossly visible change in color. The two far right reactions, have no K562 RNA and show no color change.
2.3 DNA PCR
Detection of BCR-ABL1 mRNA was favored over detection of the Ph translocation, mainly because the mRNA was much more abundant and the BCR and ABL breakpoint region is potentially vast, requiring multiple sets of primers to be used to first find the patient’s particular breakpoint. However amplification of DNA has several advantages, especially in sample stability and standardizing to a cell number since the actual number of cells can accurately be determined by a control gene copy number). In DNA PCR, after initial PCR interrogation with multiple sets of primers, the fusion PCR product can be sequenced, and breakpoint specific primers can be generated for use in subsequent assays (similar to IgH VDJ PCR quantification). This approach has a theoretical level of sensitivity down to 1:106 in many cases greater than qRT-PCR. In a preliminary small study of 38 samples, comparing nested DNA PCR to qRT-PCR, there appears to be good correlation between the two assays even in samples for MRD, and there is potentially higher sensitivity for the nested DNA PCR assay to detect BCR-ABL1, as 16 samples with undetectable mRNA transcripts, 8 were detectable by DNA qPCR [40]. In a larger study (92 patients with CML) from the same group in 2014, DNA qPCR was more precise at the level of minimal residual disease [41].
2.4 In situ flow/RT-PCR
Detection of the BCR-ABL1 fusion protein through flow based assays for labeled immunobeads have also shown potential as a possible avenue for diagnosis. In this approach, BCR-ABL1 fusion proteins bind to immunobeads coated with specific antibodies, then detector antibodies are added which bind specifically to another part of the BCR-ABL1 fusion protein. A sandwich complex comprised of immunobead-BCR-ABL1 fusion protein-detector antibody is generated which can then be detected on a flow cytometer. Early studies have shown comparable sensitivity, positive predictive values and negative predictive values but slightly lower specificity when comparing this immunobead flow based assay to molecular and cytogenetic testing [42].
3.0 Molecular Testing for Monitoring Treatment response or TKI Discontinuation
Detection of the chimeric BCR-ABL1 mRNA by qRT-PCR, is currently the most sensitive assay for monitoring of CML and can detect one CML cell in approximately 100,000 to 1,000,000 total cells [43]. To control for specimen quality of the RNA and to obtain semi-quantitative results, the quantity of BCR-ABL1 transcript is determined in relation to an endogenous control gene, such as BCR, ABL1, or GUSB. BCR-ABL1 qRT-PCR testing for CML monitoring can be performed on peripheral blood specimens enabling the most convenient and sensitive test clinical workflow. Consensus guidelines from European LeukemiaNet (ELN) and National Comprehensive Cancer Network (NCCN) recommends qRT-PCR be utilized for disease monitoring in all patients with CML [15, 16, 43].
NCCN and ELN have defined several clinically significant levels of measurable disease defined in CML (Figure 1). Complete hematologic response, the earliest milestone of response, is defined as normalization of peripheral blood counts and can be achieved in ~90% of patients by three months of initiating TKI therapy for patients in chronic phase disease. Various levels of cytogenetic response are determined by marrow metaphase chromosome analysis (using a minimum requirement of ≥20 metaphases to be analyzed) [16].
Figure 1.
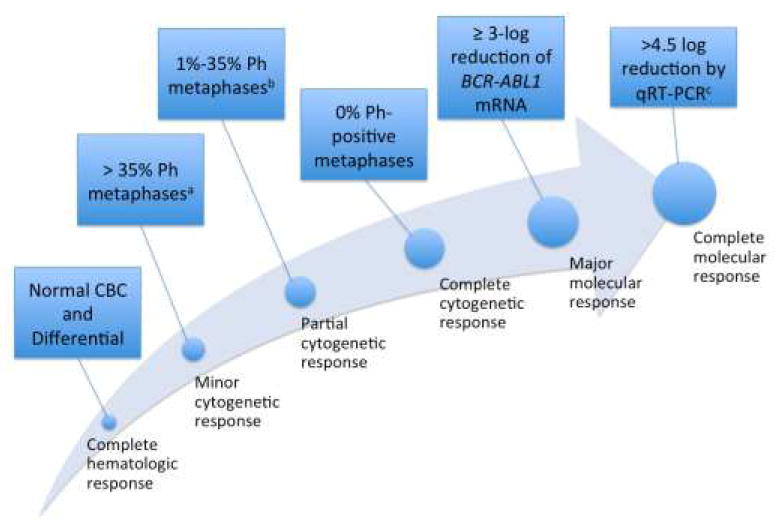
Schematic representation of the increasing “levels of response” outlines by National Comprehensive Cancer Network Response Criteria for CML [27]
CBC, complete blood count; Ph, Philadelphia chromosome; qRT-PCR, quantitative reverse transcription polymerase chain reaction.
a For cytogenetic response, ≥20 metaphases must be studied.
b Major cytogenetic response includes partial and complete responses.
c Per the National Comprehensive Cancer Network, complete molecular remission requires a sample where a ≥4.5-log depletion can be assessed.
The higher levels of response, MMR and complete molecular remission (CMR) require molecular testing by qRT-PCR. The concept of a MMR was introduced in the IRIS trial, defined as a ≥3-log reduction in the standardized baseline taken to represent 100% on the IS, and became an established response metric when achievement of MMR correlated with better long-term outcomes [10].
A rising need for more sensitive BCR-ABL detection is partially driven by interest in TKI discontinuation in patients with very low (MR 4.0 or below) or undetectable BCR-ABL1. After discontinuing imatinib, more than half of the patients show evidence of molecular relapse and usually before 6 months despite previously sustained undetectable BCR-ABL1 transcripts [11, 14], which indicates that in most patients there are expandable leukemic cells existing below the limit of detection of standard qRT-PCR methods. Although the hope is that increasingly sensitive methods of detection can better differentiate patients with a high risk of molecular relapse and several techniques designed to increase assay sensitivity for CML detection have shown promise. However, discontinuation of TKI therapy in CML is not a current recommendation as clinical trials are still underway and long term follow-up of these initial patients have not been studied.
3.1 Digital PCR
Digital PCR technology involves partitioning a specimen into hundreds or thousands (or more) of tiny droplets/chambers, so that each partition either show the presence or absence of the target template and yields a binary, or “digital,” result (positive or negative; Figure 2). The method represents a more accurate alternative method of direct quantitative detection of rare transcripts and have been shown to increase the limit of detection by 1-2 logs [44, 45]. When viewed as a signal to noise problem, finding the “needle” (e.g. the rare transcript) in the “haystack” (e.g. all the background normal signal) by digital PCR, makes good sense as the partitions are sorted so that individual bins each contain one needle (BCR-ABL1 molecule) or hay (RNA without BCR-ABL). The presence or absence of BCR-ABL1 transcripts allows for quantification of rare target sequences through application of Poisson distribution. The method has additional advantages since calibration curves are not needed for an absolute numerical value and have lower technical variation (greater similarity between repeat runs). Although still early in its application into clinical trials for CML, recent work demonstrates the sensitivity, precision, and feasibility of digital PCR are comparable with qRT-PCR in the detection of BCR-ABL1 [46, 47].
Figure 2.
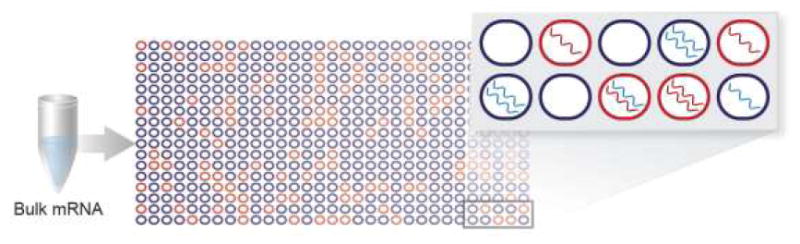
Digital PCR. Red wells indicate target (BCR-ABL1) mRNA, and blue wells indicate background (wild-type) mRNA. Individual wells may contain exclusively target or background mRNA, or they may contain a mixture of target and background mRNA.
mRNA, messenger RNA; PCR, polymerase chain reaction.
4.0 Molecular Testing When There is Resistance or Treatment Failure
4.1 Mutation detection
Resistance in CML is sub-divided into two categories. Primary resistance occurs when treatment are not initially met, whereas secondary resistance occurs when disease relapses after meeting treatment milestones (as defined by ELN and NCCN guidelines). In most cases, there is correlation between response and disease phase, with the best responses (thus, lowest rates of resistance) observed in patients with new CML diagnosis in chronic-phase having a low Sokal score, and the poorest responses observed in patients presenting with blast crisis [48-51][31-34]. In approximately 50% of patients with secondary resistance, base pair mutations in the ABL kinase domain dull the ability of TKIs in their inhibition of aberrant BCR-ABL1 kinase activity [52]. Acquisition of these mutations and loss of response are associated with increased risk of progression to advanced-phase disease.
ABL kinase domain mutation screening is limited by the sensitivity of currently available assays. Most direct nucleotide sequencing methodologies can detect an ABL tyrosine kinase domain mutation at an allelic frequency of 10% to 20% [9]. Identification of point mutations before the onset of frank resistance are hindered by the low sensitivity of these assays and up-front mutation testing is not recommended in patients with newly diagnosed CML in chronic phase disease since ABL mutations are quite rare at this time.
In patients with a rising BCR-ABL1 transcript (without justification of poor adherence), monitoring of ABL tyrosine kinase domain mutation during therapy is recommended. In patients with a > 2-fold increase in BCR-ABL1, more than 50% had detectable mutations, while in contrast, mutations were detected in less than 1% of patients with stable or decreasing BCR-ABL1 [53]. These observations came from a lab dedicated to CML, and since most labs have the overwhelming technical sophistication of such specialized centers, the NCCN guidelines suggest mutation testing in cases with BCR-ABL1 at least 10-fold increased and with a loss of MMR [16]. Therefore, mutation screening is reasonable for patients with chronic-phase disease with increasing BCR-ABL1 levels (especially those increasing to MMR), those who have not achieved treatment milestones, and patients with advanced-phase disease.
Rising BCR-ABL1 levels indicate either “bad biology” (resistance) or “bad behavior” (poor adherence to medication) [54, 55]. There are conflicting recommendations on whether there should be an early intervention in the form of changing TKIs for a rising BCR-ABL1 from ELN and NCCN. Whereas ELN recommends no change at 3 months despite rising BCR-ABL1, the NCCN recommends that there can be a change. Despite the slightly different suggestions, both agree a clinical trial is needed to see if an early change in TKI therapy would improve outcomes. Until this data is available it is our humble opinion that a change in treatment strategy should be considered when there is increasing BCR-ABL1 transcript levels with a mutation predicting relative or absolute resistance (e.g., T315I or E255V on imatinib).
4.2 Next generation sequencing
Next-generation sequencing (NGS) collectively refers to high-throughput methods that can sequence large numbers of RNA or DNA molecules and can be digitally tabulated. These platforms result in absolute numbers of sequencing reads and allow for discovery of sequences occurring at lower frequencies even when seen in a heterogeneous background. An important distinction to this technology is the massive quantity of information generated and therefore sophisticated bioinformatics and computational methods are required for analysis. Compared to conventional Sanger sequencing, with a limit of sensitivity of approximately 10% to 20%, next generation sequencing have shown sensitivities in the range of 1-15% and reveal a complex clonal architecture consisting of a dynamic mix of polyclonal and compound mutations [56]. However, confident mutation detection is limited to approximately 1%, mainly restricted by errors generated in the NGS pipeline [57, 58].
4.3 Duplex sequencing
An improvement beyond “next generation” sequencing, this novel methodology boasts more accurate and less error prone by “duplex” sequencing. This method takes advantage of the opposite DNA strand sequence as a built in control for technical errors. Simultaneously sequencing both strands together then analysis will require that true mutations show complementary bases to be in both strands while random mutations will be only seen in one strand. With this algorithm, the probability for a false positive call will require a technical error to put an exact complementary base(s) in exactly the complementary site-which is an exceedingly unlikely event, thereby reducing the error rates in several orders of magnitude [59].
5.0 To infinity and beyond: NEW METHODS ON THE HORIZON
Driven by patient care needs, scientific curiosity, or often just for the proverbial scientific “kick,” labs are always innovating to develop molecular assays that are more robust, more sensitive, and more methodologically elegant.
5.1 Extreme PCR
PCR typically takes several hours of thermocycling. However, by optimizing primer, enzyme, magnesium concentration and buffers and changing the classic PCR reaction vessels to microfluidics or to materials that allow for very rapid temperature changes, extreme PCR platforms have shown that reaction speeds of the enzymes were not the limiting factor [60]. In extreme PCR, the time required for a single traditional PCR cycle is reduced to less than one minute (Figure 3) and the complete set of PCR cycles can be reduced to less than 10 mins [60, 61].
Figure 3.
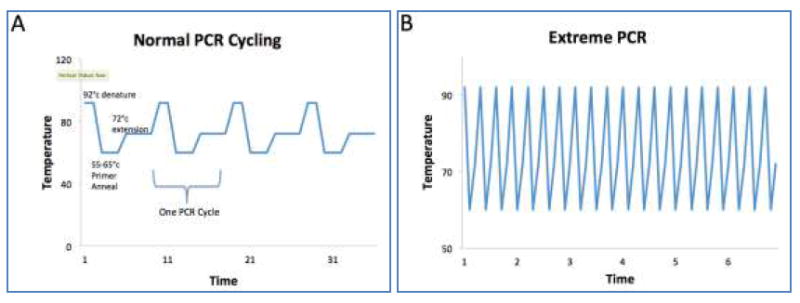
Cycling differences between traditional versus extreme PCR
A comparison of traditional PCR (Panel A) and extreme PCR (Panel B) demonstrates the key concept in this method is to push the limits of the reaction time by allowing for traditional PCR cycle steps to occur faster. Traditional PCR requires time to ramp up to 92°c for denaturation, ramp down to primer annealing temperatures between 55-66°c, and ramp up to 72°c for extension of the new DNA; making a typical PCR reaction 4-7 minutes per cycle. By comparison, extreme PCR do not have the lag time required for temperature changes and the PCR cocktails are optimized for near instant reaction speeds allowing for a PCR cycle to occur in less than one minute.
5.2 Real-time sequencing
The “next generation” of next generation sequencing platforms enables reads of the nucleic acid sequences in real time compare to contemporary sequencing techniques. Currently, two systems are available: the first uses an tiny wells embedded with an ultra-sensitive metallic light detection system into the glass of the floor and a single polymerase attached to the bottom of this well to allowing for extension of a DNA molecule in close approximation to the light sensor metallic plate. Fluorescent-labeled phosphor-linked nucleotides are added which extends the DNA strand, resulting in a momentary burst of colored light from the excited fluorochrome that can be captured and interpreted as “real-time” sequences [62, 63]. A second platform utilizes a nanopore protein embedded onto a synthetic polymer membrane (Figure 5). The nanopore protein has DNA helicase activity at one opening and the internal passage or “pore” is so small that as individual bases pass through they emit small measurable changes to the electrical field of the synthetic polymer membrane corresponding to each specific nucleotide base [64].
Figure 5.
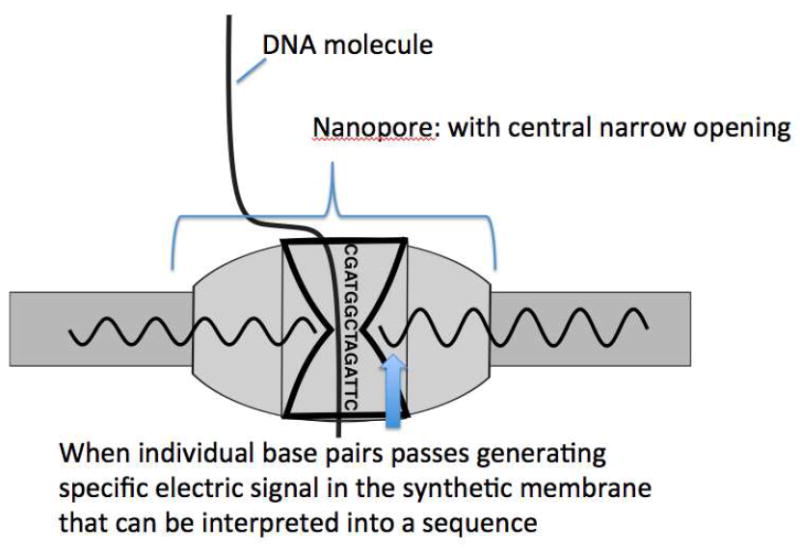
Real Time Sequencing [66]
With the nanopore technology, the enzyme is embedded into a synthetic electrical nanomembrane which senses very small changes in electrical pulses resulting from the different base pairs of the DNA molecule passing through the narrow channel “pore”.
5.3 Quantum chemistry
Nano-particles called quantum dots have been used as highly sensitive biosensors. Sharma et al. created a device capable of detecting BCR-ABL DNA molecules with chitosan–cadmium-telluride quantum dots deposited onto indium-tin-oxide coated glass substrate, then immobilized a small oligonucleotide probe targeting BCR-ABL1. BCR-ABL1 bound to quantum dot labeled probes can then be interrogated by laser, which can also quantify the amount of bound BCR-ABL. Preliminary studies show this assay has been able to detect as low as 2.56 pM concentrations of complementary DNA molecules and the device chamber can be rinsed for several reuses [65].
6.0 CONCLUSION
Molecular diagnostics has become critical in the management of patients with CML to help predict outcomes and guide therapeutic decisions. Common molecular diagnostic tools we have described in this article that are routinely employed include qRT-PCR done by most molecular laboratories, mutation testing for TKI resistance and targeted amplification based next generation sequencing, which is available in some specialized laboratories. Digital PCR and isothermal PCR may be next on the horizon for clinical practice implementation. Duplex sequencing, real-time sequencing and all of the “to infinity and beyond“ molecular diagnostic platforms we have described here, will have some time before implementation into clinical practice. Fortunately it has also evolved into a relatively non-invasive method (peripheral blood) such that marrow is typically not needed in the monitoring stages and considered relatively low in cost. CML continues to be the poster child of target therapies in this era truly going from bench to bedside, and back again.
7.0 Expert commentary
Treatment with TKI in CML is a superb example of targeted therapy, and the use of the sensitive monitoring of disease burden to determine clinical efficacy is a model for all malignancies. PCR testing of BCR-ABL1 mRNA in peripheral blood is the standard for disease monitoring in CML, and both ELN and NCCN guidelines use this assay to establish treatment milestones to guide clinical decision-making. Besides routine monitoring, the field is now facing two very different challenges. First, several studies have demonstrated a surprising poor adherence to testing recommendations, and thus the development of rapid, point of care assays are seen as a potential remedy. Moreover, the recent realization that some patients with persistently no or low disease burdens can discontinue TKI and not subsequently relapse has lead investigators to look for ways to predict which patients can or cannot be offered discontinuation as an option. The “low hanging fruit” in such research is the development of more sensitive assays, either by advances in next generation sequencing (next next generation?), and digital PCR methods.
8.0 Five-year view
Advances in molecular platforms will lead to the development of faster, more sensitive, cheaper, and more user-friendly instruments and kits. The ability of improved BCR-ABL1 detection assays with sensitivity levels below that of conventional quantitative RT-PCR will be routinely used in the context of discontinuation of TKI, making this practice safer in regard to reducing the risk of molecular relapse. Point of care devices in the future will make diagnosis and monitoring of BCR-ABL1 transcripts in CML even easier for patients, and may well develop into home testing. New and upcoming sequencing platforms which are more accurate and sensitive will be important in detecting early resistance and will have an even greater impact on the understanding of clonal competition and the development of resistance.
Key issues.
CML is the “poster child” of how targeted therapy can be highly effective and can be coupled with molecular diagnostic tools.
Disease diagnosis and staging of CML currently includes standard morphology, FISH, karyotyping, and qRT-PCR.
Future molecular diagnostic methods for the diagnosis of CML include digital droplet PCR, point of care devices, isothermal PCR, DNA PCR, and flow cytometry for BCR-ABL1 fusion protein.
Molecular testing for monitoring treatment response or TKI discontinuation requires highly sensitive assays such as qRT-PCR.
Future molecular diagnostic methods for monitoring of CML may include which may provide a more accurate quantitative result and higher sensitivity.
Molecular testing when there is resistance or treatment failure includes BCR-ABL1 mutation testing with Sanger sequencing.
Future molecular diagnostic methods for treatment resistance testing may include use of next generation platforms, or duplex sequencing.
Very novel molecular diagnostic methods with future potential application to CML include real-time PCR sequencing, extreme PCR, and quantum chemistry.
Acknowledgments
Bret A Helton, Research technician, Radich Laboratory, Fred Hutchinson Cancer Research Center. Acknowledgment for his image used in Figure 4.
Funding
This work was partially funded by NIH/NCI (P01 CA 18029-37A1) Adult Leukemia Research Center - Sub-Project: Molecular Diagnostics.
CCS Yeung has received research funding from Gilead for unrelated research work. J Radich discloses consulting for Novartis, BMS Ariad and received research funding from Novartis.
Footnotes
Declaration of Interest
The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Contributor Information
Cecilia Ching Sze Yeung, Assistant Member, Fred Hutchinson Cancer Research Center Ringgold standard institution - Clinical Research Division, Seattle, Washington 98109-9024, United States; Assistant Professor, University of Washington School of Medicine Ringgold standard institution - Pathology, Seattle, Washington 98195-6340, United States.
Daniel Egan, Fred Hutchinson Cancer Research Center Ringgold standard institution - Clinical Research Division, Seattle, Washington 98109-9024, United States.
Jerald P Radich, Fred Hutchinson Cancer Research Center, Seattle, United States.
References
Reference annotations
-
*
Of interest
-
**
Of considerable interest
- 1.Sasaki K, et al. Relative survival in patients with chronic-phase chronic myeloid leukaemia in the tyrosine-kinase inhibitor era: analysis of patient data from six prospective clinical trials. Lancet Haematol. 2015;2(5):e186–93. doi: 10.1016/S2352-3026(15)00048-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Druker BJ, et al. Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. N Engl J Med. 2006;355(23):2408–17. doi: 10.1056/NEJMoa062867. [DOI] [PubMed] [Google Scholar]
- 3.Hochhaus A, et al. Six-year follow-up of patients receiving imatinib for the first-line treatment of chronic myeloid leukemia. Leukemia. 2009;23(6):1054–61. doi: 10.1038/leu.2009.38. [DOI] [PubMed] [Google Scholar]
- 4.Hughes TP, et al. Early molecular response predicts outcomes in patients with chronic myeloid leukemia in chronic phase treated with frontline nilotinib or imatinib. Blood. 2014;123(9):1353–60. doi: 10.1182/blood-2013-06-510396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jabbour E, et al. Early response with dasatinib or imatinib in chronic myeloid leukemia: 3-year follow-up from a randomized phase 3 trial (DASISION) Blood. 2014;123(4):494–500. doi: 10.1182/blood-2013-06-511592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kantarjian H, et al. Dasatinib versus imatinib in newly diagnosed chronic-phase chronic myeloid leukemia. N Engl J Med. 2010;362(24):2260–70. doi: 10.1056/NEJMoa1002315. [DOI] [PubMed] [Google Scholar]
- 7.Radich JP, et al. A randomized trial of dasatinib 100 mg versus imatinib 400 mg in newly diagnosed chronic-phase chronic myeloid leukemia. Blood. 2012;120(19):3898–905. doi: 10.1182/blood-2012-02-410688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Saglio G, et al. Nilotinib versus imatinib for newly diagnosed chronic myeloid leukemia. N Engl J Med. 2010;362(24):2251–9. doi: 10.1056/NEJMoa0912614. [DOI] [PubMed] [Google Scholar]
- 9.Hughes T, Branford S. Molecular monitoring of BCR-ABL as a guide to clinical management in chronic myeloid leukaemia. Blood Rev. 2006;20(1):29–41. doi: 10.1016/j.blre.2005.01.008. [DOI] [PubMed] [Google Scholar]
- 10.Hughes TP, et al. Frequency of major molecular responses to imatinib or interferon alfa plus cytarabine in newly diagnosed chronic myeloid leukemia. N Engl J Med. 2003;349(15):1423–32. doi: 10.1056/NEJMoa030513. [DOI] [PubMed] [Google Scholar]
- 11*.Ross DM, et al. Safety and efficacy of imatinib cessation for CML patients with stable undetectable minimal residual disease: results from the TWISTER study. Blood. 2013;122(4):515–22. doi: 10.1182/blood-2013-02-483750. This reference is of importance as it is one of the early large clincial trials which have attempted to stop TKI treatment in CML. [DOI] [PubMed] [Google Scholar]
- 12*.Branford S, et al. Early molecular response and female sex strongly predict stable undetectable BCR-ABL1, the criteria for imatinib discontinuation in patients with CML. Blood. 2013;121(19):3818–24. doi: 10.1182/blood-2012-10-462291. This reference is of importance as it is one of the early large clincial trials which have attempted to stop TKI treatment in CML. [DOI] [PubMed] [Google Scholar]
- 13.Takahashi N, et al. Discontinuation of imatinib in Japanese patients with chronic myeloid leukemia. Haematologica. 2012;97(6):903–6. doi: 10.3324/haematol.2011.056853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14*.Mahon FX, et al. Discontinuation of imatinib in patients with chronic myeloid leukaemia who have maintained complete molecular remission for at least 2 years: the prospective, multicentre Stop Imatinib (STIM) trial. Lancet Oncol. 2010;11(11):1029–35. doi: 10.1016/S1470-2045(10)70233-3. This reference is of importance as it is one of the early large clincial trials which have attempted to stop TKI treatment in CML. [DOI] [PubMed] [Google Scholar]
- 15**.Baccarani M, et al. European LeukemiaNet recommendations for the management of chronic myeloid leukemia: 2013. Blood. 2013;122(6):872–84. doi: 10.1182/blood-2013-05-501569. This reference is of critical importance as it is the practice guidelines for the European Leukemia network. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16**.NCCN, Clinical Practice Guidelines in Oncology. Chronic Myelogenous Leukemia. 2016 doi: 10.6004/jnccn.2009.0065. This reference is of critical importance as it is the practice guidelines for the European Leukemia network. [DOI] [PubMed] [Google Scholar]
- 17.Jabbour E, et al. Frequency and clinical significance of BCR-ABL mutations in patients with chronic myeloid leukemia treated with imatinib mesylate. Leukemia. 2006;20(10):1767–73. doi: 10.1038/sj.leu.2404318. [DOI] [PubMed] [Google Scholar]
- 18.Lahaye T, et al. Response and resistance in 300 patients with BCR-ABL-positive leukemias treated with imatinib in a single center: a 4.5-year follow-up. Cancer. 2005;103(8):1659–69. doi: 10.1002/cncr.20922. [DOI] [PubMed] [Google Scholar]
- 19.Reddy EP, Aggarwal AK. The ins and outs of bcr-abl inhibition. Genes Cancer. 2012;3(5-6):447–54. doi: 10.1177/1947601912462126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Milojkovic D, et al. Responses to second-line tyrosine kinase inhibitors are durable: an intention-to-treat analysis in chronic myeloid leukemia patients. Blood. 2012;119(8):1838–43. doi: 10.1182/blood-2011-10-383000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Giles FJ, et al. Nilotinib in imatinib-resistant or imatinib-intolerant patients with chronic myeloid leukemia in chronic phase: 48-month follow-up results of a phase II study. Leukemia. 2013;27(1):107–12. doi: 10.1038/leu.2012.181. [DOI] [PubMed] [Google Scholar]
- 22.Klamova H, et al. Dasatinib in imatinib-resistant or -intolerant CML patients: data from the clinical practice of 6 hematological centers in the Czech Republic. Neoplasma. 2010;57(4):355–9. [PubMed] [Google Scholar]
- 23.Cortes JE, et al. A phase 2 trial of ponatinib in Philadelphia chromosome-positive leukemias. N Engl J Med. 2013;369(19):1783–96. doi: 10.1056/NEJMoa1306494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Khoury HJ, et al. Bosutinib is active in chronic phase chronic myeloid leukemia after imatinib and dasatinib and/or nilotinib therapy failure. Blood. 2012;119(15):3403–12. doi: 10.1182/blood-2011-11-390120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Radich JP, et al. HLA-matched related hematopoietic cell transplantation for chronic-phase CML using a targeted busulfan and cyclophosphamide preparative regimen. Blood. 2003;102(1):31–5. doi: 10.1182/blood-2002-08-2619. [DOI] [PubMed] [Google Scholar]
- 26.Quintas-Cardama A, Cortes JE. Chronic myeloid leukemia: diagnosis and treatment. Mayo Clin Proc. 2006;81(7):973–88. doi: 10.4065/81.7.973. [DOI] [PubMed] [Google Scholar]
- 27.Bennour A, et al. Molecular cytogenetic characterization of variant Philadelphia translocations in chronic myeloid leukemia: genesis and deletion of derivative chromosome 9. Cancer Genet Cytogenet. 2009;194(1):30–7. doi: 10.1016/j.cancergencyto.2009.05.010. [DOI] [PubMed] [Google Scholar]
- 28.Gorusu M, et al. On the genesis and prognosis of variant translocations in chronic myeloid leukemia. Cancer Genet Cytogenet. 2007;173(2):97–106. doi: 10.1016/j.cancergencyto.2006.10.006. [DOI] [PubMed] [Google Scholar]
- 29.Verma D, et al. Chronic myeloid leukemia (CML) with P190 BCR-ABL: analysis of characteristics, outcomes, and prognostic significance. Blood. 2009;114(11):2232–5. doi: 10.1182/blood-2009-02-204693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Swansbury J. Introduction. Cancer cytogenetics: methods and protocols. Methods Mol Biol. 2003;220:1–8. doi: 10.1385/1-59259-363-1:001. [DOI] [PubMed] [Google Scholar]
- 31.Bartley PA, et al. Rapid isolation of translocation breakpoints in chronic myeloid and acute promyelocytic leukaemia. Br J Haematol. 2010;149(2):231–6. doi: 10.1111/j.1365-2141.2009.08071.x. [DOI] [PubMed] [Google Scholar]
- 32.Ismail F, et al. The accuracy and timeliness of a Point Of Care lactate measurement in patients with Sepsis. Scand J Trauma Resusc Emerg Med. 2015;23(1):68. doi: 10.1186/s13049-015-0151-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Winn-Deen ES, et al. Development of an integrated assay for detection of BCR-ABL RNA. Clin Chem. 2007;53(9):1593–600. doi: 10.1373/clinchem.2007.085472. [DOI] [PubMed] [Google Scholar]
- 34.Juul S, et al. Droplet microfluidics platform for highly sensitive and quantitative detection of malaria-causing Plasmodium parasites based on enzyme activity measurement. ACS Nano. 2012;6(12):10676–83. doi: 10.1021/nn3038594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gingeras TR, Whitfield KM, Kwoh DY. Unique features of the self-sustained sequence replication (3SR) reaction in the in vitro amplification of nucleic acids. Ann Biol Clin (Paris) 1990;48(7):498–501. [PubMed] [Google Scholar]
- 36.Stevens WS, Scott LE, Crowe SM. Quantifying HIV for monitoring antiretroviral therapy in resource-poor settings. J Infect Dis. 2010;201(Suppl 1):S16–26. doi: 10.1086/650392. [DOI] [PubMed] [Google Scholar]
- 37.Petti CA, et al. Laboratory medicine in Africa: a barrier to effective health care. Clin Infect Dis. 2006;42(3):377–82. doi: 10.1086/499363. [DOI] [PubMed] [Google Scholar]
- 38.Sooknanan R, et al. Detection and direct sequence identification of BCR-ABL mRNA in Ph+ chronic myeloid leukemia. Exp Hematol. 1993;21(13):1719–24. [PubMed] [Google Scholar]
- 39*.Dugan LC, et al. Detection of BCR-ABL Fusion mRNA Using Reverse Transcriptase Loop-mediated Isothermal Amplification. 2011 Medium: ED; Size: PDF-file: 27 pages; size: 2.4 Mbytes. This reference is of importance as it is one of the early studies implementing isothermal PCR testing for CML. [Google Scholar]
- 40.Bartley PA, et al. Sensitive detection and quantification of minimal residual disease in chronic myeloid leukaemia using nested quantitative PCR for BCR-ABL DNA. Int J Lab Hematol. 2010;32(6 Pt 1):e222–8. doi: 10.1111/j.1751-553X.2010.01236.x. [DOI] [PubMed] [Google Scholar]
- 41.Bartley PA, et al. A DNA real-time quantitative PCR method suitable for routine monitoring of low levels of minimal residual disease in chronic myeloid leukemia. J Mol Diagn. 2015;17(2):185–92. doi: 10.1016/j.jmoldx.2014.10.002. [DOI] [PubMed] [Google Scholar]
- 42*.Kelani R, Monem F. Reconsideration of BCR-ABL protein flow cytometric immunobead assay: how potent to diagnose and monitor chronic myeloid leukemia? Int J Lab Hematol. 2015;37(5):723–8. doi: 10.1111/ijlh.12394. This reference is of importance as it is one of the early studies implementing digital PCR testing for CML. [DOI] [PubMed] [Google Scholar]
- 43.Luu MH, Press RD. BCR-ABL PCR testing in chronic myelogenous leukemia: molecular diagnosis for targeted cancer therapy and monitoring. Expert Rev Mol Diagn. 2013;13(7):749–62. doi: 10.1586/14737159.2013.835573. [DOI] [PubMed] [Google Scholar]
- 44.Goh HG, et al. Sensitive quantitation of minimal residual disease in chronic myeloid leukemia using nanofluidic digital polymerase chain reaction assay. Leuk Lymphoma. 2011;52(5):896–904. doi: 10.3109/10428194.2011.555569. [DOI] [PubMed] [Google Scholar]
- 45.Huggett JF, Cowen S, Foy CA. Considerations for digital PCR as an accurate molecular diagnostic tool. Clin Chem. 2015;61(1):79–88. doi: 10.1373/clinchem.2014.221366. [DOI] [PubMed] [Google Scholar]
- 46.Zagaria A, et al. BCR-ABL1 e6a2 transcript in chronic myeloid leukemia: biological features and molecular monitoring by droplet digital PCR. Virchows Arch. 2015;467(3):357–63. doi: 10.1007/s00428-015-1802-z. [DOI] [PubMed] [Google Scholar]
- 47.Jennings LJ, et al. Detection and quantification of BCR-ABL1 fusion transcripts by droplet digital PCR. J Mol Diagn. 2014;16(2):174–9. doi: 10.1016/j.jmoldx.2013.10.007. [DOI] [PubMed] [Google Scholar]
- 48.Kantarjian HM, et al. Imatinib mesylate (STI571) therapy for Philadelphia chromosome-positive chronic myelogenous leukemia in blast phase. Blood. 2002;99(10):3547–53. doi: 10.1182/blood.v99.10.3547. [DOI] [PubMed] [Google Scholar]
- 49.O’Brien SG, et al. Imatinib compared with interferon and low-dose cytarabine for newly diagnosed chronic-phase chronic myeloid leukemia. N Engl J Med. 2003;348(11):994–1004. doi: 10.1056/NEJMoa022457. [DOI] [PubMed] [Google Scholar]
- 50.Saglio G, et al. Dasatinib in imatinib-resistant or imatinib-intolerant chronic myeloid leukemia in blast phase after 2 years of follow-up in a phase 3 study: efficacy and tolerability of 140 milligrams once daily and 70 milligrams twice daily. Cancer. 2010;116(16):3852–61. doi: 10.1002/cncr.25123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sawyers CL, et al. Imatinib induces hematologic and cytogenetic responses in patients with chronic myelogenous leukemia in myeloid blast crisis: results of a phase II study. Blood. 2002;99(10):3530–9. doi: 10.1182/blood.v99.10.3530. [DOI] [PubMed] [Google Scholar]
- 52.Soverini S, et al. Contribution of ABL kinase domain mutations to imatinib resistance in different subsets of Philadelphia-positive patients: by the GIMEMA Working Party on Chronic Myeloid Leukemia. Clin Cancer Res. 2006;12(24):7374–9. doi: 10.1158/1078-0432.CCR-06-1516. [DOI] [PubMed] [Google Scholar]
- 53.Branford S, et al. Real-time quantitative PCR analysis can be used as a primary screen to identify patients with CML treated with imatinib who have BCR-ABL kinase domain mutations. Blood. 2004;104(9):2926–32. doi: 10.1182/blood-2004-03-1134. [DOI] [PubMed] [Google Scholar]
- 54.Ibrahim AR, et al. Poor adherence is the main reason for loss of CCyR and imatinib failure for chronic myeloid leukemia patients on long-term therapy. Blood. 2011;117(14):3733–6. doi: 10.1182/blood-2010-10-309807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Trivedi D, et al. Adherence and persistence among chronic myeloid leukemia patients during second-line tyrosine kinase inhibitor treatment. J Manag Care Spec Pharm. 2014;20(10):1006–15. doi: 10.18553/jmcp.2014.20.10.1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Soverini S, et al. Unraveling the complexity of tyrosine kinase inhibitor-resistant populations by ultra-deep sequencing of the BCR-ABL kinase domain. Blood. 2013;122(9):1634–48. doi: 10.1182/blood-2013-03-487728. [DOI] [PubMed] [Google Scholar]
- 57.Loman NJ, et al. Performance comparison of benchtop high-throughput sequencing platforms. Nat Biotechnol. 2012;30(5):434–9. doi: 10.1038/nbt.2198. [DOI] [PubMed] [Google Scholar]
- 58.Meacham F, et al. Identification and correction of systematic error in high-throughput sequence data. BMC Bioinformatics. 2011;12:451. doi: 10.1186/1471-2105-12-451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Schmitt MW, et al. Detection of ultra-rare mutations by next-generation sequencing. Proc Natl Acad Sci U S A. 2012;109(36):14508–13. doi: 10.1073/pnas.1208715109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Farrar JS, Wittwer CT. Extreme PCR: efficient and specific DNA amplification in 15-60 seconds. Clin Chem. 2015;61(1):145–53. doi: 10.1373/clinchem.2014.228304. [DOI] [PubMed] [Google Scholar]
- 61.Sheel Kumar V, Webster M. Extreme PCR: a breakthrough innovation for outbreaks? Clin Chem. 2015;61(4):674–6. doi: 10.1373/clinchem.2014.236950. [DOI] [PubMed] [Google Scholar]
- 62.Korlach J, et al. Selective aluminum passivation for targeted immobilization of single DNA polymerase molecules in zero-mode waveguide nanostructures. Proc Natl Acad Sci U S A. 2008;105(4):1176–81. doi: 10.1073/pnas.0710982105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Eid J, et al. Real-time DNA sequencing from single polymerase molecules. Science. 2009;323(5910):133–8. doi: 10.1126/science.1162986. [DOI] [PubMed] [Google Scholar]
- 64.Mikheyev AS, Tin MM. A first look at the Oxford Nanopore MinION sequencer. Mol Ecol Resour. 2014;14(6):1097–102. doi: 10.1111/1755-0998.12324. [DOI] [PubMed] [Google Scholar]
- 65.Sharma A, et al. Chitosan encapsulated quantum dots platform for leukemia detection. Biosens Bioelectron. 2012;38(1):107–13. doi: 10.1016/j.bios.2012.05.010. [DOI] [PubMed] [Google Scholar]
- 66.Rovigatti U. Cancer modelling in the NGS era - Part I: Emerging technology and initial modelling. Crit Rev Oncol Hematol. 2015;96(2):274–307. doi: 10.1016/j.critrevonc.2015.05.017. [DOI] [PubMed] [Google Scholar]