

Abstract
Introduction
Aspects of radiopharmaceutical development are illustrated through preclinical studies of [125I]-(E)-1-(2-(2,3-dihydrobenzofuran-5-yl)ethyl)-4-(iodoallyl)piperazine ([125I]-E-IA- BF-PE-PIPZE), a radioligand for sigma-1 (σ1) receptors, coupled with examples from the recent literature. Findings are compared to those previously observed for [125I]-(E)-1-(2-(2,3-dimethoxy-5-yl)ethyl)-4-(iodoallyl)piperazine ([125I]-E-IA-DM-PE-PIPZE).
Methods
Syntheses of E-IA-BF-PE-PIPZE and [125I]-E-IA-BF-PE-PIPZE were accomplished by standard methods. In vitro receptor binding studies and autoradiography were performed, and binding potential was predicted. Measurements of lipophilicity and protein binding were obtained. In vivo studies were conducted in mice to evaluate radioligand stability, as well as specific binding to σ1 sites in brain, brain regions and peripheral organs in the presence and absence of potential blockers.
Results
E-IA-BF-PE-PIPZE exhibited high affinity and selectivity for σ1 receptors (Ki = 0.43 ± 0.03 nM, σ2 / σ1 = 173). [125I]-E-IA-BF-PE-PIPZE was prepared in good yield and purity, with high specific activity. Radioligand binding provided dissociation (koff) and association (kon) rate constants, along with a measured Kd of 0.24 ± 0.01 nM and Bmax of 472 ± 13 fmol / mg protein. The radioligand proved suitable for quantitative autoradiography in vitro using brain sections. Moderate lipophilicity, Log D7.4 2.69 ± 0.28, was determined, and protein binding was 71 ± 0.3%. In vivo, high initial whole brain uptake, > 6% injected dose / g, cleared slowly over 24 h. Specific binding represented 75% to 93% of total binding from 15 min to 24 h. Findings were confirmed and extended by regional brain biodistribution. Radiometabolites were not observed in brain (1%).
Conclusions
Substitution of dihydrobenzofuranylethyl for dimethoxyphenethyl increased radioligand affinity for σ1 receptors by 16-fold. While high specific binding to σ1 receptors was observed for both radioligands in vivo, [125I]-E-IA-BF-PE-PIPZE displayed much slower clearance kinetics than [125I]-E-IA-DM-PE-PIPZE. Thus, minor structural modifications of σ1 receptor radioligands lead to major differences in binding properties in vitro and in vivo.
Keywords: Imaging, validation, radiopharmaceutical, radiotracer, radiopharmacology, sigma receptor
1. Introduction
Radiotracers serve as scientific tools for biomedical research in nearly all fields, including neuroscience, neurology, psychiatry, oncology and cardiology. The diagnostic radiopharmaceutical counterparts are invaluable for answering clinical research questions concerning human health and disease by the non-invasive nuclear imaging techniques of positron emission tomography (PET) and single photon emission computed tomography (SPECT). Radiopharmaceuticals can interrogate enzyme function, neurotransmitter function and receptor status, define organ physiology and pathophysiology, and can visualize and quantitate the abnormal brain deposits (β–amyloid, τ protein) associated with Alzheimer’s disease. Nuclear imaging studies can also reflect the competition between a radiopharmaceutical and a non-radioactive ligand at specific biological recognitions sites. Such “occupancy” studies inform on mechanisms and dosing protocols, and require the radioligand to be sensitive to this molecular interplay. Validated radiotracers are now indispensable aids for reducing the time and lowering the cost of developing new drug entities within the pharmaceutical industry [1–3].
Nuclear imaging can also elucidate modes of drug action. An enlightening example is SPECT studies of D2 / D3 receptors in human beings using [123I]-IBZM, which showed occupancy of the striatal sites by EMD 59983, an active metabolite of the “selective” sigma (σ) receptor ligand panamesine (EMD 57445). The observation suggests that the atypical antipsychotic profile of the drug is due, at least in part, to dopaminergic activity of the metabolite [4]. The role of imaging in drug development for neurodegeneration has been discussed [5] and a recent review, focused on the role of PET in the development of new drugs to treat heart failure [6], exemplifies the power of molecular imaging in this regard. The extensive biomedical advances made possible by radiopharmaceuticals and nuclear imaging are exemplified by over 150 books and review articles within the last three years. These scientific works can be general [7,8], focused on specific radionuclides [9] or on particular organ systems [10–12].
Questions to be answered through the use of radiopharmaceuticals change over time. In the 1970’s, pioneering work on visualization of neuroreceptors by autoradiography [13,14] prompted the question of whether or not neuroreceptors might also be imaged in living human beings. The question was answered in the early 1980’s by PET and SPECT brain imaging of dopamine D2 and muscarinic receptors in human beings using [11C]-N-methylspiperone ([11C]-NMSP) [15] and [123I]-quinuclidinyl benzilate ([123I]-QNB) [16], respectively. In the ensuing years, the questions have become more complex, and dependent on clinical disease state [11,17]. When advances in instrumentation, radioligand synthesis and mathematical modeling are combined [18], imaging is likely to provide answers. A challenge for the field is the following conundrum: as the specificity of a radiopharmaceutical increases, applicability for truly “personalized medicine” increases [17]; however, the overall impact on patient care decreases if the indications for use are too narrow [19,20].
There is a reticence of governmental organizations such as the United States FDA to approve radiopharmaceuticals for broad indications without compelling proof of diagnostic accuracy and clinical utility. A recent success is the 2011 approval of [123I]-ioflupane (DaTSCAN) to assist physicians in evaluation of patients presenting with neurodegenerative disorders [21]. At this time, however, the β–amyloid imaging compounds Amyvid™ and Vizamyl™ have limited approvals in the United States for a scan to exclude a diagnosis of Alzheimer’s disease (AD) [22]. Additional clinical trials will determine if the indication can be broadened to include a diagnosis of AD [23]. On the horizon, PET imaging of human histone deacetylase with [11C]-Martinostat [24] should yield valuable information for understanding the emerging field of neuroepigenetics [25].
The development of new radiopharmaceuticals is guided by the radiotracer principle, for which George de Hevesy won the 1943 Nobel Prize in Chemistry [26]. The validation process can be referred to as the radiopharmaceutical paradigm (Chart 1). Proposals to devise a new radiopharmaceutical often originate from reviewing the published literature and patent applications, from data presented at scientific meetings, or from discussions with colleagues. Inherent in these readings and conversations is relevance for the biological target, which must exist in sufficient density for imaging. Moreover, the synthesis and radiosynthesis of the chemical target must be feasible, with choice of radionuclide and physical decay characteristics being key components. Programs seeking new radiopharmaceuticals require significant capital investment, so the justification to embark on the journey is paramount [27,28]. The unmet need, or questions that could be answered, should be vetted prior to project initiation. Plans for synthesis, radiosynthesis and preclinical biological evaluation are then established. The “ADME” concept (Absorption, Distribution, Metabolism, Excretion) helps guide new drug development, and is also applied to radiopharmaceuticals. Although it may be tempting to fast track a new radiotracer into the clinic without extensive evaluation, this approach may be a failure from both scientific and ethical perspectives. Guidelines for “success” are quite stringent for radiopharmaceuticals. An extremely small mass of radioactive compound must survive chemically in vivo, reach the intended biological target, yield a target to non-target count rate sufficient for external imaging, and also be safely administered to the subject without adverse effects. When the physical half-life is short, as for [11C]-labeled ligands (t½ 20.4 min; 511 keV, β+), these obstacles are formidable.
Chart 1.

Radiopharmaceutical Paradigm
Development of a new radiotracer, with a view toward use as a radiopharmaceutical, is an iterative process (Chart 2). Synthesis and evaluation of multiple compounds is often warranted, since gaining a better understanding of the relationships between structure and activity focus the efforts toward an entity that is likely to be successful. The present article illustrates general aspects of the early preclinical validation of receptor-binding radiotracers through selected literature examples, coupled with a synopsis of our recent studies of [125I]-(E)-1-(2-(2,3-dihydrobenzofuran-5-yl)ethyl)-4-(iodoallyl)piperazine ([125I]-E-IA-BF-PE-PIPZE), a radioligand for σ1 receptors [29]. Our objective is to provide a road map for basic tactics that might prove of interest to those beginning studies in the field.
Chart 2.

Synthesis and Early Preclinical Validation Experiments
2. Sigma receptors and ligand design
2.1 Sigma receptors
Investigations of the sigma-1 (σ1, S1R) / sigma-2 (σ2, S2R) receptor pair are of current interest from a host of perspectives. This system was initially misclassified as a member of the opioid family [30], and the existence of two discrete subtypes was not recognized until the early 1990’s [31]. These receptors have been implicated in psychostimulant and alcohol abuse, depression and anxiety, sequalae of stroke and pain, amyotrophic lateral sclerosis (ALS) and multiple sclerosis, as well as cancer. The reader is referred to recent reviews describing their known and suspected roles [32–40].
Of the potential radiopharmaceuticals developed for imaging σ1 and σ2 receptors [41–43] only a few have progressed to human imaging studies. Two of these are the σ1 selective ligands TPCNE [44] and SA4503 [45] (Fig. 1). These compounds represent different chemical classes, with TPCNE having higher apparent affinity (0.67 nM) [44] than SA4503 (4.6 nM) [46] for the sites in vitro. In vivo, the pharmacokinetics of these radiotracers proved to be quite different. As a radioligand for SPECT, [123I]-TPCNE exhibited good specific binding to cerebral σ1 receptors, with no clearance from human brain over 30 h [47]. This SPECT imaging protocol exemplifies the flexibility of using I-123, which has a long physical half-life (t½ 13.2 h; 159 keV, γ). A relentless binder such as [123I]-TPCNE should give clear images and be useful for certain applications, but may not be sensitive enough to competition for use in occupancy studies [47–49]. By contrast, dynamic PET imaging of σ1 receptors using [11C]-SA4503 in human brain is usually conducted over a 90 min period [50], which is in keeping with the 20.4 min half-life of carbon-11. [11C]-SA4503 is useful for occupancy studies, allowing PET confirmation that the clinically significant drugs donepezil (Aricept ®), [51,52], and fluvoxamine [53,54] interact with the σ1 receptor in human brain. These findings suggest a σ1 receptor contribution to the primary modes of action for these drugs. One drawback to [11C]-SA4503 PET is the requirement for production using an on-site cyclotron.
Fig. 1.

Structures of TPCNE, SA4503 and the molecular hybrid, E-IA-DM-PE-PIPZE.
We hypothesized that an iodinated TPCNE / SA4503 structural hybrid might give a ligand that retained high affinity and selectivity for σ1 receptors, and also be readily reversible in vivo. For radiolabeling, we chose to use I-125, a radioisotope of iodine having a long half-life (~60 d) and soft (35 keV) γ emission. Thus, laboratory studies would be convenient for characterization of receptor binding and physicochemical parameters in vitro, and for establishing pharmacokinetics and pharmacology in small animals. Our target ligand, [125 / 127I]-E-IA-DM-PE-PIPZE (Fig. 1), did exhibit high affinity and selectivity for σ1 receptors in vitro, labeled the sites in mouse brain and periphery in vivo, and proved sensitive to in vivo competition by strong ligands [55–57] and by weak ligands, such as cocaine [58]. Extension to imaging by SPECT using I-123 or by PET using I-124 (t½ 4.2 d; 511 keV, β+) [59] may be possible.
For the most part, equivalent preclinical studies can be done using ligands labeled with In-111 (t½ 2.8 d; 171, 245 keV, γ), Tc-99m (t½ 6.0 h; 140 keV, γ), F-18 (t½ 110 min; 511 keV, β+), and even C-11, as long as experimental setups are prepared in advance. In vitro binding with C-11, however, is quite difficult. While C-14 certainly has an appropriate half-life (5730 y), and is often used for distribution, metabolism and excretion studies, the specific radioactivity (62 mCi / mmol) is too low to detect specific binding to receptors. So, if detailed in vitro binding studies are needed, tritium-labeled ligands (t½ 12.3 y; 6 keV, β−) with specific activities of 29 Ci/ mmol or higher, depending on level of tritium incorporation, are used. A notable example is [3H]-carfentanil characterization of in vitro binding in human brain tissue to aid interpretation of PET studies of μ opioid receptors using [11C]-carfentanil [60–62].
With this backdrop, a more detailed description of the radiopharmaceutical paradigm is shown in Chart 2, where a variety of experiments are listed that help define receptor-binding radiotracers. Some experiments are performed with the non-radioactive ligand, while others demand the use of the radiolabeled ligand. As shown in Chart 2 and discussed earlier, medicinal radiochemistry is an iterative process, and we wished to build on the knowledge gained from studies of [125 / 127I]-E-IA-DM-PE-PIPZE. During a parallel study of qualitative structure-activity relationships (SAR) for SA4503 analogs [63] (Fig. 2), substitution of a methylenedioxy moiety (1) improved affinity for σ1 receptors. The steric bulk of the 6- and 7-membered ring congeners 2 and 3 interfered with binding. These results indicate that a more compact substitution pattern improves affinity. YZ-067, a molecule structurally related to SA4503 (Fig. 2) but devoid of one of the methoxy groups, also displays improved affinity for σ1 receptors [64], and shows that only one oxygen substituent is required for binding. We reasoned that incorporation of both design features could be achieved with a dihydrobenzofuranyl group (Fig. 2). Accordingly, we selected E-IA-BF-PE-PIPZE as a new ligand for study. Evaluation in vitro and in vivo would provide insight to the receptor binding and pharmacokinetic effects of this structural modification. Rationales, procedures and results obtained for [125 / 127I]-E-IA-BF-PE-PIPZE are described herein, while the experimental details are available elsewhere [65].
Fig. 2.

Structures and σ1 receptor binding affinities of SA4503 and analogs, that provides a rationale for modification of E-IA-DM-PE-PIPZE to E-IA-BF-PE-PIPZE. aData taken from [46]; bData taken from [64]; cData taken from [63].
2.2 Organic synthesis
2.2.1 Target ligand
Once a candidate radioligand has been identified, the first experimental phase concerns the synthesis and chemical characterization of the non-radioactive version. E-IA-BF-PE-PIPZE was prepared as shown in Scheme 1, and characterized by standard organic chemistry techniques including 1H and 13C nuclear magnetic resonance (NMR) spectroscopy, as well as by combustion analysis. This aspect of the work should confirm key structural features, such as the trans (E) configuration of the iodoallyl moiety in the present case, and the elemental composition. Mass spectroscopy (MS), infrared (IR) spectroscopy and ultraviolet (UV) spectroscopy are other valuable tools in structure determination. Typically, for amine containing compounds, a hydrochloride or oxalate salt is prepared from the free base for long-term stability and ease in weighing. Chromatography, including normal- and reversed-phase techniques, is nearly always vital for achieving and assessing purity. In particular, high performance liquid chromatography (HPLC) is a sensitive and widely used preparative and analytical method.
Scheme 1.

Synthetic route for preparation of E-IA-BF-PE-PIPZE / [125I]-E-IA-BF-PE-PIPZE.
While E-IA-BF-PE-PIPZE has no chiral centers, many candidate ligands will [66,67], and absolute stereochemistry can have a strong impact on binding affinity, as well as metabolism and toxicity. In a pertinent example, the (R)-(+)- and (S)-(−)-enantiomers of the σ1 receptor PET radioligand [18F]-fluspidine have been prepared for detailed comparison [68]. These non-superimposable “mirror images” are shown in Fig. 3. The non-radioactive (R)-(+)-isomer showed a 4-fold higher affinity for σ1 receptors in vitro, and the pharmacokinetics of the two [18F]-labeled enantiomers were different in vivo in pig brain by PET, with higher standardized uptake values (SUVs) for the more potent (R)-(+)-isomer. However, the [18F]-labeled (S)-(−)isomer proved to have greater metabolic stability than the (R)-(+)-isomer in this species, and was considered to have greater sensitivity for occupancy studies, presumably due to lower binding affinity. Such preclinical studies illustrate the depth of analysis that might be required during radiopharmaceutical development in order to select the most appropriate candidate radioligand for clinical translation.
Fig. 3.
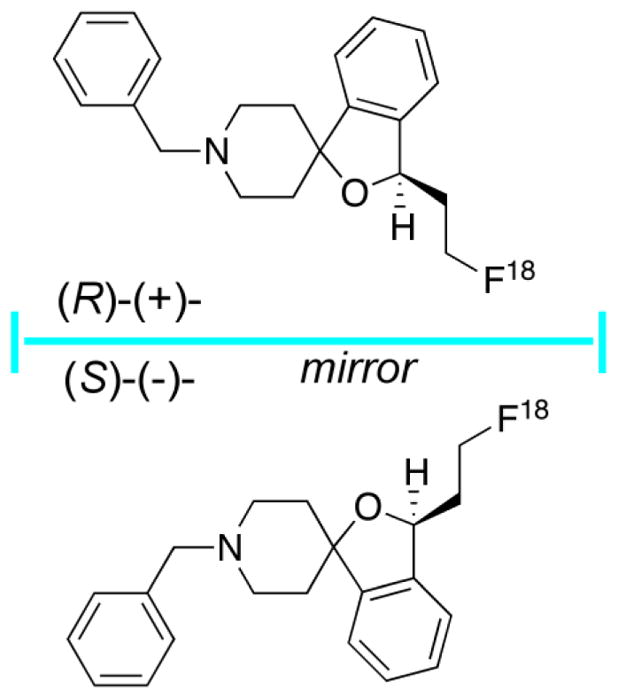
(R)-(+)- and (S)-(−)-enantiomers of the σ1 receptor PET radioligand [18F]-fluspidine.
High chemical purity of the non-radioactive target ligand is requisite prior to biological evaluations. Minimum purity levels of 95% are often noted in the literature. However, even minor contaminants might lead to erroneous conclusions and, perhaps, a false start. Consider a hypothetical case where a ligand known to have very high affinity is structurally modified for testing during the quest for a new radioligand. If the parent ligand displays a 0.02 nM affinity for the receptor, and the novel derivative has no affinity at all, just 2% contamination of the derivative with the parent during a binding assay would lead one to believe that the new “ligand” has 1.0 nM affinity.
2.2.2 Precursor
A rule in radiochemistry is to incorporate the marker element at, or very near, the end of the synthetic sequence. This leads to improved radiochemical yields, and minimizes the radiation exposure of the chemist. For short-lived radionuclides such as C-11 and F-18, specific radioactivities are also much improved, and rapid radiolabeling is central to success. Comprehensive reviews are available that detail these challenges and provide practical solutions [69–72]. The synthetic methods and special practical issues associated with incorporation of radioiodine [73–76] and radiometals [77–80] have also been addressed. New synthetic methods are routinely reported, and keeping abreast of the literature expands the radiosynthetic options available for complex targets.
Clearly, choice of precursor and radiolabeling method are crucial components of radiopharmaceutical development. The precursor used in the microscale radiosynthesis may be structurally different from that used in the macroscopic non-radioactive synthesis. A major chemical difference for the radiolabeling procedure is the precursor will be present in vast excess over the radioactive reagent, leading to pseudo-first order kinetics. The concentration of the precursor will be key to a good radiochemical yield. At concentrations lower than 10−5 – 10−6 M, a typical bimolecular reaction will be ineffective, leading to a low radiochemical yields. During radiosynthesis of [125I]-E-IA-BF-PE-PIPZE (Section 4.1), the precursor concentration is 10−3 M, and the molar ratio of precursor to [125I]-NaI is 150 to 1. In this case, the identical precursor is used for both the non-radioactive and radiolabeled target ligand (Scheme 1). While the vinylstannylated alkylating agent used for precursor synthesis was initially developed as a prosthetic group for radioiodination of small molecules [81], the presence of the iodine in E-IA-BF-PE-PIPZE and related compounds is an integral requirement for high binding affinity [55]. Aspects of radiosynthesis are discussed in more detail in Section 4.1.
3. Competition binding assays
With a target ligand in hand, the natural first question is: What is its affinity? The answer may determine the fate of the project. Radioligand binding assays are the main way to measure affinity, and detailed guidance on both theory and experimental design is available [82–88]. Binding studies entail several levels of analysis, and the first stage is to assess the affinity of the non-radioactive target ligand for the primary sites of interest. The next stage is to examine affinity for potential secondary binding sites, sometimes referred to as “off-target” sites. The goal is to obtain a pharmacological profile of the ligand’s innate and relative ability to interact with multiple sites, otherwise known as selectivity. The final stage, direct receptor binding studies using the radiolabeled ligand, is addressed in Section 6.3.
Briefly, competition for a given receptor occurs between increasing concentrations of the test ligand and a fixed concentration of a well-characterized radioligand for that receptor. Conditions to consider include tissue type, protein concentration, time, temperature and incubation buffer, and should be chosen so radioligand binding to a single receptor is known to be reversible, and at equilibrium, under the law of mass-action shown in Equation 1:
| (Eq. 1) |
As a rule of thumb, the radioligand should be used at a concentration at or below its Kd, the equilibrium dissociation constant, where 50% of the receptors are occupied (Bound) by the radioactive ligand to form a receptor-radioligand complex. The Kd is the ratio of the dissociation (koff) to association (kon) rate constants shown in Equation 2:
| (Eq. 2) |
Since Kd represents the reciprocal of affinity, lower values indicate higher affinity. The objective of competition experiments is to determine the concentration of test ligand that inhibits 50% of the radioligand binding. This half-maximal inhibitory concentration is termed the IC50, and is dependent upon the experimental conditions.
Another commonly used term is the apparent affinity, Ki, which is mathematically derived from the IC50, when receptor levels are lower than the radioligand Kd, by the Cheng-Prusoff [89] relationship shown in Equation 3:
| (Eq. 3) |
This relationship considers the radioligand Kd known for those experimental conditions and the concentration of the radioligand actually used, allowing conversion of IC50 value into an apparent dissociation constant for the test ligand as if it were radiolabeled. “Apparent” is used because the value is obtained indirectly. If the radioligand concentration used is equal to its Kd, then the Ki of the test ligand will be half of its IC50 (Eq. 3). Since Ki values incorporate key experimental variables, they are considered useful measures for normalizing and comparing IC50 data obtained under differing conditions and between laboratories.
In a typical binding assay, ten to twelve test ligand concentrations are employed that vary over four to six orders of magnitude and are centered on the suspected IC50. If the ligand is a close analog of known ligands, the range needed to bracket the IC50 could be similar. That said, minor structural perturbations could lead to major differences in affinity, so range-finding experiments might be necessary. Care should be taken that additives in test ligand formulations do not adversely affect the determinations. For instance, dimethyl sulfoxide (DMSO) might seem reasonable for increasing ligand solubility from a chemical point of view, and is often employed during drug screening procedures. However, DMSO, dimethylformamide, acetonitrile and alcohols can all be problematic, and even 1% can adversely impact some receptor binding assays [90]. In addition, if stock solutions and serial dilutions are refrigerated between assays, they should be inspected to confirm that precipitation has not occurred, which would change their actual concentration [91]. Both test ligand and radioligand solutions should be checked for stability during the course of the experiments. HPLC is a helpful tool to analyze their concentration and purity when indicated [46,92].
Competition binding data is analyzed using a semi-logarithmic plot, so test concentrations are ideally 3.16-fold serial dilutions that provide equal spacing in half-log units on the X-axis. The radioligand binding on the Y-axis is expressed as CPM, DPM or % Control, the latter referring to the proportion of radioactivity bound in the presence of a test ligand relative to the maximum amount, defined as 100%, observed in its absence. Radioligands usually bind not only to the receptor of interest, the specific binding, but also to other receptors, various tissue components and even the test tubes. These interactions are collectively deemed non-specific binding. Separation of bound radioligand from the free radioligand present in the mixture is generally accomplished by rapid filtration of an assay through glass fiber filter paper or by centrifugation. Bound radioactivity detected on the papers or in the pellets is termed total binding, and reflects both specific and non-specific contributions.
To define specific binding, the metric of interest, additional data is obtained where radioligand competes with a well-characterized, non-radioactive ligand used at a high enough concentration to fully block binding of the radioligand to the receptor. Bound radioactivity detected under these circumstances is termed non-specific, and is subtracted from the total binding observed at each concentration of test ligand to provide the specific binding data for analysis. Assays are performed in duplicate or triplicate for each condition, and are conducted 3 – 6 times to delineate the binding parameters.
If the simple single-site binding model (Eq. 1) holds true, a smooth sigmoidal curve should be observed where the IC50 for the test ligand can be calculated by non-linear regression [83] using a specialized computer program designed for radioligand binding analysis, like Prism from GraphPad Software, to solve Equation 4:
| (Eq. 4) |
where Y is the specific radioligand binding observed at a given test ligand inhibitor concentration [I], Ymax is the top plateau for specific radioligand binding in the absence of inhibitor, Ymin is the bottom plateau when radioligand binding is fully inhibited, and IC50 is the [I] that produces half-maximal inhibition. The Hill coefficient, nH, describes the steepness of the slope connecting the plateaus, and is a measure of deviation from simple model behavior where the slope should be - 1.0. This procedure gives a four-parameter logistic curve fit (top, bottom, Hill slope, IC50), and is illustrated in Fig. 4 for E-IA-BF-PE-PIPZE competition against the σ1 receptor radioligand, tritiated (+)-pentazocine ([3H]-PTZ).
Fig. 4.

Inhibition of [3H]-PTZ specific binding to σ1 receptors by E-IA-BF-PE-PIPZE. Data shown are means ± SEM for 4 assays, each performed in duplicate.
3.1 Primary sites
First considerations in determining the affinity of a novel target ligand for primary binding sites are: What receptor preparation and radioligand do I use? The receptor source should be readily available, and relevant to the study at hand. Neuroreceptor binding ligands are often screened using membranes prepared from whole rodent brains or a particular brain region, while ligands meant for oncology studies are often screened using cancer cell lines. Radioligands of choice typically display low non-specific binding and high affinity, and commercial availability adds convenience. Literature precedents should guide selection of the assay conditions. In the case of E-IA-BF-PE-PIPZE, we chose to use membranes prepared from normal CD-1® mouse brains for several reasons. The mice are readily available, and both primary sites of interest, the σ1 and σ2 receptor subtypes, are known to be present. Further, binding parameters for each subtype are defined in these membranes for “gold standard,” commercially available tritiated radioligands [58]. Finally, in vivo evaluations of [125 I]-E-IA-BF-PE-PIPZE were planned in the CD-1® mouse strain (Section 7), so the in vitro and in vivo findings would be unified.
A different radioligand was needed to evaluate binding at each subtype. [3H]-PTZ, a high affinity and selective radioligand for σ1 receptors was employed, using 1000 nM haloperidol to define non-specific binding. Haloperidol is not selective for σ1 receptors, and also displays high affinity, for example, to σ2 receptors and dopamine D2 receptors. Nonetheless, haloperidol has been routinely used in such assays for over 25 years [93–95]. The 1000 nM concentration is much greater than its Ki of 1.3 nM for σ1 sites in mouse brain membranes [58], so it fully inhibits [3H]-PTZ binding. Ligands for definition of non-specific binding should always be used at > 100 times their Ki to achieve > 99% inhibition of radioligand binding according to the law of mass-action. The competitive binding curve obtained is shown in Fig. 4. The IC50 was determined to be 0.68 ± 0.04 nM, and the Hill slope was −1.23 ± 0.04. The Cheng-Prusoff relationship (Eq. 3) provided a Ki of 0.43 ± 0.03 nM, considering the 3.0 nM concentration of [3H]-PTZ used and its known Kd of 5.5 nM under these conditions.
Tritiated ditolylguanidine ([3H]-DTG) was employed for the σ2 assay, and is a rather low-affinity, non-selective ligand that binds to both σ1 and σ2 receptors [95]. Therefore, non-radioactive PTZ (500 nM) is included in the assay to “mask” [3H]-DTG binding to σ1 sites, allowing a Kd of 28 nM to be determined for σ2 sites in mouse brain membranes [58]. The concentration of PTZ used for masking influences the subsequent σ2 receptor Ki values determined [46], and should be > 100 times the affinity of PTZ for σ1 sites. Haloperidol (σ2 Ki = 73 nM), at a higher concentration of 10000 nM, was again used to define the non-specific component. As shown in Table 1, E-IA-BF-PE-PIPZE displayed high affinity and selectivity for binding to σ1 over σ2 receptors: σ1 Ki = 0.43 ± 0.03 nM; σ2 Ki = 74.5 ± 4.44 nM; selectivity σ2Ki/σ1 Ki = 173. The selectivity of ligand binding for one receptor relative to another is conveniently assessed as a simple ratio of their Ki values. Binding methods evolve, and tritiated ligands with high affinity and excellent selectivity for σ2 receptors have been identified. One of these is the tetrahydroisoquinolinylbenzamide [3H]-RHM-1, which is useful for assays in membranes from brain [96] and cancer cells [97], and also for quantitative autoradiography (Section 9). If radioligands such as [3H]-RHM-1 become commercially available, a new “gold standard” for σ2 receptor binding assays would emerge.
Table 1.
Competition receptor binding of E-IA-BF-PIPZE to primary and secondary sitesa
| Primary Site (radioligand for assay) | Ki (nM) |
|---|---|
| Sigma-1 ([3H]-PTZ)b | 0.43 ± 0.03 |
| Sigma-2 ([3H]-DTG / PTZ)c | 74.5 ± 4.44 |
| Selectivity: σ2/σ1 | 173 |
| Secondary “off-target” sites | IC50 (nM) |
|---|---|
| Opioid | |
| Mu ([3H]-DAMGO)d | > 5000 |
| Kappa ([3H]-U69593)e | > 5000 |
| Delta ([3H]-Naltrindole)f | > 5000 |
| Monoamine Transporters | |
| SERT ([3H]-Paroxetine)g | > 10000 |
| NET ([3H]-Nisoxetine)h | > 10000 |
| DAT ([125I]-RTI-121)i | > 10000 |
(mean ± SEM, n = 4);
1.0 nM [3H]-PTZ in mouse brain membranes;
3.0 nM [3H]-ditolylguanidine ([3H]-DTG) / 500 nM (+)-PTZ in mouse brain membranes;
[3H]-DAMGO (μ, 0.6 nM) and
[3H]-U69,593 (κ, 0.6 nM) in guinea pig membranes;
[3H]-naltrindole (δ, 0.15 nM) in mouse brain membranes;
0.3 nM [3H]-paroxetine, mouse brain membranes.
0.4 nM [3H]-nisoxetine, mouse brain cortical membranes;
15 pM [125I]-RTI-121, mouse brain striatal membranes.
3.2 Secondary sites
To expand the pharmacological profile of a new receptor binding ligand, in vitro affinities for additional receptors, transporters and other sites should be assessed. Likely secondary binding interactions to “off target” sites can often be identified for a given project from previous literature reports. Binding assays are laborious, and preliminary experiments can be conducted to screen secondary sites using one high concentration of test ligand against an appropriate radioligand. If no, or minimal, inhibition of specific radioligand binding is observed in these “spot tests,” a limit on the affinity of the new ligand for that site can be set based upon the concentration tested. If significant inhibition is observed, a full characterization of binding may be warranted.
For illustration (Table 1), E-IA-BF-PE-PIPZE showed ≤50% inhibition of specific radioligand binding to the three classical opioid receptors (μ, κ, δ) when used at a single 5000 nM concentration, or to the dopamine (DAT), serotonin (SERT) and norepinephrine (NET) transporters when used at 10000 nM. The radioligands, brain membrane preparations and other assay conditions matched those previously reported [98–101]. Among the eight binding sites tested so far, E-IA-BF-PE-PIPZE shows high affinity and selectivity for the σ1 receptor. That said, various other secondary interactions are possible. For instance, SA4503 displays an appreciable 50 nM apparent affinity for the vesicular acetylcholine transporter (VAChT) in vitro, but does not label the sites in vivo [102]. Accordingly, it would be interesting to determine whether or not E-IA-BF-PE-PIPZE also interacts with the VAChT. It is not practical for most laboratories to conduct their own high throughput binding analyses, and services are commercially available to aid drug discovery by testing lead compounds for binding or functional activity at hundreds of biological recognitions sites. The US National Institutes of Mental Health (NIMH) supports the Psychoactive Drug Screening Program (PDSP) at the University of North Carolina, Chapel Hill. This resource, available to academic investigators at no cost, screens novel ligands against a wide panel of receptors, transporters and ion channels.
4. Radiosynthesis, purification and specific activity
The development of a new radiotracer is an iterative process (Chart 2). The in vitro assay results for a given candidate can lend support to further evaluation, or suggest that refinements of structure and the selection strategy are needed. When a suitable target has been identified, radiosynthesis is a next step. Experiments conducted with this candidate radioligand will contribute valuable information regarding physicochemical characteristics (Section 5), and the parameters for direct interactions with the receptor in vitro (Section 6). Moreover, this candidate is intended to advance to in vivo studies in small animals (Section 7) and to imaging trials (Section 9). Outcomes from all of these studies are critically dependent upon the quality of the radioligand.
Isolation of radiochemically pure material, identical in structure to the non-radioactive ligand, is one key criterion. Radiochemical impurities must be separated prior to evaluation. Perhaps less obvious, but just as important, the radioligand must be chemically pure. If the impurities are non-radioactive, how might they affect the results? A precursor or chemical byproduct that is similar in structure to the radioligand may also bind the receptor. Radioligands having high specific activity are required for in vivo receptor studies, and their associated mass is low. Put simply, even minor non-radioactive impurities could effectively compete with the radioligand and prevent its binding. Several major factors to consider are exemplified below for [125I]-E-IA-BF-PE-PIPZE, including method of radiosynthesis, purification and determination of specific radioactivity.
4.1 Radiosynthesis
As described in Section 2.2, choice of precursor and method for radiosynthesis depend upon project specifics and the radionuclide. Here we used a convergent approach, where the same tri-n-butyltin precursor was used to prepare both E-IA-BF-PE-PIPZE and [125I]-E-IA-BF-PE-PIPZE by stereospecific electrophilic iododestannylation (Scheme 1). The reaction conditions are quite different, however. On macroscale, precursor was treated with excess iodine, 1.2 equivalents, at room temperature for ten min in methylene chloride. For radiosynthesis on the no-carrier-added scale, a 150 to 1 molar ratio of precursor (84 μg, 150 nmol) to [125I]-NaI (2 mCi, ca. 1 nmol) was used in methanol containing 3% acetic acid, Chloramine-T was employed for in situ oxidation of the radioiodide, and the reaction was complete within two min at room temperature. Side reactions during this particular type of radioiodination include proto- and chlorodestannylation [81,103]. These chemical by-products would likely bind, possibly with high affinity, to σ1 receptors. Accordingly, they must be separated from the radioligand, along with the removal of remaining precursor and radioactive impurities.
4.2 Purification
While a variety of techniques may be used for the purification and analysis of radiotracers depending upon their chemical characteristics, HPLC is particularly powerful [104]. A typical HPLC system includes a sample injector, fluid pumps, radioactivity detector, mass detector (UV) and digital / analog signal recorders. Columns and mobile phase compositions are variable, and have to be tailored exactly to the purification of individual radiotracers. Reversed-phase conditions are often used, where a hydrophobic column is eluted with a mixture of organic and aqueous solvents. The efficiency and resolution of a separation procedure can be evaluated quantitatively [105], but most work is done qualitatively. When appropriate conditions are employed, either by referral to the literature or by previous experience, the multiple components in a crude reaction mixture can be separated, and will elute at different times.
These retention times (tR) vary with chromatographic parameters, and are directly comparable only when the same conditions (mobile phase, column, flow rate) are used. Capacity factor (k′) can be considered a more robust indicator of a compound’s retention profile. In theory, k′ does not change with mobile phase flow rate or column dimensions, as it depends only on the relative amount of time a compound is moving forward in the mobile phase compared to its time interacting with the column stationary phase [105]. Times, of course, are indicative of the mobile phase volumes. As long as conditions are fairly similar, k′ serves as a normalizing factor between laboratories and between columns. This is particularly informative when transitioning from analytical studies to preparative work on a larger column, or when establishing chromatography conditions based on a literature method. Volumes or time can be used to calculate k′ in reference to the column void (dead space), which is the amount of volume or time it takes to elute a component that does not interact with the stationary phase. This is frequently approximated by the first deflection observed in the baseline after an injection, as this minor perturbation likely represents a non-interacting component of the sample solvent. A convenient k′ calculation is dividing the difference between tR and the void volume time (t0) by the void volume time, as shown in Equation 5:
| (Eq. 5) |
A preparative HPLC chromatogram for the crude reaction mixture obtained during radiosynthesis of [125I]-E-IA-BF-PE-PIPZE is shown in Fig. 5. One major radioactive peak, associated with low mass, is observed at the tR of 17.3 min (k′ 28) expected for the radioligand from prior HPLC work using E-IA-BF-PE-PIPZE. The two major UV-active species at 4.1 min (k′ 5.8) and 8.2 min (k′ 12.7) can be tentatively assigned as the chemical by-products of proto- and chlorodestannylation, respectively, based upon the method of radiosynthesis and their relative retention profile. Prior work showed that the highly lipophilic tri-n-butyltin precursor was retained indefinitely under these conditions. Thus, purified radiotracer can be collected in eluent devoid of contaminants. Multiple HPLC conditions should be investigated to make sure that an impurity is not hidden. Direct evaporation of eluent can lead to radiolysis, and also leaves behind non-volatile salts that may have been used in the mobile phase. Consequently, solid phase extraction (SPE) techniques are popular for final isolation [104]. The goal is to retain the product on the cartridge while separating residual solvents and salts, followed by elution of the product in a small volume of ethanol. Further formulations are then readily accomplished for in vitro or in vivo studies.
Fig. 5.
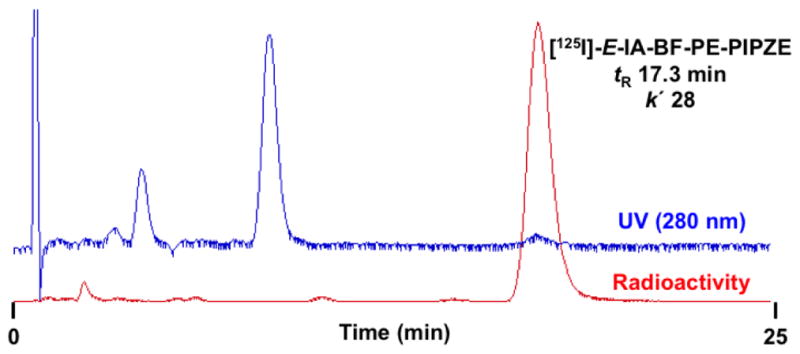
Preparative reversed-phase HPLC for radiosynthesis of [125I]-E-IA-BF-PE-PIPZE showing UV and Radioactivity traces of the crude reaction mixture. Conditions: column, NovaPak® C18, 8 × 100 mm; mobile phase, 23% organic (1:1 MeOH:CH3CN), 77% aqueous (Et3N / HOAc); flow rate: 3.8 mL / min.
Analytical HPLC is then used to verify purity, and can also be used to assess stability under various conditions. Injection of a mixture prepared from the purified radioligand and the chemically characterized non-radioactive ligand should be done to help establish structural identity by co-elution. Again, several sets of HPLC conditions should be used for studies of this kind. Analytical HPLC chromatograms of purified [125I]-E-IA-BF-PE-PIPZE alone, and when mixed with E-IA-BF-PE-PIPZE, are shown in Fig. 6. The column, flow rate and mobile phase constituents were the same for the preparative and analytical work shown here. However, the analytical runs were done using a greater proportion of the organic solvent, which decreased mobile phase polarity. This reduced the hydrophobic interaction between the solute, [125I]-E-IA-BF-PE-PIPZE, and the solid support, an octadecyl carbon chain (C18) bonded to silica, resulting in faster elution. Under these circumstances, the decrease in tR matches the decrease in k′. Since the mass associated with high specific activity radiotracers is so small, rigorous analytical HPLC studies are almost the sole means of structural characterization. If additional confirmations are needed during development efforts, mass can be added intentionally. For example, dual labeling studies using [11C]- and [13C]-iodomethane, with 13C NMR performed after HPLC isolation and radioactive decay, defined the molecular position of [11C]-incorporation for the opioid receptor PET radioligand [11C]-diprenorphine [106].
Fig. 6.

Top panel: Reversed-phase HPLC chromatogram of purified [125I]-E-IA-BF-PE-PIPZE. Bottom panel: HPLC chromatogram of purified [125I]-E-IA-BF-PE-PIPZE with E-IA-BF-PE-PIPZE added to demonstrate co-elution. Conditions: column, NovaPak® C18, 8 × 100 mm; mobile phase, 28% organic (1:1 MeOH:CH3CN), 72% aqueous (Et3N / HOAc); flow rate: 3.8 mL / min.
4.3 Specific Activity
The specific activity of a radionuclide is defined as the amount of radioactivity in the mass associated with the element. When the radionuclide is incorporated into a molecule, the mass of compound, usually in molar terms, is used as the denominator. A related term of importance in radiopharmaceutical chemistry is “effective specific activity,” where the mass used for the calculation includes not only the mass of the compound of interest, but also the mass of any other chemical species present that could interact with the biological target. For high capacity - low affinity biological systems, a low effective specific activity may be reasonable [3]. Receptors are low capacity - high affinity systems, and demand radioligands of high effective specific activity for detection. Methods chosen for radiosynthesis impact the specific activity to be expected (Section 2.2). Isotopic exchange reactions proceed smoothly for radioiodination, but give low specific activity products by definition.
Methods are available for exchange of radioiodide for bromide, which yields high specific activity [125I]- and [123I]-labeled radioligands for receptor studies [107]. Such procedures require rigorous purification to ensure that all brominated precursor has been removed, or the radioligand will be unsuitable because of a low effective specific activity. Specific activity considerations for radionuclide production and synthesis of radioiodinated ligands, as well as [11C]- and [18F]-labeled ligands, are detailed in the reviews listed in Section 2.2.
Regardless of production method or the source of radiolabeled reagents, an experimental determination of radioligand specific activity is needed from several perspectives. Adventitious introduction of carrier might occur, such as carbon dioxide from the air or fluoride from the water. In direct radioligand binding studies, specific activity is one of the inputs for determination of a receptor density (Bmax) in quantitative terms such as fmol / mg protein (Section 6). Of paramount importance, specific activities must be known with confidence for radioligands to be administered safely to animals and to human subjects. Pharmacologically, ligands for a receptor fall into two broad classifications. Agonists are ligands that activate the receptor and produce effects in dose-dependent fashion. For a full agonist, a low level of receptor occupancy engenders a maximal effect. For a partial agonist, lower effects are observed even at high receptor occupancy. By contrast, an antagonist produces no effects, but can block those of agonists. In particular, potent agonists are used for opioid receptor PET [60,61,67], including [11C]-carfentanil, which is so active pharmacologically that the non-radioactive form is used to immobilize wild animals. The main effect is respiratory depression, and the carrier considered to be sub-pharmacological for a PET scan is less than 0.03 μg / kg [108]. Fortunately, this represents an extreme. Many radioligands for receptor imaging are antagonists, and most agonist radioligands do not cause such pronounced effects.
Considering the method of synthesis and the use of no-carrier-added I-125 (2175 mCi / μmol), [125I]-E-IA-BF-PE-PIPZE should be obtained with high specific activity. This was verified by HPLC. First, a standard curve was generated to relate UV absorbance peak area to the injected mass of E-IA-BF-PE-PIPZE over a range that brackets the area of interest (Fig. 7). Deviations from Beer’s law were not problematic. Then, a sample of purified radioligand having a known amount of radioactivity, obtained using a dose calibrator, was injected. The UV peak area was determined, and compared to the standard curve to give the mass of carrier associated with the radioactivity. The specific activity was determined to be near theoretical, at 2134 mCi / μmol (79 GBq / μmol).
Fig. 7.

Top panel: Analytical HPLC chromatograms of sequential E-IA-BF-PE-PIPZE injections showing lower peak areas associated with decreasing mass. Bottom panel: Standard curve relating mass to peak area, constructed so the mass can be determined for a sample of [125I]-E-IA-BF-PE-PIPZE having known radioactivity, allowing calculation of specific radioactivity.
5. Physicochemical Properties
It is well known that molecular properties determine how effectively candidate radiopharmaceuticals reach their biological targets in vivo. A variety of analyses have categorized overall molecule features favoring oral availability or penetration through the blood-brain barrier [109–114]. For readers interested specifically in development of radiotracers for diagnostic imaging of the central nervous system (CNS), reviews by Wong and Pomper [115] and Van de Bittner et al. [116] highlight specific design features. General considerations for CNS imaging radiotracers include a molecular weight less than 500 Daltons, neutral charge at physiological pH, moderate lipophilicity and modest protein binding. Below, the topics of lipophilicity and protein binding are discussed in more detail.
5.1 Lipophilicity
The lipophilicity of a radioligand impacts absorption, distribution, metabolism and excretion (ADME), including potential to cross the blood-brain barrier and extent of non-specific binding [110,117,118]. A balance must be struck, since increasing lipophilicity improves permeability, but concomitantly enhances non-specific binding to plasma proteins and hydrophobic tissue constituents. Consequently, drug activity in general, and radioligand brain uptake in particular, tend to exhibit inverted parabolic relationships with lipophilicity. Pajouhesh and Lenz [110] noted a mean ClogP of 2.5 for marketed CNS drugs. Kessler and colleagues [119] showed this experimentally for striatal uptake of five [125I]-labeled benzamide ligands for the dopamine D2 receptor, where peak values were noted for log k′w between 2.4 – 2.8. The Log D7.4 of [125I]-E-IA-BF-PE-PIPZE (2.69 ± 0.28; n = 5) and [125I]-E-IA-DM-PE-PIPZE (2.25 ± 0.01; n = 5 [55]) are also in the optimal range. Terms and measurement techniques for this fundamental parameter are discussed below.
Traditional lipophilicity measurements (Log P) call for partitioning between an organic phase, such as n-octanol, and an aqueous phase, typically water. To increase in vivo relevance, a distribution coefficient (Log D) is more often determined between n-octanol and an aqueous phase, such as Dulbecco’s phosphate-buffered saline (DPBS) at physiological pH (Log D7.4). As inferred by the term, values of lipophilicity are expressed on the logarithmic scale, because most small organic molecules are lipophilic in nature. Thus, a Log P of 3 means for every one molecule in the aqueous layer, there are 1000 molecules in the organic layer. Measuring lipophilicity with a non-radioactive compound by partitioning can be fraught with difficulty. The mass to be partitioned must be small enough that a dynamic process is recorded. In doing so, however, the amount of a lipophilic compound remaining in the aqueous layer can fall below a reasonable detection limit, even with sophisticated instrumentation (e.g., LC-MS/MS). The converse is also true, and measurements of the organic concentration of hydrophilic compounds may be problematic. Other techniques, such as the HPLC method of Minick [120], determine surrogate values for lipophilicity; in this case, log k′w. Computer programs, such as the proprietary ClogP software from BioByte Corp. that is included as a subroutine with program ChemBioDraw, conveniently provide a calculated value based mostly on addition of the lipophilicity values ascribed to structural fragments [121]. Caution should be exercised if relying solely on computational methods, since other factors, including electronic effects, are not accounted for, and calculated values may or may not correlate well with experimental determinations [122].
On the other hand, direct measurement of a compound’s lipophilicity can be accomplished utilizing the radiolabeled compound by the “shake-flask” method. Samples of the radiolabeled compound are partitioned between equal volumes of n-octanol and aqueous buffer (pH 7.4). Each phase should be presaturated with the other. Vortexing followed by centrifugation gives two clear layers that are sampled and counted for radioactivity, allowing a distribution ratio to be calculated and converted to log scale. In practice, for lipophilic radiotracers, there may be traces of hydrophilic impurities (e.g., [125I]-iodide, [18F]-fluoride). These would partition into the aqueous layer, so sampling after a single equilibration might lead to erroneous results [123]. It is advisable to repeat the ‘washing’ process several times before sampling the layers. For hydrophilic radiotracers, it is preferable to use freshly prepared and rigorously purified samples, as ‘washing’ techniques are more difficult. To compensate for fewer counts in the aqueous layer for lipophilic radiotracers, a larger volume of aqueous over organic phase may be taken. The reverse would apply to hydrophilic radiotracers. Layers should be sampled in duplicate or triplicate, and the counts in each phase should be normalized to a “per g” or “per mL” basis before the radioactivity concentration ratio is calculated and converted to the Log D7.4. Multiple determinations should be made to ensure the findings are reproducible. Representative procedures are given in references [55] and [98,124] for lipophilic and hydrophilic radiotracers, respectively.
5.2 Protein binding
The degree to which a radiopharmaceutical associates with blood proteins serves as an indicator of the free fraction available for uptake by the brain and other organs [125–128]. These interactions are often adventitious and weak, although some are specific and strong. The latter are exemplified by the ability of transferrin to strip gallium and indium from some radiopharmaceuticals [129]. Radiopharmaceutical lipophilicity is a major contributor to the weak interactions [128], and albumin is the chief target since it makes up about half of the total protein in human serum. There are two primary, and multiple secondary, binding sites for drugs on human serum albumin [130,131]. Species differences are well known for the binding of radiopharmaceuticals to human and animal serum albumins, a factor that should be kept in mind when attempting to extrapolate results from one species to another [132].
Estimates of protein binding can be obtained directly in vivo by microdialysis using animal models, but applicability is limited by the requirement for complex survival surgery techniques. A variety of in vitro methods, each with experimental caveats, can be used to assess serum or plasma protein binding. These include equilibrium dialysis, ultrafiltration, ultracentrifugation and precipitation assays [126,131,133]. The 2,2,2-trichloroacetic acid (TCA) protein precipitation assay is a simple way to evaluate plasma protein binding. For example, [125I]-E-IA-BF-PE-PIPZE was incubated at 37 °C for 30 min in commercially available mouse plasma, followed by addition of cold 10% TCA / 5% HCl. The strong acid causes proteins to precipitate, and centrifugation permits separation of the supernatant, containing unbound radiotracer, from the pellet containing protein bound radiotracer. A radioactivity balance should be measured to verify good recovery. In this instance, a 99% radioactivity balance was obtained, and the extent of protein binding was 71 ± 0.25% (n = 9). The experimental details have been fully described for studies of [125I]-E-IA-DM-PE-PIPZE [55]. A caveat to the TCA method is the potential for physical trapping of free radiopharmaceutical during the process of protein precipitation. In the equilibrium dialysis and ultrafiltration techniques, adsorption of the radiotracer to the filtration membranes, as well as the precise membrane porosity, can be problematic. Recently, a gel filtration HPLC method has been developed to circumvent these issues during PET studies needing fast analysis of plasma protein binding [134].
6. Direct receptor binding
Binding assays using the radiolabeled version of a novel ligand give direct, sensitive and quantitative measures of specific interactions with the recognition site of interest. They complement the indirect measurements described in Section 3 by providing proof that the radioligand binds to the biological target, along with key information regarding rates of interaction, radioligand affinity and target site density. This information helps identify radioligands suited to in vivo studies and imaging trials. As noted previously, abundant resources are available regarding binding theory and practice [82–88]. Variations are required for assessing agonist radioligand internalization and efflux or recognition site trafficking using living cancer cells or cloned receptors in stably transfected cells [135,136]. Here we describe the three major types of direct binding experiments, namely kinetic, saturation and competition studies, as applied to [125I]-E-IA-BF-PE-PIPZE and [125I]-E-IA-DM-PE-PIPZE. For these radioligands, simple single-site binding models proved appropriate for analysis.
6.1 Kinetics
One aspect of direct radioligand binding is determining the time it takes to reach a steady state, where the binding observed is no longer increasing noticeably. This is sometimes referred to as “pseudo-equilibrium,” since it represents the approach to a true equilibrium process where association of the radioligand to the receptor has become equal to the dissociation of the receptor-radioligand complex (Eq. 1). In practice, kinetic techniques are a variation of those described in Section 3.3. Total radioligand binding is measured at various times after incubation starts, and parallel experiments are conducted using a known inhibitor to define non-specific binding. Some non-specific binding sites may also be saturable, so the non-radioactive inhibitor used should ideally be from a different structural class. Specific radioligand binding is then plotted against time. Fig. 8A shows data for [125I]-E-IA-BF-PE-PIPZE (0.16 nM) in mouse brain membranes at 37 °C, where a plateau in binding, the steady-state, is achieved near 180 min. Excellent specific binding, 85 – 97 % of the total, was observed between 30 and 240 min.
Fig. 8.

Association (Panel A) and dissociation (Panel B) kinetics for [125I-E-IA-BF-PE-PIPZE (0.16 nM) binding in mouse brain membranes, 0.15 mg protein / mL, at 37 °C. Data shown are from representative experiments performed in duplicate. Haloperidol (1.0 μM) was used to define non-specific binding, and also to initiate radioligand dissociation (Panel B) following the initial 180 min incubation to reach steady-state binding.
The observed rate of association (kobs) has units of inverse time and depends upon the radioligand concentration. Steady-state is reached more quickly for higher concentrations of radioligand. Ideally, several concentrations should be tested. The process is usually treated as pseudo first-order as shown in Equation 6:
| (Eq. 6) |
where Yt is radioligand binding at time t, and Ymax is the binding plateau at infinite time. For [125I]-E-IA-BF-PE-PIPZE, a kobs of 0.016 ± 0.002 min−1 was determined using program Prism. Since kobs reflects rates of association (kon) and dissociation (koff) [83,85], determining the bimolecular kon, with units of nM−1 min−1, requires knowledge of the dissociation rate constant koff to solve Equation 7:
| (Eq. 7) |
The koff is unimolecular with units of min−1, and depends only upon the concentration of the receptor-radioligand complex. It can be determined by allowing the radioligand and receptor to reach equilibrium, followed by addition of a non-radioactive inhibitor to prevent radioligand from rebinding to the receptor after dissociation occurs. Loss of binding is then determined as a function of time, and koff calculated according to monophasic exponential decay Equation 8:
| (Eq. 8) |
where Yt is the specific binding at a given time t after dissociation has been initiated, and Ymax is the specific binding observed at the equilibrium starting point. Fig. 8B shows this experiment for [125I]-E-IA-BF-PE-PIPZE (0.16 nM) in mouse brain membranes at 37 °C, which gives a koff of 0.0033 ± 0.0003 min−1. With the known koff and radioligand concentration of 0.16 nM, Equation 7 yields a kon of 0.078 nM−1 min−1. Further, according to Equation 2, the kinetically determined equilibrium dissociation constant Kd = koff / kon = 0.042 nM. Thus, [125I]-E-IA-BF-PE-PIPZE proved a very high affinity radioligand having a slow dissociation rate.
6.2 Saturation (Kd and Bmax)
Saturation studies are conducted, ideally under equilibrium conditions, to measure a thermodynamic radioligand affinity (Kd) for a receptor and the density of that receptor (Bmax) in a given preparation. In short, the amount of radioligand-receptor complex is driven to the point that the maximal level of binding can be estimated as if no more complex could be formed. Two procedural variations are used. The first, and arguably most accurate, is called “hot” saturation, where increasing concentrations of radioligand alone provide the mass needed. The second procedure, designed to reduce the amount of radioactivity required, uses both the radioactive and non-radioactive versions of the same ligand, and is called “cold” saturation.
6.2.1 “Hot” saturation
In practice, a receptor preparation (membranes, cells, tissue sections, etc.) is incubated with increasing concentrations of radioligand until steady-state binding is achieved. Incubation time is often based upon observed association kinetics (e.g., Fig. 8A). The radioligand-receptor complex (Bound) increases as a function of the radioligand added. Under ideal conditions, the concentration of added radioligand is not appreciably depleted by binding (< 10%), and can be used to approximate the unbound radioligand concentration, termed Free. Non-specific binding for each concentration is determined in parallel using a competing non-radioactive ligand. Nonlinear regression [83,85] then provides a numerical solution to the one-site mass-action Equation 9:
| (Eq. 9) |
where Y is specific radioligand binding (Bound) at a given radioligand concentration [Radioligand], which approximates the Free. The dissociation constant, Kd is typically given in nM, and the maximal receptor density, Bmax, is usually expressed as fmol per mg protein or as the number of binding sites per cell, but can also be expressed in nM units.
The plot takes the form of a hyperbola, with Free on the X-axis, Bound on the Y-axis, and Bmax approached asymptotically. In a typical design for a single-site binding model, six to eight radioligand concentrations are used that are centered on the suspected Kd, and span from one-tenth to ten times that value. Representative data for [125I]-E-IA-BF-PE-PIPZE is shown in Fig. 9A using six concentrations, spaced on arithmetic scale, ranging from 0.0032 nM to 1.6 nM. These were chosen anticipating the Kd to be between the kinetic Kd of 0.042 nM (Section 6.1) and the apparent affinity, Ki of 0.43 nM (Section 3.1), previously determined by competition binding against [3H]-PTZ. Non-linear regression provided a Kd of 0.24 ± 0.01 nM and a Bmax of 477 ± 13 fmol / mg protein for four trials. Specific binding represented 95% of total binding at the Kd concentration. As expected, non-specific binding increased linearly with radioligand concentration. Under these conditions, < 3% of radioligand added was involved in total binding, so Free was estimated accurately. If greater than 10% total binding is observed, special forms of Equation 9 are available to correct for radioligand depletion. The Bmax was consistent with published σ1 receptor densities for this receptor preparation [55,58]. Such comparisons offer confidence that future studies, especially those using an uncharacterized receptor preparation, would also yield meaningful data.
Fig. 9.

Panel A: Saturation binding of [125I]-E-IA-BF-PE-PIPZE (0.016 – 1.6 nM) to mouse brain membranes (0.15 mg protein / mL) at 37 °C with a 180 min incubation and haloperidol (1.0 μM) to define non-specific binding. Panel B: Traditional Rosenthal plot for visualization of the relationship between bound radioligand, and the ratio of bound radioligand to free radioligand. Data are from a representative experiment performed in duplicate, and replicated four times to give Kd 0.24 ± 0.01 nM and Bmax 472 ± 13 fmol / mg protein by non-linear regression.
In theory, kinetic and thermodynamic Kd values should match. However, a 5.7-fold higher affinity (Kd 0.042 nM) was calculated for [125I]-E-IA-BF-PE-PIPZE using rate constants compared to saturation binding (Kd 0.24 nM). Such discrepancies are frequently observed [93,137–140]. Agreement can be improved in some cases by correcting for radioligand depletion, either mathematically or by using low receptor concentrations [137,138]. In many cases, the discrepancy rises from failure to conduct saturation assays at equilibrium. According to theory, the incubation time required to reach 97% of equilibrium is 5-fold greater than the dissociation half-life [84]. The half-life for [125I]-E-IA-BF-PE-PIPZE dissociation can be calculated as 210 min (t½ = 0.693 / koff), indicating that 18 h of incubation would be required for true equilibrium. This is not very practical, and also might lead to appreciable receptor or radioligand degradation, particularly at 37 °C. Consequently, saturation binding is often performed, either knowingly or unknowingly, under pseudo-equilibrium conditions where Kd values are overestimated and affinity is underestimated.
Fig. 9B linearizes the saturation binding data by a traditional Rosenthal - Scatchard plot of Bound / Free on the Y-axis against specific radioligand binding Y (Bound) on the X-axis. The slope of the connecting line is proportional to the negative reciprocal of Kd, the X-intercept represents Bmax, and the Y-intercept defines Bmax / Kd as shown by algebraic transformation of Equation 9 to give Equation 10:
| (Eq. 10) |
This transformation is useful for visualizing changes in Kd and Bmax under differing experimental conditions, or for detecting a curvilinear plot that might suggest binding to more than one type of site, but is less accurate than non-linear regression for calculating numerical values. In Fig. 9B, Bound / Free is plotted as the ratio of the concentration of radioligand specifically bound to that which is free, a unitless measure. This common way to display the plot shows fractional radioligand binding at a glance. For the slope of the line to actually equal −1 / Kd in units of nM, however, Bound / Free must be expressed as (fmol / mg protein) / [Radioligand (nM)]. This also gives a unitless Y-intercept, calculated as 1942 for this experiment (not shown). Although appropriate for graphical Kd calculation, such plots are not particularly useful upon visual inspection. When Bound / Free and Bound are represented yet another way, using tissue concentrations in nM rather than fmol / mg protein, the maximal value of Bound / Free becomes meaningful again, and is termed Binding Potential (BP) as discussed in Section 8. Assuming protein represents 5% of brain tissue, the Bound / Free value of 1942 can be converted to a BP of 97 for [125I]-E-IA-BF-PE-PIPZE in this single trial.
6.2.2 “Cold” saturation
For radioligands having very high specific activity, addition of non-radioactive ligand may be required to achieve the necessary mass for “hot” saturation without using an extraordinary amount of radioactivity. For example, [111In]-labeled analogs of naltrindole were intentionally brought down to 1500 mCi / μmol prior to saturation binding studies of δ opioid receptors in vitro [98]. An alternative approach is “cold” saturation [141–143]. In this technique, a single low concentration of radioligand competes for the receptor against increasing concentrations of the non-radioactive version. Since the hot and cold ligands vying for the receptor are structurally identical, the assay is also known as homologous competition binding where the radioligand Kd equals the ligand Ki. As previously described [142], substitution into the Cheng-Prusoff relationship (Eq. 3) allows expression of Kd as IC50 – [Radioligand]. Substitution of this definition of Kd into the mass-action Equation 9, followed by rearrangement, provides a convenient way to obtain Bmax using non-linear regression to solve Equation 11:
| (Eq. 11) |
“Cold” saturation studies are employed for practical purposes in order to limit the radioactivity required. This applies to direct binding studies of high specific activity radioligands, and to work with expensive commercially available radioligands. An example of the latter is characterization of binding parameters for [3H]-CP55940, a cannabinoid receptor type 2 (CB2) radioligand of high affinity (Kd 1.5 nM). Subsequently, economical competition binding studies against the tritiated radioligand were performed to identify appropriate target ligands to be labeled with fluorine-18 for CB2 receptor PET [144]. Homologous competition is routinely used for characterizing moderate affinity radioligands, such as the σ2 selective [3H]-DTG (Kd 28 nM) [46,58], where “hot” saturation binding would be prohibitively expensive. We used “cold” saturation to determine Kd and Bmax for [125I]-E-IA-DM-PE-PIPZE, a radioligand having a relatively high σ1 receptor affinity (Kd 3.79 nM) [55]. In this case, “hot” saturation studies would have required the preparation of stock solutions having up to a 400 nM concentration of radioligand for dilution into the binding assays. For perspective, 800 μCi would be required to prepare 2 mL of stock, considering the specific radioactivity of 2000 mCi / μmol. By contrast, the homologous competition studies required only a 0.015 nM fixed concentration. The main issue with “cold” saturation studies is ambiguity in the parameter estimates unless conditions are carefully controlled [83]. For instance, the concentration of radioligand used should be well below its Kd. Thus, “hot” saturation studies are inherently more accurate, and are preferable when feasible.
6.3 Competition
In Section 3, competition binding was discussed from the viewpoint of characterizing the relative ability of a candidate ligand, in non-radioactive form, to interact with primary sites of interest, as well as potential secondary sites, using a panel of well-characterized radioligands. Together, those results provide a pharmacological binding profile for the novel ligand, and may strongly suggest that the radiolabeled version will bind selectively to the chosen receptor. The objective of the competition studies described here is to confirm or refute that hypothesis by obtaining a pharmacological profile of the binding sites actually labeled by the novel radioligand. Methodologically, the experiments are equivalent, except a panel of well-characterized non-radioactive ligands is tested against the novel radioligand.
Inhibitory potencies for a panel of eight σ receptor ligands against [125I]-E-IA-BF-PE-PIPZE in mouse brain membranes are given in Table 2. The radioligand was used at 0.1 nM, < 50% of its thermodynamic Kd of 0.24 nM, and the incubation was 180 min at 37 °C based on time to reach steady-state (Fig. 8A). Specific radioligand binding represented 85% – 90% of the total. The IC50 values were converted to Ki using the Cheng-Prusoff relationship (Eq. 3). The ligands fully inhibited radioligand binding in concentration-dependent fashion (not shown). Hill slopes were near unity, which is consistent with radioligand binding to a single class of sites. Inhibition by non-radioactive E-IA-BF-PE-PIPZE shows the saturability of the binding sites, a fundamental concept in receptor theory. The (+)- and (−)-enantiomers of SKF10,047 were included as a test for binding site stereoselectivity [145]. Appropriate σ1 receptor enantioselectivity was observed, with much higher affinity noted for (+)-SKF10,047. Interestingly, the nomenclature sigma (σ) receptor has its origins from the first letter “S” of (+)-SKF10,047, considered the prototypical ligand over 40 years ago when the sites were thought to be a novel opioid receptor [30,38].
Table 2.
Pharmacological profile of sites labeled by [125I]-E-IA-BF-PIPZE is consistent with selective radioligand binding to σ1 receptors in membranes from whole mouse braina,b
| Compound | IC50 (nM) | Ki (nM) | Hill (nH) |
|---|---|---|---|
| Haloperidol | 1.2 ± 0.2 | 0.9 ± 0.2 | 0.9 ± 0.1 |
| E-IA-BF-PIPZE | 1.7 ± 0.2 | 1.1 ± 0.1 | 1.0 ± 0.1 |
| (+)-Pentazocine | 13.7 ± 1.4 | 8.1 ± 0.8 | 0.9 ± 0.1 |
| BD1063 | 13.6 ± 0.7 | 9.6 ± 0.5 | 1.3 ± 0.1 |
| (+)-SKF10,047 | 43.2 ± 3.5 | 28.8 ± 2.4 | 1.1 ± 0.1 |
| DTG | 209 ± 14 | 114 ± 7 | 1.0 ± 0.1 |
| (−)-SKF10,047 | 7091 ± 519 | 5005 ± 366 | 1.3 ± 0.3 |
| Br-Mach | 9851 ± 807 | 6390 ± 524 | 1.1 ± 0.2 |
Values are means ± SEM (n = 3 – 6).
0.1 nM [125I]-E-IA-BF-PIPZE; 37 °C, 180 min; 50 mM TRIS, pH 8, 0.01% bovine serum albumin; 1.0 μM haloperidol defined non-specific binding.
Qualitatively, ligands known to have high affinity for the σ1 receptor (haloperidol, (+)-pentazocine, BD1063, (+)-SKF10,047) competed effectively for [125I]-E-IA-BF-PE-PIPZE binding, while those known to have poor affinity, such as (−)-SKF10,047 did not. A ligand developed by the Mach group, 5-bromo-N-[4-(6,7-dimethoxy-3,4-dihydro-1H-isoquinolin-2-yl)-butyl)]-2,3-dimethoxybenzamide, that has 1500-fold selectivity for σ2 over σ1 receptors [146,147] was also included. For simplicity, we refer to this ligand in-house as “Br-Mach,” and found negligible affinity for sites labeled by [125I]-E-IA-BF-PE-PIPZE. Quantitatively, a strong Pearson correlation (r = 0.99, p = 0.0001; Fig. 10) of pKi values exists between the data of Table 2 and that previously reported for six of the inhibitors against [3H]-PTZ binding in mouse brain membranes [58]. Based upon the pharmacology observed, the sites labeled by [125I]-E-IA-BF-PE-PIPZE in vitro can be assigned as σ1 receptors.
Fig 10.
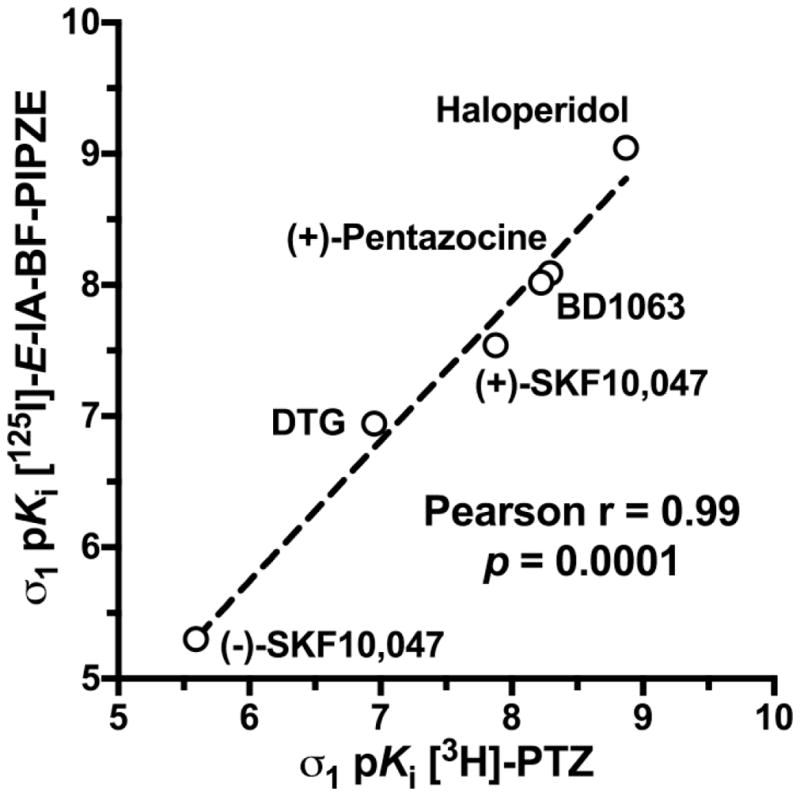
Pearson correlation of ligand inhibitory potencies, as pKi values, determined in CD-1® mouse brain membranes for [125I]-E-IA-BF-PIPZE (Table 2) with published data for [3H]-PTZ [58].
7. In vivo evaluation
Good in vitro values for affinity and selectivity, along with suitable physicochemical properties are necessary, but not sufficient, criteria for a receptor-binding radiotracer to be useful in vivo. Animal studies of several different types are necessary to establish suitability for basic science studies, and even more are required for development as a radiopharmaceutical. Generally, animal work with a candidate receptor-binding radiotracer begins with rodent studies to determine tissue distribution, receptor specificity and metabolism. Key goals are to provide an ADME profile, and to confirm a receptor-mediated mechanism of localization. The maximum amount of information should be obtained from the least number of animals. There is international support for the applying the “three R’s” (Replacement, Refinement, Reduction) to animal experimentation [148]. Scientific journals, such as the British Journal of Pharmacology, are adapting manuscript guidelines to specify inclusion of all aspects of animal work, from protocol design through welfare [149]. Protocols for experimentation must conform to relevant national regulations and local guidelines. In the United States, animal research must be in compliance with the Guide for the Care and Use of Laboratory Animals [150], and be approved prior to conduct by a local Institutional Animal Care and Use Committee (IACUC). The animal studies described herein were governed by these requirements.
Requirements for animal research protocols to be viewed as acceptable evolve over time. Presently, literature searching must be documented to indicate that new knowledge will be gained, unnecessary duplication of prior studies will be avoided, and alternatives to animal testing have been considered. Types of experiments, statistical rationales (power analysis) for animal numbers, methods to relieve pain and distress, definitions of humane endpoints, veterinary oversight and methods of euthanasia must be delineated. Refinements in techniques, as well as husbandry and environmental enrichment, must be addressed. Radioligands and drugs to be administered must be given with their source and purity, as well as amounts to be used and routes of administration. Purchase and housing should be coordinated with the experimental schedule to maintain a close window for age and weight in order to minimize confounders. Modern protocols not only ensure animal welfare, but also consider protections for those who perform studies. These occupational health considerations include use of personal protective equipment and immunizations as appropriate, as well as provisions to reduce exposures that might lead to allergies or to the transmission of infectious diseases from laboratory animals to human beings (zoonoses). Reference texts are available for mouse [151] and rat [152] handling and techniques, and periodicals are devoted to state-of-the-art animal experimentation. In addition to these reference compilations, most institutions have, and require, hands-on training so those new to the field can gain proficiency.
7.1 Pharmacokinetics
Pharmacokinetic studies are performed to determine the temporal biodistribution of the candidate radioligand. The results impact future animal study designs, and perhaps, choice of radionuclide and imaging modality. For instance, the pharmacokinetics of a [11C]-labeled radioligand might suggest that a longer-lived [18F]-labeled version would be better in PET imaging scenarios. The first step in this endeavor is deciding how to administer the radiotracer to the animal. Numerous routes are available, including intravenous (i.v.), intraperitoneal (i.p.), intrathecal (IT), intratumoral (i.t.), intramuscular (i.m.) and subcutaneous (s.c.) [148]. The choice depends upon study goals and radiotracer characteristics. For instance, IT administration delivers proteins to the brain [153], while the i.t. approach delivers nanoparticles directly to a solid tumor [154]. Volumes and formulations should be compatible with the size and physiology of the animal. For a small molecular weight radioligand that targets the CNS, bolus i.v. injection is preferred, since the dose is delivered directly to the bloodstream, avoiding first-pass metabolism.
To minimize natural variations among species and genotypes, many laboratories standardize radiotracer comparisons to one species. We work primarily with the CD-1® mouse strain. For mouse, a lateral tail vein is used, and doses should be formulated so a low volume (ca. 100 – 200 μL) is injected to avoid perturbation of total blood volume. Vehicles are usually sterile 0.9% saline containing a few percent of relatively innocuous additives (e.g., ethanol, propylene glycol). Due to exquisite sensitivity of detection, only a few μCi of radioactive tracer, a sub μg / kg quantity at a high specific activity, is required for each animal. The experiment is designed to measure the biodistribution of the radiotracer as a function of time in brain, peripheral organs and bodily fluids. For each time to be investigated, groups of animals are dosed and then euthanized. Tissues and fluids are harvested, weighed and counted for radioactivity along with standard dilutions of the injected dose (%ID). Concentrations of radioactivity are expressed as either %ID / g or as %ID / organ. Further normalization to body weight for each animal would provide a standardized uptake value (SUV).
“Time-activity” curves that graphically illustrate uptake and clearance of radioactivity for [125I]-E-IA-BF-PE-PIPZE are given in Fig. 11. The biodistribution was followed to 24 h because SPECT using I-123, with a 13.2 h half-life, could accommodate this time frame. Initial brain uptake (Panel A) can be considered high, with 6.14 ± 0.94 %ID / g (mean ± SD) at 5 min. Peak uptake of 6.56 ± 1.23 %ID / g was reached at 15 min, with a decline to 4.48 ± 0.22 %ID / g evident at 4 h. By 24 h, brain uptake dropped to 0.76 ± 0.091 %ID / g. Statistically speaking, brain uptake did not change significantly (ANOVA) over the first 4 h of the study.
Fig. 11.

Temporal biodistribution in brain and selected peripheral organs after administration of [125I-E-IA-BF-PE-PIPZE (2.5 μCi, i.v.) to male CD-1® mice. Non-specific binding defined by BD1063 (5.0 μmol / kg, i.v.) pretreatment at each time. Values are %ID / g (means ± SEM, n = 4). Non-visible error bars are contained within the symbols.
Good brain uptake not only confirms passage across the blood-brain barrier, but also indicates that [125I]-E-IA-BF-PE-PIPZE is not likely to be a substrate for efflux transporters, such as the P-glycoprotein (PGP) pump. Both factors are important considerations. For example, [3H]-PTZ, the standard for in vitro binding assays of σ1 receptors, is quickly pumped out of brain by PGP [155], which precludes use in vivo. If low uptake of a candidate radioligand is observed in whole brain, in vitro methods exist to test for PGP interactions. These include the Pgp-Glo™ Assay System, a kit complete with recombinant human PGP membranes, and the use of MDR-1 MDCK cells, available from the Laboratory of Cell Biology at the United States National Cancer Institute (NCI) [156]. For an in vivo test, paired wild type and PGP-deficient CF-1™ mice are available from Charles River Laboratories. Considerable substrate dependent differences in PGP activity are well known between human beings and various animal species, presenting further complications. Efflux transporters are targets for imaging in their own right [157]. Expert reviews on these topics in relation to radiopharmaceutical development are available from Pike [8,158].
The pharmacokinetics of [125I]-E-IA-BF-PE-PIPZE in heart, lung and muscle are shown in Fig. 11, Panels B – D. Radioligand uptake was 50% higher in heart than brain, and fully 5-fold higher in lung compared to brain. Activity cleared slowly from heart (r2 0.90; t½ 119 min) and lung (r2 0.88; t½ 104 min) according to monexponential decay. Muscle showed much lower uptake, with a pharmacokinetic profile similar to brain. Blood levels were low throughout the study, and decreased from 0.31 ± 0.03 %ID / g at 5 min to 0.08 ± 0.01 %ID / g at 24 h (not shown), indicating good extraction of radiotracer from whole blood into the organs. Various other tissues were also examined (Fig. 12). For instance, radioactivity was observed in the eye at 5 min (2.01 ± 0.13 %ID / g) that did not clear significantly (ANOVA) over the first 120 min. Such data is of most interest when internal radiation absorbed dose estimates are required (Section 10). In that regard, the present study was not designed to assess dosimetry. Such studies have additional design features [159]. For example, animals often void when euthanized, so urine is collected on filter papers that are combined with the harvested urinary bladder for absorbed dose calculations.
Fig. 12.

Effects of pretreatments with various σ receptor ligands (2.5 μmol / kg, i.v.) on the uptake of [125I-E-IA-BF-PE-PIPZE at 60 min in brain and selected peripheral organs of male CD-1® mice. Values are %ID / g (means ± SD, n = 4).
7.2 Pharmacology
Pharmacokinetic studies describe total radiotracer uptake and clearance, but do not provide the fraction of radioactivity associated with specific binding to a receptor. Similar to an in vitro binding assay, non-specific binding must be defined by a parallel study, using separate groups of animals, where radioligand competes for receptor against an excess of non-radioactive ligand known to bind that receptor. If timing and dosing are appropriate, specific radioligand binding will be blocked, and residual tissue radioactivity will represent the non-specific component. Subtraction of non-specific for a given tissue from the total determined in the first experiment then provides estimates of receptor-specific radioligand binding as %ID / g. Appropriate protocols for many receptors are available, but novel systems may require timing and dose-ranging studies to make sure the blocker and radioligand are present simultaneously to compete for the receptor. Supplemental pharmacology studies, using a panel of ligands, may be included to assess potential off-target interactions. Animals receiving macroscopic amounts of drugs should be monitored for adverse effects or abnormal behaviors.
In the present case, we treated mice with the σ1 receptor antagonist BD1063 (5 μmol / kg, i.v.) 5 min before administration of [125I]-E-IA-BF-PE-PIPZE (Fig. 11). This serves to define non-specific binding based on prior studies [55–58]. BD1063 greatly reduced uptake and enhanced clearance of [125I]-E-IA-BF-PE-PIPZE. As defined by BD1063, specific radioligand binding to σ1 receptors at 15 min was 75% in brain, 87% in heart, 87% in lung and 61% in muscle (Fig. 11). The proportion of specific binding increased over time, as non-specific binding washed out more rapidly than the specific binding. For all tissues, clearance of non-specific binding as %ID / g was monoexponential (r2 0.90 – 0.95) and rapid (t½ 6.8 min – 11.4 min), consistent with the absence of a receptor-mediated mechanism of retention.
Additional σ receptor ligands were then tested as potential inhibitors in pretreatment experiments analyzed at a single time, 60 min (Fig. 12). These were given at 2.5 μmol / kg (i.v.), and included E-IA-BF-PE-PIPZE to test for saturability, BD1063 (σ1), haloperidol (σ1 / σ2) and the σ2 receptor-selective Br-Mach. No adverse effects or abnormal behaviors were observed. Significance was tested at the α = 0.05 significance level (ANOVA) against the saline-treated control group (post hoc Dunnett’s test). [125I]-E-IA-BF-PE-PIPZE uptake was reduced significantly by E-IA-BF-PE-PIPZE (53% – 94%), haloperidol (47% – 90%) and BD1063 (53% – 90%) in brain and peripheral organs, with the exception of the liver. Expression of σ1 receptors by all of these organs has been confirmed in prior in vitro or in vivo studies [42,55,140,145]. The liver showed increased uptake upon inhibition of σ1 receptor binding in the other organs. This is accompanied by a 2-fold increase of radioactivity in the blood at 60 min compared to controls (e.g., for BD1063, 0.76 ± 0.04 %ID / g vs. 0.38 ± 0.09 %ID / g). We hypothesize a portion of the circulating radioligand is then metabolically trapped by the liver (Section 7.4).
Qualitatively, BD1063 and haloperidol provided similar levels of inhibition, despite their structural differences. Saturation by cold ligand appeared somewhat greater. This may reflect identical access of the cold and hot ligands to all tissue compartments; coupled, perhaps, with a small component of saturable but non-specific binding to unkown sites or structures. Br-Mach did not inhibit radioligand uptake in spleen, kidney, lung or liver, but a low decrease was noted for brain (14%), heart (15%) and muscle (24%). Although statistically significant, this minimal level of inhibition is not likely to be biologically meaningful when taken with the other data [160]. Together, these pharmacology studies show a σ1 receptor-mediated mechanism of localization for [125I]-E-IA-BF-PE-PIPZE in mouse brain and most peripheral organs.
7.3 Regional distribution in brain
A useful neuroreceptor radioligand must show in vivo binding in discrete brain regions that mirrors the relative distribution of the sites. Biodistribution studies that include anatomical dissection of the brain into regions allow correlation of uptake or specific binding to that known from prior in vitro or in vivo studies. For some systems, an appropriate regional distribution is readily apparent. For example, striatum and cerebellum are definitive target and non-target regions, respectively, for dopamine receptor and dopamine transporter studies. By contrast, σ1 receptors are heterogeneously distributed throughout mammalian brain, but densities are rather similar between the regions. There is no sizeable internal reference region to serve as a “tissue blank” representing non-specific uptake. This presents some technical limitations, but there are also advantages, because many questions can be addressed simply by examining whole brain.
Fig. 13 displays [125I]-E-IA-BF-PE-PIPZE uptake and pharmacology at 60 min for nine regions of mouse brain. As expected from prior studies of σ1 receptors, the highest control levels were in pons / medulla and superior / inferior colliculi, while the lowest levels were in striatum and hippocampus. These data were collected during the pharmacology studies described above in Section 7.2, and the effects of the pretreatments matched those observed in whole brain. A saturating dose of E-IA-BF-PE-PIPZE reduced uptake by over 92% across all regions, while effective inhibition was noted for haloperidol (68% – 90%) and BD1063 (86% – 90%) (ANOVA, post hoc Dunnett’s test). As in whole brain, minimal but statistically significant, inhibition was observed for Br-Mach in most brain regions.
Fig. 13.

Effects of pretreatments with various σ receptor ligands (2.5 μmol / kg, i.v.) on the uptake of [125I-E-IA-BF-PE-PIPZE at 60 min in male CD-1® mouse brain regions. Values are %ID / g (means ± SD, n = 4).
The σ1 receptor-specific binding of [125I]-E-IA-BF-PE-PIPZE in these 9 brain regions gave a robust Pearson correlation (Pearson r = 0.86, p = 0.003) with in vivo data previously obtained using [125I]-E-IA-DM-PE-PIPZE (Section 8, Fig. 16). In a related experiment, specific binding data obtained by in vitro autoradiography using [125I]-E-IA-BF-PE-PIPZE in brain sections was correlated well with ex vivo data from (+)-[3H]-SKF10,047 (Section 9, Fig. 18). Thus, correlations and cross-correlations confirm selective labeling of the σ1 receptor in vivo by [125I]-E-IA-BF-PE-PIPZE throughout the mouse brain.
Fig. 16.
[125I]-E-IA-BF-PE-PIPZE specific binding in vivo correlates (Pearson r = 0.86, p = 0.003) with [125I]-E-IA-DM-PE-PIPZE specific binding [58] in vivo for 9 regions of male CD-1® mouse brain. Values are %ID / g (means, n = 4).
Fig. 18.

Pearson correlation of quantitative autoradiography data obtained using [125I]-E-IA-BF-PE-PIPZE in vitro in CD-1® mouse brain sections with autoradiography data reported for [3H](+)-SKF10,047 ex vivo in CD-1® mouse brain [178]. PAG = periaqueductal gray; Cb = cerebellum; TCtx = temporal cortex; FCtx = frontal cortex; CA1, CA2, CA3 = fields of the hippocampus; DG = dentate gyrus; CCtx = cingulate cortex; ECtx = entorhinal cortex; CPu = caudate putamen.
7.4 Metabolism (radiometabolites)
Only radioactivity is measured in the in vitro and in vivo experiments described thus far. An initial assumption is the signal represents unchanged radiotracer, but the chemical nature of that radioactivity should be assessed experimentally. In vivo, most radioligands are transformed by active enzymes that are binding proteins in their own right. Thus, enzymes exhibit substrate selectivity, and there are also species differences in metabolism. Two well-known cases use metabolism of the radiopharmaceutical as a design element: the Tc-99m complex derived from N,N′-1,2-ethylenedylbis-L-cysteine diethyl diester (Neurolite®) and [18F]-2-fluorodeoxyglucose ([18F]-FDG). In the case of Neurolite®, the neutral lipophilic complex for imaging of brain blood perfusion passively crosses the blood-brain-barrier, and endogenous esterases convert one of the pendant ester groups to a charged carboxylate anion. This metabolism provides a trapping mechanism to permit imaging by SPECT. Otherwise, Neurolite® would passively diffuse out of the brain. In the case of [18F]-FDG, glucose transport proteins actively carry the radiopharmaceutical into the cell for phosphorylation by hexokinase. The second enzymatic step is blocked by the presence of the carbon-fluorine bond, allowing accumulation of the charged species. At the opposite end of that spectrum, unstable [99mTc]-labeled complexes show high stomach activity from pertechnetate anion (99mTcO4−) and unstable [18F]-labeled compounds show extensive bone uptake from [18F]-F−.
Metabolism is an unwanted feature of most receptor-binding ligands, and occurs largely in the periphery rather than the brain. For instance, radioligands labeled with C-11 in N- or O-methyl positions often suffer metabolic loss of the radiolabel during neuroreceptor brain imaging by PET. The downside may be limited to loss of the radiolabel, since the radiolabeled metabolites are polar and do not cross the blood-brain barrier. If lipophilic radiometabolites are produced, and do cross the blood-brain barrier, the specific to non-specific binding signal would be reduced and confound analysis. Such metabolites might also bind to the receptor of interest, but with different kinetics, or even bind to an altogether different site. Depending on the situation, the molecular position for radiolabeling might need to be altered. Complexity further increases when considering the suitability of a radioligand for studies of CNS receptors as well as the peripheral sites located on normal organs or, perhaps, tumors.
In the early stages of preclinical validation, analytical methods can be used to examine radioligand metabolism in organs and bodily fluids harvested from small animals, such as the mouse. For a radiometabolite protocol to provide meaningful data, a high percentage of the radioactivity must be recovered and processed for analysis. While multiple methods can be used, HPLC is one of the most powerful techniques. Higher levels of radiotracer are administered than for pharmacokinetic studies, but the amount of radioactivity in some of the samples may still be lower than the detection limit of an in-line radioactivity detector. Thus, fractions of HPLC eluent may need to be collected, counted and then graphed to produce a chromatogram.
For illustration, Fig. 14 shows the HPLC radiochromatograms obtained by extracting the brain, lung and liver of a CD-1® mouse 60 min after administration of 100 μCi (i.v.) of [125I]-E-IA-BF-PE-PIPZE. The peak at 8.8 min corresponds to unmetabolized radioligand using conditions similar to those of Fig. 6. Each organ was counted for radioactivity, and then homogenized in methanol, acetonitrile and water containing DTG (100 μM). This mixture was selected to precipitate proteins, solubilize the radioactivity, and help ensure HPLC compatibility. DTG was included to aid displacement of any radioligand that might be σ receptor bound. Mixtures were centrifuged, and a radioactivity balance obtained for extracts and pellets with respect to the original sample. Organ extracts were diluted with water and then filtered (0.45 μm) for HPLC analysis. The urine sample required only dilution with HPLC mobile phase and filtration. Details for a similar procedure have been reported for [125I]-E-IA-DM-PE-PIPZE.
Fig. 14.

Reversed-phase HPLC radiochromatograms of extracts from brain (A), lung (B) and liver (C), as well as a urine sample (D) 60 min after administration of [125I]-E-IA-BF-PE-PIPZE (100 μCi, i.v.) to a male CD-1® mouse. The peak at 8.8 min corresponds to unmetabolized [125I]-E-IA-BF-PE-PIPZE.
High percentages of radioactivity were recovered for brain, heart and lung. About 99% of the radioactivity detected in brain tissue was from intact radioligand. Thus, biodistribution data for brain accurately reflect the radioligand and not radiometabolites. Similar results were noted for lung and heart (not shown). For liver, only 80% of the radioactivity was extractable. Of that, only 40% was associated with unchanged radioligand. The remainder was accounted for by multiple more polar radiometabolites. The liver data contrasts with that from [125I]-E-IA-DM-PE-PIPZE, where the extractable activity was 90% unchanged radioligand and σ1 receptor-specific binding was readily detected. The large proportion of radiometabolites in liver reduces the chances of observing specific binding for [125I]-E-IA-BF-PE-PIPZE, and prompted our speculation of radiometabolite trapping as a contributor to the increased liver uptake upon blockade of σ1 receptors (Fig. 12). Urinary excretion of radioactivity from [125I]-E-IA-BF-PE-PIPZE consisted entirely of polar metabolites (99%).
8. Comparisons of [125I]-E-IA-BF-PE-PIPZE to [125I]-E-IA-DM-PE-PIPZE
Our scientific objective (Section 2.1) was to determine how substitution of the dimethoxyphenethyl group of E-IA-DM-PE-PIPZE with the dihydrobenzofuranylethyl moiety of E-IA-BF-PE-PIPZE would affect in vitro and in vivo binding to σ receptors, with a view toward their use in SPECT imaging trials. As we had thought, in vitro σ receptor binding proved quite sensitive to this modification, with E-IA-BF-PE-PIPZE displaying 15- to 20-fold higher apparent affinities for both σ1 and σ2 receptors than E-IA-DM-PE-PIPZE (Table 3). Since binding was enhanced to the same extent at both sites, comparable σ2 / σ1 selectivity was observed for the two ligands based upon Ki ratios. Neither ligand exhibited appreciable affinity for opioid receptors or monamine transporters in vitro, indicating some overall selectivity for σ1 receptors. Direct binding also showed a 16-fold higher measured affinity of [125I]-E-IA-BF-PE-PIPZE for sites in mouse brain membranes, with a comparable site density for the two radioligands (Table 3). Sites labeled by [125I]-E-IA-BF-PE-PIPZE (Table 2) and [125I]-E-IA-DM-PE-PIPZE [55] were confirmed as σ1 receptors by the pharmacology observed for competitive binding against well-known non-radioactive σ receptor ligands.
Table 3.
In vitro and in vivo comparisons of E-IA-DM-PIPZE and E-IA-BF-PIPZE
| In Vitro Metric | E-IA-DM-PIPZE* | E-IA-BF-PIPZE |
|---|---|---|
| Sigma-1, Ki (nM) | 6.7 ± 0.9 | 0.43 ± 0.03 |
| Sigma-2, Ki (nM) | 1447 ± 220 | 74.5 ± 4.44 |
| Selectivity: σ2/σ1 | 216 | 173 |
| In Vitro Metric | [125I]-E-IA-DM-PIPZE* | [125I]-E-IA-BF-PIPZE |
|---|---|---|
| Kd (nM) | 3.79 ± 0.21 | 0.24 ± 0.01 |
| Bmax (fmol/mg protein) | 599 ± 28 | 472 ± 13 |
| Log D7.4 | 2.25 ± 0.01 | 2.69 ± 0.28 |
| Protein Binding | 48.2 ± 1.0% | 71.2 ± 0.3% |
| In Vivo Metric | [125I]-E-IA-DM-PIPZE | [125I]-E-IA-BF-PIPZE |
|---|---|---|
| Calculated Binding Potential (Bmax / Kd) | 7.9 | 98 |
| Specific / Non-Specific Whole Brain, 30 min | 5.0 ** (3.95 / 0.79) | 5.5 ** (4.90 / 0.89) |
| Specific / Non-Specific Whole Brain, 60 min | 4.8 ** (2.18 / 0.45) | 9.0 ** (5.38 / 0.60) |
| Specific / Non-Specific Whole Brain, 240 min | Not Determined | 12.2 ** (4.14 / 0.34) |
| Radiometabolites Whole Brain, 60 min | 8% | 1% |
Both lipophilicity and protein binding increased for [125I]-E-IA-BF-PE-PIPZE with respect to [125I]-E-IA-DM-PE-PIPZE (Table 3). Log D7.4 measurements showed a 0.44 log unit difference, indicating 2.8-fold higher lipophilicity for [125I]-E-IA-BF-PE-PIPZE. Protein binding also increased, by a substantial 50%. We had anticipated the direction of these changes in the physicochemical parameters, considering that the replacement of an oxygen atom with a methylene group reduces the possibilities for hydrogen bonding and increases the strength of hydrophobic interactions.
At this juncture in the development process, the in vitro information has considerable worth for predicting in vivo behavior and molecular imaging potential. In 1979, Eckelman and colleagues [161] posed the concept of using binding models and in vitro data to better understand the target to non-target ratios that might be observed in vivo for reversible receptor-binding radiopharmaceuticals. Briefly, imaging radiotracers are used in vivo at high specific radioactivity, so concentrations are well below their in vitro Kd. Thus, specific radioligand binding approaches zero for the Rosenthal - Scatchard transformation (Equation 10, Y -> 0; Section 6), which reduces to Equation 12:
| (Eq. 12) |
Consequently, the maximal Bound / Free ratio is also approached, which is defined in the mass-action binding model as Bmax / Kd. This gives an upper limit to the target to non-target ratios that might be observed for a given system in vivo based solely upon radioligand affinity and target site density [161–163]. The relationship further demonstrates that targets with lower density are more difficult to image and require radioligands of higher affinity. G-protein coupled receptors (GPCR) provide one example. As discussed by Pike [8], most successful radioligands for this class are antagonists that can bind to both the coupled and non-coupled states. Selective labeling of the coupled state, a small proportion of total sites in vivo, is much more difficult, in part because of the lower Bmax.
For translation to the in vivo situation, Bmax should be in nM units, and based on tissue rather than protein. Protein represents about 5% of brain tissue, so simple division by a factor of 20 converts a Bmax in fmol / mg protein to units of nM in brain tissue. In 1984, Mintun and colleagues [164] used compartmental modeling of neuroreceptor PET data to arrive at much the same conclusion, and introduced the term Binding Potential (BP) to represent the in vivo Bmax / Kd. Imaging determinations of BP are widely used, and are actually proportional to Bmax / Kd depending upon how the analyses are conducted. By expert consensus [165], “BP without subscript refers to the “true” in vitro measurement of Bmax/Kd,” while BPND refers to the equilibrium ratio of specifically bound to nondisplaceable radioligand in tissue by compartmental modeling of in vivo imaging data.
Using BP provides a convenient way to gauge potential in vivo outcomes for candidate recognition site ligands, and gives an estimation of the maximal specific to non-specific binding that could be observed. For instance, a BP of 7.9 for [125I]-E-IA-DM-PE-PIPZE and a BP of 98 for [125I]-E-IA-BF-PE-PIPZE can be calculated from their respective Bmax / Kd ratios (Table 3). For these radioligands, the BP differential reflects their affinities, since target site densities are the same for both. By definition (vide supra) and in practice, the specific to non-specific binding signal observed in vivo is less than the calculated BP value. Reviews by Patel and Gibson [163] and Pike [8] show that successful neuroreceptor imaging agents have a BP of 5 or greater. The BP method is useful for comparisons and predictions, but suffers inherent limitations considering that critical factors, including plasma protein binding and metabolism, are not addressed [162].
The in vivo behavior of [125I]-E-IA-BF-PE-PIPZE has been treated in Section 7. Here we compare [125I]-E-IA-BF-PE-PIPZE and [125I]-E-IA-DM-PE-PIPZE [55,58], with emphasis on aspects relevant to CNS imaging of σ1 receptors. Both radioligands passed the first critical requirement by crossing the blood-brain barrier (Fig. 15) in accord with their molecular size and Log D7.4 (Table 3). While their uptakes in whole mouse brain were comparable at the earliest times, the pharmacokinetics diverged within 30 min. Brain uptake for [125I]-E-IA-BF-PE-PIPZE showed a stable plateau, and did not change significantly (ANOVA) over the first 120 min. By contrast, [125I]-E-IA-DM-PE-PIPZE cleared by 75% from 15 min to the 120 min, with data fit by a monexponential decay (r2 = 0.99; t½=36 min) [55].
Fig. 15.
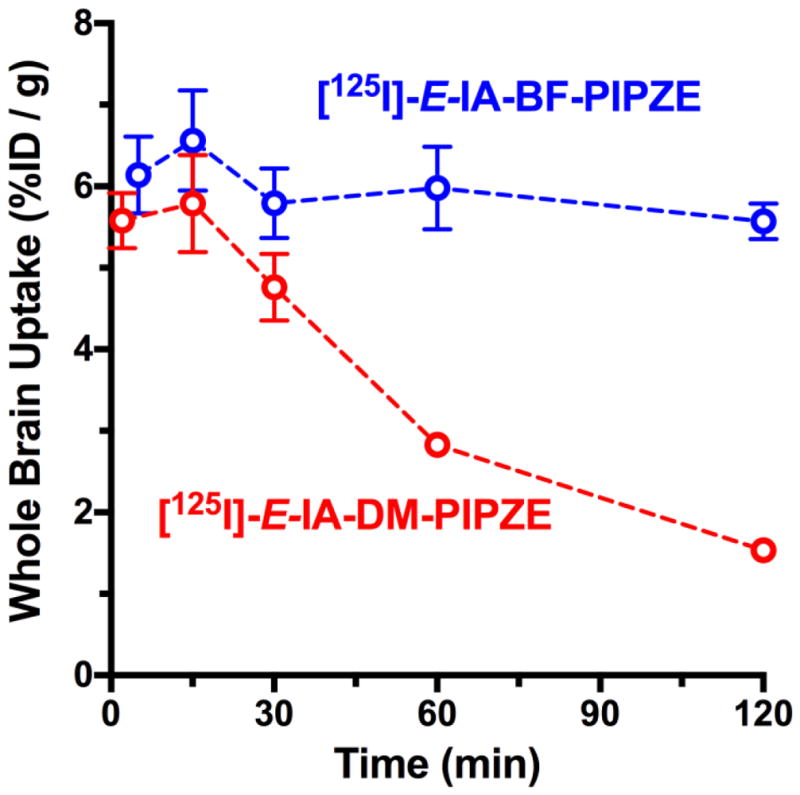
Pharmacokinetic comparison of whole brain uptake of [125I]-E-IA-DM-PE-PIPZE [55] and [125I]-E-IA-BF-PE-PIPZE in male CD-1® mice. Values are %ID / g (means ± SEM, n = 4; 2.5 μCi / mouse, i.v.).
For both radioligands, the majority of brain uptake represented specific binding to σ1 receptors. At 30 min and 60 min, specific binding was 80% to 90% of total. Explicit comparisons of specific to non-specific binding ratios, along with the %ID /g data used for the calculations, are given in Table 3. The ratio was near 5 for [125I]-E-IA-DM-PE-PIPZE at both 30 min and 60 min, and the washout of both specific binding and non-specific binding proved similar. For [125I]-E-IA-BF-PE-PIPZE, the specific to non-specific binding ratios roughly doubled over this time period because the specific binding signal was more strongly retained. Levels of non-specific binding for [125I]-E-IA-BF-PE-PIPZE were marginally higher than for [125I]-E-IA-DM-PE-PIPZE, in keeping with its higher lipophilicity. Consistent with receptor binding as the mechanism of localization, the higher target to non-target ratio observed for [125I]-E-IA-BF-PE- PIPZE reflects the 16-fold higher σ1 receptor affinity compared to [125I]-E-IA-DM-PE-PIPZE.
The pharmacokinetics for these two radioligands proved complementary. Their regional cerebral distributions are well correlated (Fig. 16), and consistent with labeling of σ1 receptors in vivo. Levels of radiometabolites were low in brain tissue, with only 1% noted for [125I]-E-IA-BF-PE-PIPZE (Fig. 14) and 8% for [125I]-E-IA-DM-PE-PIPZE (Table 3). This is an important consideration during imaging trials, and for selection of an appropriate approach to modeling (Section 9). The specific to non-specific ratios of 5 observed in vivo for [125I]-E-IA-DM-PE-PIPZE are in good agreement with the maximum value of 7.9 predicted from the in vitro BP calculations. For [125I]-E-IA-BF-PE-PIPZE, however, the specific to non-specific binding ratios ranging from 5.5 at 30 min to 12 at 240 min were much below the in vitro BP of 98. Since levels of non-specific binding in brain tissues were fairly similar for these radioligands, a plausible explanation is the higher plasma protein binding observed for [125I]-E-IA-BF-PE-PIPZE in vitro (Table 3) indeed occurs in vivo. Like non-specific binding, this is partly a function of lipophilicity, and reduces the free fraction of radioligand in plasma that is available to cross the blood-brain barrier. Thus, an upper limit would be imposed on the [125I]-E-IA-BF-PE-PIPZE specific binding attained.
The dissociation of [125I]-E-IA-BF-PE-PIPZE from σ1 receptors in vitro is quite slow (Section 6). In vivo, when no competing ligand prevents rebinding, the effective off-rate may be even slower. Generally, low koff influences drug efficacy in vivo because slower dissociation implies a longer duration of action at the recognition site [166]. From a radiopharmaceutical viewpoint, a low koff helps ensure high enough bound radioligand for detection by imaging. If sensitivity to recognition site occupancy is an objective, the koff may need to be relatively higher. As discussed by Eckelman [162], a Kd in the pM range may indicate an off-rate so slow that imaging outcomes are blood flow dependent, while a Kd greater than 10 nM may indicate an off-rate too fast for visualization of specific binding. Overall, in vivo binding and recognition site imaging reflects a complex interplay of many factors. Characteristics observed in the mouse suggest that [123I]-E-IA-BF-PE-PIPZE would provide higher quality SPECT images of σ1 receptors due to temporal signal stability, a higher count rate and a greater target to non-target ratio. On the other hand, [123I]-E-IA-DM-PE-PIPZE would be likely to provide adequate SPECT images, along with greater sensitivity for occupancy studies. Thus, the properties of these two radioligands should be complementary.
In relation to the field, we imagine that [123I]-E-IA-BF-PE-PIPZE (Kd 0.24 nM) might prove similar to [123I]-TPCNE (Ki 0.67 nM), which exhibited irreversible kinetics in rat brain by dissection [44] and in human brain by SPECT [47]. An irreversible model best described the human brain kinetics, and the authors suggested that outcomes might be blood flow dependent. [123I]-E-IA-DM-PE-PIPZE (Kd 3.79 nM) might prove to be a SPECT counterpart to [11C]-SA4503 (Kd 4.5nM, [167]). [11C]-SA4503 displays reversible kinetics in rat brain by PET [52,168,169], similar to those of [125I]-E-IA-DM-PE-PIPZE in mouse brain. [11C]-SA4503 PET is validated for σ1 receptor occupancy measurements in human beings, with data analysis by a 2-tissue 3-compartment model yielding BPND [51,53,170]. Considering the σ1 receptor Bmax (636 fmol / mg protein, ca. 31.8 nM) for human frontal cortex [171], and the Kd or Ki of these 4 ligands, the in vitro BP predictions are 7.1 for SA4503, 8.4 for E-IA-DM-PE-PIPZE, 47 for TPCNE and 132 for E-IA-BF-PE-PIPZE. Though SA4503 and E-IA-DM-PE-PIPZE have comparable BP, SA4503 is a σ1 receptor agonist [45,172] while E-IA-DM-PE-PIPZE shows antagonist character [58]. For σ1 receptors, structurally distinct from GPCR [38], agonist and antagonist radioligands appear to be equally suitable for in vivo studies. Behavioral or biochemical assays, distinct from the types of studies described herein, are usually required to make functional differentiations.
9. Preclinical Imaging
Imaging is the culmination of the considerable efforts devoted to synthesis, radiosynthesis and a variety of in vitro and in vivo studies. Preclinical imaging in rodents or non-human primates (NHP) provides great insight into the potential of a novel receptor-binding radiotracer to translate into a radiopharmaceutical. As mentioned in the Introduction, autoradiography provided support and accelerated progress in non-invasive receptor imaging by PET and SPECT early on. High-resolution autoradiography continues to be combined with PET or SPECT imaging to provide a complete picture. At a higher level, questions regarding receptor-related sex differences can be addressed through preclinical imaging, and receptor imaging in animal models can help to test hypotheses regarding disease.
9.1 Autoradiography
Autoradiography is widely used in biomedical research and pharmaceutical development. It is an efficient, high-resolution technique for quantitative localization of radioactivity in thin slices taken from brain, peripheral organs, solid tumors or, in the case of a small rodent, even the whole body [173]. The present context will be limited to detection of receptor distributions in brain, which was pioneered by Michael Kuhar and colleagues [13,14]. In brief, radioligand binding to receptors is visualized in thin (10 – 20 μm) sections prepared from frozen brain using a cryostat microtome. These sections are thaw-mounted onto microscope slides, and then apposed to X-ray film or a phosphorimaging plate. Qualitative images of the radioactivity distribution are obtained by development of the film or inspection of the phosphorimage. Quantitative data for brain regions of interest are then obtained by comparison to calibrated standards of radioactivity. Multiple brain regions, including those of small size, can be examined simultaneously and with confidence because of the high sensitivity and high spatial resolution. This is a great advantage for autoradiography studies as compared to anatomical dissection methods in vivo, or to homogenate binding assays in vitro. The spatial resolution achieved does depend upon the decay characteristics of the radionuclide employed. Detailed reviews of theory and practice are available [174–176]. The method can be applied in vitro or ex vivo, as follows.
9.1.1 In vitro autoradiography
In the in vitro case, slide-mounted brain tissue sections are incubated in a buffered solution of the radioligand in the presence and absence of a blocker to define non-specific binding. Kinetic, saturation and competition binding studies can be performed [175], using techniques that are essentially the same as described in Section 6. After incubation, the slides are washed using ice-cold buffer to remove the non-specifically bound material. Chilling lowers koff, so repetitive washes can be used to maximize the specific binding signal. As with other in vitro binding assays, this method obviates the need to consider delivery of the radiotracer or metabolism. Gibson [88] suggests that comparison of radioligands using a “nonwash” autoradiographic procedure in vitro can be a useful predictor of the receptor-binding signal to noise ratio that might be observed in vivo.
In vitro autoradiograms for [125I]-E-IA-BF-PE-PIPZE binding to σ1 receptors in mouse brain sections in the presence and absence of BD1063 are shown in Fig. 17. Procedurally, the brain sections were incubated at 37 °C for 180 min with the radioligand (0.1 nM), chilled and then washed three times for 15 min in ice-cold buffer. Slides were dried, and then apposed to film for 24 h. The relationship between film optical densities and radioactivity was calibrated using radioactivity standards and a MCID™ digital image analysis system. The total binding image for [125I]-E-IA-BF-PE-PIPZE displays a heterogeneous distribution of radioactivity throughout mouse brain, with the lowest levels of radioactivity in white matter structures, such as the corpus callosum (Fig. 17). In the presence of BD1063, a low and homogeneous level of activity, just above film background, was detected. Thus, the total binding image is almost entirely σ1 receptor-specific. A mouse brain atlas [177] was used to identify discrete anatomical regions for further analysis. Specific binding of [125I]-E-IA-BF-PE-PIPZE in vitro, measured in fourteen regions, was then plotted against the specific binding data known for those same regions of mouse brain from ex vivo autoradiography using the σ1 receptor radioligand (+)-[3H]-SKF10,047 [178]. As shown in Fig. 18, excellent correlation (Pearson r = 0.92, p < 0001) was observed, confirming specific labeling of σ1 receptors by [125I]-E-IA-BF-PE-PIPZE.
Fig. 17.

Selective labeling of cerebral σ1 receptors in horizontal sections (20 μm) from CD-1® mouse brain using [125I]-E-IA-BF-PE-PIPZE (0.1 nM). Left image: Total binding. Right image: Non-specific binding defined by inclusion of BD1063 (1.0 μM). Calibrated pseudo-color palette (fmol / mg tissue) is shown on the far right.
9.1.2 Ex vivo autoradiography
For ex vivo autoradiography, the radioligand is administered (i.v.) to the animal, usually a rodent. After an appropriate time, based upon prior studies, the animal is euthanized, the brain is removed, quick-frozen and sliced into thin sections. These sections are dried, transferred to glass slides and data analysis performed as described for the in vitro case. An ex vivo autoradiography study of σ1 receptors in mouse brain using [125I]-E-IA-DM-PE-PIPZE is shown in Fig. 19 [58]. Specific binding to σ1 receptors was 80% of the total binding observed for saline-treated controls. The similarity between the regional distributions of σ1 receptors obtained ex vivo and in vitro is readily apparent.
Fig. 19.

Visualization of σ1 receptors in horizontal sections (20 μm) from CD-1® mouse brain 60 min after administration of [125I]-E-IA-DM-PE-PIPZE (150 μCi, i.v.). Left image: Total binding, saline-treated control. Center image: Reduced binding in the presence of (−)-cocaine (100 μmol / kg, i.p.). Right image: Non-specific binding defined by BD1063 (2.5 μmol / kg, i.v.). Calibrated pseudo-color palette (fmol / mg tissue) is shown on the far right. Figure and legend adapted, with permission, from [58].
The in vivo properties of [125I]-E-IA-DM-PE-PIPZE are amenable to occupancy studies, and a behaviorally active dose of (−)-cocaine (100 μmol / kg, i.p.) inhibited radioligand uptake by 50% (Fig. 19). Thus, (−)-cocaine interacts significantly with σ1 receptors in vivo despite weak affinity, Ki ca.1500 nM, for the sites. This finding supports the notion that (−)-cocaine’s psychostimulant effects are partly due to agonist interactions with σ1 receptors. In further support of this mode of action, [125I]-E-IA-DM-PE-PIPZE was used to establish a good correlation (r2 = 0.88) between the ability of a potent antagonist, PD144418, to occupy cerebral σ1 receptors and to attenuate (−)-cocaine induced locomotor hyperactivity in mice [56]. Non-radioactive E-IA-DM-PE-PIPZE also partially attenuates (−)-cocaine induced locomotor hyperactivity, suggesting antagonist character [58]. At present, such behavioral studies are a principal way to assign the functional properties of σ1 receptor ligands [38,64,179,180]. Dopamine transporter (DAT) inhibition is recognized as the main contributor to (−)-cocaine’s behavioral effects and abuse liability. In fact, PET studies relating DAT occupancy by (−)-cocaine to behavioral effects in human beings, including the dose needed for a “high,” are hallmarks for the field [48,181–183].
9.2 PET or SPECT imaging studies in rodents or non-human primates
Preclinical imaging studies in animals provide unique information in their own right, which is also relevant to future nuclear imaging studies in human beings [184]. Data regarding pharmacokinetics, drug occupancy and endogenous neurotransmitter release, as well as effects of sex, aging or disease can be obtained [185–187]. Having images of the highest quality is essential, which takes into account not only the radioligand, but also the instrumentation, and the protocols for image reconstruction and registration with anatomical features. There has been much progress in PET and SPECT imaging of conscious animals that includes the use of behavioral training and special restraints [188–191], software development to reduce motion artifacts [191–193], and specialized miniature tomographs, such as the RatCAP, that can be mounted on the head of an awake animal [194]. For the near future, however, small animals will ordinarily be under general anesthesia during image acquisition. Anesthetics necessarily modulate synaptic transmission and exert profound effects on systemic physiology [195–197]. As a consequence, significant differences may be observed between the results from imaging or biodistribution studies performed under various forms of anesthesia, and from the results obtained for awake, freely moving animals.
After high quality images are acquired, uptake has to be quantified accurately and precisely, and converted to time-activity curves. Mathematical models are then designed and validated that incorporate critical information, some of which must be obtained experimentally during a scan. For instance, the amount of radiotracer in plasma that is available for transport into the brain, an “input function,” may well need to be determined, so the radioligand distribution throughout the various “compartments” (specific binding, non-specific binding, etc.) can be modeled over time to provide outcomes such as BP. An excellent recent review has summarized three decades of progress in quantitative imaging by PET [18]. Advances in taking imaging physics and instrumentation technology from the human scale to the microscale needed for small animals have also been reviewed [198,199]. In one account, a microPET instrument exhibiting a resolution of approximately 1.5 mm was used to study binding of [11C]-SA4503 to the σ1 receptor in young and aged rats. Age-related changes were observed in thalamus, midbrain, pons, and medulla, but not cortex by analysis of distribution volume and BP [169]. In another recent study, high-quality PET imaging, ex vivo autoradiography and a knockout mouse model were employed to show clear differences between the high uptake of the σ1 receptor radioligand [18F]-FTC-146 in discrete brain regions of wild type vs. σ1 receptor knockout mouse (Fig. 20) [200].
Fig. 20.

Panel A: PET / MR coronal images using [18F]-FTC-146 in wild type (WT) and σ1 receptor knockout (S1R-KO) mouse brain, with and without blocking by BD1047 (1 mg / kg). Ctx = cortex; cp = caudate putamen; hc = hippocampus; cer = cerebellum. Panel B: Pharmacokinetics of [18F]-FTC-146 in whole brain (WT and S1R-KO) without and with blocking. Figure and legend adapted, with permission, from [200].
Preclinical brain imaging studies in NHP, such as Rhesus macaques or Papio anubis baboons are also of much interest because these animals are closer to the human being than rodents [201]. The marmoset, a “New World” monkey, is also being investigated for imaging research because it has a more developed brain than a rodent, is small in size (ca. 250 – 400 g) and is readily accommodated by small animal scanners [202,203]. Marmosets may become more prevalent in biomedical research considering ease of handling, lower costs and a favorable reproductive cycle. Both normal NHP imaging, as well as imaging using specific NHP models of neuropathology [201] and drug abuse [204,205] have been described. Due to the limited availability of NHP, a multi-institutional collaboration might be necessary to conduct studies.
9.3 Sex as a biological variable in the validation process
Disease can manifest differently between men and women, and one sex or the other may show a different susceptibility or outcome. While the majority of preclinical and clinical investigations have used male animals and men exclusively, study designs that investigate both sexes are on the rise, and are providing interesting information. The United States National Institutes of Health (NIH) has now adopted a policy that requires consideration of sex as a biological variable in almost all biomedical research applications [206,207]. Much debate has ensued, with strong arguments presented for, and against, this particular approach to improving health outcomes [208–212]. Nevertheless, radiotracer techniques have long provided unique, quantitative and sensitive ways to identify and address potential sex differences. Over twenty-five years ago, differences in the biodistribution of [18F]-labeled opioid receptor ligands were observed as a consequence of divergent metabolic pathways between male and female rats [213]. In 1999, Zubieta and colleagues [214] observed both sex- and age-dependent differences in mu-opioid receptor BP for several brain regions of healthy subjects by PET, and discussed the implications. There are many other examples of such characterizations in the radiopharmaceutical literature.
A sampling of recent articles shows that clinical radiopharmaceutical imaging continues to inform. For instance, a large [18F]-FDG PET study consisting of 493 men and 470 women spanning 5 decades (32 – 87 years) showed lower metabolism in parietal cortex for men compared to women, while lower metabolism was observed in ventrolateral prefrontal cortex for women compared to men [215]. In a multi-institutional study of nonischemic heart failure, three radiotracers, [15O]-H2O, [11C]-palmitate and [11C]-glucose, were used to evaluate myocardial blood flow and metabolism [216]. Findings indicated that sex, but not other variables such as age, was associated with the higher blood flow and fatty acid metabolism observed in women, while glucose metabolism did not differ appreciably between the sexes. Highlights regarding neurochemical systems include work by London and colleagues [217], who used [18F]-fallypride and PET to investigate midbrain dopamine D2-type receptor status in male and female cigarette smokers. Significantly higher midbrain BP was found for the female smokers, which supports certain clinical observations on sex-associated aspects of nicotine dependence. PET studies of [11C]-OMAR, a radioligand targeting the cannabinoid CB1 receptor [218], showed higher brain uptake across regions in men than women [219], augmenting observations of sex-specific differences gained by [18F]-MK-9470 and PET [220]. Findings on the endocannabinoid system may have important implications, particularly considering the adoption of laws supporting medical and casual marijuana use [221].
Radiotracer work in the preclinical arena using mouse models of disease is also yielding insight into sex differences. In the inbred SJL mouse model of multiple sclerosis, a [11C]-labeled sphingosine-1-phosphate receptor 2 ligand showed higher cerebellar uptake in female over male animals [222]. Results were consistent across biodistribution, ex vivo autoradiography and PET imaging experiments. The findings are in accord with recent postmortem observations of a cerebellar increase of this receptor in female compared to male patients having multiple sclerosis [223], and may translate to imaging trials to further define sex-associated aspects of the disease. A recent study from Mach’s group compared σ2 receptor levels in the CNS for male and female mice using the APP/PS1 model of Alzheimers Disease as well as wild type C57BL/6J controls [224]. Quantitative in vitro autoradiographic imaging using the σ2-selective ligand [3H]-RHM-1 [96] showed that site densities were higher in three brain regions (piriform and motor cortices, striatum) of wild type females compared to wild type males. Further, σ2 receptor densities in these same brain regions of female APP/PS1 mice were significantly lower compared to those of wild type females. Differences were not observed between male wild type and APP/PS1 mice. These results are provocative, and indicate an avenue for further research is exploration of sex-associated alterations of CNS σ2 receptors in Alzheimers Disease. Such studies illustrate why incorporation of sex as a biological variable at an early stage of preclinical radiotracer development may well become the routine.
10. Conclusions
Many criteria must be fulfilled for a new radiotracer to progress from basic science studies to adoption as a radiopharmaceutical for clinical use. While poor results from a single critical experiment may be sufficient to terminate development efforts, good results from many experiments are necessary for success. For any new radioligand, there are regulatory hurdles that must be overcome before translation to imaging studies in human beings. Radiation absorbed dose estimates and acute toxicology studies are specific prerequisites prior to human imaging. A whole body biodistribution study in animals can provide important quantitative data for estimating dosimetry in human beings using standard calculations [225–227]. Caution should be exercised, however. For instance, for [11C]-SA4503, critical organs were spleen for human beings but liver for the mouse [159]. Moreover, the effective dose in the human was 1.9 times higher than for the mouse. Due to such differences, there are calls to calculate dosimetry directly in human beings through microdosing, and eliminate the possibility of species differences [1,228,229].
The regulatory requirements to perform a “first in human” study (Phase 0 clinical trial) are detailed in multiple resources, including expert reviews [230–232], governmental regulations [Europe, FDA] and examples for specific radiopharmaceuticals [233–235]. A robust risk-benefit ratio is a key component of the approval process, and required for informed consent by an individual. A good understanding of ligand pharmacology, especially the implications of using antagonist or agonist radioligands [8,239,240], is necessary during development and for translation. For neuroreceptor work, antagonist radioligands are advantageous.
In summary, studies similar to those described here are often used for preclinical validation of novel receptor-binding radiotracers. Variations in the paradigm presented are, of course, dependent upon the specific project, resources available and results obtained. We have presented some aspects of the theory, mechanics and rationale behind these interdisciplinary studies, drawing mainly upon work with σ1 receptor radioligands that are in various stages of development. More generally, the quest for receptor-binding radiopharmaceuticals having clinical impact has been a major focus for the field, and undoubtedly will continue to drive future research.
Acknowledgments
We thank Emily A. Fergason-Cantrell, Lisa D. Watkinson and Terry L. Carmack for assistance with in vitro and in vivo studies. We are grateful to Dr. Frederick T. Chin, Stanford University, Stanford, CA USA for sharing the data for Figure 20. We thank the National Institute on Drug Abuse (DA028477: Development of Anti-Cocaine Medications; DA039818: Radioligands for Sigma-2 Receptor Studies in the CNS by PET) for partial support. We also acknowledge resources provided by Harry S. Truman Memorial Veterans’ Hospital. This work does not represent the views of the United States Department of Veterans Affairs.
Footnotes
Dedicated to the memory of Professor Henry N. Wagner, Jr., MD
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bergström M, Grahnén A, Långström B. Positron emission tomography microdosing: a new concept with application in tracer and early clinical drug development. Eur J Clin Pharmacol. 2003;59:357–366. doi: 10.1007/s00228-003-0643-x. [DOI] [PubMed] [Google Scholar]
- 2.Gee AD. Neuropharmacology and drug development: imaging in clinical neuroscience. Br Med Bull. 2003;65:169–177. doi: 10.1093/bmb/65.1.169. [DOI] [PubMed] [Google Scholar]
- 3.Matthews PM, Rabiner EA, Passchier J, Gunn RN. Positron emission tomography molecular imaging for drug development. Br J Clin Pharmacol. 2012;73:175–186. doi: 10.1111/j.1365-2125.2011.04085.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Grunder G, Muller MJ, Andreas J, Heydari N, Wetzel H, Schlosser R, et al. Occupancy of striatal D2-like dopamine receptors after treatment with the sigma ligand EMD 57445, a putative atypical antipsychotic. Psychopharmacol. 1999;146:81–86. doi: 10.1007/s002130051091. [DOI] [PubMed] [Google Scholar]
- 5.Potter WZ. Biomarkers of brain structure and function for neurodegenerative disorders: are they adequate for go / no go decisions in drug development? Clin Pharmacol Ther. 2015;98:472–474. doi: 10.1002/cpt.196. [DOI] [PubMed] [Google Scholar]
- 6.Papadimitriou L, Smith-Jones PM, Sarwar CMS, Marti CN, Yaddanapudi K, Skopicki HA, et al. Utility of positron emission tomography for drug development for heart failure. Am Heart J. 2016;175:142–152. doi: 10.1016/j.ahj.2016.02.016. [DOI] [PubMed] [Google Scholar]
- 7.Welch MJ, Eckelman WC, editors. Targeted Molecular Imaging. Boca Raton: CRC Press, Taylor and Francis Group; 2012. [Google Scholar]
- 8.Pike VW. Considerations in the development of reversibly binding PET radioligands for brain imaging. Curr Med Chem. 2016;23:1818–1869. doi: 10.2174/0929867323666160418114826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brust P, van den Hoff J, Steinback J. Development of 18F-labeled radiotracers for neuroreceptor imaging with positron emission tomography. Neurosci Bull. 2014;30:777–811. doi: 10.1007/s12264-014-1460-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gropler RJ. Recent advances in metabolic imaging. J Nucl Cardiol. 2013;20:1147–1172. doi: 10.1007/s12350-013-9786-z. [DOI] [PubMed] [Google Scholar]
- 11.Dierckx RA, Otte A, deVries EFJ, van Waarde A, Luiten PGM, editors. PET and SPECT of neurobiological systems. Heidelberg, Berlin: Springer; 2014. [Google Scholar]
- 12.Noble G, Gean E, Nandi A, Frolov B, Zaidi E, Lee H, et al. Advances in CNS imaging agents: Focus on PET and SPECT tracers in experimental and clinical use. CNS Drugs. 2015;29:313–330. doi: 10.1007/s40263-015-0237-z. [DOI] [PubMed] [Google Scholar]
- 13.Pert CB, Kuhar MJ, Snyder SH. Opiate receptor: Autoradiographic localization in rat brain. Proc Natl Acad Sci USA. 1976;73:3729–3733. doi: 10.1073/pnas.73.10.3729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kuhar MJ, Yamamura HI. Light autoradiographic localisation of cholinergic muscarinic receptors in rat brain by specific binding of a potent antagonist. Nature. 1975;253:560–561. doi: 10.1038/253560a0. [DOI] [PubMed] [Google Scholar]
- 15.Wagner HN, Jr, Burns HD, Dannals RF, Wong DF, Långström B, Duelfer T, et al. Imaging dopamine receptors in the human brain by positron tomography. Science. 1983;221:1264–1266. doi: 10.1126/science.6604315. [DOI] [PubMed] [Google Scholar]
- 16.Eckelman WC, Reba RC, Rzeszotarski WJ, Gibson RE, Hill T, Holman BL, et al. External imaging of cerebral muscarinic acetylcholine receptors. Science. 1984;223:291–293. doi: 10.1126/science.6608148. [DOI] [PubMed] [Google Scholar]
- 17.Eckelman WC, Reba RC, Kelloff GJ. Targeted imaging: An important biomarker for understanding disease progression in the era of personalized medicine. Drug Discov Today. 2008;13(17/18) doi: 10.1016/j.drudis.2008.05.009. [DOI] [PubMed] [Google Scholar]
- 18.Gunn RN, Slifstein M, Searle GE, Price JC. Quantitative imaging of protein targets in the human brain with PET. Phys Med Biol. 2015;60:R363–R411. doi: 10.1088/0031-9155/60/22/R363. [DOI] [PubMed] [Google Scholar]
- 19.Lever SZ. The evolution of radiopharmaceuticals for diagnosis and therapy. J Cell Biochem. 2002;39(Suppl):60–64. doi: 10.1002/jcb.10405. [DOI] [PubMed] [Google Scholar]
- 20.Schwaiger M, Wester H-J. How many PET tracers do we need? J Nucl Med. 2011;52:36S–41S. doi: 10.2967/jnumed.110.085738. [DOI] [PubMed] [Google Scholar]
- 21. [accessed July 5, 2016]; http://us.datscan.com.
- 22. [accessed July 6, 2016]; http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm372261.htm.
- 23.Johnson KA, Minoshima S, Bohnen NI, Donohoe KJ, Foster NL, Herscovitch P, et al. Appropriate use criteria for amyloid PET: A report of the amyloid imaging task force, the Society of Nuclear Medicine and Molecular Imaging, and the Alzheimer’s Association. Alzheimer’s & Dementia. 2013;9:e1–e16. doi: 10.1016/j.jalz.2013.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wey H-Y, Gilbert T, Zürcher NR, She A, Bhanot A, Taillon BD, et al. Insights into neuroepigentics through human histone deacetylase PET imaging. Sci Transl Med. 2016;351:351ra106. doi: 10.1126/scitranslmed.aaf7551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sweatt JD. The emerging field of neuroepigenetics. Neuron. 2013;80:624–632. doi: 10.1016/j.neuron.2013.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nobel Media AB. [accessed July 6, 2016];2014 http://www.nobelprize.org/nobel_prizes/chemistry/laureates/1943/
- 27.Nunn AD. Molecular imaging and personalized medicine: an uncertain future. Cancer Biother Radiopharm. 2007;22:722–739. doi: 10.1089/cbr.2007.0417. [DOI] [PubMed] [Google Scholar]
- 28.Zimmermann RG. Why are investors not interested in my radiotracer? The industrial and regulatory constraints in the development of radiopharmaceuticals. Nuc Med Biol. 2013;40:155–166. doi: 10.1016/j.nucmedbio.2012.10.012. [DOI] [PubMed] [Google Scholar]
- 29.Lever SZ, Fan K-H, Fergason-Cantrell E, Carmack T, Watkinson L, Lever JR. [I-125]-(E)-1-(2-(2,3-dihydrobenzofuran-5-yl)ethyl)-4-(iodoallyl)piperazine: a new pharmacological tool for the evaluation of sigma-1 receptors. J Labelled Comp Radiopharm. 2015;58:S74. [Google Scholar]
- 30.Martin WR, Eades CG, Thompson JA, Huppler RE, Gilbert PE. The effects of morphine and nalorphine-like drugs in the nondependent and morphine-dependent chronic spinal dog. J Pharmacol Exp Ther. 1976;197:517–532. [PubMed] [Google Scholar]
- 31.Quirion R, Bowen WD, Itzhak Y, Junien JL, Musacchio JM, Rothman RB, et al. A proposal for the classification of sigma binding sites. Trends Pharmacol Sci. 1992;13:85–86. doi: 10.1016/0165-6147(92)90030-a. [DOI] [PubMed] [Google Scholar]
- 32.Mach RH, Zeng C, Hawkins WG. The σ2 receptor: A novel protein for the imaging and treatment of cancer. J Med Chem. 2013;56:7137–7160. doi: 10.1021/jm301545c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rousseaux CG, Greene SF. Sigma receptors [σRs]: biology in normal and diseased states. J Recept Signal Transduct. 2016;36:327–388. doi: 10.3109/10799893.2015.1015737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nguyen L, Kaushal N, Robson MJ, Matsumoto RR. Sigma receptors as potential therapeutic targets for neuroprotection. Eur J Pharmacol. 2014;743:42–47. doi: 10.1016/j.ejphar.2014.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Maurice T, Su T-P. The pharmacology of sigma-1 receptors. Pharmacol Ther. 2009;124:195–206. doi: 10.1016/j.pharmthera.2009.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hayashi T, Tsai S-Y, Mori T, Fujimoto M, Su T-P. Targeting ligand-operated chaperone sigma-1 receptors in the treatment of neuropsychiatric disorders. Expert Opin Ther Targets. 2011;15:557–577. doi: 10.1517/14728222.2011.560837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.van Waarde A, Rybczynska AA, Ramakrishnan NK, Ishiwata K, Elsinga PH, Dierckx AJO. Potential applications for sigma receptor ligands in cancer diagnosis and therapy. BBA-Biomembranes. 2015;1848:2703–2714. doi: 10.1016/j.bbamem.2014.08.022. [DOI] [PubMed] [Google Scholar]
- 38.Matsumoto RR, Nguyen L, Kaushal N, Robson MJ. Chapter nine – Sigma (σ) receptors as potential therapeutic targets to mitigate psychostimulant effects. Adv Pharmacol. 2014a;69:323–386. doi: 10.1016/B978-0-12-420118-7.00009-3. [DOI] [PubMed] [Google Scholar]
- 39.Romero L, Merlos M, Vela JM. Chapter Seven-antinociception by sigma-1 receptor antagonists: Central and peripheral effects. Adv Pharmacol. 2016;75:179–215. doi: 10.1016/bs.apha.2015.11.003. [DOI] [PubMed] [Google Scholar]
- 40.Mavlyutov TA, Guo L-W, Epstein ML, Ruoho AE. Role of the sigma-1 receptor in Amyotrophic Lateral Sclerosis (ALS) J Pharmacol Sci. 2015;127:10–16. doi: 10.1016/j.jphs.2014.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mach RH, Wheeler KT. Development of molecular probes for imaging sigma-2 receptors in vitro and in vivo. Cent Nerv Syst Agents Med Chem. 2009;9:230–245. doi: 10.2174/1871524910909030230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Banister SD, Manoli M, Kassiou M. The development of radiotracers for imaging sigma (σ) receptors in the central nervous system (CNS) using positron emission tomography (PET) J Labelled Comp Radiopharm. 2013;56:215–224. doi: 10.1002/jlcr.3010. [DOI] [PubMed] [Google Scholar]
- 43.Brust P, Deuther-Conrad W, Lehmkuhl K, Jia H, Wünsch B. Molecular imaging of σ1 receptors in vivo: Current status and perspectives. Curr Med Chem. 2014;21:35–69. doi: 10.2174/09298673113209990214. [DOI] [PubMed] [Google Scholar]
- 44.Waterhouse RN, Mardon K, Giles KM, Collier TL, O’Brien JC. Halogenated 4-(phenoxymethyl)piperidines as potential radiolabeled probes for sigma-1 receptors: in vivo evaluation of [123I]-1-(iodopropen-2-yl)-4-[(4-cyanophenoxy)methyl]piperidine. J Med Chem. 1997;40:1657–1667. doi: 10.1021/jm960720+. [DOI] [PubMed] [Google Scholar]
- 45.Matsuno K, Nakazawa M, Okamoto K, Kawashima Y, Mita S. Binding properties of SA4503, a novel and selective σ1 receptor agonist. Eur J Pharmacol. 1996;306:271–279. doi: 10.1016/0014-2999(96)00201-4. [DOI] [PubMed] [Google Scholar]
- 46.Lever JR, Gustafson JL, Xu R, Allmon RL, Lever SZ. σ1 and σ2 receptor binding affinity and selectivity of SA4503 and fluoroethyl SA4503. Synapse. 2006;59:350–358. doi: 10.1002/syn.20253. [DOI] [PubMed] [Google Scholar]
- 47.Stone JM, Arstad E, Erlandsson K, Waterhouse RN, Ell PJ, Pilowsky LS. [123I]TPCNE - a novel SPET tracer for the sigma-1 receptor: first human studies and in vivo haloperidol challenge. Synapse. 2006;60:109–117. doi: 10.1002/syn.20281. [DOI] [PubMed] [Google Scholar]
- 48.Gatley SJ, Volkow ND, Fowler JS, Ding Y-S, Logan J, Wang G-J, et al. Positron emission tomography and its use to image the occupancy of drug binding sites. Drug Develop Res. 2003;59:194–207. [Google Scholar]
- 49.Olsson H, Farde L. Potentials and pitfalls using high affinity radioligands in PET and SPET determinations on regional drug induced D2 receptor occupancy – A simulation study based on experimental data. NeuroImage. 2001;14:936–945. doi: 10.1006/nimg.2001.0879. [DOI] [PubMed] [Google Scholar]
- 50.Sakata M, Kimura Y, Naganawa M, Oda K, Ishii K, Chihara K, et al. Mapping of human cerebral sigma1 receptors using positron emission tomography and [11C]SA4503. NeuroImage. 2007;35:1–8. doi: 10.1016/j.neuroimage.2006.11.055. [DOI] [PubMed] [Google Scholar]
- 51.Ishikawa M, Sakata M, Ishii K, Kimura Y, Oda K, Toyohara J, et al. High occupancy of σ1 receptors in human brain after single oral administration of donepezil: A positron emission tomography study using [11C]SA4503. Int J Neuropsychopharmacol. 2009;12:1127–1131. doi: 10.1017/S1461145709990204. [DOI] [PubMed] [Google Scholar]
- 52.Ramakrishnan NK, Visser AK, Schepers M, Luurtsema G, Nyakas CJ, Elsinga PH, et al. Dose-dependent sigma-1 receptor occupancy by donepezil in rat brain can be assess with 11C-SA4503 and microPET. Psychopharmacology. 2014;231:3997–4006. doi: 10.1007/s00213-014-3533-2. [DOI] [PubMed] [Google Scholar]
- 53.Ishikawa M, Ishiwata K, Ishii K, Kimura Y, Sakata M, Naganawa M, et al. High occupancy of sigma-1 receptors in the human brain after single oral administration of fluvoxamine: a positron emission tomography study using [11C]SA4503. Biol Psychiatry. 2007;62:878–883. doi: 10.1016/j.biopsych.2007.04.001. [DOI] [PubMed] [Google Scholar]
- 54.Hindmarch I, Hashimoto K. Cognition and depression: the effects of fluvoxamine, a sigma-1 receptor agonist, reconsidered. Hum Psychopharm Clin. 2010;25:193–200. doi: 10.1002/hup.1106. [DOI] [PubMed] [Google Scholar]
- 55.Lever SZ, Xu R, Fan K-H, Fergason-Cantrell EA, Carmack TL, Watkinson LD, et al. Synthesis, Radioiodination and In Vitro Sigma Receptor Binding Studies of N-1-Allyl-N′-4-phenethylpiperazine Analogs. Nucl Med Biol. 2012;39:401–414. doi: 10.1016/j.nucmedbio.2011.10.001. [DOI] [PubMed] [Google Scholar]
- 56.Lever JR, Miller DK, Fergason-Cantrell EA, Green CL, Watkinson LD, Carmack TL, et al. Relationship between cerebral sigma-1 receptor occupancy and attenuation of cocaine’s motor stimulatory effects in mice by PD144418. J Pharmacol Exp Ther. 2014;351:153–163. doi: 10.1124/jpet.114.216671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lever JR, Miller DK, Green CL, Fergason-Cantrell EA, Watkinson LD, Carmack TL, et al. A selective sigma-2 receptor ligand antagonizes cocaine-induced hyperlocomotion in mice. Synapse. 2014;68:73–84. doi: 10.1002/syn.21717. [DOI] [PubMed] [Google Scholar]
- 58.Lever JR, Fergason-Cantrell EA, Watkinson LD, Carmack TL, Lord SA, Xu R, et al. Cocaine occupancy of sigma1 receptors and dopamine transporters in mice. Synapse. 2016;70:98–111. doi: 10.1002/syn.21877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Braghirolli AMS, Waissmann W, da Silva JB, dos Santos GR. Production of iodine-124 and its applications in nuclear medicine. Appl Radiat Isot. 2014;90:138–148. doi: 10.1016/j.apradiso.2014.03.026. [DOI] [PubMed] [Google Scholar]
- 60.Dannals RF, Ravert HT, Frost JJ, Wilson AA, Burns HD, Wagner HN., Jr Radiosynthesis of an opiate receptor binding radiotracer: [11C]carfentanil. Int J Appl Radiat Isot. 1985;36:303–306. doi: 10.1016/0020-708x(85)90089-4. [DOI] [PubMed] [Google Scholar]
- 61.Frost JJ, Wagner HN, Jr, Dannals RF, Ravert HT, Links JM, Wilson AA, et al. Imaging opiate receptors in the human brain by positron tomography. J Comput Assist Tomogr. 1985;9:231–236. doi: 10.1097/00004728-198503000-00001. [DOI] [PubMed] [Google Scholar]
- 62.Titeler M, Lyon RA, Kuhar MJ, Frost JF, Dannals RF, Leonhardt S, et al. μ opiate receptors are selectively labelled by [3H]carfentanil in human and rat brain. Eur J Pharmacol. 1989;167:221–228. doi: 10.1016/0014-2999(89)90582-7. [DOI] [PubMed] [Google Scholar]
- 63.Xu R, Lord SV, Peterson RM, Fergason-Cantrell EA, Lever JR, Lever SZ. Ether modifications to 1-[2-(3,4-dimethoxyphenyl)ethyl]-4-(3-phenylpropyl)piperazine (SA4503): Effects on binding affinity and selectivity for sigma receptors and monoamine transporters. Bioorg Med Chem. 2015;23:222–230. doi: 10.1016/j.bmc.2014.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Matsumoto RR, Potelleret FH, Mack A, Pouw B, Zhang Y, Bowen WD. Structure-activity comparison of YZ-069, a novel σ ligand, and four analogs in receptor binding and behavioral studies. Pharmacol Biochem Be. 2004;77:775–781. doi: 10.1016/j.pbb.2004.01.014. [DOI] [PubMed] [Google Scholar]
- 65.Fan K-H. PhD thesis. University of Missouri; Dec, 2010. Synthesis and evaluation of sigma receptor ligands. [Google Scholar]
- 66.Wilson AA, Dannals RF, Ravert HT, Wanger HN., Jr Synthesis and biological evaluation of [125I]- and [123I]-4-Iododexetimide, a potent muscarinic cholinergic receptor antagonist. J Med Chem. 1989;32:1057–1062. doi: 10.1021/jm00125a021. [DOI] [PubMed] [Google Scholar]
- 67.Ravert HT, Scheffel U, Mathews WB, Musachio JL, Dannals RF. [11C]-GR89696, a potent kappa opiate receptor radioligand; in vivo binding of the R and S enantiomers. Nucl Med Biol. 2002;29:47–53. doi: 10.1016/s0969-8051(01)00285-2. [DOI] [PubMed] [Google Scholar]
- 68.Brust P, Deuther-Conrad W, Becker G, Patt M, Donat CK, Stittsworth S, et al. Distinctive in vivo kinetics of the new σ1 receptor ligands (R)-(+)- and (S)-(−)-18F-Fluspidine in porcine brain. J Nucl Med. 2014;55:1730–1736. doi: 10.2967/jnumed.114.137562. [DOI] [PubMed] [Google Scholar]
- 69.Tu Z, Mach RH. C-11 radiochemistry in cancer imaging applications. Curr Top Med Chem. 2010;10:1060–1095. doi: 10.2174/156802610791384261. [DOI] [PubMed] [Google Scholar]
- 70.Dollé F. Carbon-11 and fluorine-18 chemistry devoted to molecular probes for imaging the brain with positron emission tomography. J Labelled Comp Radiopharm. 2013;56:65–67. doi: 10.1002/jlcr.3037. [DOI] [PubMed] [Google Scholar]
- 71.Cole EL, Stewart MN, Littich R, Hoareau R, Scott PJ. Radiosyntheses using fluorine-18: the art and science of late stage fluorination. Curr Top Med Chem. 2014;14:875–900. doi: 10.2174/1568026614666140202205035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Honer M, Gobbi L, Martarello L, Comley RA. Radioligand development for molecular imaging of the central nervous system with positron emission tomography. Drug Discov Today. 2014;12:1936–1944. doi: 10.1016/j.drudis.2014.08.012. [DOI] [PubMed] [Google Scholar]
- 73.Bolton R. Radiohalogen incorporation into organic systems. J Labelled Comp Radiopharm. 2002;45:485–528. [Google Scholar]
- 74.Eersels JLH, Travi MJ, Herscheid JDM. Manufacturing 123I-labelled radiopharmaceuticals. Pitfalls and solutions. J Labelled Comp Radiopharm. 2005;48:241–257. [Google Scholar]
- 75.Coenen HH, Mertens J, Maziére B. Radioiodination reactions for pharmaceuticals - compendium for effective synthesis strategies. Netherlands: Springer; 2006. [Google Scholar]
- 76.Wager KM, Jones GB. Radio-iodination methods for the production of SPECT imaging agents. Curr Radiopharm. 2010;3:37–45. [Google Scholar]
- 77.Lever SZ, Lydon JD, Cutler CS, Jurisson SS. Radioactive Metals in Imaging and Therapy. In: Ward MD, editor. Comprehensive Coordination Chemistry II: From Biology to Nanotechnology. Amsterdam: Elsevier, Ltd; 2003. pp. 883–911. [Google Scholar]
- 78.Liu S. The role of coordination chemistry in the development of target-specific radiopharmaceuticals. Chem Soc Rev. 2004;33:445–461. doi: 10.1039/b309961j. [DOI] [PubMed] [Google Scholar]
- 79.Wadas TJ, Wong EH, Weisman GR, Anderson CJ. Coordinating radiometals of copper, gallium, indium, yttrium, and zirconium for PET and SPECT imaging of disease. Chem Rev. 2010;110:2858–2902. doi: 10.1021/cr900325h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Notni J, Wester HJ. A Practical Guide on the Synthesis of Metal Chelates for Molecular Imaging and Therapy by Means of Click Chemistry. Chemistry. 2016 doi: 10.1002/chem.201600928. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 81.Musachio JL, Lever JR. Vinylstannylated alkylating agents as prosthetic groups for radioiodination of small molecules: design, synthesis and application to iodoallyl analogues of spiperone and diprenorphine. Bioconjugate Chem. 1992;3:167–175. doi: 10.1021/bc00014a012. [DOI] [PubMed] [Google Scholar]
- 82.Bylund DB, Toews ML. Radioligand binding methods practical guide and tips. Am J of Physiol. 1993;265:L421–L429. doi: 10.1152/ajplung.1993.265.5.L421. [DOI] [PubMed] [Google Scholar]
- 83.Motulsky H, Christopoulos A. Fitting models to biological data using linear and non-linear regression: a practical guide to curve fitting. Oxford: Oxford University Press; 2004. [Google Scholar]
- 84.Hulme EC, Trevethick MA. Ligand binding assays at equilibrium: validation and interpretation. Br J Pharmacol. 2010;161:1219–1237. doi: 10.1111/j.1476-5381.2009.00604.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Leach K, Valant C, Sexton PM, Christopoulos A. Measurement of Ligand–G Protein-Coupled Receptor Interactions. In: Poyner DR, Wheatley M, editors. G Protein-Coupled Receptors: Essential Methods. Oxford: Wiley-Blackwell; 2010. [DOI] [Google Scholar]
- 86.Motulsky HJ, Neubig RR. Analyzing binding data. Curr Protoc Neurosci. 2010;52:7.5.1–7.5.65. doi: 10.1002/0471142301.ns0705s52. [DOI] [PubMed] [Google Scholar]
- 87.Maguire JJ, Kuc RE, Davenport AP. Radioligand binding assays and their analysis. Methods Mol Biol. 2012;897:31–77. doi: 10.1007/978-1-61779-909-9_3. [DOI] [PubMed] [Google Scholar]
- 88.Gibson RE. In vitro approaches to site-specific imaging agents. In: Welch MJ, Eckelman WC, editors. Targeted molecular imaging. Boca Raton: CRC Press, Taylor & Francis Group; 2012. pp. 3–15. [Google Scholar]
- 89.Cheng Y, Prusoff WH. Relationship between the inhibition constant (Ki) and the concentration of inhibitor which causes 50 per cent inhibition (IC50) of an enzymatic reaction. Biochem Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- 90.Tjernberg A. DMSO-related effects in protein characterization. J Biomol Screen. 2006;11:131–137. doi: 10.1177/1087057105284218. [DOI] [PubMed] [Google Scholar]
- 91.Kaushal N, Robson MJ, Vinnakota H, Narayanan S, Avery BA, McCurdy CR, et al. Synthesis and pharmacological evaluation of 6-acetyl-3-(4-(4-(4-fluorophenyl)piperazin-1-yl)butyl)benzo [d]oxazol-2(3H)-one (SN79), a cocaine antagonist in rodents. AAPS J. 2011;13:336–346. doi: 10.1208/s12248-011-9274-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Fan K-H, Lever JR, Lever SZ. Effect of structural modification in the amine portion of substituted aminobutyl-benzamides as ligands for binding σ1 and σ2 receptors. Biorg Med Chem. 2011;19:1852–1859. doi: 10.1016/j.bmc.2011.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.DeHaven-Hudkins DL, Fleissner LC, Ford-Rice FY. Characterization of the binding of [3H](+)-pentazocine to sigma recognition sites in guinea pig brain. Eur J Pharmacol. 1992;227:371–378. doi: 10.1016/0922-4106(92)90153-m. [DOI] [PubMed] [Google Scholar]
- 94.Bowen WD, de Costa BR, Hellewell SB, Walker JM, Rice KC. [3H]-(+)-Pentazocine: a potent and highly selective benzomorphan-based probe for sigma-1 receptors. Mol Neuropharmacol. 1993;3:117–126. [Google Scholar]
- 95.Chu UB, Ruoho AE. Sigma receptor binding assays. Curr Protoc Pharmacol. 2015;71:1.34.1–21. doi: 10.1002/0471141755.ph0134s71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Xu J, Tu Z, Jones A, Vangveravong S, Wheeler KT, Mach RH. [3H]N- [4-(3,4-dihydro-6,7-dimethoxyisoquinolin-2(1H)-yl)butyl]-2-methoxy-5-methylbenzamide: A novel sigma-2 receptor probe. Eur J Pharmacol. 2005;524:8–17. doi: 10.1016/j.ejphar.2005.09.063. [DOI] [PubMed] [Google Scholar]
- 97.Kashiwagi H, McDunn JE, Simon PO, Jr, Goedegebuure PS, Xu J, Jones L, et al. Selective sigma-2 ligands preferentially bind to pancreatic adenocarcinomas: applications in diagnostic imaging and therapy. Mol Cancer. 2007;6:48. doi: 10.1186/1476-4598-6-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Duval RA, Allmon RL, Lever JR. Indium-labeled macrocyclic conjugates of naltrindole: high affinity radioligands for in vivo studies of peripheral δ opioid receptors. J Med Chem. 2007;50:2144–2156. doi: 10.1021/jm0700013. [DOI] [PubMed] [Google Scholar]
- 99.Boja JW, Cadet JL, Kopajtic TA, Lever J, Seltzman HH, Wyrick CD, et al. Selective labeling of the dopamine transporter by the high affinity ligand 3β-(4-[125I]Iodophenyl)tropane-β-carboxylic acid isopropyl ester. Mol Pharm. 1995;47:779–786. [PubMed] [Google Scholar]
- 100.Hirano K, Kimura R, Sugimoto Y, Yamada J, Uchida S, Kato Y, et al. Relationship between brain serotonin transporter binding, plasma concentration and behavioural effect of selective serotonin reuptake inhibitors. Br J Pharmacol. 2005;144:695–702. doi: 10.1038/sj.bjp.0706108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Tejani-Butt SM, Brunswick DJ, Frazer A. [3H]nisoxetine: a new radioligand for norepinephrine uptake sites in brain. Eur J Pharmacol. 1990;191:239–243. doi: 10.1016/0014-2999(90)94155-q. [DOI] [PubMed] [Google Scholar]
- 102.Ishiwata K, Kawamura K, Yajima K, QingGeLeTu, Mori H, Shiba K. Evaluation of (+)-p [11C]methylvesamicol for mapping sigma1 receptors: a comparison with [11C]SA4503. Nucl Med Biol. 2006;33:543–548. doi: 10.1016/j.nucmedbio.2006.01.008. [DOI] [PubMed] [Google Scholar]
- 103.Lever JR, Scheffel U, Stathis M, Seltzman HH, Wyrick CD, Abraham P, et al. Radiosynthesis and in vivo studies of a selective ligand for the dopamine transporter: 3β-(4-[125I]iodophenyl)tropan-2β-carboxylic acid isopropyl ester ([125I]RTI-121) Nucl Med Biol. 1996;23:277–284. doi: 10.1016/0969-8051(95)02074-8. [DOI] [PubMed] [Google Scholar]
- 104.Zheng QH, Mock BH. Purification of carbon-11 PET radiotracers from unlabeled precursors by preparative HPLC and SPE. Biomed Chromatogr. 2005;19:671–676. doi: 10.1002/bmc.494. [DOI] [PubMed] [Google Scholar]
- 105.Ornaf RM, Dong MW. Key Concepts of HPLC in Pharmaceutical Analysis. In: Ahuja S, Dong D, editors. Handbook of Pharmaceutical Analysis by HPLC. Amsterdam: Elsevier, Ltd; 2005. pp. 19–45. [Google Scholar]
- 106.Lever JR, Dannals RF, Wilson AA, Ravert HT, Wagner HN., Jr Synthesis of carbon-11 labeled diprenorphine: a radioligand for positron emission tomographic studies of opiate receptors. Tetrahedron Lett. 1987;28:4015–4018. [Google Scholar]
- 107.Kinter CM, Lever JR. Synthesis of radioiodinated naltrindole analogues: ligands for studies of delta opioid receptors. Nucl Med Biol. 1995;22:599–606. doi: 10.1016/0969-8051(94)00149-e. [DOI] [PubMed] [Google Scholar]
- 108.Zubieta JK, Bueller JA, Jackson LR, Scott DJ, Xu Y, Koeppe RA, et al. Placebo effects mediated by endogenous opioid activity on μ-opioid receptors. J Neurosci. 2005;25:7754–7762. doi: 10.1523/JNEUROSCI.0439-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility in drug discovery and development settings. Adv Drug Del Rev. 1997;23:3–25. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 110.Pajouhesh H, Lenz GR. Medicinal chemical properties of successful central nervous system drugs. NeuroRx®. 2005;2:541–553. doi: 10.1602/neurorx.2.4.541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Besnard J, Ruda G, Setola V, Abecassis K, Rodriguiz RM, Huang X-P, et al. Automated design of ligands to polypharmacological profiles. Nature. 2012;492:215–220. doi: 10.1038/nature11691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Wager TT, Hou X, Verhoest PR, Villabos A. Moving beyond Rules: The development of a central nervous system multiparameter optimization (CNS MPO) approach to enable alignment of druglike properties. ACS Chem Neurosci. 2012;1:435–449. doi: 10.1021/cn100008c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Bickerton GR, Paolini GV, Besnard J, Muresan S, Hopkins AL. Quantifying the chemical beauty of drugs. Nat Chem. 2012;4:90–98. doi: 10.1038/nchem.1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Doak BC, Zheng J, Dobritzsch D, Kihlberg J. How beyond rule of 5 drugs and clinical candidates bind to their targets. J Med Chem. 2016;59:2312–2327. doi: 10.1021/acs.jmedchem.5b01286. [DOI] [PubMed] [Google Scholar]
- 115.Wong DF, Pomper MG. Predicting the success of a radiopharmaceutical for in vivo imaging of central nervous system neuroreceptor systems. Mol Imaging Biol. 2003;3:350–362. doi: 10.1016/j.mibio.2003.09.011. [DOI] [PubMed] [Google Scholar]
- 116.Van de Bittner GC, Ricq EL, Hooker JM. A philosophy for CNS radiotracer design. Acc Chem Res. 2014;47:3127–3134. doi: 10.1021/ar500233s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Waterhouse RN. Determination of lipophilicity and its use as a predictor of blood-brain barrier penetration of molecular imaging agents. Mol Imaging Biol. 2003;5:376–389. doi: 10.1016/j.mibio.2003.09.014. [DOI] [PubMed] [Google Scholar]
- 118.Rankovic Z. CNS drug design: balancing physicochemical properties for optimal brain exposure. J Med Chem. 2015;58:2584–2608. doi: 10.1021/jm501535r. [DOI] [PubMed] [Google Scholar]
- 119.Kessler RM, Ansari MS, de Paulis T, Schmidt DE, Clanton JA, Smith HE, et al. High affinity dopamine D2 receptor radioligands. 1. Regional rat brain distribution of iodinated benzamides. J Nucl Med. 1991;32:1593–1600. [PubMed] [Google Scholar]
- 120.Minick DJ, Frenz JH, Patrick MA, Brent DA. A comprehensive method for determining hydrophobicity constants by reversed-phase high-performance liquid chromatography. J Med Chem. 1988;31:1923–1933. doi: 10.1021/jm00118a010. [DOI] [PubMed] [Google Scholar]
- 121.Hansch C. A quantitative approach to biochemical structure–activity relationships. Acc Chem Res. 1969;2:232–239. [Google Scholar]
- 122.Mannhold R, Poda GI, Ostermann C, Tetko IV. Calculation of molecular lipophilicity: State-of-the-art and comparison of log P methods on more than 96,000 compounds. J Pharm Sci. 2009;98:861–893. doi: 10.1002/jps.21494. [DOI] [PubMed] [Google Scholar]
- 123.Wilson AA, Jin L, Garcia A, DaSilva JN, Houle S. An admonition when measuring the lipophilicity of radiotracers using counting techniques. Appl Radiat Isot. 2001;54:203–208. doi: 10.1016/s0969-8043(00)00269-4. [DOI] [PubMed] [Google Scholar]
- 124.Srivastava S, Fergason-Cantrell EA, Nahas RI, Lever JR. Synthesis and opioid receptor binding of indium (III) and [111In]-labeled macrocyclic conjugates of diprenorphine: novel ligands designed for imaging studies of peripheral opioid receptors. Tetrahedron. 2016 doi: 10.1016/j.tet.2016.08.015. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Urien S, Riant P, Albengres E, Brioude R, Tillement JP. In vitro studies on the distribution of probucol among human plasma lipoproteins. Mol Pharm. 1984;26:322–327. [PubMed] [Google Scholar]
- 126.Jeghers O, Piepsz A, Ham HR. What does protein binding of radiopharmaceuticals mean exactly? Eur J Nuc Med. 1990;17:101–102. doi: 10.1007/BF00811433. [DOI] [PubMed] [Google Scholar]
- 127.Berridge MS. The importance of kinetic enhancement. J Nucl Med. 2009;50:1203–1204. doi: 10.2967/jnumed.108.060905. [DOI] [PubMed] [Google Scholar]
- 128.Zoghbi SS, Anderson KB, Jenko KJ, Luckenbaugh DA, Innis RB, Pike VW. On quantitative relationships between drug-like compound lipophilicity and plasma free fraction in monkey and human. J Pharm Sci. 2012;101:1028–1039. doi: 10.1002/jps.22822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Moerlein SM, Welch MJ. The chemistry of gallium and indium as related to radiopharmaceutical production. Int J Nuc Med Biol. 1981;8:277–287. doi: 10.1016/0047-0740(81)90034-6. [DOI] [PubMed] [Google Scholar]
- 130.Ghuman J, Zunszain PA, Petitpas I, Bhattacharya AA, Otagiri M, Curry S. Structural basis of the drug-binding specificity of human serum albumin. J Mol Biol. 2005;353:38–52. doi: 10.1016/j.jmb.2005.07.075. [DOI] [PubMed] [Google Scholar]
- 131.Zhivkova ZD. Studies on drug – human serum albumin binding: The current state of the matter. Curr Pharm Design. 2015;21:1817–1830. doi: 10.2174/1381612821666150302113710. [DOI] [PubMed] [Google Scholar]
- 132.Basken NE, Mathias CJ, Lipka AE, Green MA. Species dependence of [64Cu]Cu-Bis(thiosemicarbazone) radiopharmaceutical to serum albumins. Nuc Med Biol. 2008;35:281–285. doi: 10.1016/j.nucmedbio.2007.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Zeitlinger MA, Derendorf H, Moulton JW, Cars O, Craig WA, Andes D, et al. Protein Binding: Do we ever learn? Antimicrob Agents Chemother. 2011;55:3067–3074. doi: 10.1128/AAC.01433-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Amini N, Nakao R, Schou M, Halldin C. Determination of plasma protein binding of positron emission tomography radioligands by high-performance frontal analysis. J Pharm Biomed Anal. 2014;98:140–143. doi: 10.1016/j.jpba.2014.05.024. [DOI] [PubMed] [Google Scholar]
- 135.Bigott-Hennkens HM, Dannoon S, Lewis MR, Jurisson SS. In vitro receptor binding assays: general methods and considerations. Q J Nucl Med Mol Imaging. 2008;52:245–253. [PubMed] [Google Scholar]
- 136.Marie N, Lecoq I, Jauzac P, Allouche S. Differential sorting of human delta-opioid receptors after internalization by peptide and alkaloid agonists. J Biol Chem. 2003;278:22795–22804. doi: 10.1074/jbc.M300084200. [DOI] [PubMed] [Google Scholar]
- 137.Seeman P, Ulpian C, Wreggett KA, Wells JW. Dopamine receptor parameters detected by [3H]spiperone depend on tissue concentration: analysis and examples. J Neurochem. 1984;43:221–35. doi: 10.1111/j.1471-4159.1984.tb06700.x. [DOI] [PubMed] [Google Scholar]
- 138.Hoffman BJ, Scheffel U, Lever JR, Karpa MD, Hartig PR. N1-Methyl-2–125I-lysergic acid diethylamide, a preferred ligand for in vitro and in vivo characterization of serotonin receptors. J Neurochem. 1987;48:115–124. doi: 10.1111/j.1471-4159.1987.tb13135.x. [DOI] [PubMed] [Google Scholar]
- 139.Sullivan SK, Hoare SRJ, Fleck BA, Zhu Y-F, Heise CE, Struthers RS, et al. Kinetics of nonpeptide antagonist binding to the human gonadotropin-releasing hormone receptor: Implications for structure-activity relationships and insurmountable antagonism. Biochem Pharmacol. 2006;72:838–849. doi: 10.1016/j.bcp.2006.07.011. [DOI] [PubMed] [Google Scholar]
- 140.Lever JR, Litton TP, Fergason-Cantrell EA. Characterization of pulmonary sigma receptors by radioligand binding. Eur J Pharm. 2015;762:118–126. doi: 10.1016/j.ejphar.2015.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.DeBlasi A, O’Reilly K, Motulsky HJ. Calculating receptor number from binding experiments using same compound as radioligand and competitor. Trends Pharmacol Sci. 1989;10:227–229. doi: 10.1016/0165-6147(89)90266-6. [DOI] [PubMed] [Google Scholar]
- 142.Jeffries WB, Waugh D, Abel PW. Analysis of data from “cold saturation” radioligand binding experiments. Methods Mol Biol. 1997;73:331–342. doi: 10.1385/0-89603-399-6:331. [DOI] [PubMed] [Google Scholar]
- 143.Bylund DB, Murrin LC. Radioligand saturation binding experiments over large concentration ranges. Life Sci. 2000;67:2897–2911. doi: 10.1016/s0024-3205(00)00877-8. [DOI] [PubMed] [Google Scholar]
- 144.Rühl T, Deuther-Conrad W, Fischer S, Günther R, Hennig L, Krautscheid H, et al. Cannabinoid receptor type 2 (CB2)-selective N-aryl-oxadiazolyl-propionamides: synthesis, radiolabelling, molecular modelling and biological evaluation. Org Med Chem Lett. 2012;2:32. doi: 10.1186/2191-2858-2-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Walker JM, Bowen WD, Walker FO, Matsumoto RR, De Costa B, Rice KC. Sigma receptors: biology and function. Pharmacol Rev. 1990;42:355–402. [PubMed] [Google Scholar]
- 146.Mach RH, Huang Y, Freeman RA, Wu L, Vangveravong S, Luedtke RR. Conformationally-flexible benzamide analogues as dopamine D3 and σ2 receptor ligands. Bioorg Med Chem Lett. 2004;14:195–202. doi: 10.1016/j.bmcl.2003.09.083. [DOI] [PubMed] [Google Scholar]
- 147.Rowland DJ, Tu Z, Xu J, Ponde D, Mach RH, Welch MJ. Synthesis and in vivo evaluation of 2 high-affinity 76Br-labeled σ2-receptor ligands. J Nucl Med. 2006;47:1041–1048. [PubMed] [Google Scholar]
- 148.Turner PV, Pekow C, Clark JM, Vergara P, Bayne K, White WJ, et al. Roles of the international council for laboratory animal science (ICLAS) and international association of colleges of laboratory animal medicine (IACLAM) in the global organization and support of 3Rs advances in laboratory animal science. J Am Assoc Lab Anim Sci. 2015;55:174–180. [PMC free article] [PubMed] [Google Scholar]
- 149.McGrath JC, McLachlan EM, Zeller R. Transparency in research involving animals: The Basel declaration and new principles for reporting research in BJP manuscripts. Br J Pharmacol. 2015;172:242702432. doi: 10.1111/bph.12956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.National Research Council. Guide for the Care and Use of Laboratory Animals. Washington DC: National Academy Press; 2011. [Google Scholar]
- 151.Hedrich HJ, Bullock G, editors. The laboratory mouse. Amsterdam, Boston: Elsevier Academic Press; 2004. [Google Scholar]
- 152.Sharp P, Villano J. The laboratory rat. Boca Raton: Taylor & Francis; 2013. [Google Scholar]
- 153.Calias P, Banks WA, Begley D, Scarpa M, Dickson P. Intrathecal delivery of protei therapeutics to the brain: A critical reassessment. Pharmacol Ther. 2014;144:114–122. doi: 10.1016/j.pharmthera.2014.05.009. [DOI] [PubMed] [Google Scholar]
- 154.Shukla R, Chanda N, Zambre A, Upendran A, Katti K, Kulkarni RR, et al. Laminin receptor specific therapeutic gold nanoparticles (198AuNP-EGCg) show efficacy in treating prostate cancer. Proc Natl Acad Sci USA. 2012;109:12426–12431. doi: 10.1073/pnas.1121174109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Kawamura K, Kobayashi T, Matsuno K, Ishiwata K. Different brain kinetics of two sigma1 receptor ligands, [3H](+)-pentazocine and [11C]SA4503, by P-glycoprotein modulation. Synapse. 2003;48:80–86. doi: 10.1002/syn.10190. [DOI] [PubMed] [Google Scholar]
- 156.Tournier N, Valette H, Peyronneau M-A, Goutal S, Saba W, Kuhnast B, et al. Transport of selected PET radiotracers by human P-glycoprotein (ABCB1) and breast cancer resistance protein (ABCG2): an in vitro screening. J Nucl Med. 2011;52:415–423. doi: 10.2967/jnumed.110.079608. [DOI] [PubMed] [Google Scholar]
- 158.Sander K, Galante E, Gendron T, Yiannaki E, Patel N, Kalber TL, et al. Development of fluorine-18 labeled metabolically activated tracers for imaging of drug efflux transporters with positron emission tomography. J Med Chem. 2015;58:6058–6080. doi: 10.1021/acs.jmedchem.5b00652. [DOI] [PubMed] [Google Scholar]
- 158.Pike VW. PET radiotracers: crossing the blood-brain barrier and surviving metabolism. Trends Pharmacol Sci. 2009;30:431–440. doi: 10.1016/j.tips.2009.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Sakata M, Oda K, Toyohara J, Ishii K, Nariai T, Kishiwata K. Direct comparison of radiation dosimetry of six PET tracers using human whole-body imaging and murine biodistribution studies. Ann Nucl Med. 2013;27:285–296. doi: 10.1007/s12149-013-0685-9. [DOI] [PubMed] [Google Scholar]
- 160.Motulsky H. Intuitive Biostatistics. New York: Oxford University Press; 1995. [Google Scholar]
- 161.Eckelman WC, Reba RC, Gibson RE, Rzeszotarski WJ, Vieras F, Mazaitis JK, et al. Receptor-binding radiotracers: A class of potential radiopharmaceuticals. J Nucl Med. 1979;20:350–357. [PubMed] [Google Scholar]
- 162.Eckelman WC. Discussion of targeting proteins in vivo: in vitro guidelines. Nuc Med Biol. 2006;33:449–451. doi: 10.1016/j.nucmedbio.2006.03.008. [DOI] [PubMed] [Google Scholar]
- 163.Patel S, Gibson R. in vivo site-directed radiotracers: a mini-review. Nuc Med Biol. 2008;35:805–815. doi: 10.1016/j.nucmedbio.2008.10.002. [DOI] [PubMed] [Google Scholar]
- 164.Mintun MA, Raichle ME, Kilbourn MR, Wooten GF, Welch MJ. A quantitative model for the in vivo assessment of drug binding sites with positron emission tomography. Ann Neurol. 1984;15:217–227. doi: 10.1002/ana.410150302. [DOI] [PubMed] [Google Scholar]
- 165.Innis RB, Cunningham VJ, Delforge J, Fujita M, Gjedde A, Gunn RN, et al. Consensus nomenclature for in vivo imaging of reversibly binding radioligands. J Cereb Blood Flow Metab. 2007;27:1533–1539. doi: 10.1038/sj.jcbfm.9600493. [DOI] [PubMed] [Google Scholar]
- 166.Vauquelin G. Effects of target binding kinetics on in vivo drug efficacy: koff, kon and rebinding. Br J Pharmacol. 2016;173:2319–2334. doi: 10.1111/bph.13504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Ishiwata K, Kobayashi T, Kawamura K, Matsuno K. Age-related changes of the binding of [3H]SA4503 to sigma1 receptors in the rat brain. Ann Nucl Med. 2003;17:73–77. doi: 10.1007/BF02988264. [DOI] [PubMed] [Google Scholar]
- 168.Ramakrishnan NK, Schepers M, Luurtsema G, Nyakas CJ, Elsinga PH, Ishiwata K, et al. Cutamesine overcomes REM sleep deprivation-induced memory loss: relationship to sigma-1 receptor occupancy. Mol Imaging Biol. 2015;17:364–372. doi: 10.1007/s11307-014-0808-2. [DOI] [PubMed] [Google Scholar]
- 169.Ramakrishnan NK, Visser AKD, Rybczynska AA, Nyakas CJ, Luiten PGM, Kwizera C, et al. Sigma-1 agonist binding in the aging rat brain: a microPET study with [11C]SA4503. Mol Imaging Biol. 2016;18:588–597. doi: 10.1007/s11307-015-0917-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Ishiwata K, Oda K, Sakata M, Kimura Y, Kawamura K, Oda K, Sasaki T, Naganawa M, Chihara K, Okubo Y, Ishii K. A feasibility study of [11C]SA4503-PET for evaluating sigmal receptor occupancy by neuroleptics: the binding of haloperidol to sigma1 and dopamine D2-like receptors. Ann Nucl Med. 2006;20:569–573. doi: 10.1007/BF03026824. [DOI] [PubMed] [Google Scholar]
- 171.Kornhuber J, Schoppmeyer K, Bendig C, Riederer P. Characterization of [3H]pentazocine binding sites in post-mortem human frontal cortex. J Neural Transm. 1996;103:45–53. doi: 10.1007/BF01292615. [DOI] [PubMed] [Google Scholar]
- 172.Fujimoto M, Hayashi T, Urfer R, Mita S, Su T-P. Sigma-1 receptor chaperones regulate the secretion of brain-derived neurotrophic factor. Synapse. 2012;66:630–639. doi: 10.1002/syn.21549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Solon EG. Autoradiography techniques and quantification of drug distribution. Cell Tissue Res. 2015;360:87–107. doi: 10.1007/s00441-014-2093-4. [DOI] [PubMed] [Google Scholar]
- 174.Kuhar MJ, De Souza EB, Unnerstall JR. Neurotransmitter receptor mapping by autoradiography and other methods. Annu Rev Neurosci. 1986;9:27–59. doi: 10.1146/annurev.ne.09.030186.000331. [DOI] [PubMed] [Google Scholar]
- 175.Frey KA, Albin RL. Receptor binding techniques. Curr Protoc Neurosci. 2001:1.4.1–1.4.14. doi: 10.1002/0471142301.ns0104s00. [DOI] [PubMed] [Google Scholar]
- 176.Manuel I, Barreda-Gómez G, González de San Román E, Veloso A, Fernández JA, Giralt MT, et al. Neurotransmitter receptor localization: from autoradiography to imaging mass spectrometry. ACS Chem Neurosci. 2015;6:362–373. doi: 10.1021/cn500281t. [DOI] [PubMed] [Google Scholar]
- 177.Franklin KBJ, Paxinos G. The mouse brain in stereotaxic coordinates. San Diego: Academic Press; 1997. [Google Scholar]
- 178.Bouchard P, Roman F, Junien J-L, Quirion R. Autoradiographic evidence for the modulation of in vivo sigma receptor labeling by neuropeptide Y and calcitonin gene-related peptide in the mouse brain. J Pharmacol Exp Ther. 1996;276:223–230. [PubMed] [Google Scholar]
- 179.Sage AS, Oelrichs CE, Davis DC, Fan K-H, Nahas RI, Lever SZ, et al. Effects of N-phenylpropyl-N′-substituted piperazine sigma receptor ligands on cocaine-induced hyperactivity in mice. Pharmacol Biochem Behav. 2013;110:201–207. doi: 10.1016/j.pbb.2013.07.006. [DOI] [PubMed] [Google Scholar]
- 180.Rodvelt KR, Lever SZ, Lever JR, Blount LR, Fan K-H, Miller DK. SA 4503 attenuates cocaine-induced hyperactivity and enhances methamphetamine substitution for a cocaine discriminative stimulus. Pharmacol Biochem Behav. 2011;97:676–682. doi: 10.1016/j.pbb.2010.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Volkow ND, Wang G-J, Fowler JS, Gatley SJ, Ding Y-S, Logan J, et al. Relationship between psychostimulant-induced “High” and dopamine transporter occupancy. Proc Natl Acad Sci USA. 1996;93:10388–10392. doi: 10.1073/pnas.93.19.10388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Volkow ND, Wang G-J, Fischman MW, Foltin RW, Fowler JS, Abumrad NN, et al. Relationship between subjective effects of cocaine and dopamine transporter occupancy. Nature. 1997;386:827–830. doi: 10.1038/386827a0. [DOI] [PubMed] [Google Scholar]
- 183.Fowler JS, Volkow ND, Logan J, Gatley SJ, Pappas N, King P, et al. Measuring dopamine transporter occupancy by cocaine in vivo: Radiotracer considerations. Synapse. 1998;28:111–116. doi: 10.1002/(SICI)1098-2396(199802)28:2<111::AID-SYN1>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 184.Cunha L, Horvath I, Ferreira S, Lemos J, Costa P, Vieira D, et al. Preclinical imaging: an essential ally in modern biosciences. Mol Diagn Ther. 2014;18:153–173. doi: 10.1007/s40291-013-0062-3. [DOI] [PubMed] [Google Scholar]
- 185.Jang B-S. MicroSPECT and microPET imaging of small animals for drug development. Toxicol Res. 2013;29:1–6. doi: 10.5487/TR.2013.29.1.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Zimmer ER, Leuzy A, Bhat V, Gauthier S, Rosa-Neto P. In vivo tracking of tau pathology using positron emission tomography (PET) molecular imaging in small animals. Transl Neurodegener. 2014;3:6. doi: 10.1186/2047-9158-3-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Finnema SJ, Scheinin M, Shahid M, Lehto J, Borroni E, Bang-Andersen B, et al. Application of cross-species PET imaging to assess neurotransmitter release in brain. Psychopharmacology. 2015;232:4129–4157. doi: 10.1007/s00213-015-3938-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Momosaki S, Hatano K, Kawasumi Y, Kato T, Hosoi R, Kobayashi K, et al. Rat-PET study without anesthesia: anesthetics modify the dopamine D1 receptor binding in rat brain. Synapse. 2004;54:207–213. doi: 10.1002/syn.20083. [DOI] [PubMed] [Google Scholar]
- 189.Itoh T, Abe K, Zoghbi SS, Inoue O, Hong J, Imaizumi M, et al. PET measurement of the in vivo affinity of 11C-(R)-rolipram and the density of its target, phosphodiesterase-4, in the brains of conscious and anesthetized rats. J Nucl Med. 2009;50:749–756. doi: 10.2967/jnumed.108.058305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Mizuma H, Shukuri M, Hayashi T, Watanabe Y, Onoe H. Establishment of in vivo brain imaging method in conscious mice. J Nucl Med. 2010;51:1068–1075. doi: 10.2967/jnumed.110.075184. [DOI] [PubMed] [Google Scholar]
- 191.Baba JS, Endres CJ, Foss CA, Nimmagadda S, Jung H, Goddard JS, et al. Molecular imaging of conscious, unrestrained mice with AwakeSPECT. J Nucl Med. 2013;54:969–976. doi: 10.2967/jnumed.112.109090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Kyme AZ, Zhou VW, Meikle SR, Fulton RR. Real-time 3D motion tracking for small animal brain PET. Phys Med Biol. 2008;53:2651–2666. doi: 10.1088/0031-9155/53/10/014. [DOI] [PubMed] [Google Scholar]
- 193.Weisenberger AG, Lee S, Smith MF. Motion-tracking technique in unrestrained small-animal single-photon emission computed tomography. Rev Neurosci. 2011;22:657–663. doi: 10.1515/RNS.2011.049. [DOI] [PubMed] [Google Scholar]
- 194.Schulz D, Southekal S, Junnarkar SS, Pratte JF, Purschke ML, Stoll SP, et al. Simultaneous assessment of rodent behavior and neurochemistry using a miniature positron emission tomograph. Nat Methods. 2011;8:347–352. doi: 10.1038/nmeth.1582. [DOI] [PubMed] [Google Scholar]
- 195.Colby LA, Morenko BJ. Clinical considerations in rodent bioimaging. Comp Med. 2004;54:623–630. [PubMed] [Google Scholar]
- 196.Hildebrandt IJ, Su H, Weber WA. Anesthesia and other considerations for in vivo imaging of small animals. ILAR J. 2008;49:17–26. doi: 10.1093/ilar.49.1.17. [DOI] [PubMed] [Google Scholar]
- 197.Tremoleda JL, Kerton A, Gsell W. Anaesthesia and physiological monitoring during in vivo imaging of laboratory rodents: considerations on experimental outcomes and animal welfare. EJNMMI Res. 2012;2:44. doi: 10.1186/2191-219X-2-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Koba W, Jelicks LA, Fine EJ. MicroPET/SPECT/CT imaging of small animal models of disease. Am J Pathol. 2013;182:319–324. doi: 10.1016/j.ajpath.2012.09.025. [DOI] [PubMed] [Google Scholar]
- 199.Vaquero JJ, Kinahan P. Positron emission tomography: current challenges and opportunities for technological advances in clinical and preclinical imaging systems. Ann Rev Biomed Eng. 2015;17:385–414. doi: 10.1146/annurev-bioeng-071114-040723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Shen B, James ML, Andrews L, Lau C, Chen S, Palner M, et al. Further validation to support clinical translation of [18F]FTC-146 for imaging sigma-1 receptors. EJNMMI Research. 2015;5:49–58. doi: 10.1186/s13550-015-0122-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Huang L, Merson TD, Bourne JA. In vivo whole brain, cellular and molecular imaging in nonhuman primate models of neuropathology. Neurosci Biobehav Rev. 2016;66:104–118. doi: 10.1016/j.neubiorev.2016.04.009. [DOI] [PubMed] [Google Scholar]
- 202.Ando K, Obayashi S, Nagai Y, Oh-Nishi A, Minamimoto T, Higuchi M, et al. PET analysis of dopaminergic neurodegeneration in relation to immobility in the MPTP-treated common marmoset, a model for Parkinson’s disease. PLoS One. 2012;7:e46371. doi: 10.1371/journal.pone.0046371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Yokoyama C, Yamanaka H, Onoe K, Kawasaki A, Nagata H, Hirakami K, et al. Mapping of serotonin transporters by positron emission tomography with [11C]DASB in conscious common marmosets: Comparison with rhesus monkey. Synapse. 2010;64:594–601. doi: 10.1002/syn.20766. [DOI] [PubMed] [Google Scholar]
- 204.Nader MA, Banks M. Environmental modulation of drug taking: Nonhuman primate models of cocaine abuse and PET neuroimaging. Neuropharmacology. 2014;76:510–517. doi: 10.1016/j.neuropharm.2013.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Howell LL, Murnane KS. Nonhuman primate positron emission tomography neuroimaging in drug abuse research. J Pharmacol Exp Ther. 2011;337:324–334. doi: 10.1124/jpet.108.136689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Clayton JA, Collins FS. Policy: NIH to balance sex in cell and animal studies. Nature. 2014;509:282–283. doi: 10.1038/509282a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.National Institutes of Health. [accessed June 17, 2016];Consideration of sex as a biological variable in NIH-funded research. NOT-OD-15–102. http://grants.nih.gov/grants/guide/notice-files/NOT-OD-15-102.html.
- 208.Klein SL, Schiebinger L, Stefanick ML, Cahill L, Danska J, de Bries GJ, et al. Opinion: Sex inclusion in basic research drives discovery. Proc Natl Acad Sci USA. 2015;112:5257–5258. doi: 10.1073/pnas.1502843112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Richardson SS, Reiches M, Shattuck-Heidorn H, LaBonte ML, Consoli T. Opinion: Focus on preclinical sex differences will not address women’s and men’s health disparities. Proc Natl Acad Sci USA. 2015;112:13419–13420. doi: 10.1073/pnas.1516958112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Clayton JA. Studying both sexes: a guiding principle for biomedicine. FASEB J. 2016;30:519–524. doi: 10.1096/fj.15-279554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Maney DL. Perils and pitfalls of reporting sex differences. Philos Trans R Soc B. 2016;371:20150119. doi: 10.1098/rstb.2015.0119. http://dx.doi.org/10.1098/rstb.2015.0119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Voskuhl R. Preclinical studies of sex differences: a clinical perspective. Biol Sex Differ. 2016;7:7. doi: 10.1186/s13293-016-0061-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Chesis PL, Griffeth LK, Mathias CJ, Welch MJ. Sex-dependent differences in N-(3-[18F]fluoropropyl)-N-nordiprenorphine biodistribution and metabolism. J Nucl Med. 1990;31:192–201. [PubMed] [Google Scholar]
- 214.Zubieta J-K, Dannals RF, Frost JJ. Gender and age influences on human brain mu-opioid receptor binding measured by PET. Am J Psychiatry. 1999;156:842–848. doi: 10.1176/ajp.156.6.842. [DOI] [PubMed] [Google Scholar]
- 215.Kakimoto A, Ito S, Okada H, Nishizawa S, Minoshima S, Ouchi Y. Age-related sex-specific changes in brain metabolism and morphology. J Nucl Med. 2016;57:221–225. doi: 10.2967/jnumed.115.166439. [DOI] [PubMed] [Google Scholar]
- 216.Kadkhodayan A, Lin CH, Coggan AR, Kisrieva-Ware Z, Schechtman KB, Novak E, et al. Sex affects myocardial blood flow and fatty acid substrate metabolism in humans with nonischemic heart failure. J Nucl Cardiol. 2016 Apr 5; doi: 10.1007/s12350-016-0467-6. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Okita K, Petersen N, Robertson CL, Dean AC, Mandelkern MA, London ED. Sex differences in midbrain dopamine D2-type receptor availability and association with nicotine dependence. Neuropsychopharmacol. 2016 Jul 13; doi: 10.1038/npp.2016.105. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Wong DF, Kuwabara H, Horti AG, Raymont V, Brasic J, Guevara M, et al. Quantification of cerebral cannabinoid receptors subtype 1 (CB1) in healthy subjects and schizophrenia by the novel PET radioligand [11C]OMAR. NeuroImage. 2010;52:1505–1513. doi: 10.1016/j.neuroimage.2010.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Normandin MD, Zheng MQ, Lin KS, Mason NS, Lin SF, Ropchan J, et al. Imaging the cannabinoid CB1 receptor in humans with [11C]OMAR: assessment of kinetic analysis methods, test-retest reproducibility, and gender differences. J Cereb Blood Flow Metab. 2015;35:1313–1322. doi: 10.1038/jcbfm.2015.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Van Laere K, Goffin K, Casteels C, Dupont P, Mortelmans L, de Hoon J, et al. Gender-dependent increases with healthy aging of the human cerebral cannabinoid-type 1 receptor binding using [18F] MK-9470 PET. NeuroImage. 2008;39:1533–1541. doi: 10.1016/j.neuroimage.2007.10.053. [DOI] [PubMed] [Google Scholar]
- 221.D’Souza DC, Cortes-Briones JA, Ranganathan M, Thumauer H, Creatura G, Surti T, et al. Rapid changes in cannabinoid 1 receptor availability in cannabis-dependent male subjects after abstinence from cannibis. Biol Psychiatry:CNNI. 2016;1:60–67. doi: 10.1016/j.bpsc.2015.09.008. [DOI] [PubMed] [Google Scholar]
- 222.Yue X, Jin H, Liu H, Rosenberg AJ, Klein RS, Tu Z. A potent and selective C-11 labeled PET tracer for imaging sphingosine-1-phosphate receptor 2 in the CNS demonstrates sexually dimorphic expression. Org Biomol Chem. 2015;13:7928–7939. doi: 10.1039/c5ob00951k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Cruz-Orengo L, Daniels BP, Dorsey D, Basak SA, Grajales-Reyes JG, McCandless EE, et al. Enhanced sphingosine-1-phosphate receptor 2 expression underlies female CNS autoimmunity susceptibility. J Clin Invest. 2014;124:2571–2584. doi: 10.1172/JCI73408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Sahlholm K, Liao F, Holtzman DM, Xu J, Mach RH. Sigma-2 receptor binding is decrease in female, but not male, APP/PS1 mice. Biochem Biophys Res Comm. 2015;460:439–445. doi: 10.1016/j.bbrc.2015.03.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Stabin M, Xu XG. Basic principles in the radiation dosimetry of nuclear medicine. Semin Nucl Med. 2014;44:162–171. doi: 10.1053/j.semnuclmed.2014.03.008. [DOI] [PubMed] [Google Scholar]
- 226.Eckerman KF, Endo A. MIRD: Radionuclide Data and Decay Schemes. 2. Reston: Society of Nuclear Medicine and Molecular Imaging; 2008. [Google Scholar]
- 227.Society of Nuclear Medicine and Molecular Imaging. [Accessed August 15, 2016];Committee on Medical Internal Radiation Dose (MIRD) http://www.snmmi.org/ClinicalPractice/content.aspx?ItemNumber=4212.
- 228.Zanotti-Fregonara P, Innis RB. Suggested pathway to assess radiation safety of (11C)C-labeled PET tracers for first-in-human studies. Eur J Nucl Med Mol Imaging. 2012;39:544–547. doi: 10.1007/s00259-011-2005-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Zanotti-Fregonara P, Lammertsma AA, Innis RB. Suggested pathway to assess radiation safety of 18F-labeled PET tracers for first-in-human studies. Eur J Nucl Med Mol Imaging. 2013;40:1781–1783. doi: 10.1007/s00259-013-2512-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.VanBrocklin HF. Radiopharmaceuticals for drug development: United States regulatory perspective. Current Radiopharmaceuticals. 2008;1:2–6. [Google Scholar]
- 231.Hung JC. Bringing new PET drugs to clinical practice - A regulatory perspective. Theranostics. 2013;3:885–893. doi: 10.7150/thno.5513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Saha GB. Basics of PET Imaging: Physics, chemistry and regulations. 3. Switzerland: Springer International Publishing; 2016. [Google Scholar]
- 233.Elsinga P, Todde S, Penuelas I, Meyer G, Farstad B, Faivre-Chauvet A, et al. Guidance on current good radiopharmacy practice (cGRPP) for the small-scale preparation of radiopharmaceuticals. Eur J Nucl Med Mol Imaging. 2010;37:1049–1062. doi: 10.1007/s00259-010-1407-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.US FDA. [accessed July 13, 2016];Guidance for industry, investigators and reviewers – exploratory IND studies. http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm078933.pdf.
- 235.Medicines and Healthcare product Regulatory Agency. [accessed August 15, 2016];Clinical trials for medicines: apply for authorisation in the UK. 2014 https://www.gov.uk/guidance/clinical-trials-for-medicines-apply-for-authorisation-in-the-uk.
- 236.Barret O, Thomas D, Tavares A, Alagille D, Papin C, Waterhouse R, et al. In vivo assessment and dosimetry of 2 novel PDE10A PET radiotracers in Humans: 18F-MNI-659 and 18F-MNI-654. J Nucl Med. 2014;55:1297–1304. doi: 10.2967/jnumed.113.122895. [DOI] [PubMed] [Google Scholar]
- 237.Hillmer AT, Wooten DW, Bajwa AK, Higgins AT, Lao PJ, Betthauser TJ, et al. First-inHuman evaluation of 18F-Mefway, a PET radioligand specific to serotonin-1A receptors. J Nucl Med. 2014;55:1973–1979. doi: 10.2967/jnumed.114.145151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Toyohara J, Sakata M, Hatano K, Yanai S, Endo S, Ishibashi K, et al. Preclinical and first-in-man studies of [11C]CB184 for imaging the 18kDa translocator protein by positron emission tomography. Ann Nucl Med. 2016:1–10. doi: 10.1007/s12149-016-1094-7. article in press. [DOI] [PubMed] [Google Scholar]
- 239.Cescato R, Waser B, Fani M, Reubi JC. Evaluation of 177Lu-DOTA-sst2 antagonist versus 177Lu-DOTA-sst2 agonist binding in human cancers in vitro. J Nucl Med. 2011;52:1886–1890. doi: 10.2967/jnumed.111.095778. [DOI] [PubMed] [Google Scholar]
- 240.Skinbjerg M, Sibley DR, Javitch JA, Abi-Dargham A. Imaging the high-affinity state of the dopamine D2 receptor in vivo: Fact or fiction? Biochem Pharmacol. 2012;83:193–198. doi: 10.1016/j.bcp.2011.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]