

Abstract
Diazo groups have broad and tunable reactivity. That and other attributes endow diazo compounds with the potential to be valuable reagents for chemical biologists. The presence of diazo groups in natural products underscores their metabolic stability and anticipates their utility in a biological context. The chemoselectivity of diazo groups, even in the presence of azido groups, presents many opportunities. Already, diazo compounds have served as chemical probes and elicited novel modifications of proteins and nucleic acids. Here, we review advances that have facilitated the chemical synthesis of diazo compounds, and we highlight applications of diazo compounds in the detection and modification of biomolecules.
Graphical Abstract
Azido groups dominate the current landscape of chemoselective reactions in chemical biology. Yet, diazo groups have attributes that are even more desirable than those of azido groups. For example, diazo groups (R1R2C=N2) are smaller than analogous azido groups (R1R2HC–N3), and diazo groups display a broader range of reactivity.1,2
The simplest diazo compound, diazomethane, is a yellow gas that was discovered by von Pechmann in 18943,4 and is a common reagent in synthetic organic chemistry. Diazomethane and other diazoalkanes are, however, highly toxic5–7 and explosively reactive,8,9 and have little utility in the context of chemical biology. The problem arises from their high basicity, as protonation of the α carbon of a diazo group leads to the formation of a diazonium species (R1R2HC–N2+) poised for a rapid SN2 reaction that releases nitrogen gas.
Recent advances in synthetic methodology provide ready access to “stabilized” diazo compounds that are compatible with living systems. The stability arises from diminished basicity due to delocalization of the electrons on the α carbon to another functional group. Such stabilized diazo compounds have the potential for widespread application in chemical biology.
Here, we review the use of diazo compounds in chemical biology. We begin with an overview of natural products and amino acids that contain a diazo group. That is followed by a summary of methods for the chemical synthesis of diazo compounds. We then highlight the remarkable versatility of diazo compounds in the context of chemical biology, and we end with a brief prospectus for the future.
NATURAL PRODUCTS
In contrast to azido groups,10 diazo groups are found in many natural products.11 Isotopic labeling studies and genome mining have provided insight into their biosynthesis.12–15 No enzyme is known to catalyze the formation of an N–N bond, though a gene cluster that encodes a nitrous acid-producing enzyme could be a source.16 Intrinsic antitumor and antibiotic activities endow some natural diazo compounds with potential clinical utility, but mechanisms of action in vivo are unclear. As the isolation and synthesis of diazo-containing natural products has been reviewed extensively elsewhere,17,18 we summarize only key findings and recent advances. We focus, in particular, on the kinamycins and lomaiviticins, two classes of natural products with unusual structures and intriguing mechanisms of reactivity (Figures 1A and 1B).
Figure 1.

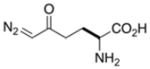
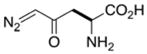
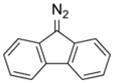
Structure and reactivity of some natural products that contain diazo groups. (A) Kinamycin D, lomaiviticin A, and lomaiviticin B. (B) Putative mechanism for the generation of a reactive vinylogous radical from lomaiviticin A.34 (C) Solution structure of the complex of lomaiviticin A with a G-C-T-A-T-A-G-C duplex.39 Displaced A·T basepairs are depicted in yellow. Phosphorous atoms are depicted in orange. Hydrogen atoms are not shown. Arrows point to the two diazo groups. Image was created with Protein Data Bank entry 2n96 and the program PyMOL from Schrödinger (New York, NY). (D) Amino acids that contain diazo groups.
The kinamycins were isolated from Streptomyces murayamaensis in 1970 and displayed antimicrobial activity against gram-positive bacteria.19 Initially, the compounds were thought to contain a cyanamide group due to their infrared absorption near ~2155 cm−1, but were later established to have a diazo moiety.20 The complex architecture of these molecules, which consist of a 4-ring carbocyclic skeleton that contains several stereogenic centers, challenged synthetic chemists until routes were developed a decade ago.21–23
Like the kinamycins, the lomaiviticins are analogs of 9-diazofluorene (Figure 1A). Lomaiviticins A and B were isolated in 2001 from the marine ascidian symbiont Salinispora pacifica and displayed antitumor activity at sub-micromolar concentrations.24 Lomaiviticins C–E were isolated in 2012 from Salinispora pacifica and demonstrated similar potency.25 Although synthetic routes to the lomaiviticins are unrealized to date, progress has been made towards intermediates and analogues.26–30
Diazofluorene analogues have long been used to investigate possible mechanisms of DNA cleavage in vitro. Using 9-diazofluorene, Arya and Jebaratnam were among the first to suggest that a diazo group could mediate DNA cleavage.31 Kinafluorenone, which contains a ketone oxygen in lieu of a diazo group, displayed no antibiotic activity and thus supported the hypothesis that the diazo moiety is the active pharmacophore.32 A variety of reactive intermediates that elicit cytotoxicity have been proposed, including a covalent adduct,33,34 ortho-quinone methide,34,35 acylfulvene,36 or vinyl radical33–35,37,38 (Figure 1B). Certain lomaiviticins, such as (−)-lomaiviticin A, are nearly a hundred-fold more toxic to cancer cells than are kinamycins,38 despite similar reactive intermediates being accessible from both kinamycins and lomaiviticins. (−)-Lomaiviticin A is especially potent, exhibiting cytotoxic activity at nanomolar–picomolar concentrations.
To reveal the basis for the superior cytotoxicity of (−)-lomaiviticin A, Herzon and coworkers performed a thorough comparison of (−)-lomaiviticin A, (−)-lomaiviticin C, and (−)-kinamycin C.38 They found that the reduction of (−)-lomaiviticin A in vitro proceeds more rapidly than does that of (−)-kinamycin C. Moreover, only (−)-lomaiviticin A causes double-stranded breaks in DNA and activates the double-strand break repair pathway in cells. This combination of attributes likely accounts for the superior potency of (−)-lomaiviticin A. Further, these authors provided evidence that DNA cleavage is instigated by a vinylic carbon radical (Figure 1B) and is independent of iron and reactive oxygen species. A solution structure of (−)-lomaiviticin A in complex with DNA revealed that both subunits of lomaiviticin A intercalate into DNA at AT-rich sequences and cause base pairs to be twisted out of the duplex (Figure 1C).39 The α carbon of the diazo group lies in close proximity to the DNA strand, facilitating hydrogen abstraction by an incipient radical.
One challenge in the investigation and application of lomaiviticins is their limited availability. Smaller analogues that are easier to synthesize provide a partial solution.40 One such analogue, a monomeric lomaiviticin aglycon, is capable of inducing DNA damage, albeit at higher concentrations than does (−)-lomaiviticin A. Both (−)-lomaiviticin A and this monomeric lomaiviticin aglycon activate homologous recombination and the non-homologous end-joining repair of DNA in cells.41 Dysfunctional DNA-repair pathways underlie many human cancers,42 rendering lomaiviticins as a potential treatment strategy. In support of this strategy, cell lines with defective DNA-repair pathways (e.g., BRCA2- and PTEN-deficient cells), are more sensitive to (−)-lomaiviticin A and monomeric lomaiviticin aglycon than are isogenic cell lines with intact damage repair pathways.
AMINO ACIDS
Some natural amino acids contain diazo groups.43,44 Notable examples include azaserine and 6-diazo-5-oxo-norleucine (DON), which are nearly isosteric to glutamine (Figure 1D).45 Both amino acids were isolated initially from Streptomyces cultures and exhibit antibiotic and tumor inhibitory properties.43,46 These diazo compounds effectively inhibit amidotransferases involved in the biosynthesis of pyrimidines and purines.47–49 DON entered early-stage clinical trials based on its beneficial activity against various carcinomas, lymphomas, and Hodgkin’s disease.50 The ability of DON to inhibit amidotransferases revealed the mechanism by which γ-glutamyl transferase acts in tandem with aminopeptidase M to transfer the glutamyl group of glutathione to amino acids and peptides.51–53 DON was also used to determine the catalytic nucleophile and characterize the substrate specificity of glutaminase–asparaginases from various organisms.54,55
Likewise, diazo-containing analogs of asparagine have found utility in medicine as well as enzymology. 5-Diazo-4-oxo-norvaline (DONV; Figure 1D) inhibits the growth of asparagine-dependent tumors by interfering with the synthesis and utilization of asparagine.44,56 DONV is also a specific inhibitor of L-asparaginase, which is used routinely in the treatment of leukemia.57 Clinical assays that aim to determine the blood concentration of asparagine in patients treated with L-asparaginase suffer from degradation of asparagine in the serum sample due to L-asparaginase. The addition of DONV to the assay mixture improves the reliability of asparagine detection.57
PREPARATION
The synthesis of diazo compounds has become facile. Common methods include (i) diazo transfer,58,59 (ii) diazotization,60,61 (iii) hydrazone decomposition62,63 or hydrazone oxidation,64,65 (iv) rearrangement of N-alkyl N-nitroso compounds,8,66 (v) 1,3-disubstituted acyl (or aryl) triazine fragmentation,67,68 and (vi) elaboration of other diazo compounds (Figure 2).69–73 Most of these routes have been reviewed extensively for their merits in the context of synthetic chemistry.74,75 Nevertheless, the preparation of diazo compounds for applications in chemical biology entails additional challenges because of restrictions on the compatibility of ancillary functional groups and on solubility.
Figure 2.

Preparation of diazo compounds by (i) diazo transfer,58,59 (ii) diazotization,60,61 (iii) hydrazone decomposition62,63 or hydrazone oxidation,64,65 (iv) rearrangement of N-alkyl N-nitroso compounds,8,66 (v) 1,3-disubstituted acyl or aryl triazine fragmentation,67,68 and (vi) elaboration of other diazo compounds. Diazo compounds can be accessed from azides via acyl triazenes in a process mediated by a phosphinoester.78,79
Diazo transfer is a simple and effective way to introduce the diazo group when the pKa of a proton on the acceptor carbon is low enough to be extracted with a mild base, as is necessary in the stabilized diazo compounds useful in chemical biology. For example, 1,8-diazabicycloundec-7-ene (DBU) can generate α-diazocarbonyl groups after a diazo transfer reaction using sulfonyl azide reagents (e.g., p-acetamidobenzenesulfonyl azide and imidazolesulfonyl azide).59,76,77 The electronic delocalization that enables diazo transfer also stabilizes the ensuing diazo compound.
Recently, our group reported on a general method to prepare a stabilized diazo group directly from a parent azide.78,79 Fragmentation of acyl triazines uses a phosphinoester to convert an azido group into its corresponding diazo group. The reactivity underlying this loss of NH, or “deimidogenation”, was derived from insight on the mechanism of the Staudinger ligation.80–84 In the Staudinger ligation as well as the Staudinger reaction,85,86 the incipient phosphazide quickly extrudes molecular nitrogen to generate an iminophosphorane. A highly reactive acylating group subverts nitrogen extrusion by trapping the phosphazide (Figure 2). The ensuing triazenophosphonium intermediate hydrolyzes quickly in water to form an acyl triazene, which is a known precursor to a diazo group.67,68
Azide deimidogenation benefits from the extraordinary chemoselectivity of phosphine for an azide. This approach has a high tolerance for other functional groups, including ketones, esters, aldehydes, thiols, α-chloroesters, epoxides, and disulfide bonds. Chemoselectivity was demonstrated by converting an azido group into a diazo group in aqueous solution containing an enzyme, which was not modified covalently and retained full catalytic activity.79 Notably, appropriate azides for deimidogenation (that is, azides with an electron-withdrawing group on the α carbon) are readily accessible via SN2 reactions with inorganic azide.87
Finally, diazo compounds that contain sensitive functional groups can be prepared by the late-stage installation of a prefabricated diazo group. This strategy typically relies on acyl transfer. In 1962, Westheimer and coworkers introduced the concept of photoaffinity labeling by acylating chymotrypsin with p-nitrophenyl diazoacetate and then forming an intramolecular crosslink upon photolysis.88 Most recent late-stage installations have employed an N-hydroxysuccinimide (NHS) ester containing a pendant α-diazocarbonyl group. Badet and coworkers developed a clever synthetic route to the simplest reagent of this class, N-hydroxysuccinimidyl diazoacetate.89 Such NHS esters have been used to install diazo groups on small molecules90,91 as well as biomolecules of varying complexity, including biotin,92 mannosamine,93 heparan-sulfate fragments,94 lysozyme,93 and bovine serum albumin (BSA).95
CYCLOADDITIONS
The archetypal reaction for the diazo group is the 1,3-dipolar cycloaddition. Soon after the synthesis of ethyl diazoacetate by Curtius,60 Buchner observed its reaction with an α,β-unsaturated carboxylic ester to form a pyrazole.96 Over the last century the reactivity of diazo groups in cycloadditions has engaged theoretical, synthetic, and biological chemists, and these explorations have been reviewed for their use and merits in synthetic chemistry.97,98 Here, we focus on recent work that is relevant to biological systems.
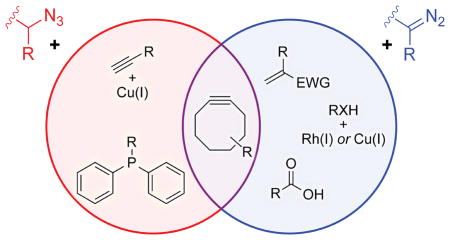
Copper-catalyzed azide–alkyne cycloadditions (CuAAC)99,100 and strain-promoted azide–alkyne cycloadditions (SPAAC)101–103 are two of the most enabling advances in the field of chemical biology.83,104,105 The diazo group shares the ability of the azido group to undergo cycloadditions with alkynes, forming a pyrazole rather than a triazole.95,106,107 The reactivity of diazo groups is remarkably predictable and tunable108—the diazo compounds can react with a strained alkyne at much higher or much lower rates than analogous azides (Figure 3A).106,107,109 Because a diazo group can be generated directly from an azido group78,79 and reacts with strained alkynes in common use, the diazo group fits easily into extant methodology.
Figure 3.

Diazo compounds in dipolar cycloadditions with strained alkynes. (A) Relative rate constants of diazo compounds and analogous azides with various cyclooctynes.92,109 (B) Labeling of a diazo-modified lysozyme with a cyclooctyne.93 (C) Labeling of a metabolized diazo sugar displayed on the surface of human cells with a cyclooctyne.92
In addition to reacting with strained alkynes, diazo groups undergo uncatalyzed cycloadditions with unstrained dipolarophiles, including terminal alkenes and alkynes. Moreover, diazo compounds can react chemoselectively with certain alkenes and alkynes in the presence of an azide. In essence, a diazo group is more electron-rich, and thus a better nucleophile in normal-electron-demand cycloadditions with electron-deficient dipolarophiles.110–113 Detailed insight is attainable from computational analyses. Distortion energies account for a majority (80%) of the activation energy for 1,3-dipolar cycloadditions. Due to their increased nucleophilicity and higher HOMO energy, diazo compounds have lower distortion energies than do their azide analogues.110,113 The reactions can occur at ambient temperature in aqueous cosolvent with reaction rates similar to or greater than those of SPAACs with azides. Notably, a diazo group can react chemoselectively with the naturally occurring amino acid dehydroalanine (Dha), which contains an electronically activated alkene.110 Selective biotinylation of activated alkenes could enable enrichment and isolation of compounds from a complex lysate, facilitating discovery of new natural products.
PROBES
The diazo group is found in the natural products of microorganisms (vide supra). In contrast, its absence in higher organisms enables its utility there as a chemical reporter. The reactivity of the diazo group with many common SPAAC dipolarophiles spawned the use of a diazo group as a chemical reporter for cell-surface glycosylation.
Leeper and coworkers prepared an N-diazoacetyl galactosamine and incubated this synthetic sugar with LL2 cells.93 Treatment with a biotin-bearing cyclooctyne and subsequent addition of an avidin fluorophore produced some increase in fluorescence of cells incubated with the diazo-bearing glycan compared to untreated cells. In the same study, an α-diazo NHS ester was reacted with a lysine residue on lysozyme to append the diazo group. Following modification, the appendage was used to attach a fluorophore to the protein via a cycloaddition between the diazo group and a cyclooctyne (Figure 3B).
Our group demonstrated the suitability of a diazoacetamide derivative of N-acetyl mannosamine as a chemical reporter of glycosylation on the surface of CHO K1, Jurkat, HEK293T, and HeLa cells (Figure 3C).92 The degree of labeling was determined by SPAAC between the diazo group and a biotin-bearing cyclooctyne, followed by treatment with an avidin fluorophore. Metabolic incorporation of the diazo-bearing sugar was evidenced through live-cell microscopy and flow cytometry, and labeling was abolished by treatment with a sialidase. Diazo and alkynyl sugars could be labeled independently on the cell surface. Notably, such dual labeling was not possible on cells displaying azido and alkynyl sugars due to the reactivity of the azide in both CuAAC and SPAAC reactions.
Diazo compounds have long been incorporated into biomolecules as photoaffinity probes.114,115 Upon irradiation with ultraviolet light, the diazo group fragments into molecular nitrogen and a carbene, which can undergo either an insertion reaction or a Wolff rearrangement116,117 followed by nucleophilic attack on the ensuing ketene, both of which crosslink the diazo compound to proximal functional groups. This strategy has been used to map the architecture of chymotrypsin (vide supra),88 reveal antibody combining sites,118 examine the structure of lipid membranes,119 and identify isoprenoid-binding sites on proteins.120
PROTEIN ALKYLATION
The ability of diazo reagents to alkylate oxygen, nitrogen, sulfur, and even carbon exemplifies their diverse reactivity.1,121–124 When applied to protein modification, these reactions are typically catalyzed by acid or transition metals. Despite the apparent promiscuity of this mode of reactivity, even highly reactive compounds such as diazomethane have historically found utility in elucidating structural and functional aspects of proteins.125 Stabilized diazo reagents enable O-alkylation of carboxyl groups and were valuable tools in classical protein chemistry and enzymology.126,127 Later, the discovery of diazo-containing amino-acid analogues led medicinal chemists and structural biologist to employ these compounds as covalent inhibitors of metabolic enzymes.45 Modern applications of diazo chemistry in chemical biology aim to capitalize on the versatility of diazo compounds to access linkages that cannot be achieved by other methods. Maintaining chemoselectivity in the presence of water and other biological nucleophiles has been a primary challenge in developing diazo compounds as useful tools for protein chemistry.128,129
The earliest uses of diazo reagents for protein labeling sought to characterize structural features of proteins. In 1914, Geake and Nierenstein used diazomethane to alkylate caseinogen so as to characterize the structure of amino-acid side chains (Table 1).125 By comparing the methylated and unmethylated protein, they identified and quantified side chains that contain amino or hydroxyl groups. Later studies addressed large-scale structural characterization of proteins, such as quantification of the number of peptide chains in a protein and identification of carboxyl groups in the binding region of the anti-hapten antibody.130,131
Table 1.
Diazo compounds that esterify proteins.
| Diazo Compound | Protein | Year | Reference |
|---|---|---|---|
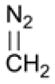
|
caseinogen insulin β-lactoglobulin lysozyme |
1914 1958 |
125 130 |
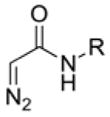
|
polyclonal antibody chymotrypsinogen ribonuclease A pepsin acid proteases prorenin O-sulfotransferase |
1960 1961 1965 1966–1968 1972–1973 1980 2015 |
131 126 127 137,139,140 146–149 151 94 |
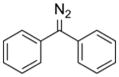
|
pepsin | 1966 | 135 |
|
phosphoribosyl pyrophosphate amidotransferase glutaminase A glutamyl transpeptidase |
1963 1973 1978 |
47 48 52–54 |
|
asparaginase | 1977 | 56 |
|
myoglobin subtilisin Yes kinase |
2004 2015 |
54 165 |
|
|
β-lactoglobulin | 2007 | 174 |
|
ribonuclease A red fluorescent protein |
2015 | 128 |
|
ribonuclease A | 2015 | 129 |
The last 100 years have seen many attempts to limit the promiscuity of the diazo reagent by using stabilized α-diazo amides (Table 1). Doscher and Wilcox used α-diazoacetamide to label chymotrypsin in work that laid the foundation for modern protein-labeling endeavors.126 They demonstrated that, although the rate of esterification was much greater than the rate of diazo-compound hydrolysis, the large excess of water molecules limits the efficiency of esterification. The authors suggested that employing a mixed aqueous–organic solvent could increase esterification efficiency by both limiting diazo hydrolysis and increasing the pKa of enzymic carboxyl groups. This idea was later explored, and did indeed increase the efficiency of protein esterification.128 Although α-diazoacetamide was more selective than diazomethane, it still S-alkylated sulfhydryl groups.
In 1917, Staudinger and Gaule became the first to use a diazo compound, diphenyldiazomethane, to form an ester.132 The mechanism of this reaction was established in elegant work by Roberts and coworkers in 1951 (Figure 4A).133,134 The heightened reactivity of carboxyl groups versus carboxylates inspired subsequent esterification experiments. Riehm and Scheraga used α-diazo acetoglycinamide to esterify the carboxyl groups in ribonuclease A.127 They found that one aspartic acid residue was esterified preferentially, and proposed that this residue resides in a solvent-accessible area of local negative charge, which would raise its pKa value and lead to its selective esterification. Shortly thereafter, Delpierre and Fruton used an α-diazoketone to label a single residue in the active site of pepsin, causing near-complete inhibition of the enzyme.135 These workers proposed that this residue was in a privileged environment that enabled its selective labeling, as was posited for the aspartic acid in ribonuclease A,127 though neither of these speculations has been explored further. Instead, the inhibition of pepsin using α-diazoketones gave rise to a breadth of studies characterizing the active site of pepsin and comparing pepsin to its zymogen form (i.e., pepsinogen), in which the active-site residue is inaccessible to solvent and thus does not react with the diazo reagent.136–144 The combination of covalent labeling using a diazo reagent with Edman degradation (which was invented concurrently) provided a robust method for determining the identity of a catalytically important residue and its surrounding sequence.145 Using these techniques, novel acid proteases were classified based on their propensity to be inactivated by a diazo compound.146–152 Nonetheless, with the advent of site-directed mutagenesis, the use of diazo compounds to characterize proteins became rare.
Figure 4.

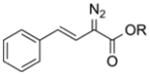
Diazo compounds for covalent modification of proteins. (A) Putative mechanism for the esterification of carboxylic acids with a diazo compound,134 and its application to the bioreversible labeling of a protein.128,129 Diazo compound I is optimized for protein esterification.129 (B) Putative mechanism of a diazo carbenoid insertion reaction, and its application to the site-specific modification of a proximal amino-acid residue.161
BIOREVERSIBLE PROTEIN MODIFICATION
The abundance and promiscuity of cellular esterases has been utilized in prodrug strategies in which chemotherapeutic agents are masked as esters and converted to their active forms upon cellular uptake.153–155 Our group envisioned a similar strategy for proteins in which carboxyl moieties are esterified by a diazo compound to install a molecular tag, such as a pharmacokinetic-enhancing, cell-type–targeting, or cell-penetrating moiety. Upon cellular uptake, the ester-linked tags are removed by endogenous esterases to recreate the native protein (Figure 4A). This strategy would be especially valuable for the delivery of proteins whose activities decrease significantly upon irreversible modification.156
In an initial study, structurally and electronically diverse diazo compounds were screened for their reactivity and selectivity in an aqueous environment.128 Of these compounds, only 9-diazofluorene esterified a panel of carboxylic acids efficiently in the presence of water. This diazo compound was used to label two model proteins, ribonuclease A and red fluorescent protein. The nascent esters were hydrolyzed upon treatment with a HeLa-cell extract, regenerating wild-type protein.
Later, a more systematic study investigated the rate and selectivity of a series of structurally similar but electronically diverse α-diazo amides.129 A Hammett analysis of these compounds, which were derived from phenylglycine, revealed that electron-donating or electron-withdrawing groups on the aryl ring had a dramatic effect on the rate of esterification. Still, the compounds were similar in their selectivity for ester formation over hydrolysis of the diazo reagent. The comparable selectivity among the compounds in this study supports the proposed mechanism in which the diazonium and carboxylate species, formed as intermediates, are held together in a solvent cage as an intimate ion pair (Figure 4A),134 and the ratio of ester to alcohol product is determined by the diffusion out of this solvent cage rather than the reactivity of the diazo compound.133,134,157 An α-diazo(p-methylphenyl)-glycinamide (I) demonstrated the fastest rate while maintaining selectivity, and esterifies proteins more efficiently than any known diazo reagent. The amide of compound I allows for facile incorporation of an amine of interest.
PEPTIDE AND PROTEIN MODIFICATION WITH CARBENOIDS
An early example of asymmetric catalysis employed a chiral transition-metal catalyst to generate a carbenoid from a diazo compound.158 Carbenoids generated similarly can access a broad scope of insertion reactions and are hence powerful reagents for modifying peptides and proteins. In a seminal study, Francis and coworkers used vinylic α-diazo esters to modify tryptophan residues in horse heart myoglobin.76 Then, Ball and coworkers employed metallopeptides to combine proximity-driven and transition metal-driven catalysis.159,160 In this system, the rhodium catalyst is displayed on a peptide, which is designed to bind a second peptide or protein of interest by forming a coiled-coil (Figure 4B).161 The catalyst on the metallopeptide is oriented such that the incipient carbenoid is generated proximal to the target residue, focusing its high reactivity and enabling modification of many types of amino acids.162 For example, although tryptophan can be modified by the addition of a diazo compound and rhodium acetate catalyst alone, employing a metallopeptide to orient the catalyst enables modification of the phenyl group of phenylalanine, imidazolyl group of histidine, and guanidinium group of arginine.
In a proof-of-concept study, Popp and Ball alkylated the aromatic amino-acid side chains by tethering the dirhodium center to a lysine-rich K3 peptide, which binds to and reacts with a glutamate-rich E3 peptide at a specific tryptophan residue.161 In a follow-up investigation, the scope of the E3/K3 system was extended to the alkylation of a broad range of functional groups, including a carboxamide.162 This system has since been used to modify maltose-binding protein fused to the E3 peptide,163 as well as for the site-selective modification of the native Fyn protein using a peptide ligand bearing the rhodium catalyst.164,165
NUCLEIC ACID ALKYLATION
Natural nucleobases can be modified in situ with diazo compounds. Gillingham and coworkers used rhodium(II) to catalyze the conversion of a diazo ester into a carbenoid that inserted into exocyclic N–H bonds (Figure 5A).166 Because this reactivity does not extend to double-helical regions, the strategy can target hairpins and single-stranded regions (Figure 5B). This selectivity is useful, for example, in studies on the mechanism of RNA interference, which entails 3′ overhangs.
Figure 5.

Covalent modification of nucleic acids using diazo compounds. (A) Representative alkylation of DNA by a diazo compound. Alkylation occurs on solvent-accessible nucleobases.166 (B) One-pot N–H insertion and azide–alkyne cycloaddition with a copper(I) catalyst.168 (C) Photoreversible O-alkylation of a phosphoryl group in RNA by a diazo coumarin.169
Rhodium(II) has been used most widely as a catalyst for the generation of carbenoids in chemical biology.167 Gillingham and coworkers showed, however, that copper(I)-carbenoid chemistry for N–H insertion is likewise effective.168 Their work demonstrated novel synergy of the diazo group with “copper-click” chemistry by combining N–H insertion with CuAAC in a one-pot single-catalyst process (Figure 5B).
An alternative strategy for nucleic-acid modification involves O-alkylation of the phosphoryl group. Okamoto and coworkers employed this method to modify an mRNA using a photolabile derivative of coumarin bearing a diazo moiety (Figure 5C).169 The ensuing “caged” mRNA, which encoded green fluorescent protein, was delivered to zebrafish embryos, where its translation could be modulated spatially and temporally by uncaging using ultraviolet light. Photolabile diazo groups have also been used to control RNA interference, in which a double-stranded precursor to an siRNA is inactivated upon modification with the diazo reagent and then uncaged with ultraviolet light.170 Diazo compounds have been employed to label and detect nucleic acids on microarrays without disrupting base pairing.171 Recently, Gillingham and coworkers reported on a diazo compound that modifies the phosphoryl groups of nucleic acids selectively in the presence of carboxylic acids.172 Their methodology could be useful for the labeling and detection of phosphorylated peptides and proteins as well.
OUTLOOK
Diazo compounds were discovered over 120 years ago. Recent advances in chemical synthesis have enabled the facile preparation of stabilized diazo compounds that are compatible with living systems. Like azido groups, diazo groups are chemoselective. Unlike azido groups, diazo groups have reactivity with natural and nonnatural functional groups that is tunable. The ability to tune their reactivity by delocalization of the electrons on the α carbon renders diazo compounds as attractive reagents in physiological contexts. Moreover, the versatility of diazo-group reactivity is extraordinary. Their ability to react rapidly, selectively, and autonomously with nonnatural functional groups (e.g., strained alkynes) as well as natural carboxyl groups, phosphoryl groups, and even the alkene in dehydroalanine residues anoints diazo groups as special. Accordingly, we envision an expansion in the use of diazo compounds to probe biological phenomena and to treat human disease, and even foresee an era of “diazophilia”.173
Acknowledgments
We are grateful to C. L. Jenkins for comments on the manuscript. K.A.M. was supported by Molecular Biosciences Training Grant T32 GM007215 (NIH). Work on diazo compounds in the Raines laboratory is supported by Grant R01 GM044783 (NIH).
KEYWORDS
- bioreversible esterification
O-alkylation of carboxylic acids to form an ester that is a substrate for a cellular esterase, for example, using a tuned diazo compound
- carbenoid
reactive intermediate, often generated from the metal-catalyzed decomposition of a diazo compound, that contains a divalent carbon with an unshared electron pair
- chemical reporter
non-natural functional group appended to a biomolecule of interest for detection or derivatization
- deimidogenation
loss of an NH moiety as in the phosphinoester-mediated conversion of an azido group to a diazo group
- diazo compound
compound that contains the functional group: –C=N+=N−
- 1,3-dipolar cycloaddition
chemical reaction between a 1,3-dipole (such as a diazo group) and a dipolarophile (such as an alkyne or alkene) to form a five-membered ring
- lomaiviticin
diazofluorene-based natural product with antiproliferative activity
Footnotes
The authors declare no competing financial interest.
References
- 1.Regitz M, Maas G. Diazo Compounds: Properties and Synthesis. Academic Press; London: 1986. [Google Scholar]
- 2.Doyle MP, McKervey MA. Recent advances in stereoselective synthesis involving diazocarbonyl intermediates. Chem Commun. 1997:983–989. [Google Scholar]
- 3.von Pechmann H. Ueber Diazomethan. Ber Dtsch Chem Ges. 1894;27:1888–1891. [Google Scholar]
- 4.von Pechmann H. Ueber Diazomethan. Ber Dtsch Chem Ges. 1895;28:855–861. [Google Scholar]
- 5.Lewinn EB. Diazomethane poisoning: Report of a fatal case with autopsy. Am J Med Sci. 1949;218:556–562. [PubMed] [Google Scholar]
- 6.Schoental R. Carcinogenic action of diazomethane and of nitroso-N-methyl urethane. Nature. 1960:420–421. doi: 10.1038/188420b0. [DOI] [PubMed] [Google Scholar]
- 7.Lewis CE. Diazomethane poisoning: Report of a case suggesting sensitization reaction. J Occup Environ Med. 1964;6:91–92. [PubMed] [Google Scholar]
- 8.de Boer TJ, Backer HJ. Diazomethane. Org Syn. 1956;36:16–18. [Google Scholar]
- 9.Sammakia T. e-EROS Encyclopedia of Reagents for Organic Synthesis. John Wiley & Sons; New York, NY: 2001. Diazomethane. [Google Scholar]
- 10.We are aware of only one natural azide metabolite β-azidoalanine which forms in microbes that are fed inorganic azide (Ciesla Z, Filutowicz M, Klopotowski T. Involvement of the L-cysteine biosynthetic pathway in azide-induced mutagenesis in Salmonella typhimurium. Mutat Res. 1980;70:261–268. doi: 10.1016/0027-5107(80)90017-2.
- 11.Köpke T, Zaleski JM. Diazo-containing molecular constructs as potential anticancer agents: From diazo[b]fluorene natural products to photoactivatable diazo-oxochlorins. Anticancer Agents Med Chem. 2008;8:292–304. doi: 10.2174/187152008783961941. [DOI] [PubMed] [Google Scholar]
- 12.Gould SJ. Biosynthesis of the kinamycins. Chem Rev. 1997;97:2499–2509. doi: 10.1021/cr9600215. [DOI] [PubMed] [Google Scholar]
- 13.Kersten RD, Lane AL, Nett M, Richter TKS, Duggan BM, Dorrestein PC, Moore BS. Bioactivity-guided genome mining reveals the lomaiviticin biosynthetic gene cluster in Salinispora tropica. Chem Bio Chem. 2013;14:955–962. doi: 10.1002/cbic.201300147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Janso JE, Haltli BA, Eustaquio AS, Kulowski K, Waldman AJ, Zha L, Nakamura H, Bernan VS, He H, Carter GT, Koehn FE, Balskus EP. Discovery of the lomaiviticin biosynthetic gene cluster in Salinispora pacifica. Tetrahedron. 2014;70:4156–4164. doi: 10.1016/j.tet.2014.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Waldman AJ, Pechersky Y, Wang P, Wang JX, Balskus EP. The cremeomycin biosynthetic gene cluster encodes a pathway for diazo formation. Chem Bio Chem. 2015;16:2172–2175. doi: 10.1002/cbic.201500407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sugai Y, Katsuyama Y, Ohnishi Y. A nitrous acid biosynthetic pathway for diazo group formation in bacteria. Nat Chem Biol. 2016;12:73–75. doi: 10.1038/nchembio.1991. [DOI] [PubMed] [Google Scholar]
- 17.Narawat CC, Moody CJ. Natural products containing a diazo group. Nat Prod Rep. 2011;28:1426–1444. doi: 10.1039/c1np00031d. [DOI] [PubMed] [Google Scholar]
- 18.Herzon SB, Woo CM. The diazofluorene antitumor antibiotics: Structural elucidation, biosynthetic, synthetic, and chemical biological studies. Nat Prod Rep. 2012;29:87–118. doi: 10.1039/c1np00052g. [DOI] [PubMed] [Google Scholar]
- 19.Ito S, Matsuya T, Omura S, Otani M, Nakagawa A, Takeshima H, Iwai Y, Ohtani M, Hata T. A new antibiotic, kinamycin. J Antibiot. 1970;23:315–317. doi: 10.7164/antibiotics.23.315. [DOI] [PubMed] [Google Scholar]
- 20.Gould SJ, Tamayo N, Melville CR, Cone MC. Revised structures for the kinamycin antibiotics: 5-Diazobenzo[b]fluorenes rather than benzo[b]carbazole cyanamides. J Am Chem Soc. 1994;116:2207–2208. [Google Scholar]
- 21.Lei X, Porco JA. Total synthesis of the diazobenzofluorene antibiotic (–)-kinamycin C. J Am Chem Soc. 2006;128:14790–14791. doi: 10.1021/ja066621v. [DOI] [PubMed] [Google Scholar]
- 22.Kumamoto T, Kitani Y, Tsuchiya H, Yamaguchi K, Seki H, Ishikawa T. Total synthesis of (±)-methyl-kinamycin C. Tetrahedron. 2007;63:5189–5199. [Google Scholar]
- 23.Nicolau KC, Li H, Nold AL, Pappo D, Lenzen A. Total synthesis of kinamycins C, F, and J. J Am Chem Soc. 2007;129:10356–10357. doi: 10.1021/ja074297d. [DOI] [PubMed] [Google Scholar]
- 24.He H, Ding W-D, Bernan VS, Richardson AD, Ireland CM, Greenstein M, Ellestad GA, Carter GT. Lomaiviticins A and B, potent antitumor antibiotics from Micromonospora lomaivitiensis. J Am Chem Soc. 2001;123:5362–5363. doi: 10.1021/ja010129o. [DOI] [PubMed] [Google Scholar]
- 25.Woo CM, Beizer NE, Janso JE, Herzon SB. Isolation of lomaiviticins C–E, transformation of lomaiviticin C to lomaiviticin A, complete structure elucidation of lomaiviticin A, and structure–activity analyses. J Am Chem Soc. 2012;134:15285–15288. doi: 10.1021/ja3074984. [DOI] [PubMed] [Google Scholar]
- 26.Nicolau KC, Denton RM, Lenzen A, Edmonds DJ, Li A, Milburn RR, Harrison RR. Stereocontrolled synthesis of model core systems of lomaiviticins A and B. Angew Chem, Int Ed. 2006;45:2076–2081. doi: 10.1002/anie.200504466. [DOI] [PubMed] [Google Scholar]
- 27.Zhang W, Baranczak A, Sulikowski GA. Stereocontrolled assembly of the C3/C3′ dideoxy core of lomaiviticin A/B and congeners. Org Lett. 2008;10:1939–1941. doi: 10.1021/ol800460a. [DOI] [PubMed] [Google Scholar]
- 28.Nicolau KC, Nold AL, Li H. Synthesis of the monomeric unit of the lomaiviticin aglycon. Angew Chem, Int Ed. 2009;121:5974–5977. doi: 10.1002/anie.200902509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee HG, Ahn JY, Lee AS, Shair MD. Enantioselective synthesis of the lomaiviticin aglycon full carbon skeleton reveals remarkable remote substituent effects during the dimerization event. Chem Eur J. 2010;16:13058–13062. doi: 10.1002/chem.201002157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Herzon SB, Lu L, Woo CM, Gholap SL. 11-Step enantioselective synthesis of (−)-lomaiviticin aglycon. J Am Chem Soc. 2011;133:7260–7263. doi: 10.1021/ja200034b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Arya DP, Jebaratnam DJ. DNA cleaving ability of 9-diazofluorenes and diaryl diazomethanes: Implications for the mode of action of the kinamycin antibiotics. J Org Chem. 1995;60:3268–3269. [Google Scholar]
- 32.Cone MC, Melville CR, Gore MP, Gould SJ. Kinafluorenone, a benzo[b]fluorene isolated from the kinamycin producer Streptomyces murayamensis. J Org Chem. 1993;58:1058–1061. [Google Scholar]
- 33.Laufer RS, Dmitrienko GI. Diazo group electrophilicity in kinamycins and lomaiviticin A: Potential insights into the molecular mechanism of antibacterial and antitumor activity. J Am Chem Soc. 2002;124:1854–1855. doi: 10.1021/ja0167809. [DOI] [PubMed] [Google Scholar]
- 34.Ballard TE, Melander C. Kinamycin-mediated DNA cleavage under biomimetic conditions. Tetrahedron Lett. 2008;49:3157–3161. [Google Scholar]
- 35.Feldman KS, Eastman KJ. Studies on the mechanism of action of prekinamycin, a member of the diazoparaquinone family of natural products: Evidence for both sp2 radical and orthoquinone methide intermediates. J Am Chem Soc. 2006;128:12562–12573. doi: 10.1021/ja0642616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mulcahy SP, Woo CM, Ding W, Ellestad GA, Herzon SB. Characterization of a reductively-activated elimination pathway relevant to the biological chemistry of the kinamycins and lomaiviticins. Chem Sci. 2012;3:1070–1074. [Google Scholar]
- 37.O’Hara KA, Wu X, Patel D, Liang H, Yalowich JC, Chen N, Goodfellow V, Adedayo O, Dmitrienko GI, Hasinoff BB. Mechanism of the cytotoxicity of the diazoparaquinone antitumor antibiotic kinamycin F. Free Radic Biol Med. 2007;43:1132–1144. doi: 10.1016/j.freeradbiomed.2007.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Colis LC, Woo CM, Hegan DC, Li Z, Glazer PM, Herzon SB. The cytotoxicity of (−)-lomaiviticin A arises from induction of double-strand breaks in DNA. Nat Chem. 2014;6:504–510. doi: 10.1038/nchem.1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Woo CM, Li Z, Paulson EK, Herzon SB. Structural basis for DNA cleavage by the potent antiproliferative agent (−)-lomaiviticin A. Proc Nat Acad Sci USA. 2015;113:2851–2856. doi: 10.1073/pnas.1519846113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Woo CM, Ranjan N, Arya DP, Herzon SB. Analysis of diazofluorene DNA binding and damaging activity: DNA cleavage by a synthetic monomeric diazofluorene. Angew Chem, Int Ed. 2014;53:9325–9328. doi: 10.1002/anie.201404137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Colis LC, Hegan DC, Kaneko M, Glazer PM, Herzon SB. Mechanism of action studies of lomaiviticin A and the monomeric lomaiviticin aglycon. Selective and potent activity toward DNA double-strand break repair-deficient cell lines. J Am Chem Soc. 2015;137:5741–5747. doi: 10.1021/ja513117p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jeggo PA, Pearl LH, Carr AM. DNA repair, genome stability and cancer: A historical perspective. Nat Rev Cancer. 2016;16:35–42. doi: 10.1038/nrc.2015.4. [DOI] [PubMed] [Google Scholar]
- 43.Dion HW, Fusari SA, Jakubowski ZL, Zora JG, Bartz QR. 6-Diazo-5-oxo-L-norleucine, a new tumor-inhibitory substance. II. Isolation and characterization. J Am Chem Soc. 1956;78:3075–3077. [Google Scholar]
- 44.Handschumacher RE, Bates CJ, Chang PK, Andrews AT, Fischer GA. 5-Diazo-4-oxo-L-norvaline: Reactive asparagine analog with biological specificity. Science. 1968;161:62–63. doi: 10.1126/science.161.3836.62. [DOI] [PubMed] [Google Scholar]
- 45.Pinkus LM. Glutamine binding sites. Methods Enzymol. 1977;46:414–427. doi: 10.1016/s0076-6879(77)46049-x. [DOI] [PubMed] [Google Scholar]
- 46.Fusari SA, Haskell TH, Frohardt RP, Bartz QR. Azaserine, a new tumor-inhibitory substance. Structural studies. J Am Chem Soc. 1954;76:2881–2883. [Google Scholar]
- 47.Hartman SC. The interaction of 6-diazo-5-oxo-L-norleucine with phosphoribosyl pyrophosphate amidotransferase. J Biol Chem. 1963;238:3036–3047. [PubMed] [Google Scholar]
- 48.Hartman SC, McGrath TF. Glutaminase A of Escherichia coli. J Biol Chem. 1973;248:8506–8510. [PubMed] [Google Scholar]
- 49.Clark VM, Shapiro RA, Curthoys NP. Comparison of the hydrolysis and the covalent binding of 6-diazo-5-oxo-L-[6-14C]norleucine by rat renal phosphate-dependent glutaminase. Arch Biochem Biophys. 1982;213:232–239. doi: 10.1016/0003-9861(82)90457-x. [DOI] [PubMed] [Google Scholar]
- 50.Rahman A, Smith FP, Luc PV, Woolley PV. Phase I study and clinical pharmacology of 6-diazo-5-oxo-L-norleucine (DON) Invest New Drugs. 1985;3:369–374. doi: 10.1007/BF00170760. [DOI] [PubMed] [Google Scholar]
- 51.Inoue M, Horiuchi S, Morino Y. Affinity labeling of rat-kidney γ-glutamyl transpeptidase. Eur J Biochem. 1977;73:335–342. doi: 10.1111/j.1432-1033.1977.tb11323.x. [DOI] [PubMed] [Google Scholar]
- 52.Tate SS, Meister A. Affinity labeling of γ-glutamyl transpeptidase and location of the γ-glutamyl binding site on the light subunit. Proc Natl Acad Sci USA. 1977;74:931–935. doi: 10.1073/pnas.74.3.931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Horiuchi S, Inoue M, Morino Y. γ-Glutamyl transpeptidase: Sidedness of its active site on renal brush-border membrane. Eur J Biochem. 1978;87:429–437. doi: 10.1111/j.1432-1033.1978.tb12392.x. [DOI] [PubMed] [Google Scholar]
- 54.Holcenberg JS, Ericsson L, Roberts J. Amino acid sequence of the diazooxonorleucine binding site of Acinetobacter and Pseudomonas 7A glutaminase–asparaginase enzymes. Biochemistry. 1978;17:411–417. doi: 10.1021/bi00596a005. [DOI] [PubMed] [Google Scholar]
- 55.Ortlund E, Lacount MW, Lewinski K, Lebioda L. Reactions of Pseudomonas 7A glutaminase–asparaginase with diazo analogues of glutamine and asparagine result in unexpected covalent inhibitions and suggests an unusual catalytic triad Thr-Tyr-Glu. Biochemistry. 2000;39:1199–1204. doi: 10.1021/bi991797d. [DOI] [PubMed] [Google Scholar]
- 56.Peterson RG, Richards FF, Handschumacher RE. Structure of peptide from active site region of Escherichia coli L-asparaginase. J Biol Chem. 1977;252:2072–2076. [PubMed] [Google Scholar]
- 57.Asselin BL, Lorenson MY, Whitin JC, Coppola DJ, Kende AS, Blakley RL, Cohen HJ. Measurement of serum L-asparagine in the presence of L-asparaginase requires the presence of an L-asparaginase inhibitor. Cancer Res. 1991;51:6568–6573. [PubMed] [Google Scholar]
- 58.Regitz M. New methods of preparative organic chemistry. Angew Chem, Int Ed. 1967;6:733–749. doi: 10.1002/anie.196701491. [DOI] [PubMed] [Google Scholar]
- 59.Baum JS, Shook DA, Davies HML, Smith HD. Diazotransfer reactions with p-acetamidobenzenesulfonyl azide. Synth Commun. 1987;17:1709–1716. [Google Scholar]
- 60.Curtius T. Ueber die Einwirkung von salpetriger Säure auf salzsauren Glycocolläther. Ber Dtsch Chem Ges. 1883;16:2230–2231. [Google Scholar]
- 61.Womack EB, Nelson AB. Ethyl diazoacetate. Org Syn. 1955;24:56–57. [Google Scholar]
- 62.Bamford WR, Stevens TS. 924. The decomposition of toluene-p-sulphonylhydrazones by alkali. J Chem Soc. 1952:4735–4740. [Google Scholar]
- 63.Fulton JR, Aggarwal VK, de Vicente J. The use of tosylhydrazone salts as a safe alternative for handling diazo compounds and their applications in organic synthesis. Eur J Org Chem. 2005;2005:1479–1492. [Google Scholar]
- 64.Holton TL, Schechter H. Advantageous syntheses of diazo compounds by oxidation of hydrazones with lead tetraacetate in basic environments. J Org Chem. 1995;60:4725–4729. [Google Scholar]
- 65.Furrow ME, Myers AG. A general procedure for the esterification of carboxylic acids with diazoalkanes generated in situ by the oxidation of N-tert-butyldimethylsilylhydrazones with (difluoroiodo)benzene. J Am Chem Soc. 2004;126:12222–12223. doi: 10.1021/ja0459779. [DOI] [PubMed] [Google Scholar]
- 66.Morandi B, Carreira EM. Iron-catalyzed cyclopropanation in 6 M KOH with in situ generation of diazomethane. Science. 2012;335:1471–1474. doi: 10.1126/science.1218781. [DOI] [PubMed] [Google Scholar]
- 67.Baumgarten RJ. Preparation of ethyl diazoacetate via a triazene intermediate. J Org Chem. 1967;32:484–485. [Google Scholar]
- 68.Schroen M, Bräse S. Polymer-bound diazonium salts for the synthesis of diazoacetic esters. Tetrahedron. 2005;61:12186–12192. [Google Scholar]
- 69.Fink J, Regitz M. Electrophilic diazoalkane substitution. Synthesis. 1985;1985:569–585. [Google Scholar]
- 70.Ye T, McKervey MA. Synthesis of chiral N-protected α-amino-β-diketones from α-diazoketones derived from natural amino acids. Tetrahedron. 1992;48:8007–8022. [Google Scholar]
- 71.Zhao Y, Wang J. Nucleophilic addition to C=O and C=N bonds by nucleophiles containing a diazo group. Synlett. 2005;2005:2886–2892. [Google Scholar]
- 72.Meyer ME, Ferreira EM, Stoltz BM. 2-Diazoacetoacetic acid, an efficient and convenient reagent for the synthesis of α-diazo-β-ketoesters. Chem Commun. 2006:1316–1318. doi: 10.1039/b517719g. [DOI] [PubMed] [Google Scholar]
- 73.Liu Y, Zhang Y, Jee N, Doyle MP. Construction of highly functionalized diazoacetoacetates via catalytic Mukaiyama–Michael reactions. Org Lett. 2008;10:1605–1608. doi: 10.1021/ol800298n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Maas G. New syntheses of diazo compounds. Angew Chem, Int Ed. 2009;48:8186–8195. doi: 10.1002/anie.200902785. [DOI] [PubMed] [Google Scholar]
- 75.Ford A, Miel H, Ring A, Slattery CN, Maguire AR, McKervey MA. Modern organic synthesis with α-diazocarbonyl compounds. Chem Rev. 2015;115:9981–10080. doi: 10.1021/acs.chemrev.5b00121. [DOI] [PubMed] [Google Scholar]
- 76.Antos JM, Francis MB. Selective tryptophan modification with rhodium carbenoids in aqueous solution. J Am Chem Soc. 2004;126:10256–10257. doi: 10.1021/ja047272c. [DOI] [PubMed] [Google Scholar]
- 77.Goddard-Borger ED, Stick RV. An efficient, inexpensive, and shelf-stable diazotransfer reagent: Imidazole-1-sulfonyl azide hydrochloride. Org Lett. 2007;9:3797–3800. doi: 10.1021/ol701581g. [DOI] [PubMed] [Google Scholar]
- 78.Myers EL, Raines RT. A phosphine-mediated conversion of azides into diazo compounds. Angew Chem, Int Ed. 2009;48:2359–2363. doi: 10.1002/anie.200804689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chou H, Raines RT. Conversion of azides into diazo compounds in water. J Am Chem Soc. 2013;135:14936–14939. doi: 10.1021/ja407822b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Nilsson BL, Kiessling LL, Raines RT. Staudinger ligation: A peptide from a thioester and azide. Org Lett. 2000;2:1939–1941. doi: 10.1021/ol0060174. [DOI] [PubMed] [Google Scholar]
- 81.Saxon E, Bertozzi CR. Cell surface engineering by a modified Staudinger reaction. Science. 2000;287:2007–2010. doi: 10.1126/science.287.5460.2007. [DOI] [PubMed] [Google Scholar]
- 82.Soellner MB, Nilsson BL, Raines RT. Reaction mechanism and kinetics of the traceless Staudinger ligation. J Am Chem Soc. 2006;128:8820–8828. doi: 10.1021/ja060484k. [DOI] [PubMed] [Google Scholar]
- 83.Sletten EM, Bertozzi CR. From mechanism to mouse: A tale of two bioorthogonal reactions. Acc Chem Res. 2011;44:666–676. doi: 10.1021/ar200148z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.McGrath NA, Raines RT. Chemoselectivity in chemical biology: Acyl transfer reactions with sulfur and selenium. Acc Chem Res. 2011;44:752–761. doi: 10.1021/ar200081s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Staudinger H, Meyer J. Über neue organische Phosphorverbindungen III. Phosphinmethylenderivate und Phosphinimine. Helv Chim Acta. 1919;2:635–646. [Google Scholar]
- 86.Staudinger H, Hauser E. Über neue organische Phosphorverbindungen IV Phosphinimine. Helv Chim Acta. 1921;4:861–886. [Google Scholar]
- 87.Caution! Sodium azide is nearly as toxic to mammals as is sodium cyanide. For example, the LD50 values for acute dermal toxicity in rabbits are 20 mg/kg and 10.4 mg/kg, respectively (MSDS).
- 88.Singh A, Thornton ER, Westheimer FH. The photolysis of diazoacetylchymotrypsin. J Biol Chem. 1962;237:PC3006–PC3008. [PubMed] [Google Scholar]
- 89.Ouihia A, René L, Guilhem J, Pascard C, Badet B. A new diazoacylating reagent: Preparation, structure, and use of succinimidyl diazoacetate. J Org Chem. 1993;58:1641–1642. [Google Scholar]
- 90.Doyle MP, Kalinin AV. Highly enantioselective intramolecular cyclopropanation reactions of N-allylic-N-methyldiazoacetamides catalyzed by chiral dirhodium(II) carboxamidates. J Org Chem. 1996;61:2179–2184. [Google Scholar]
- 91.Fuerst DE, Stoltz BM, Wood JL. Synthesis of C(3) benzofuran-derived bisaryl quaternary centers: Approaches to diazonamide A. Org Lett. 2000;2:3521–3523. doi: 10.1021/ol006578v. [DOI] [PubMed] [Google Scholar]
- 92.Andersen KA, Aronoff MR, McGrath NA, Raines RT. Diazo groups endure metabolism and enable chemoselectivity in cellulo. J Am Chem Soc. 2015;137:2412–2415. doi: 10.1021/ja5095815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Josa-Culleré L, Wainman YA, Brindle KM, Leeper FJ. Diazo group as a new chemical reporter for bioorthogonal labelling of biomolecules. RSC Adv. 2014;4:52241–52244. [Google Scholar]
- 94.Zhou W, Hsieh P-H, Xu Y, O’Leary TR, Huang X, Liu J. Design and synthesis of active heparan sulfate-based probes. Chem Commun. 2015;51:11019–11021. doi: 10.1039/c5cc02008e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Friscourt F, Fahrni CJ, Boons G-J. Fluorogenic strain-promoted alkyne–diazo cycloadditions. Chem Eur J. 2015;21:13996–14001. doi: 10.1002/chem.201502242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Buchner E. Einwirkung von Diazoessigäther auf die Aether ungesättigter Säuren. Ber Dtsch Chem Ges. 1888;21:2637–2647. [Google Scholar]
- 97.Huisgen R. 1,3-Dipolar cycloadditions. Past and future. Angew Chem, Int Ed. 1963;2:565–598. [Google Scholar]
- 98.Maas G. Diazoalkanes. In: Padwa A, Pearson WH, editors. Synthetic Applications of 1,3-Dipolar Cycloaddition Chemistry Toward Heterocycles and Natural Products. John Wiley & Sons, Inc; New York, NY: 2002. pp. 539–621. [Google Scholar]
- 99.Rostovtsev VV, Green LG, Fokin VV, Sharpless KB. A stepwise Huisgen cycloaddition process: Copper(I)-catalyzed regioselective “ligation” of azides and terminal alkynes. Angew Chem, Int Ed. 2002;41:2596–2599. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 100.Tornøe CW, Christensen C, Meldal M. Peptidotriazoles on solid phase: [1,2,3]-Triazoles by regiospecific copper(I)-catalyzed 1,3-dipolar cycloadditions of terminal alkynes to azides. J Org Chem. 2002;67:3057–3064. doi: 10.1021/jo011148j. [DOI] [PubMed] [Google Scholar]
- 101.Wittig G, Krebs A. Zur Existenz niedergliedriger Cycloalkine, I. Chem Ber. 1961;94:3260–3275. [Google Scholar]
- 102.Agard NJ, Prescher JA, Bertozzi CR. A strain-promoted [3 + 2] azide–alkyne cycloaddition for covalent modification of biomolecules in living systems. J Am Chem Soc. 2004;126:15046–15047. doi: 10.1021/ja044996f. [DOI] [PubMed] [Google Scholar]
- 103.Baskin JM, Prescher JA, Laughlin ST, Agard NJ, Chang PV, Miller IA, Lo A, Codelli JA, Bertozzi CR. Copper-free click chemistry for dynamic in vivo imaging. Proc Natl Acad Sci USA. 2007;104:16793–16797. doi: 10.1073/pnas.0707090104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Debets MF, van der Doelen CWJ, Rutjes FPJT, van Delft FL. Azide: A unique dipole for metal-free bioorthogonal ligations. Chem Bio Chem. 2010;11:1168–1184. doi: 10.1002/cbic.201000064. [DOI] [PubMed] [Google Scholar]
- 105.Patterson DM, Nazarova LA, Prescher JA. Finding the right (bioorthogonal) chemistry. ACS Chem Biol. 2014;9:592–605. doi: 10.1021/cb400828a. [DOI] [PubMed] [Google Scholar]
- 106.Moran J, McKay CS, Pezacki JP. Strain-promoted 1,3-dipolar cycloadditions of diazo compounds with cyclooctynes. Can J Chem. 2010;89:148–151. doi: 10.1039/b921630h. [DOI] [PubMed] [Google Scholar]
- 107.Sanders BC, Friscourt F, Ledin PA, Mbua NE, Arumugam S, Guo J, Boltje TJ, Popik VV, Boons G-J. Metal-free sequential [3 + 2]-dipolar cycloadditions using cyclooctynes and 1,3-dipoles of different reactivity. J Am Chem Soc. 2011;133:949–957. doi: 10.1021/ja1081519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Bihlmaier W, Huisgen R, Reissig H-U, Voss S. Reactivity sequences of dipolarophiles towards diazocarbonyl compounds—MO perturbation treatment. Tetrahedron Lett. 1979:2621–2624. [Google Scholar]
- 109.McGrath NA, Raines RT. Diazo compounds as highly tunable reactants in 1,3-dipolar cycloaddition reactions with cycloalkynes. Chem Sci. 2012;3:3237–3240. doi: 10.1039/C2SC20806G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Aronoff MR, Gold B, Raines RT. 1,3-Dipolar cycloadditions of diazo compounds in the presence of azides. Org Lett. 2016;18:1538–1541. doi: 10.1021/acs.orglett.6b00278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Aronoff MR, Gold B, Raines RT. Rapid cycloaddition of a diazo group with an unstrained dipolarophile. Tetrahedron Lett. 2016;57:2347–2350. doi: 10.1016/j.tetlet.2016.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Gold B, Aronoff MR, Raines RT. 1,3-Dipolar cycloaddition with diazo groups: Noncovalent interactions overwhelm strain. Org Lett. 2016;18:4466–4469. doi: 10.1021/acs.orglett.6b01938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Gold B, Aronoff MR, Raines RT. Decreasing distortion energies without strain: Diazo-selective 1,3-dipolar cycloadditions. J Org Chem. 2016;81:5998–6006. doi: 10.1021/acs.joc.6b00948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Bayley H, Knowles JR. Photoaffinity labeling. Methods Enzymol. 1977;46:69–114. doi: 10.1016/s0076-6879(77)46012-9. [DOI] [PubMed] [Google Scholar]
- 115.Chowdhry V, Westheimer FH. Photoaffinity labeling of biological systems. Annu Rev Biochem. 1979;48:293–325. doi: 10.1146/annurev.bi.48.070179.001453. [DOI] [PubMed] [Google Scholar]
- 116.Wolff L. Ueber Diazoanhydride. Justus Liebigs Ann Chem. 1902;325:129–195. [Google Scholar]
- 117.Kirmse W. 100 Years of the Wolff rearrangement. Eur J Org Chem. 2002:2193–2256. [Google Scholar]
- 118.Converse CA, Richards FF. Two-stage photosensitive label for antibody combining sites. Biochemistry. 1969;8:4431–4436. doi: 10.1021/bi00839a031. [DOI] [PubMed] [Google Scholar]
- 119.Gupta CM, Costello CE, Khorana HG. Sites of intermolecular crosslinking of fatty acyl chains in phospholipids carrying a photoactivable carbene precursor. Proc Natl Acad Sci USA. 1979;76:3139–3143. doi: 10.1073/pnas.76.7.3139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Kale TA, Distefano MD. Diazotrifluoropropionamido-containing prenylcysteines: Syntheses and applications for studying isoprenoid–protein interactions. Org Lett. 2003;5:609–612. doi: 10.1021/ol026752a. [DOI] [PubMed] [Google Scholar]
- 121.Padwa A, Weingarten MD. Cascade processes of metallocarbenoids. Chem Rev. 1996;96:223–269. doi: 10.1021/cr950022h. [DOI] [PubMed] [Google Scholar]
- 122.Doyle MP, McKervey MA, Ye T. Modern Catalytic Methods for Organic Synthesis with Diazo Compounds: From Cyclopropanes to Ylides. John Wiley & Sons; New York, NY: 1998. [Google Scholar]
- 123.Davies HML, Beckwith REJ. Catalytic enantioselective C–H activation by means of metal-carbenoid-induced C–H insertion. Chem Rev. 2003;103:2861–2904. doi: 10.1021/cr0200217. [DOI] [PubMed] [Google Scholar]
- 124.Candelas NR, Alfonso CA. Developments in the photochemistry of diazo compounds. Curr Org Chem. 2009;13:763–787. [Google Scholar]
- 125.Geake A, Nierenstein M. The action of diazomethane on caseinogen. Biochem J. 1914;8:287–292. doi: 10.1042/bj0080287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Doscher MS, Wilcox PE. Chemical derivatives of α-chymotrypsinogen: IV. A comparison of the reactions of α-chymotrypsinogen and of simple carboxylic acids with diazoacetamide. J Biol Chem. 1961;236:1328–1337. [PubMed] [Google Scholar]
- 127.Riehm JP, Scheraga HA. Structural studies of ribonuclease. XVII. A reactive carboxyl group in ribonuclease. Biochemistry. 1965;4:772–782. doi: 10.1021/bi00880a023. [DOI] [PubMed] [Google Scholar]
- 128.McGrath NA, Andersen KA, Davis AKF, Lomax JE, Raines RT. Diazo compounds for the bioreversible esterification of proteins. Chem Sci. 2015;6:752–755. doi: 10.1039/c4sc01768d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Mix KA, Raines RT. Optimized diazo scaffold for protein esterification. Org Lett. 2015;17:2359–2361. doi: 10.1021/acs.orglett.5b00840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Chibnall AC, Rees MW. Studies on the amide and C-terminal residues in proteins. 1. The characterization of the C-terminal residue. Biochem J. 1958;68:105–111. doi: 10.1042/bj0680105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Grossberg AL, Pressman D. Nature of the combining site of antibody against a hapten bearing a positive charge. J Am Chem Soc. 1960;82:5478–5482. [Google Scholar]
- 132.Staudinger H, Gaule A. Diphenylendiazomethan. Ber Dtsch Chem Ges. 1917;49:1951–1960. [Google Scholar]
- 133.Roberts JD, Watanabe W, McMahon RE. The kinetics and mechanism of the reaction of diphenyldiazomethane and benzoic acid in ethanol. J Am Chem Soc. 1951;73:760–765. [Google Scholar]
- 134.Roberts JD, Watanabe W, McMahon RE. The kinetics and mechanism of the reaction of diphenyldiazomethane with 2,4-dinitrophenol in ethanol. J Am Chem Soc. 1951;73:2521–2523. [Google Scholar]
- 135.Delpierre GR, Fruton JS. Specific inactivation of pepsin by a diazo ketone. Proc Natl Acad Sci USA. 1966;56:1817–1822. doi: 10.1073/pnas.56.6.1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Delpierre GR, Fruton JS. Inactivation of pepsin by diphenyldiazomethane. Proc Natl Acad Sci USA. 1965;54:1161–1167. doi: 10.1073/pnas.54.4.1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Rajagopalan TG, Stein WH, Moore S. The inactivation of pepsin by diazoacetylnorleucine methyl ester. J Biol Chem. 1966;241:4295–4297. [PubMed] [Google Scholar]
- 138.Hamilton GA, Spona J, Crowell LD. The inactivation of pepsin by an equimolar amount of 1-diazo-4-phenylbutanone-2. Biochem Biophys Res Commun. 1967;26:193–198. doi: 10.1016/0006-291x(67)90233-1. [DOI] [PubMed] [Google Scholar]
- 139.Ong EB, Perlmann GE. Specific inactivation of pepsin by benzyloxycarbonyl-L-phenyldiazomethane. Nature. 1967;215:1492–1494. doi: 10.1038/2151492b0. [DOI] [PubMed] [Google Scholar]
- 140.Bayliss RS, Knowles JR. An active site peptide from pepsin. Chem Commun. 1968:196–198. [Google Scholar]
- 141.Fry KT. A reactive aspartyl residue of pepsin. Biochem Biophys Res Commun. 1968;30:489–495. doi: 10.1016/0006-291x(68)90078-8. [DOI] [PubMed] [Google Scholar]
- 142.Stepanov VM, Vaganova TI. Identification of the carboxyl group of pepsin reacting with diazoacetamide derivatives. Biochem Biophys Res Commun. 1968;31:825–830. doi: 10.1016/0006-291x(68)90637-2. [DOI] [PubMed] [Google Scholar]
- 143.Lundblad RL, Stein WH. On the reaction of diazoacetyl compounds with pepsin. J Biol Chem. 1969;244:154–160. [PubMed] [Google Scholar]
- 144.Bayliss RS, Knowles JR, Wybrandt GB. An aspartic acid residue at the active site of pepsin. Biochem J. 1969;113:377–386. doi: 10.1042/bj1130377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Edman P, Högfeldt E, Sillén LG, Kinell P. Method for the determination of the amino acid sequence in peptides. Acta Chem Scand. 1950;4:283–293. [Google Scholar]
- 146.Takahashi K, Mizobe F, Chang W. Inactivation of acid proteases from Rhizopus chinensis, Aspergillus saitoi and Mucor pusillus, and calf rennin by diazoacetylnorleucine methyl ester. J Biochem. 1972;71:161–164. doi: 10.1093/oxfordjournals.jbchem.a129739. [DOI] [PubMed] [Google Scholar]
- 147.Kovaleva GG, Shimanskaya MP, Stepanov VM. The site of diazoacetyl inhibitor attachment to acid proteinase of Aspergillus awamori—An analog of penicillopepsin and pepsin. Biochem Biophys Res Commun. 1972;49:1075–1081. doi: 10.1016/0006-291x(72)90322-1. [DOI] [PubMed] [Google Scholar]
- 148.Murao S, Oda K, Matsushita Y. New acid proteases from Scytalidium lignicolum M-133. Agr Biol Chem. 1972;36:1647–1650. [Google Scholar]
- 149.Mizobe F, Takahashi K, Ando T. The structure and function of acid proteases. J Biochem. 1973;73:61–68. [PubMed] [Google Scholar]
- 150.Takahashi K, Chang W. Specific chemical modifications of acid proteinases in the presence and absence of pepstatin. J Biochem. 1973;73:675–677. doi: 10.1093/oxfordjournals.jbchem.a130129. [DOI] [PubMed] [Google Scholar]
- 151.Johnson RL, Poisner AM. Inactivation of amniotic prorenin by ethyl diazoacetylglycinate. Biochem Biophys Res Commun. 1980;95:1404–1409. doi: 10.1016/s0006-291x(80)80053-2. [DOI] [PubMed] [Google Scholar]
- 152.Oda K, Sugitani M, Fukuhara K, Murao S. Purification and properties of a pepstatin-insensitive carboxyl proteinase from a Gram-negative bacterium. Biochem Biophys Acta. 1987;923:463–469. doi: 10.1016/0304-4165(87)90055-9. [DOI] [PubMed] [Google Scholar]
- 153.Testa B, Mayer JM. Hydrolysis in Drug and Prodrug Metabolism. Verlag Helvetica Chimica Acta; Zürich, Switzerland: 2003. [Google Scholar]
- 154.Liederer BM, Borchardt RT. Enzymes involved in the bioconversion of ester-based prodrugs. J Pharm Sci. 2006;95:1177–1195. doi: 10.1002/jps.20542. [DOI] [PubMed] [Google Scholar]
- 155.Tian L, Yang Y, Wysocki LM, Arnold AC, Hu A, Ravichandran B, Stenerson SM, Looger LL, Lavis LD. Selective esterase–ester pair for targeting small molecules with cellular specificity. Proc Natl Acad Sci USA. 2012;109:4756–4761. doi: 10.1073/pnas.1111943109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Harris JM, Chess RB. Effect of pegylation on pharmaceuticals. Nat Rev Drug Discov. 2003;2:214–221. doi: 10.1038/nrd1033. [DOI] [PubMed] [Google Scholar]
- 157.Szele I, Tencer M, Zollinger H. 163. Reactions of alkenediazonium salts. Part 1. 2,2-Diethoxyethenediazonium hexachloroantimonate: A diazonium, a carbenium, or an oxonium salt? Helv Chim Acta. 1983;66:1691–1703. [Google Scholar]
- 158.Nozaki H, Moriuti S, Takaya H, Noyori R. Asymmetric induction in carbenoid reaction by means of a dissymmetric copper chelate. Tetrahedron Lett. 1966;7:5239–5244. [Google Scholar]
- 159.Ball ZT. Designing enzyme-like catalysts: A rhodium(II) metallopeptide case study. Acc Chem Res. 2013;46:560–570. doi: 10.1021/ar300261h. [DOI] [PubMed] [Google Scholar]
- 160.Ball ZT. Molecular recognition in protein modification with rhodium metallopeptides. Curr Opin Chem Biol. 2015;25:98–102. doi: 10.1016/j.cbpa.2014.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Popp BV, Ball ZT. Structure-selective modification of aromatic side chains with dirhodium metallopeptide catalysts. J Am Chem Soc. 2010;132:6660–6662. doi: 10.1021/ja101456c. [DOI] [PubMed] [Google Scholar]
- 162.Popp BV, Ball ZT. Proximity-driven metallopeptide catalysis: Remarkable side-chain scope enables modification of the Fos bZip domain. Chem Sci. 2011;2:690–695. [Google Scholar]
- 163.Chen Z, Popp BV, Bovet CL, Ball ZT. Site-specific protein modification with a dirhodium metallopeptide catalyst. ACS Chem Biol. 2011;6:920–925. doi: 10.1021/cb2001523. [DOI] [PubMed] [Google Scholar]
- 164.Chen ZC, Coughlin JM, Stagg LJ, Arold ST, Ladbury JE, Ball ZT. Catalytic protein modification and dirhodium metallopeptides: Specificity in designed and natural systems. J Am Chem Soc. 2012;134:10138–10145. doi: 10.1021/ja302284p. [DOI] [PubMed] [Google Scholar]
- 165.Vohidov F, Coughlin JM, Ball ZT. Rhodium(II) metallopeptide catalyst design enables fine control in selective functionalization of natural SH3 domains. Angew Chem, Int Ed. 2015;54:4587–4591. doi: 10.1002/anie.201411745. [DOI] [PubMed] [Google Scholar]
- 166.Tishinov K, Schmidt K, Häussinger D, Gillingham DG. Structure-selective catalytic alkylation of DNA and RNA. Angew Chem, Int Ed. 2012;51:12000–12004. doi: 10.1002/anie.201205201. [DOI] [PubMed] [Google Scholar]
- 167.Gillingham D, Fei N. Catalytic X–H insertion reactions based on carbenoids. Chem Soc Rev. 2013;42:4918–4931. doi: 10.1039/c3cs35496b. [DOI] [PubMed] [Google Scholar]
- 168.Tishinov K, Fei N, Gillingham D. Cu(I)-catalysed N–H insertion in water: A new tool for chemical biology. Chem Sci. 2013;4:4401–4406. [Google Scholar]
- 169.Ando H, Futara T, Tsien RY, Okamoto H. Photo-mediated gene activation using caged RNA/DNA in zebrafish embryos. Nat Genet. 2001;28:317–325. doi: 10.1038/ng583. [DOI] [PubMed] [Google Scholar]
- 170.Shah S, Jain PK, Kala A, Karunakaran D, Friedman SH. Light-activated RNA interference using double-stranded siRNA precursors modified using a remarkable regiospecificity of diazo-based photolabile groups. Nucleic Acids Res. 2009;37:4508–4517. doi: 10.1093/nar/gkp415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Laayoun A, Kotera M, Sothier I, Trévisiol E, Bernal-Méndez E, Bourget C, Menou L, Lhomme J, Troesch A. Aryldiazomethanes for universal labeling of nucleic acids and analysis on DNA chips. Bioconjugate Chem. 2003;14:1298–1306. doi: 10.1021/bc0341371. [DOI] [PubMed] [Google Scholar]
- 172.Fei N, Sauter B, Gillingham D. The pKa of Brønsted acids controls their reactivity with diazo compounds. Chem Commun. 2016;52:7501–7504. doi: 10.1039/c6cc03561b. [DOI] [PubMed] [Google Scholar]
- 173.Caution! Unlike stabilized diazo compounds (e.g., diazo compound I in Figure 4A), unstabilized diazo compounds (e.g., diazomethane) are dangerous and should never be used in the context of chemical biology. See: refs. 5–9.
- 174.Bao Z, Wang S, Shi W, Dong S, Ma H. Selective modification of Trp19 in β-lactoglobulin by a new diazo fluorescence probe. J Proteome Res. 2007;6:3835–3841. doi: 10.1021/pr070284n. [DOI] [PubMed] [Google Scholar]