

Abstract
Gliomas are aggressive brain tumors that are resistant to conventional chemotherapy and radiotherapy. Much of this resistance is attributed to endogenous nitric oxide (NO). Recent studies revealed that 5-aminolevulinic acid (ALA)-based photodynamic therapy (PDT) has advantages over conventional treatments for glioblastoma. In the present study, we used an in vitro model to assess whether NO from glioblastoma cells can interfere with ALA-PDT. Human U87 and U251 cells expressed significant basal levels of neuronal NO synthase (nNOS) and its inducible counterpart (iNOS). After an ALA/light challenge, iNOS level increased 3–4 fold over 24 h, whereas nNOS remained unchanged. Elevated iNOS resulted in a large increase in intracellular NO. Extent of ALA/light-induced apoptosis increased substantially when an iNOS inhibitor or NO scavenger was present, implying that iNOS/NO was acting cytoprotectively. Moreover, cells surviving a photochallenge exhibited a striking increase in proliferation, migration, and invasion rates, iNOS/NO again playing a dominant role. Also observed was a large iNOS/NO-dependent increase in matrix metalloproteinase-9 activity, decrease in tissue inhibitor of metalloproteinase-1 expression, and increase in survivin and S100A4 expression, each effect being consistent with accelerated migration/invasion as a prelude to metastasis. Our findings suggest introduction of iNOS inhibitors as pharmacologic adjuvants for glioblastoma PDT.
Graphical abstract
Glioblastoma cells exploit endogenous nitric oxide (NO) for survival and expansion. In this study, we showed that glioblastoma cells sensitized with 5-aminolevulinic acid (ALA)-induced protoporphyrin-IX upregulated inducible nitric oxide synthase (iNOS) and NO after irradiation. NO signaled for greater resistance to apoptosis as well as greater growth, migration, and invasiveness of surviving cells. By limiting these negative effects, as demonstrated in this study, iNOS inhibitors should improve PDT efficacy for these dreadful brain malignancies.
INTRODUCTION
Introduced about 40 years ago as a novel approach for selectively eradicating solid tumors, photodynamic therapy (PDT) consists of three components: (i) systemic or topical application of a photosensitizing agent (PS) or metabolic precursor, (ii) photoexcitation of PS by long wavelength (near infra-red) laser light, and (iii) molecular oxygen (1–3). Tumor targeting of the PS and its photoexcitation by laser light delivered via fiber optics make this approach highly site-specific. Moreover, the PS is typically innocuous until photoactivated, whereupon it gives rise to signaling or cytotoxic reactive oxygen species (ROS), singlet oxygen (1O2) being one of the most prominent (2,3). The first US FDA-approved PDT sensitizer was Photofrin®, a porphyrin derivative administered systemically which targets many types of solid tumors (2,3). 5-Aminolevulinic acid (ALA)-based PDT is a variant approach in which ALA or an ester derivative thereof is metabolized to protoporphyrin IX (PpIX), the active PS, which accumulates initially in mitochondria (4,5). ALA-PDT has been used for a variety of malignancies, including glioblastoma multiforme (GBM) (6).
GBM is the most common, aggressive, and lethal type of human brain malignancy, resulting in more than ten thousand patient deaths per year in the USA (7–9). Despite recent advances in surgical techniques and adjuvant application of radiation or chemotherapy, the GBM prognosis remains dismal. PDT has exhibited clear advantages over conventional GBM treatments, one clinical trial showing that average patient survival time after diagnosis (~25 weeks) could be more than doubled by administering Photofrin®-PDT (10,11). Similar promising results have been obtained using ALA-based PDT (6,12,13). However, our understanding of how GBM tumors might resist or adapt to PDT oxidative stress by activating pro-survival and/or expansion (proliferative/invasive/metastatic) pathways is still rudimentary. There is now compelling evidence that many types of malignant cells, including glioblastoma cells, can exploit low flux nitric oxide (NO) produced by inducible nitric oxide synthase (iNOS) as a pro-survival and tumor-expanding signaling molecule (14–17). For example, Eyler et al. (18) reported that glioma stem cells from human brain tumors expressed significantly more iNOS and NO than normal counterparts and proliferated more rapidly than the latter. Importantly, the tumor initiation/progression potency of these stem cells in mice was substantially reduced by selective inhibition or knockdown of iNOS (18). In a related study ca. five years ago, Kostourou et al. (19) showed that rat glioma C6 cells exploited iNOS/NO for optimal proliferation in vitro and for angiogenesis and tumor progression in vivo. More recently, Kim et al. (20) reported that fractionated ionizing radiation increased iNOS/NO expression in two glioma cell lines and that this played a key role in post-irradiation expansion on stem-like cells and also resistance of the latter to chemotherapeutic agents.
In the present study, we asked whether iNOS/NO would also be upregulated in glioblastoma cells subjected to a non-ionizing ALA-PDT challenge. Not only was this observed, but the induced iNOS/NO proved to be directly involved in (i) the elevated resistance to photokilling displayed by these cells and (ii) the accelerated proliferation, migration, and invasion exhibited by cells that had escaped photokilling. This is the first report on how endogenous iNOS/NO may limit the effectiveness of PDT for glioblastomas and other brain malignancies. Our findings provide a strong rationale for considering use of iNOS inhibitors as adjuvants for improving clinical outcomes of PDT for brain tumors.
MATERIALS AND METHODS
Chemicals, Reagents, and Antibodies
The non-specific NOS inhibitor L-NG-nitroarginine methyl ester (L-NAME), iNOS-specific inhibitor N-[3-(aminomethyl)benzyl]acetamidine (1400W), NO scavenger 2-(4-carboxyphenyl)-4,4,5,5-tetramethylimidazoline-1-oxyl-3-oxide (cPTIO), and NO fluorophore probe 4,5-diaminofluorescein diacetate (DAF-2DA) were obtained from Cayman Chemicals (Ann Arbor, MI). Cayman Chemicals also supplied monoclonal antibodies against human iNOS and nNOS, and an Annexin V-FITC plus propidium iodide (PI) kit for detecting apoptosis vs. necrosis. Monoclonal antibodies against human MMP-9, TIMP-1, and TIMP-2 were obtained from EMD Millipore (Bellerica, MA), while those against S100A4, survivin, and β-actin were from Cell Signaling Technology (Danvers, MA). All other reagents, including ALA, 3-(4,5-dimethylthiazolyl-2-yl)-2,5-diphenyl tetrazolium bromide (MTT), rhodamine-123 (Rh123), fetal bovine serum (FBS), growth media, and other cell culture materials were from Sigma Aldrich (St. Louis, MO).
Cell Culture
Human glioblastoma U87-MG and U251-MG cells were obtained from the American Type Culture Collection (ATCC) in Manassas, VA. From here on, these cells are referred to as U87 and U251, respectively. Cells were cultured in Minimal Essential Medium (MEM) with Earle’s salts supplemented with 10% FBS, 1% pyruvate, penicillin (100 units/ml), and streptomycin (0.1 mg/ml). Culture dishes were incubated at 37 °C in a humidified atmosphere under 5% CO2. Proliferating cells received fresh medium every third day and were passaged fewer than 6 times for all experiments. Additional details were similar to those described previously for other cancer cell lines (21,22).
Cell sensitization in the absence vs. presence of NOS inhibitors or NO scavenger
For experiments in which phototoxicity was to be evaluated, Glioblastoma cells at ~60% confluency in 35-mm dishes in serum- and phenol red-free MEM were metabolically sensitized with PpIX by incubating with 1.0 mM ALA in serum-free MEM lacking phenol red for 30 min in the dark. When an NOS inhibitor (L-NAME or 1400W) or NO scavenger (cPTIO) was used, it was introduced 30 min before ALA and kept at the same concentration in the medium during irradiation and post-irradiation incubation. After ALA treatment, cells were switched to fresh phenol red-free medium either lacking or containing a NOS inhibitor or NO trap and then irradiated.
Cell irradiation
Culture dishes were irradiated at room temperature on a translucent plastic platform over a bank of four GE 40W cool-white fluorescent lamps, which emitted broad-band visible light. The light fluence rate (irradiance) at the culture dish bottom surfaces was ~1.1 mW/cm2, as measured with YSI radiometer (Yellow Springs, OH). A typical irradiation period was 15 min, corresponding to a delivered light fluence of ~1 J/cm2. It is important to note that only a portion of this fluence would have resulted in PpIX excitation because the source irradiance from 500–700 nm, for example, was ~77% of the total output (supplier information), whereas PpIX absorption over this range was only ~12%. Immediately after irradiation, the cells were overlaid with fresh medium containing 1% FBS with or without a NOS inhibitor or NO scavenger, and placed in the 37 °C incubator. At specified post-irradiation times, parameters such as viable cell fraction, apoptotic fraction, and proliferation, migration, and invasion rates of surviving cells were determined. ALA-only and light-only controls were also prepared and analyzed alongside.
Immunoblot analyses
The expression status of iNOS and nNOS in U87 cells before and after an ALA/light challenge was assessed by Western blotting. Irradiated cells, along with controls, were recovered by gentle scraping, centrifuged, and washed with cold PBS. The cells were suspended in lysis buffer containing a mixture of protease inhibitors and homogenized (22). After centrifugation, the supernatant fraction was analyzed for total protein by BCA assay, and then subjected to SDS-PAGE, using appropriate proportions of acrylamide and bis-acrylamide. Separated proteins were transferred to a polyvinylidene difluoride membrane and, after blocking, the membrane was exposed overnight at 4 °C to a given primary antibody at dilution recommended by the supplier. After washing, the blot was treated with a peroxidase-conjugated secondary antibody and then analyzed using SuperSignal West Pico chemiluminescence detection (Thermo Scientific, Rockford, IL).
Detection of NOS-generated NO in photostressed cells
The fluorophore DAF-2DA was used to probe for pre-existing and photostress-induced NO in glioblastoma cells. DAF-2DA enters cells and is hydrolyzed to DAF-2, which is trapped inside (23). Weakly fluorescent DAF-2 does not react with NO directly, but rather with its autoxidation product, N2O3, which nitrosates DAF-2 to a strongly fluorescent triazole (24,25). A stock solution of DAF-2DA in DMSO was prepared immediately before an analysis and kept in the dark. At designated times after irradiation in the absence vs. presence of a NOS inhibitor, cells in serum-free medium were incubated with 20 μM DAF-2DA for 20 min in the dark. After washing, the cells were analyzed for DAF-2 traizole fluorescence, using a Nikon Eclipse TS100 microscope set at 488 nm excitation and 610 nm emission.
Determination of cell viability and extent of apoptosis
Glioblastoma cells were treated with ALA as described and irradiated. After 20–24 h of post-irradiation dark incubation, the viable cell fraction was determined by MTT assay (26,27) and expressed relative to that of an ALA-only dark control. When included in a reaction system, a NOS inhibitor or NO trap was present before, during, and after irradiation. Early stage apoptosis (typically assessed 4 h after irradiation) was determined by Annexin V-FITC fluorescence assay, which detects externalization of plasma membrane phosphatidylserine (28). Propidium iodide (PI) was included to detect any necrosis. A 96-well plate reader format (Biotek SynergyMX) was used, with 485 nm excitation and 535 nm emission. Camptothecin, which induces complete apoptosis, was used a reference standard. Other details were as specified by the reagent supplier.
Determination of surviving cell proliferation
The proliferation rate of cells that survived an ALA/light challenge was evaluated by MTT assay. Twenty-four hours after an ALA/light challenge in the absence vs. presence of a NOS inhibitor or NO scavenger, followed by a wash to remove dead or dying cells, attached surviving cells were overlaid with 10% FBS-containing medium, and placed in the incubator. At increasing time intervals up to 48 h, the medium was removed and replaced with 1.0 ml of fresh medium containing MTT (0.5 mg/ml). After a 4 h period in the incubator, the cells were solubilized in 1.0 ml of acidified isopropanol and immediately thereafter, formazan A563 was measured, which reflected the number of live proliferating cells.
Evaluation of surviving cell migration
A gap closure or “wound-healing” assay was used to analyze the migration rate of glioblastoma cells that survived ALA/light-induced stress and how this rate was affected by NOS inhibition. Cells were grown to ~90% confluency on 35-mm dishes. After ALA treatment, a linear scratch was made across the monolayer, using a 200 μl pipet tip. Cells were then irradiated, washed, and returned to the incubator. The gap zone was photographed before and after irradiation, using a Nikon Eclipse TS100 microscope equipped with a CoolSnap ES Photometrics camera and MetaVue software (Molecular Devices, Sunnyvale, CA). Extent (%) of gap closure in the absence vs. presence of a NOS inhibitor was determined using the following equation: 100 x [time-0 gap − time-t gap]/time-0 gap. A non-irradiated control was analyzed similarly. Data were obtained from at least four replicate experiments for each experimental condition.
Evaluation of surviving cell invasiveness
A 96-place trans-well device (Model MBA96) from NeuroProbe (Gaithersburg, MD) was used for assessing the effects of photodynamic stress on glioblastoma cell invasiveness. In preparation for these measurements, cells were grown to ~60% confluency in 10-cm dishes. The invasion chamber was prepared for use by adding 225 μl of 10% FBS-containing medium to each lower well, the FBS serving as a chemoattractant. A matrigel-infused polycarbonate filter with 8 μm pores was then inserted over each lower well, after which the entire unit was pre-warmed to 37 °C. Meanwhile, the cells were sensitized with ALA-induced PpIX, irradiated, and then gently scraped into serum-free medium. The recovered cells in serum-free medium were carefully layered into the upper wells of the invasion chamber (225 μl per well). Immediately thereafter, the entire chamber unit was placed in the 37 °C incubator. After a given incubation period, typically 24 h, the medium in upper wells was gently aspirated off and cells remaining on top of each filter were wiped off. Cells that had traversed to the bottom of each filter were detached by centrifugation into the FBS-containing medium (400g; 10 min) and allowed to adhere during ~6 h on incubation. The recovered invasive cells were first photographed and then quantified by MTT assay.
Determination of matrix metalloproteinase activity
Gelatin zymography was used to determine the activity of matrix metalloproteinase-9 (MMP-9) in glioblastoma cells that had been subjected to ALA/light treatment. This in-gel assay is based on electrophoretic separation of the ~92 kDa pro-enzyme from the ~82 kDa active enzyme and assessing the latter’s ability to degrade gelatin in its vicinity, thereby creating a void in the zymograph (29). U87 or U251 cells grown to ~90% confluency in 10-cm dishes were sensitized as described above and irradiated. For determining iNOS/NO involvement in enzyme activation, the inhibitor 1400W was included in the cell medium and maintained at a given concentration throughout. Irradiated cells, along with dark controls, were kept in the incubator for 24 h, after which the medium from each type was collected, concentrated using a 0.5 ml centrifugal filter (Amicon, Billerica, MA), and analyzed for protein concentration. A sample of the concentrate (~15 μg protein) was mixed with 2x sample buffer (Life Technologies, Madison, WI) and subjected to SDS-PAGE, using 10% acrylamide co-polymerized with gelatin (0.1 mg/ml), 175 V, and a running time of 90 min. The gel was then incubated with renaturing buffer (30 min), followed by developing buffer (24 h at 37 °C), after which it was stained and photographed. ImageLab 4.0 software was used for band quantification. Additional details were as described previously (30).
Statistical analysis
The two-tailed Student’s t-test was used for judging the significance of apparent differences between data plotted as means ± SEM of values from at least three replicate experiments. P values <0.05 are considered to be statistically significant.
RESULTS
Subcellular location of PpIX in ALA-treated U87 cells
Glioblastoma U87 cells were metabolically sensitized with PpIX by incubating with ALA in serum-free medium. Under the conditions used, the PpIX was localized primarily in mitochondria because porphyrin fluorescence observed by confocal microscopy overlapped that of the mitochondrial marker Rh123 123 (Fig. 1). Similar results were obtained with ALA-treated U251 cells (not shown). Consequently, mitochondria would have been the principal sites of photodynamic action upon irradiation of sensitized U87 and U251 cells.
Figure 1.

Localization of PpIX in ALA-treated U87 cells. Cells grown to ~40 % confluence on cover slips were dark-incubated with 1 mM ALA in serum-free medium for 30 min. After a switch to ALA-free medium, the cells were treated with 1 μM Rhodamine-123 (Rh123) for 20 min, washed and immediately examined by confocal fluorescence microscopy. Excitation and emission wavelengths for PpIX were 488 nm and 620–650 nm, respectively; those for Rh123 were 505 nm and 530–540 nm, respectively. Scale bar: 25 μm.
Cell photokilling in the absence vs. presence of NOS inhibitors and a NO scavenger
U87 cells sensitized with ALA-induced PpIX underwent a progressive decrease in MTT-assessed viability after exposure to increasing fluences of broad band visible light (Fig. 2a). As can be seen, a fluence of 4 J/cm2 resulted in ~45% loss of viability measured 20 h after irradiation. ALA alone or light alone had no significant effect. The extent of cell photokilling was substantially increased when the iNOS inhibitor 1400W or NO scavenger cPTIO was present throughout, suggesting that iNOS/NO was acting cytoprotectively (Fig. 2a). Similar results were obtained with U251 cells (Fig. 2b). In this case, 1400W and L-NAME, a non-specific NOS inhibitor at 40-times the 1400W concentration, exerted approximately the same stimulatory effects. Thus, both cell types appeared to exploit iNOS and NO for defense against a moderately cytotoxic ALA/light challenge.
Figure 2.

Viability loss of glioblastoma cells after a photodynamic challenge. (a) ALA-treated U87 cells were exposed to the indicated light fluences in the absence (○) or presence of 25 μM 1400W (□) or 25 μM cPTIO (△). After irradiation, cells were switched to serum-containing medium, and after 20 h of dark incubation, examined for viability by MTT assay. (b) U251 cells were sensitized identically to U87 cells, irradiated in the absence (○) vs. presence of 25 μM 1400W (□) or 1 mM L-NAME (△), and then examined for viability after 20 h in the dark. Also represented in (a) and (b) are ALA-only or light-only controls (x). Plotted values in (a) and (b) are means ± SEM (n=3); W, cP, and N denote 1400W, cPTIO, and L-NAME, respectively.
We used Annexin V-FITC fluorescence in a 96-well microplate reader assay to determine whether cell photokilling occurred via intrinsic apoptosis, as would be expected, since PpIX was localized to mitochondria. ALA-treated U87 cells were exposed to a 1 J/cm2 light fluence and after 4 h of dark incubation, examined for extent of apoptosis, using camptothecin (CPT) as a standard for assessing complete apoptosis. As shown in Fig. 3, cells given light alone were ~10% apoptotic, which was no more than that observed in untreated cells. ALA/light treatment increased apoptosis to ~29%. However, when 1400W or L-NAME was present, ~42% of the cells were apoptotic, a significant 45% increase over ALA/light alone (Fig. 3). These findings are again consistent with iNOS playing an important role in cell resistance to photokilling. That L-NAME at a much higher concentration than 1400W produced no greater stimulation of apoptotic death suggests that NO from other NOS isoforms, e.g. nNOS, was not as important as that from iNOS. This point was considered further in subsequent experiments.
Figure 3.

Apoptotic photokilling of U87 cells and effects of NOS inhibitors. U87 cells at 40–45% confluence were treated with 1 mM ALA without or with a NOS inhibitor (25 μM 1400W or 1 mM L-NAME), and then irradiated (1 J/cm2). After 4 h of follow-up dark incubation, the cells were treated with Annexin V-FITC and Propidium Iodide (PI) according to supplier recommendations. Camptothecin (CPT, 25 μM) served as a maximum apoptosis standard. Extent of staining was quantified using a plate reater with excitation/emission wavelengths of 485/535 nm for Annexin V-FITC and 560/595 nm for PI. Less than 5% PI staining was detected for any of the treatment conditions.
iNOS and nNOS protein levels in ALA/light-challenged cells
We asked whether the NO-dependent resistance described in Fig. 2 was due to background iNOS or whether photostress-induced enzyme might have been involved, as observed previously for other cancer lines (30–33). Using Western blot analysis, we found that U87 cells expressed significant basal iNOS, but that its level increased progressively during post-irradiation incubation, reaching ~3-times and ~2-times the basal level at 5 h and 20 h, respectively (Fig. 4Aa). The level in an ALA-only dark control (DC) was essentially the same as that in untreated cells. As expected for their phenotype, U87 cells expressed significant basal levels of nNOS, similar to iNOS, but in contrast to the latter, nNOS was not upregulated to any significant extent after ALA/light treatment (Fig. 4Ab). Like U87 counterparts, sensitized U251 cells exhibited a continuous post-irradiation increase in iNOS, which reached >4-times the DC level at 20 h (Fig. 4B). When photostressed, therefore, both glioblastoma cell types added significantly to their pre-existing content of iNOS, which evidently enhanced their resistance to photokilling.
Figure 4.

Western blots of iNOS and nNOS in glioblastoma cells after a photodynamic challenge. (A) ALA-treated U87 cells were irradiated (1 J/cm2), switched from serum-free to 1% serum-containing medium and at the indicated post-irradiation times, subjected to Western analysis for iNOS (a) or nNOS (b). A 20 h dark control (DC) was also analyzed. (B) ALA-treated U251 cells were irradiated similarly to U87 and Western-analyzed for iNOS at the indicated post-irradiation times. Intensity relative to β-actin and normalized to DC is indicated below each NOS band in (A) and (B).
Elevated NO levels in photostressed U87 cells
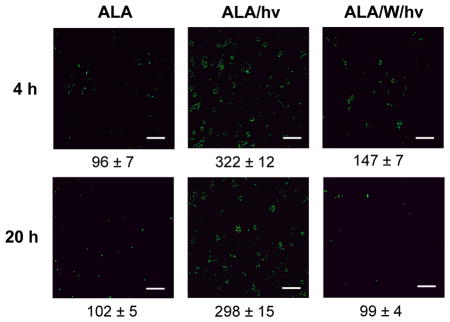
To establish whether the observed elevation of iNOS after a photodynamic challenge (Fig. 4) resulted in an elevation of NO level, we used the fluorophore DAF-2, which can detect intracellular NO after its conversion to dinitrogen trioxide, N2O3 (25). ALA-treated U87 cells were analyzed 4 h and 20 h after exposure to a 1 J/cm2 light fluence. As shown in Fig. 5, a dark control (ALA-only) exhibited relatively low mean fluorescence after 4 h of incubation, and this did not change significantly after 20 h. Essentially the same fluorescence signal was observed for cells not treated with ALA (results not shown). When present in the control system, the non-specific NOS inhibitor L-NAME reduced fluorescence intensity by ~80% at 4 h and 20 h, suggesting that pre-existing iNOS and nNOS (Fig. 4) were supplying NO in these cells. After ALA-treated cells were irradiated, a striking 2.4-fold and 1.9-fold increase in DAF-2-derived fluorescence was observed at 4 h and 20 h, respectively (Fig. 5). This increase was blunted substantially by L-NAME (64% at 4 h; 54% at 20 H) and by 1400W (56% at 4 h; 67% at 20 h) (Fig. 5). Low affinity/specificity L-NAME (used at 40-times the concentration of 1400W) would have inhibited both nNOS and iNOS. However, only iNOS was upregulated by photostress (Fig. 4A) and since iNOS generates NO at a much higher rate than nNOS (34), the surge in NO generation observed after ALA/light treatment (Fig. 5) was probably due to upregulated iNOS.
Figure 5.

Images of NO accumulation in U87 cells after a photodynamic challenge. ALA-treated U87 cells were irradiated in the absence vs. presence of a NOS inhibitor as specified in Fig. 2. An ALA-only dark control without or with L-NAME (1 mM) was included. At the indicated post-irradiation times, the cells were treated with DAF-2DA (20 μM) for 20 min in the dark, and then examined by fluorescence microscopy using 488 nm excitation and 650 nm emission. Image intensities were quantified using ImageJ software. Means ± SEM of values from three separate experiments are shown.
Accelerated proliferation of cells withstanding an ALA/light challenge: role of iNOS
We asked whether glioma cells that exploited iNOS/NO to resist photokilling (Fig. 2) might also take advantage of it to increase growth and migratory aggressiveness. The first property to be examined was the proliferation rate of remaining live cells 24 h after a standard ALA/light challenge. ALA-treated U87 cells were either not irradiated (control) or irradiated (1 J/cm2) in the absence vs. presence of 1400W, after which their viability/growth status was monitored over a 48 h dark incubation period. As shown in Fig. 6a, U87 cell viability decreased steadily throughout the first 24 h post-irradiation interval and this decrease was enhanced by 1400W, in agreement with the increasing fluence phototoxicity results described in Fig. 2a. Ongoing growth of a control over the same 24 h interval was slowed somewhat by 1400W, suggesting a slight stimulatory effect of constitutive NO. During a subsequent 24 h post-irradiation period, cells that had withstood the photostress exhibited a striking growth spurt over control growth, and this was strongly attenuated by 1400W (Fig. 6a). Calculated growth rates relative to the control rate for the 24–48 h post-irradiation period are shown in Fig. 6b. As can be seen, ALA/light treatment stimulated growth of surviving cells ~2-fold over this interval and in a manner almost entirely dependent on iNOS. The NO trap cPTIO also blunted the growth spurt significantly (results not shown), thereby establishing that NO played a key signaling role. It is important to point out that when surviving U87 cells at 24 h post-irradiation were normalized to the same starting titer as control cells, the growth spurt in the former persisted (results not shown).
Figure 6.

Loss of U87 cell viability and subsequent proliferation of surviving cells after a photodynamic challenge: effects of iNOS inhibition. (a) U87 cells were plated and allowed to grow to ~40% confluency. Two dark controls were used, one with ALA alone (x), and one with ALA plus 25 μM 1400W (◇). Experimental conditions included cells treated with ALA and light (○) and ALA, light and 1400W (△). At the indicated times, cells were subjected to MTT assay to determine viability relative to the time-zero point. (b) Quantified growth rates for each condition over the 24–48 h time period. Means ± SEM of values from at least 3 replicate experiments are plotted.
Enhanced migration of cells withstanding a photochallange: iNOS/NO involvement
Knowing that glioblastoma cells are inherently more prone to migration than normal neuronal cells (9), we asked whether this property would be enhanced in cells surviving a photodynamic insult. To investigate this, we used a “wound-healing” assay, better known now as a “gap-closure” assay (35). In this approach, cell departure from the main population and movement into a scratch-voided zone is assessed over a given time period. For our experiments, this was a period of dark incubation beginning 24 h after cells were irradiated. As shown by the photographs in Fig. 7, ALA/light-stressed U87 cells migrated ~45% faster than ALA-only controls in 24 h, and 1400W reduced this by nearly 30%, thereby establishing at least partial iNOS/NO dependency. By comparison, 1400W reduced control cell migration by only ~15%, suggesting some iNOS/NO dependency, but much less than that in photostressed cells, which had upregulated iNOS (Figs. 4A and 5). Clearly, therefore, accelerated migration was as another manifestation of increased aggressiveness of residual U87 cells in the aftermath of a photodynamic challenge.
Figure 7.

Migration of U87 cells after ALA/light treatment in the absence vs. presence of an iNOS inhibitor. Cells were grown to ~90% confluency, sensitized, and irradiated in the absence or presence of 1400W (25 μM). After irradiation, a 200-μl pipet tip was used to make a scratch across each cell layer. Immediately thereafter and 24 h later, the cells were photographed. Extent of gap closure at each time point was determined using the expression given in the Methods section. Numbers are means ± SD of values from at least 4 experiments.
Increased invasiveness of cells withstanding a photochallenge: iNOS/NO involvement
It was important to learn whether increased mobilization of photostress-surviving glioblastoma cells would also be manifested in greater invasiveness. To address this, we used a Boyden Chamber-like device with multiple trans-well units to assess the ability of U87 and U251 cells to traverse an ECM-like basement membrane, moving from serum-free medium (upper chamber) to serum-containing medium (lower chamber). Cell movement from the upper to lower membrane surface over a 24 h period was used as a measure of invasiveness. As shown in Fig. 8a, U87 cells exposed to ALA and kept in the dark exhibited a significant level of invasiveness, which was no different from that of untreated cells (not shown). In the presence of 1400W, the ALA-only control showed a small (~15%) decrease in invasion, which was barely significant. After irradiation (1 J/cm2), ALA-treated cells exhibited a striking 50% increase in invasion relative to the dark control, which was highly significant (Fig. 8a). A strong iNOS/NO dependency of this response was demonstrated by 1400W’s large inhibitory effect (~50%), reducing the response to about the control level with 1400W (Fig. 8a). U251 cells that escaped ALA/light-induced killing exhibited a similar iNOS/NO-dependent increase in invasiveness (Fig. 8b) and again, this greatly overshadowed the dependency shown by control cells.
Figure 8.

Invasiveness of photostressed U87 cells (a) and U251 cells (b) after a photodynamic challenge in the absence vs. presence of 1400W. Cells were plated in 10-cm dishes and allowed to grow to ~60% confluency, and then sensitized with PpIX by ALA treatment. The cells were then either irradiated (1 J/cm2) or not (dark controls) in the absence or presence of 1400W (25 μM). Immediately thereafter, irradiated and control cells were recovered by gentle scraping into serum-free medium. The cells were then placed in a pre-warmed invasion chamber according to supplier recommendations. After a 24 h incubation period, cells were spun down from the chamber into a 96 well plate, and then photographed and quantified by MTT assay. Plotted data are means ± SEM of values from at least three replicate experiments.
NO-dependent MMP-9 activation in photostressed glioblastoma cells
By catalyzing the degradation of extracellular matrix (ECM) components such as collagen, laminin, and fibronectin, zinc-containing matrix metalloproteinases (MMPs) play a central role in cancer cell mobilization leading to metastasis. Since type-9 matrix metalloproteinase (MMP-9) is known to play a significant role in glioma cell migration/invasion (36,37), we assessed the effects of ALA/light treatment on its expression/activity and whether NO played a role in any changes thereof. As shown by the Western blot in Fig. 9a, sensitized, but non-irradiated U87 cells expressed significant pro-MMP-9, the level of which was identical to that in untreated cells (not shown). The protein is referred to as the uncleaved (inactive) zymogen because it exists predominately as such within cells (38,39). After irradiation of sensitized cells, no change in pro-MMP-9 level was observed during dark incubation for at least 4 h, but a small increase occurred thereafter, amounting to ~20% at 24 h (Fig. 9a). Thus, there was only a modest long-term induction of total protein after cells were photostressed. However, when gel zymography was used to examine the activity status of extracellular (exported) enzyme, much more striking post-irradiation effects were observed. As shown in Fig. 9b, two zones depleted of gelatin appeared in each lane, the upper one representing pro-MMP-9 (~92 kDA) and the lower one, active MMP-9 (~82 kDa). The lower band was significantly stronger by ~80% in the ALA/light-treated sample than in the ALA-only control (Fig. 9b plot), indicating that much more enzyme activation had occurred in the former. This activation was substantially blunted by L-NAME (>90%) and by 1400W (~70%), implying that the NO played a strong role in MMP-9 hyper-activation. Since there was very little photostress-induction of pro-MMP-9, the activation must have represented pre-existing protein for the most part. There is evidence that low level NO can activate pro-MMP-9 by disrupting the restraining Zn(II)-cysteine thiolate bond in the zymogen (40), and this might explain our findings. The seemingly greater activity in the upper (pro-enzyme) zone of the ALA/hν lane (Fig. 9b) appears to be an artifact that arose after zymogram development, e.g. during treatment with renaturing buffer (29).
Figure 9.

Effects of an ALA/light challenge on MMP-9 expression, MMP-9 activity, and TIMP-1 expression in U87 cells: role of iNOS/NO. (a) MMP-9 expression. Whole cell lysates were collected at the given time points and subjected to Western analysis to assess level of intracellular MMP-9 protein. (b) MMP-9 activity. Conditioned medium from irradiated cells and dark controls was collected and concentrated. After protein determination, samples were subjected to SDS-PAGE on a gel co-polymerized with gelatin. The gel was then incubated in a renaturing buffer (30 min), followed by developing buffer (24 h), after which it was stained and photographed. Bands were quantified using ImageLab software. Plotted values are means ± SEM of values from 3 separate experiments. (c) TIM-1 expression. Western blots of TIMP-1 levels in cells irradiated without or with 1400W present. Numbers below lanes indicate band intensities relative to β-actin and normalized to the dark control (DC).
Altered expression of survival/progression regulatory proteins in photostressed cells
Glycoproteins known as tissue inhibitors of metalloproteinases (TIMPs) regulate the activity of many different MMPs, and TIMP-1 is known to be highly specific for MMP-9 (40,41). We asked whether photostress would affect TIMP-1 expression in U87 cells and, if so, whether NO was implicated. As shown by the immunoblots in Fig. 9c, TIMP-1 was progressively down-regulated after an ALA/light challenge, reaching half its control level after 24 h. 1400W strongly inhibited this decline, particularly over the first 6 h, thus implicating iNOS/NO in the TIMP-1 suppression. This result correlates with the NO-mediated increase in MMP-9 activity after irradiation (Fig. 9b).
Survivin is an inhibitor of apoptosis protein which also plays a role in cancer cell proliferation, migration, and invasion (42,43). There is evidence for survivin induction by PDT-like stress (32,44) and also for NO involvement in its overexpression (32,45). Upon examining survivin status in U87 cells, we found that the protein was upregulated >4-fold 6 h after ALA/light, and 1400W inhibited this by nearly 70% (Fig. 10a).
Figure 10.

Influence of iNOS/NO on expression of selected pro-growth/migration/invasion effector proteins in photostressed U87 cells. Whole cell lysates were collected at the indicated time points after ALA/light treatment. After protein determination, samples were subjected to Western blot analysis. Separate experiments were carried out in the presence of 1400W to assess iNOS involvement in protein expression. Blots were probed for (a) survivin and (b) S100A4; β-actin was also probed for as a loading standard. Images are representative of at least 3 separate experiments with similar results.
S100A4 (formerly known as metastasin-1) is a calcium-binding protein of ~11 kDa that has been studied extensively as a key regulator of, among other things, tumor cell migration, invasion and metastasis (46–48). We examined S100A4 expression status in U87 cells after ALA/light treatment in the absence vs. presence of 1400W. As shown by the immunoblot in Fig. 10b, there was a remarkably strong upregulation of S100A4 over a 24 h post-irradiation period; the dark control signal was too weak to quantify accurately, so the relative increase at 24 h, though very large, is indeterminate. Equally striking is the almost total inhibition of this strong signal by 1400W, which suggests that iNOS-derived NO is a key mediator of S100A4 expression. This is the first known evidence for NO control of S100A4 expression, including that in a PDT setting.
Although much remains to be learned about how NO modulates expression of these effector proteins, this modulation helps to explain the enhanced aggressiveness described in Figs. 6–8.
DISCUSSION
Malignant gliomas are the most prevalent primary brain tumors and among these, glioblastoma (GBM), a Grade-4 astrocytoma, is the most aggressive and resistant to a variety of therapies (7–9,49). Average patient survival after even the most advanced surgical treatments or surgery combined with radiation or chemotherapy still remains only 12–18 months (49,50). Although a Phase-II trial involving Bevacizumab (Avastin), an anti-VEGF antibody that inhibits neovascularization has shown some early promise, long term benefits have not been forthcoming (51). Some attempts at targeted toxin gene therapy have also been made, but again with disappointing long term results (52). A better understanding of how glioblastoma develops and progresses, the signaling mechanisms involved, and how they might be targeted is clearly needed. There is growing evidence that besides nNOS, glioblastomas express iNOS, and that iNOS-derived NO at a sub-micromolar level plays a major role in tumor survival and progression (18,19,53,54). Similar pro-survival/expansion effects of iNOS/NO have also been demonstrated for a variety of other malignant tumors, including breast, prostate, and colon (14–17). In all of these cases, including GBM, pharmacologic intervention with highly specific iNOS inhibitors has been advocated, either by itself or as an adjunct to surgery, radiotherapy, or chemotherapy. However, iNOS inhibitors might interfere with the anti-tumor effects of vascular macrophages and other immune cells. These potential off-target complications should be seriously considered as clinical use of these inhibitors is contemplated.
PDT has emerged as one of the most effective approaches for treating GBM and other difficult brain malignancies (10–13). Pioneering clinical trials in Australia revealed that hematoporphyrin derivative-based PDT could prolong the average lifetime of GBM patients after initial diagnosis from 4–6 months to more than a year (55). In more recent clinical trials involving Photofrin® as the sensitizer and higher light fluences, more promising outcomes were obtained, although much room for improvement was admitted (10,11,56). Among the various sensitization approaches used for anti-glioma PDT, ALA-induced PpIX has received considerable attention (12,56). It was reported that ALA-PDT for GBM in rat models caused highly selective damage to the tumor per se, with little effect on peripheral normal tissue, although the basis of this selectivity was not assessed (12). Nevertheless, ALA-PDT continues to be an attractive option when compared with other sensitization approaches.
Although PDT for glioblastoma has distinct advantages over chemotherapy or radiotherapy, e.g. fewer off-target effects and longer patient survival, the question of how tumor iNOS/NO might impact PDT efficacy has not been addressed up to now. In the present study, we showed for the first time that glioblastoma U87 and U251 cells can upregulate iNOS by a substantial amount (3–4-fold) several hours after being exposed to an ALA/light challenge. This upregulation added to a preexisting pool of iNOS in these cells and persisted for at least 20 h. After 24 h, iNOS gradually decayed to its background level (not shown) concurrent with cell division. As expected, these cells also expressed significant nNOS, but in contrast to iNOS, the nNOS level did not increase after ALA/light treatment. Using the fluorophore DAF-2, which detects pre-existing NO in cells, we found a marked NO elevation in U87 cells 20 h after ALA/light exposure and 1400W sharply reduced this, indicating that stress-induced iNOS was responsible. As has been reported for other anti-glioma interventions (54), we found that 1400W, L-NAME, or cPTIO strongly increased the extent of U87 and U251 viability loss or apoptosis after ALA/light treatment, suggesting that upregulated NO had increased cell resistance to photostress. Since nNOS was not stress-induced, the cytoprotective NO must have derived mainly from iNOS, which in any event, generates NO at a substantially higher rate than nNOS.
In addition to exhibiting stress-induced resistance to photokilling, U87 cells surviving a photodynamic insult were found to proliferate much more rapidly than non-stressed controls. This striking finding was somewhat unexpected because the background growth rate of these cells is typically quite high, and greater than that of most breast and prostate cancer lines (54). As in the case of hyperresistance, elevated iNOS/NO was found to play a prominent role in the accelerated U87 proliferation by virtue of the inhibitory effects of 1400W and cPTIO. Of added importance were our findings that U87 cells that survived a photochallenge migrated and invaded more rapidly than controls, and again, these responses were primarily dependent on iNOS activity and iNOS-generated NO. Similar findings were obtained with U251 cells, suggesting that these responses may be generally applicable to glioblastoma-derived cells. Consistent with increased invasiveness, we found that MMP-9, which facilitates cell translocation via degradation of ECM components, exhibited an iNOS/NO-dependent activity boost in photostressed U87 cells, whereas TIMP-1, which inhibits MMP-9 activity, was down-regulated. Moreover, we found that survivin and S100A4, both of which inhibit apoptosis and stimulate migration/invasion (43,43,47,48), were upregulated after ALA/light-imposed stress. Thus, we discovered that glioblastoma cells which resisted or withstood photodynamic inactivation, underwent a large NO-dependent increase in growth, migration, and invasion aggressiveness. Although iNOS-derived low flux NO has been widely reported to support malignant glioma cell survival, progression, and drug resistance, our findings in this study are the first to demonstrate such responses in the context of PDT.
It is of interest to note that Etminan et al. (57) showed that irradiation of ALA-treated U373 glioma spheroids not only induced apoptotic cell death, but almost completely inhibited the migratory ability of remaining cells, which were found to be ~50% viable. Despite this residual viability, these cells showed no signs of proliferation over several days, which could explain their inability to migrate. In contrast to this, we found that surviving U87 cells actually migrated more rapidly than controls. These contrasting results could relate to the fact that Etminan et al. (57) used a glioma cell type, experimental configuration (spheroids), and sensitization procedure that was different from ours. Of added interest is a study by Zhang et al. (58), which showed that low dose Photofrin-PDT on normal brains of nude mice caused no obvious cytotoxicity, but did induce greater VEGF expression and endothelial cell proliferation. Both of these responses could be conducive to tumor progression if occurring in a glioma-bearing animal, and suggest that the adverse pro-growth/migration adaptations we describe may be applicable in vivo.
The question of how NO might affect PDT efficacy was first addressed by Henderson et al. (59) and Korbelik et al. (60) in studies on syngeneic mouse tumors subjected to Photofrin-based PDT. These studies (involving breast, cutaneous, and RIF carcinomas) demonstrated that PDT efficacy could be greatly improved by administering NOS inhibitors (L-NAME, L-NNA), and that the extent of improvement correlated with background NO production, tumors with relatively high output responding best (60). An effect on the tumor micro-vasculature was deduced, viz. that vasoconstriction by PDT was being opposed by NO-mediated vasorelaxation. However, whether NO might also interfere with PDT by acting at the level of tumor cells per se was not rigorously addressed until our own more recent studies on human breast and prostate cancer lines (22,30–33). This work, extended to brain cancer in the present study, revealed for the first time that photostress-induced NO can signal for both hyper-resistance and greater growth and migratory aggressiveness. Recent studies by Rapozzi et al. (61,62), using a different PDT model system (melanoma cells sensitized with pheophorbide-a), have supported our findings regarding the cytoprotective effects of iNOS/NO. Interestingly, low level NO generated by low dose PDT was cytoprotective through anti-apoptotic activation of the NF-κB/Snail/RKIP pathway. On the other hand, high level NO from high dose PDT proved to be cytotoxic due to pro-apoptotic activation of this pathway (62). We have not yet determined whether increasing the induction of iNOS/NO via greater photodynamic pressure might produce a similar NO-enhanced pro-apoptotic response in glioblastoma cells. It is worth mentioning that some investigators have proposed using high level NO from chemical donors (either alone or in combination with conventional treatments) to eradicate malignant tumors. Non-specific NO donors such as glyceryl trinitrite, sodium nitroprusside, S-nitrosoglutathione, and diazeniumdiolates (NONOates) have been tested along these lines with varying degrees of success, a key question being off-target effects (63,64). The issue of target specificity has been addressed with a donor such as JS-K, a pro-drug that releases NO upon activation by glutathione-S-transferases, which are typically expressed at relatively high levels in malignant cells (65). Although non-targeted and tumor-targeted NO donors have yet to be tested on gliomas, concerns are apparent from this and our earlier studies (30,33), viz. that any persistent NO release in relatively low fluxes might actually enhance tumor aggressiveness. Such concerns might be diminished with a nitrosyl phthalocyanine ruthenium complex recently developed to enhance PDT efficacy (66). This complex, which sensitizes both singlet oxygen and NO generation upon photoactivation, was tested on melanoma cells and found to be more cytotoxic than a non-NO control, suggesting that rapid and complete NO liberation in high fluxes is effective, and without concerns about long-lasting NO release at relatively low levels.
CONCLUSIONS AND PERSPECTIVES
PDT is increasingly being considered as the modality of choice for treating difficult brain malignancies such as GBM (11,56). We have demonstrated in this study that glioblastoma cells exposed to an ALA-PDT-like challenge exploit iNOS/NO to resist photokilling and to promote a more aggressive phenotype in surviving cells. Although we have identified some contributors to this altered phenotype (TIMP-1, surviving, S100A4), how NO modifies their expression or activity/expression of other important effectors remains to be investigated. Of special interest are recent studies involving other cancer lines and non-PDT treatments, which showed that S-nitrosation by endogenous NO can inhibit activation of pro-apoptotic caspase-3 (67) or stimulate activation of epidermal growth factor receptor (68). If occurring in vivo, the negative effects we describe would favor tumor persistence and progression. This draws attention to iNOS as a target for intervention with specific inhibitors. Agents such as L-NIL (69) and GW274150 (70) have already been tested in clinical trials unrelated to cancer or PDT, and with no negative side effects. Therefore, future pharmacologic use of such inhibitors as PDT adjuvants for gliomas and other brain lesions is advocated.
Acknowledgments
This research was supported in part by a grant from the Advancing a Healthier Wisconsin Research and Education Program, and by USPHS grant CA70823 from the National Cancer Institute. The authors are grateful to Harry Whelan for helpful insights about PDT for GBM.
References
- 1.Henderson BW, Dougherty TJ. How does photodynamic therapy work? Photochem Photobiol. 1992;55:145–157. doi: 10.1111/j.1751-1097.1992.tb04222.x. [DOI] [PubMed] [Google Scholar]
- 2.Dougherty TJ, Gomer CJ, Henderson BW, Jori G, Kessel D, Korbelik M, Moan J, Peng Q. Photodynamic Therapy. J Natl Cancer Inst. 1998;90:889–905. doi: 10.1093/jnci/90.12.889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Agostinis P, Berg K, Cengel KA, Foster TH, Girotti AW, Gollnick SO, Hahn SM, Hamblin MR, Juzeniene A, Kessel D, Korbelik M, Moan J, Mroz P, Nowis D, Piette J, Wilson BC, Golab J. Photodynamic therapy of cancer: an update. CA Cancer J Clin. 2011;61:250–281. doi: 10.3322/caac.20114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Peng Q, Berg K, Moan J, Kongshaug M, Nesland JM. 5-Aminolevulinic acid-based photodynamic therapy: principles and experimental research. Photochem Photobiol. 1997;65:235–251. doi: 10.1111/j.1751-1097.1997.tb08549.x. [DOI] [PubMed] [Google Scholar]
- 5.Yang X, Palasuberniam P, Kraus K, Chen B. Aminolevulinic acid-based tumor detection and therapy: molecular mechanisms and strategies for enhancement. Int J Mol Sci. 2015;16:25865–25880. doi: 10.3390/ijms161025865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bechet D, Mordon SR, Guillemin F, Heyob-Barberi MA. Photodynamic therapy of malignant brain tumours: a complementary approach to conventional therapies. Cancer Treat Rev. 2014;40:229–241. doi: 10.1016/j.ctrv.2012.07.004. [DOI] [PubMed] [Google Scholar]
- 7.Behin A, Hoang-Suan K, Carpentier AF, Delattre JY. Primary brain tumours in adults. Lancet. 2003;361:323–331. doi: 10.1016/S0140-6736(03)12328-8. [DOI] [PubMed] [Google Scholar]
- 8.Laperriere N, Zuraw L, Cairncross G. Radotherapy for newly diagnosed malignant glioma in adults: a systematic review. Radiother Oncol. 2002;64:259–273. doi: 10.1016/s0167-8140(02)00078-6. [DOI] [PubMed] [Google Scholar]
- 9.Dirks PB. Brain tumor stem cells: bringing order to the chaos of brain cancer. J Clin Oncol. 2008;26:2916–2924. doi: 10.1200/JCO.2008.17.6792. [DOI] [PubMed] [Google Scholar]
- 10.Whelan HT. High-grade glioma/glioblastoma multiforme: is there a role for photodynamic therapy? J Natl Compr Canc Netw. 2012;10:S31–S34. doi: 10.6004/jnccn.2012.0171. [DOI] [PubMed] [Google Scholar]
- 11.Quirk BJ, Brandal G, Donlon S, Vera JC, Mang TS, Foy AB, Lew SM, Girotti AW, Jogal S, LaViolette PS, Connelly JM, Whelan HT. Photodynamic therapy (PDT) for malignant brain tumors--where do we stand? Photodiagnosis Photodyn Ther. 2015;12:530–544. doi: 10.1016/j.pdpdt.2015.04.009. [DOI] [PubMed] [Google Scholar]
- 12.Olzowy B, Hundt CS, Stocker S, Bise K, Reulen HJ, Stummer W. Photoirradiation therapy of experimental malignant glioma with 5-aminolevulinic acid. J Neurosurg. 2002;97:970–976. doi: 10.3171/jns.2002.97.4.0970. [DOI] [PubMed] [Google Scholar]
- 13.Hirschberg H, Uzal FA, Chighvinadze D, Zhang MJ, Peng Q, Madsen SJ. Disruption of the blood-brain barrier following ALA-mediated photodynamic therapy. Lasers Surg Med. 2008;40:535–542. doi: 10.1002/lsm.20670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jenkins CD, Charles IG, Thomsen LL, Moss DW, Holmes LS, Baylis SA, Rhodes P, Westmore K, Emson PC, Moncada S. Role of nitric oxide in tumor growth. Proc Nat Acad Sci USA. 1995;92:4392–4396. doi: 10.1073/pnas.92.10.4392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lechner M, Lirk P, Rieder J. Inducible nitric oxide synthase (iNOS) in tumor biology: the two sides of the same coin. Seminars Cancer Biol. 2005;15:277–289. doi: 10.1016/j.semcancer.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 16.Brian K, Ghassemi G, Sotolongo A, Siu A, Shauger L, Kots A, Murad F. NOS-2 signaling and cancer therapy. IMBMB Life. 2012;64:678–687. doi: 10.1002/iub.1057. [DOI] [PubMed] [Google Scholar]
- 17.Burke AJ, Sullivan FJ, Giles FJ, Glynn SA. The yin and yang of nitric oxide in cancer progression. Carcinogenesis. 2013;34:503–512. doi: 10.1093/carcin/bgt034. [DOI] [PubMed] [Google Scholar]
- 18.Eyler CE, Wu Q, Yan Y, MacSwords MM, Chandler-Militello D, Misuraca KL, Lathia JD, Forrester MT, Lee J, Stamler JS, Goldman SA, Bredel M, McLendon KE, Sloan AE, Hjelmeland AB, Rich JN. Glioma stem cell proliferation and tumor growth are promoted by nitric oxide synthase-2. Cell. 2011;146:53–66. doi: 10.1016/j.cell.2011.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kostourou V, Cartwright JE, Johnstone AP, Boult JKR, Cullis ER, Whitley GSJ, Robinson SP. The role of tumour-derived iNOS in tumor progression and angiogenesis. Br J Cancer. 2011;104:83–90. doi: 10.1038/sj.bjc.6606034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim RK, Suh Y, Cui YH, Hwang E, Lim EJ, Yoo YC, Lee GH, Yi JM, Kang SG, Lee SJ. Fractionated radiation-induced nitric oxide promotes expansion of glioma stem-like cells. Cancer Sci. 2014;104:1172–1177. doi: 10.1111/cas.12207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hurst R, Korytowski W, Kriska T, Esworthy RS, Chu FF, Girotti AW. Hyperresistance to cholesterol hydroperoxide-induced peroxidative injury and apoptotic death in a tumor cell line that overexpresses glutathione peroxidase isotype-4. Free Radic Biol Med. 2001;31:1051–1065. doi: 10.1016/s0891-5849(01)00685-2. [DOI] [PubMed] [Google Scholar]
- 22.Bhowmick R, Girotti AW. Signaling events in apoptotic photokilling of 5-aminolevulinic acid-treated tumor cells: inhibitory effects of nitric oxide. Free Radic Biol Med. 2009;47:731–740. doi: 10.1016/j.freeradbiomed.2009.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nagano T. Bioimaging probes for reactive oxygen species and reactive nitrogen species. J Clin Biochem Nutr. 2009;45:111–124. doi: 10.3164/jcbn.R09-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wardman P. Fluorescent and luminescent probes for measurement of oxidative and nitrosative species in cells and tissues: progress, pitfalls, and prospects. Free Radic Biol Med. 2007;43:995–1022. doi: 10.1016/j.freeradbiomed.2007.06.026. [DOI] [PubMed] [Google Scholar]
- 25.Lancaster JR. The use of diaminofluorescein for nitric oxide detection: conceptual and methodological distinction between NO and nitrosation. Free Radic Biol Med. 2010;49:1145. doi: 10.1016/j.freeradbiomed.2010.06.015. [DOI] [PubMed] [Google Scholar]
- 26.Mossmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods. 1983;65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- 27.Kriska T, Korytowski W, Girotti AW. Hyperresistance to photosensitized lipid peroxidation and apoptotic killing in 5-aminolevulinate-treated tumor cells overexpressing mitochondrial GPx4. Free Radic Biol Med. 2002;33:1389–1402. doi: 10.1016/s0891-5849(02)01078-x. [DOI] [PubMed] [Google Scholar]
- 28.Vermes I, Haanen C, Steffens-Nakken H, Reutelingsperger C. A novel assay for apoptosis. Flow cytometric detection of phosphatidylserine expression on early apoptotic cells using fluorescein labelled Annexin V. J Immunol Methods. 1995;184:39–51. doi: 10.1016/0022-1759(95)00072-i. [DOI] [PubMed] [Google Scholar]
- 29.Vandooren J, Geurts N, Martens E, Vanden Steen PE, Opdenakker G. Zymography methods for visualizing hydrolytic enzymes. Nat Methods. 2013;10:211–220. doi: 10.1038/nmeth.2371. [DOI] [PubMed] [Google Scholar]
- 30.Fahey JM, Girotti AW. Accelerated migration and invasion of prostate cancer cells after a photodynamic therapy-like challenge: role of nitric oxide. Nitric Oxide. 2015;49:47–55. doi: 10.1016/j.niox.2015.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bhowmick R, Girotti AW. Rapid upregulation of cytoprotective nitric oxide in breast tumor cells subjected to a photodynamic therapy-like oxidative challenge. Photochem Photobiol. 2011;87:378–386. doi: 10.1111/j.1751-1097.2010.00877.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bhowmick R, Girotti AW. Cytoprotective signaling associated with nitric oxide upregulation in tumor cells subjected to photodynamic therapy-like oxidative stress. Free Radic Biol Med. 2013;57:39–49. doi: 10.1016/j.freeradbiomed.2012.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bhowmick R, Girotti AW. Pro-survival and pro-growth effects of stress-induced nitric oxide in a prostate cancer photodynamic therapy model. Cancer Lett. 2014;343:115–122. doi: 10.1016/j.canlet.2013.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Alderton WK, Cooper CE, Knowles RG. Nitric oxide synthases: structure, function and inhibition. Biochem J. 2001;357:593–615. doi: 10.1042/0264-6021:3570593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kramer N, Walzl A, Unger C, Rosner M, Krupitza G, Henstschlager M, Dolznig H. In vitro cell migration and invasion assays. Mutation Res. 2013;752:10–24. doi: 10.1016/j.mrrev.2012.08.001. [DOI] [PubMed] [Google Scholar]
- 36.Kachra Z, Beaulieu E, Delbecchi L, Mousseau N, Berthelet F, Moumdjian R, Del Maestro R, Béliveau R. Expression of matrix metalloproteinases and their inhibitors in human brain tumors. Clin Exp Metastasis. 1999;17:555–566. doi: 10.1023/a:1006760632766. [DOI] [PubMed] [Google Scholar]
- 37.Veeravalli KK, Rao JS. MMP-9 and uPAR regulated glioma cell migration. Cell Adh Migr. 2012;6:509–512. doi: 10.4161/cam.21673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stamenkovic I. Matrix metalloproteinases in tumor invasion and metastasis. Semin Cancer Biol. 2000;10:415–433. doi: 10.1006/scbi.2000.0379. [DOI] [PubMed] [Google Scholar]
- 39.Bjorklund M, Koivunen E. Gelatinase-mediated migration and invasion of cancer cells. Biochim Biophys Acta. 2005;1755:37–69. doi: 10.1016/j.bbcan.2005.03.001. [DOI] [PubMed] [Google Scholar]
- 40.O’Sullivan S, Medina C, Ledwidge M, Radomski MW, Gilmer JF. Nitric oxide-matrix metalloproteinase-9 interactions: biology and pharmacological significance. Biochim Biophys Acta. 2014;1843:603–617. doi: 10.1016/j.bbamcr.2013.12.006. [DOI] [PubMed] [Google Scholar]
- 41.Olsen MW, Gervasi DC, Mobasheri S, Friedman R. Kinetic analysis of the binding of human matrix metalloproteinase-2 and -9 to tissue inhibitor of metalloproteinase (TIMP-1) and TIMP-2. J Biol Chem. 1997;272:29975–29983. doi: 10.1074/jbc.272.47.29975. [DOI] [PubMed] [Google Scholar]
- 42.Pennati M, Folini M, Zaffaroni N. Targeting survivin in cancer therapy: fulfilled promises and open questions. Carcinogenesis. 2007;28:1133–1139. doi: 10.1093/carcin/bgm047. [DOI] [PubMed] [Google Scholar]
- 43.Altieri DC. Survivin, cancer networks, and pathway-directed drug discovery. Nat Rev Cancer. 2008;8:61–70. doi: 10.1038/nrc2293. [DOI] [PubMed] [Google Scholar]
- 44.Ferrario A, Rucker N, Wong S, Luna M, Gomer CJ. Survivin, a member of the inhibitor of apoptosis family, is induced by photodynamic therapy and is a target for improving treatment response. Cancer Res. 2007;67:4989–4995. doi: 10.1158/0008-5472.CAN-06-4785. [DOI] [PubMed] [Google Scholar]
- 45.Fetz V, Bier C, Habtemichael N, Schuon R, Schweitzer A, Kunkel M, Engels K, Kovács AF, Schneider S, Mann W, Stauber RH, Knauer SK. Inducible NO synthase confers chemoresistance in head and neck cancer by modulating survivin. Int J Cancer. 2009;124:2033–2041. doi: 10.1002/ijc.24182. [DOI] [PubMed] [Google Scholar]
- 46.Garrett SC, Varney KM, Weber DJ, Bresnick AR. S100A4, a mediator of metastasis. J Biol Chem. 2006;281:677–680. doi: 10.1074/jbc.R500017200. [DOI] [PubMed] [Google Scholar]
- 47.Boyle K, Maelandsmo GM. S100A4 and metastasis: a small actor playing many roles. Am J Pathol. 2010;176:528–535. doi: 10.2353/ajpath.2010.090526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jin T, Zhang Z, Yang XF, Luo JS. S100A4 expression is closely linked to genesis and progression of glioma by regulating proliferation, apoptosis, migration and invasion. Asian Pac J Cancer Prev. 2015;16:2883–2887. doi: 10.7314/apjcp.2015.16.7.2883. [DOI] [PubMed] [Google Scholar]
- 49.Furnari FB, Fenton T, Bachoo RM, Mukasa A, Stommel JM, Stegh A, Hahn WC, Ligon KL, Louis DN, Brennan C, Chin L, DePinho RA, Cavenee WK. Malignant astrocytic glioma: genetics, biology, and paths to treatment. Genes Dev. 2007;21:2683–2710. doi: 10.1101/gad.1596707. [DOI] [PubMed] [Google Scholar]
- 50.Wen PY, Kesari S. Malignant gliomas in adults. New Engl J Med. 2008;359:492–507. doi: 10.1056/NEJMra0708126. [DOI] [PubMed] [Google Scholar]
- 51.Friedman HS, Prados MD, Wen PY, Mikkelsen T, Schiff D, Abrey LE, Yung WK, Paleologos N, Nicholas MK, Jensen R, Vredenburgh J, Huang J, Zheng M, Cloughesy T. Bevacizumab alone and in combination with irinotecan in recurrent glioblastoma. J Clin Oncol. 2009;27:4733–4740. doi: 10.1200/JCO.2008.19.8721. [DOI] [PubMed] [Google Scholar]
- 52.Rainov NG, Soling A. Clinical studies with targeted toxins in malignant glioma. Rev Recent Clin Trials. 2009;1:119–131. doi: 10.2174/157488706776876454. [DOI] [PubMed] [Google Scholar]
- 53.Lam-Himlin D, Espey MG, Perry G, Smith MA, Castellani RJ. Malignant glioma progression and nitric oxide. Neurochem Int. 2006;49:764–768. doi: 10.1016/j.neuint.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 54.Jahani-Asi A, Bonni A. iNOS: a potential therapeutic target for malignant glioma. Curr Mol Med. 2013;13:1241–1249. doi: 10.2174/1566524011313080002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Stylli SS, Kaye AH, MacGregor L, Howes M, Rajendra P. Photodynamic therapy of high grade glioma: long-term survival. J Clin Neurosci. 2005;12:389–398. doi: 10.1016/j.jocn.2005.01.006. [DOI] [PubMed] [Google Scholar]
- 56.Bechet D, Mordon SR, Guillemin F, Barberi-Heyob MA. Photodynamic therapy of malignant brain tumours: a complementary approach to conventional therapies. Cancer Treatment Rev. 2014;40:229–241. doi: 10.1016/j.ctrv.2012.07.004. [DOI] [PubMed] [Google Scholar]
- 57.Etminan N, Peters C, Ficnar J, Anlasik S, Bunemann E, Slotty PJ, Hanggi D, Steiger HJ, Sorg RV, Stummer W. Modulation of migratory activity and invasiveness of human glioma spheroids following 5-aminolevulinic acid-based photodynamic treatment. J Neurosurg. 2011;115:281–288. doi: 10.3171/2011.3.JNS10434. [DOI] [PubMed] [Google Scholar]
- 58.Zhang X, Jiang F, Shang ZG, Kalkanis SN, Hong X, deCarvalho AC, Chen J, Yang H, Rubin AM, Chopp M. Low dose photodynamic therapy increases endothelial cell proliferation and VEGF expression in nude mice brane. Lasers in Med Sci. 2005;20:74–79. doi: 10.1007/s10103-005-0348-8. [DOI] [PubMed] [Google Scholar]
- 59.Henderson BW, Sitnik-Busch TM, Vaughan LA. Potentiation of photodynamic therapy antitumor activity in mice by nitric oxide synthase inhibition is fluence rate-dependent. Photochem Photobiol. 1999;70:64–71. [PubMed] [Google Scholar]
- 60.Korbelik M, Parkins CS, Shibuya H, Cecic I, Stratford MRL, Chaplin DJ. Nitric oxide production by tumor tissue: impact on the response to photodynamic therapy. Br J Cancer. 2000;82:1835–1843. doi: 10.1054/bjoc.2000.1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rapozzi V, Della Pietra E, Zorzet S, Zacchigna M, Bonavida B, Xodo LE. Nitric oxide-mediated activity in anti-cancer photodynamic therapy. Nitric Oxide. 2013;30:26–35. doi: 10.1016/j.niox.2013.01.002. [DOI] [PubMed] [Google Scholar]
- 62.Rapozzi V, Della Pietra E, Bonavida B. Dual Roles of nitric oxide in the regulation of tumor cell response and resistance to photodynamic therapy. Redox Biol. 2015;6:311–317. doi: 10.1016/j.redox.2015.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bonavida B, Baritaki S, Huerta-Yepez S, Bega MI, Jazirehi AR, Berenson J. Nitric oxide donors are a new class of anti-cancer therapeutics for the reversal of resistance and inhibition of metastasis. In: Bonavida B, editor. Nitric Oxide (NO) and Cancer. Springer; New York: 2010. pp. 459–477. [Google Scholar]
- 64.Reynolds MM, Witzeling SD, Damodaran VB, Medeiros TN, Knodle RD, Edwards MA, Lookian PP, Brown MA. Applications for nitric oxide in halting proliferation of tumor cells. Biochem Biophys Res Commun. 2013;431:647–651. doi: 10.1016/j.bbrc.2013.01.041. [DOI] [PubMed] [Google Scholar]
- 65.Kiziltepe T, Hideshima T, Ishitsuka K, Ocio EM, Raje N, Catley L, Li CQ, Trudel LJ, Yasui H, Vallet S, Kutok JL, Chauhan D, Mitsiades CS, Saavedra JE, Wogan GN, Keefer LK, Shami PJ, Anderson KC. JS-K, a GST-activated nitric oxide generator, induces DNA double-strand breaks, activates DNA damage response pathways, and induces apoptosis in vitro and in vivo in human multiple myeloma cells. Blood. 2007;110:709–718. doi: 10.1182/blood-2006-10-052845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Heinrich TA, Tedesco AC, Fukuto JM, da Silva RS. Production of reactive oxygen and nitrogen species by light irradiation of a nitrosyl phthalocyanine ruthenium complex as a strategy for cancer treatment. Dalton Trans. 2014;43:4201–4205. doi: 10.1039/c3dt52217b. [DOI] [PubMed] [Google Scholar]
- 67.Godoy LC, Anderson CT, Chowdhury R, Trudel LJ, Wogan GN. Endogenously produced nitric oxide mitigates sensitivity of melanoma cells to cisplatin. Proc Natl Acad Sci U S A. 2012;109:20373–20378. doi: 10.1073/pnas.1218938109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Switzer CH, Glynn SA, Cheng RY, Ridnour LA, Green JE, Ambs S, Wink DA. S-nitrosylation of EGFR and Src activates an oncogenic signaling network in human basal-like breast cancer. Mol Cancer Res. 2012;10:1203–1215. doi: 10.1158/1541-7786.MCR-12-0124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hansel TT, Kharitonov SA, Donnelly LE, Erin EM, Currie MG, Moore WM, Manning PT, Recker DP, Barnes PJ. A selective inhibitor of inducible nitric oxide synthase inhibits exhaled breath nitric oxide in healthy volunteers and asthmatics. FASEB J. 2003;17:1298–1317. doi: 10.1096/fj.02-0633fje. [DOI] [PubMed] [Google Scholar]
- 70.Singh D, Richards D, Knowles RG, Schwartz S, Woodcock A, Langley S, O’Connor BJ. Selective inducible nitric oxide synthase inhibition has no effect on allergen challenge in asthma. Am J Respir Crit Care Med. 2007;176:988–993. doi: 10.1164/rccm.200704-588OC. [DOI] [PubMed] [Google Scholar]