

Abstract
The factor inhibiting hypoxia inducible factor-1α (FIH) is a nonheme Fe(II)/αKG oxygenase using a 2-His-1-Asp facial triad. FIH activates O2 via oxidative decarboxylation of α-ketoglutarate (αKG) to generate an enzyme-based oxidant which hydroxylates the Asn803 residue within the C-terminal transactivation domain (CTAD) of HIF-1α. Tight coupling of these two sequential reactions requires a structural linkage between the Fe(II) and the substrate binding site to ensure that O2 activation occurs after substrate binds. We tested the hypothesis that the facial triad carboxylate (Asp201) of FIH linked substrate binding and O2 binding sites. Asp201 variants of FIH were constructed and thoroughly characterized in vitro using steady-state kinetics, crystallography, autohydroxylation, and coupling measurements. Our studies revealed each variant activated O2 with a catalytic efficiency similar to that of wild-type (WT) FIH (kcat/KM(O2) = 0.17 μM−1 min−1), but led to defects in the coupling of O2 activation to substrate hydroxylation. Steady-state kinetics showed similar catalytic efficiencies for hydroxylation by WT-FIH (kcat/KM(CTAD) = 0.42 μM−1 min−1) and D201G (kcat/KM(CTAD) = 0.34 μM−1 min−1); hydroxylation by D201E was greatly impaired, while hydroxylation by D201A was undetectable. Analysis of the crystal structure of the D201E variant revealed steric crowding near the diffusible ligand site supporting a role for sterics from the facial triad carboxylate in the O2 binding order. Our data support a model in which the facial triad carboxylate Asp201 provides both steric and polar contacts to favor O2 access to the Fe(II) only after substrate binds, leading to coupled turnover in FIH and other αKG oxygenases.
Graphical Abstract Synopsis
The facial triad carboxylate structurally links O2 and substrate binding in the factor-inhibiting hypoxia inducible factor-1α (FIH), an α-ketoglutarate dependent oxygenase.
Introduction
The Fe(II)/αKG dioxygenases catalyze a wide range of oxidations including hydroxylation of proteins, demethylation of RNA and DNA, and desaturations of antibiotic precursors [1, 2]. Enzymes in this superfamily bind the nonheme Fe(II) cofactor with a 2-His-1-Carboxylate facial triad, completing the coordination environment with a bidentate alpha-ketoglutrate (αKG) and an exchangeable ligand site that is occupied by H2O in resting enzyme (Scheme 1) [1, 3]. Binding primary substrate near the cofactor increases the reactivity of Fe toward O2 [4, 5], leading to a sequential mechanism with ordered O2 activation, resulting in tight coupling between the two half reactions. The linkage between substrate binding and O2 reactivity is thought to involve changes in contacts within the second coordination sphere of the Fe cofactor [6–12]. FIH, an Fe(II)/αKG oxygenase responsible for O2 sensing in humans, is one of the few αKG oxygenases that is active with a substituted facial triad carboxylate residue [13]. This affords a unique opportunity to understand how local contacts to the facial triad carboxylate ligand affect the coupling between O2 reactivity and substrate binding.
Scheme 1.

Consensus Mechanism for FIH
The consensus mechanism for FIH and other Fe(II)/αKG oxygenases (Scheme 1) involves the sequential half reactions of O2 activation followed by substrate hydroxylation [1, 14–18]. Substrate binding leads to release of the aquo ligand in FIH and other Fe(II)/αKG oxygenases as shown by variable temperature and variable field magneto circular dichroism (VTVH-MCD) spectroscopy [19–21], as part of a process that has been termed triggering [22]or priming [23]. O2 is proposed to bind as a ferric superoxide at the open coordination site, then attack the C-2 carbonyl of αKG, passing through a transition state resembling a peroxohemiketal [17, 24, 25] to form succinate and a ferryl as the active oxidant. The ferryl oxidant abstracts an H-atom to form a substrate radical and ferric hydroxide, which then rebounds to form the ferrous cofactor and hydroxylated product [22, 26–31]. As primary substrate does not directly coordinate the Fe(II), the structural linkage between substrate binding and increased O2 reactivity are crucial to tight coupling in this family of enzymes.
As the primary substrate for FIH is the C-terminal transactivation domain (CTAD) of the hypoxia inducible factor, the Fe active site is exposed to solvent until the primary substrate is bound as shown by crystal structures of (Fe+αKG)FIH and (Fe+αKG)FIH/CTAD [32, 33]. MCD spectroscopy indicates that (Fe+αKG)FIH is six coordinate[20] and crystal structures show the facial triad carboxylate, Asp201, is well positioned to hydrogen bond to the distal aquo ligand [32–34]. In structures where the aquo ligand is resolved, the distal O-atom of Asp201 is within hydrogen bonding distance to the aquo ligand (Odistal---OH2 = 2.52Å – 2.85Å) [35, 36]. Although O2 activation occurs slowly in the absence of CTAD [37, 38], CTAD binding alters the local contacts to Asp201 and stimulates O2 reactivity [10, 13, 33, 34], suggesting that changes in the chemical environment near the diffusible ligand site connects CTAD binding to O2 reactivity.
We test the facial triad carboxylate as the nexus connecting substrate binding to O2 reactivity in FIH. Previously, variants of Asp201 were shown to hydroxylate CTAD with varying yields, but the role of these variants in linking O2 activation with substrate binding and hydroxylation was not explored[13]. In this work, three facial-triad variants (Glu, Gly, Ala) were prepared and purified to perturb the chemical environment near the diffusible ligand. Steady-state kinetics, crystallography, and assays to measure the coupling between O2 consumption and CTAD hydroxylation indicated that the identity of the facial triad residue had little effect on O2 activation, but played a deciding role in coupling the O2 activation to substrate hydroxylation. Overall, these data indicate that the facial triad carboxylate is not essential to activate O2, but rather serves to ensure that O2 activation is coupled with substrate hydroxylation.
Methods
Materials
All reagents were purchased from commercial vendors and were used as received, with the exception of the 39-residue CTAD peptide. The sequence of the 39-residue CTAD peptide (theoretical molecular weight = 42544.6 Da; pI=3.95) followed that of the C-terminal activation domain of human HIF-1α, (HIF-1α788–826), with the exception of a Cys800→Ala substitution to prevent peptide oxidation (Asn803 underlined), DESGLPQLTSYDAEVNAPIQGSRNLLQGEELLRALDQVN. CTAD was purchased as a desalted peptide from EZBiolab (Carmel, IN, USA) with free N and C-termini, then purified as previously described using reversed-phase HPLC to obtain peptide of >95% purity [4].
Protein Expression and Purification
The QuikChange mutagenesis kit (Stratagene) was used to make mutations in the pET28a-FIH construct [38]. The DNA sequence of each mutated plasmid was sequenced (Genewiz, NJ, USA) to confirm the desired point mutation. WT-FIH and each variant was overexpressed in BL21-DE3 E. coli with an N-terminal His6 tag and purified as previously described [4]. Three non-native residues (NH2-GlySerHis-) remained on the N-terminus of FIH following thrombin cleavage. Purified FIH was buffer exchanged into 50 mM HEPES pH 7.00. The purity (>95%) of each variant was assessed by SDS-PAGE.
Steady-State Kinetics
The standard reaction conditions used 50 mM HEPES pH 7.00 (37.0 °C) containing 100 mM NaCl, ascorbic acid (2 mM), αKG (100 μM), FeSO4 (25 μM). Assays in which CTAD was the varied substrate (15–275 μM) used saturating αKG (100 μM). Assays in which αKG was the varied substrate (0–200 μM) contained ascorbate (2 mM), FeSO4 (25 μM) and a fixed CTAD concentration (80 μM). Initial rate measurements were made as previously described using a Bruker MicroFlexII MALDI-TOF-MS and fit to the Michaelis-Menten equation [4].
Steady-State Kinetics varying O2
Assays in which the O2 concentration was varied were performed as previously described [25]. Briefly, HEPES buffer (50 mM, pH 7.00) was stirred to equilibrate the solution under an atmosphere of N2 and O2 at 37.0°C within an AtmosBag (Sigma-Aldrich). The concentration of dissolved O2 was measured in buffer using a Clarke electrode (YSI) prior to each reaction. The reaction mixture (45 μL) containing ascorbic acid (2mM), αKG (100 μM), FeSO4 (25 μM), NaCl (100 mM) and CTAD (80 μM), was incubated at 37.0 °C for two minutes prior to addition of FIH to initiate the reaction. The FIH stock was gently pipetted down the side of the microcentrifuge tube to equilibrate the solution with the N2/O2 mixture before initiating turnover. Reaction aliquots were quenched in 75% acetonitrile/0.2% TFA saturated with 3,5-dimethoxy-4-hydroxycinnamic acid prior to analysis on a Bruker microFlex MALDI-TOF-MS to quantify product formation. Initial rates were determined as previously described[4] and fit to the Michaelis-Menten equation.
Succinate Quantification
Reactions to determine the production of succinate and CTADOH concentrations were run at 37.0° C in 50 mM Tris, pH 7.00 and analyzed as previously reported [11]. Briefly, aliquots (20 μL) from a common reaction containing αKG (100 μM), FeSO4 (100 μM), CTAD (100 μM) and FIH (100 μM) were quenched in 500 mM H2SO4 containing 500 μM succinate (5 μL). From each quenched aliquot, HPLC-UV/Vis was used to quantitate the succinate concentration produced from the reaction and MALDI-TOF-MS was used to quantitate the CTADOH concentration. The coupling ratio (C) was determined by taking the ratio of the concentration of succinate and the concentration of CTADOH from matching quenched reactions.
UV-Vis absorption spectroscopy
All reagents, except the buffer, were made anaerobic by argon sparging. The buffer was made anaerobic by multiple cycles of vacuum/N2 flush. Samples of (Fe+αKG)FIH, where the active-site occupancy is denoted within parenthesis, were prepared in an anaerobic glove box as solutions containing FIH (400 μM), FeSO4 (375 μM) and αKG (400 μM) in 50 mM HEPES pH 7.00. An Avantes AvaSpec-2048 Fiber Optic spectrometer with a 2048 pixel CCD Detector Array was used to record all spectra in a 1 cm path-length cuvette. An (Fe)FIH spectrum was subtracted from each (Fe+αKG)FIH spectrum to record the changes in spectral features due to formation of the Fe+αKG chromophore.
Autohydroxylation
Autohydroxylation assays were performed as previously described [37, 38]. All reagents except buffer were degassed individually by argon flush before being handled in a glove box. The buffer was made anaerobic using multiple cycles of vaccum and N2 flush. Samples containing FIH (l00μM), FeSO4 (100μM) and αKG (500μM) in 50 mM HEPES pH 7.00 were prepared in a masked cuvette and sealed. Removal of the cap exposed the anaerobic samples to air, allowing for slow oxidation of the samples. Experiments were performed at room temperature and all spectra were collected using an Agilent HP 8453 diode-array UV-Vis Spectrophotometer.
Crystallography and data refinement
Crystals of (Fe+αKG)D201E, (Zn+αKG)D201E and (Zn+αKG)D201E/CTAD were grown anaerobically using conditions similar to those used in the literature[13]. Crystals were grown anaerobically for (Fe+αKG)FIH via hanging drop vapor diffusion method by mixing 2 μl of a 20 mg/ml protein solution containing 0.55 mM FeSO4, AND 2mM αKG in 50 mM HEPES pH 7.00 with 1 μL of the reservoir buffer containing 0.1 M HEPES, 1.2 M (NH4)2SO4, and 3 % PEG-400, pH 7.50. Crystals grew within 5 days at 20 °C inside the glovebox, then were harvested quickly outside the glovebox to minimize O2 diffusion and flash frozen in reservoir solution containing 24% glycerol. Crystals of the (Zn+αKg)D201E and (Zn+αKG)D201E/CTAD were grown aerobically in a similar manner using 0.55 mM ZnCl2 in the place of FeSO4. A 19 residue CTAD peptide corresponding to HIF-1α788–807 was used for co-crystalization studies. This 19-mer peptide was purchased from EZBiclabs as a >95% pure peptide with free N and C termini with sequence DESGLPQLTSYDAEVNAPI (Asn803 underlined).
Data were collected at the Brookhaven National Synchrotron Radiation facility at 100 K using radiation of λ = 1.08 A. 240 frames were collected for (Fe+αKG)D201E and (Zn+αKG)D201E at a crystal to detector distance of 225 mm with 0.5° oscillation and 10 sec exposure per image. 136 frames were collected for (Zn+αKG)D201E/CTAD at a crystal to detector distance of 250 with 0.5° oscillation and 15 second exposure per image. The data sets were processed using HKL2000 [39]. Coordinates from PDBID 3D8C [13] were employed for molecular replacement using Phaser [40] then refined using COOT [41] and Refmac5 [42] in the CCP4 software package [43]. During refinement, 5% of the data was withheld and used to obtain an Rfree value.
Results and discussion
UV- Vis Absorption Spectroscopy
The electronic environment for the Fe(II) cofactor in the D201X variants was assessed by UV-Vis absorption spectroscopy. The expected metal-to-ligand charge transfer (MLCT) electronic transition for each of the (Fe+αKG)FIH D201X variants was observed as a broad absorption peak centered near 500 nm (Figure 1) indicating that each variant bound Fe(II) along with αKG. Both variants with a carboxylate residue (X = Asp, Glu) exhibited a similar peak wavelength (λmax = 500 nm); however, the variants lacking a facial triad carboxylate (X = Gly, Ala) exhibited a blue-shifted peak wavelength (λmax = 485 nm). The blue shift in the MLCT transition for the D201X (X = Gly, Ala) variants was consistent with a stronger Lewis acidity for the Fe(II) in these variants than for X = Asp or Glu variants, which we attribute to stabilization of the Fe t2g(π) orbitals due to removal of the π-donor carboxylate ligand for D201G and D201A. Increased Lewis acidity of the cofactor might have been expected to increase the intrinsic reactivity for the D201(G/A) variants, however the kinetics studies (below) did not bear this out.
Figure 1.

Anaerobic UV-Vis spectra of WT-FIH and the D201X variants for the (Fe+αKG) form of WT-FIH (line), D201E (dashed), D201A (dotted) and D201G (dot/dash); FIH (400 μM), FeSO4, (375 μM), αKG (400 μM).
Steady-state kinetics with varied CTAD and αKG
The relative activity levels for CTAD hydroxylation by the Asp201 variants was previously shown to follow the trend WT > D201G ≫ D201E = D201A = 0, with a large amount of succinate formed due to uncoupling by the Glu and Ala variants [13]. We observed a similar trend, with the notable exception that the D201E variant was competent to hydroxylate CTAD before rapidly inactivating. This led us to suspect that the facial triad carboxylate residue strongly impacted the coupling between CTAD binding and O2 activation, and that the D201E variant might autohydroxylate as seen for WT-FIH after prolonged incubation [37, 38].
The initial steady-state characterization of the Asp201 variants with CTAD as the varied substrate was performed under standard assay conditions using saturating concentrations of FeSO4 (25 μM), αKG (100 μM), and ascorbate (2 mM) in 50 mM HEPES, 100 mM NaCl, pH 7.00 (Figure 2). These assays revealed WT-like hydroxylation activity for D201G (kcat = 23 ± 2 min−1, kcat/KM(CTAD) = 0.34 ± 0.05 μM−1 min−1) and diminished activity for D201E (kcat = 1.6 ± 0.2 min−1, kcat/KM(CTAD) = 0.022 ± 0.002 μM−1 min−1), with D201A activity below our detection level (Table 1). These significant decreases in these kinetic parameters for D201E indicated that CTAD hydroxylation was impeded in this variant.
Figure 2.

Steady-state kinetics of FIH variants with CTAD as the varied substrate. WT (■) D201G (●) Inset: D201E (▼) D201A (◆). All assays contained saturating concentrations of FeSO4 (25 μM), αKG (100 μM), and ascorbate (2 mM), with an ambient O2 concentration (217 μM) and were performed in 50 mM HEPES containing 100 mM NaCl, pH 7.00, 37°C.
Table 1.
Steady-state kinetic parameters of WT-FIH, D201G and D201Ea
| kcatapp (min−1) b | kcat/KM(CTAD)app (μM−1 min−1) b | KM(CTAD)app (μM) b | KM(αKG)app (μM) c | |
|---|---|---|---|---|
| WT FIH | 33 ± 2 | 0.42 ± 0.08 | 78 ± 17 | 20 ± 5 |
| D201E | 1.6 ± 0.2 | 0.022 ± 0.002 | 70 ± 15 | 26 ± 8 |
| D201G | 23 ± 2 | 0.34 ± 0.05 | 71 ± 12 | 23 ± 6 |
| D201A | d | d | d | d |
Based on KM(O2), each variant had a different degree of O2 saturation. WT, 52%; D201E, 75%; D201G, 65%.
Reactions contained ascorbate (2 mM), αKG (100 μM), FeSO4 (25 μM) and CTAD (0–260 μM) in 50 mM HEPES containing 100 mM NaCl, pH 7.00, 37.0 °C.
Reactions contained ascorbate (2 mM), αKG (0–200 μM), FeSO4 (25 μM) and CTAD (80 μM) in 50 mM HEPES containing 100 mM NaCl, pH 7.00, 37.0 °C.
No hydroxylation activity detected; Estimated hydroxylation detection limit <0.005 min−1, if active.
Comparison of the Michaelis constant for αKG revealed that KM(αKG) was insensitive to the identity of the residue at position 201 (Table 1). This was consistent with the limited second-sphere contacts between αKG and the residues of the facial triad, and suggested minimal impact of the facial triad carboxylate upon the αKG chemical environment.
Steady-State Kinetics Varying O2
While O2 activation was quite slow for WT-FIH (kcat/KM(O2)app=0.17 μM−1 min−1) the kinetic parameters for the O2-reaction of these facial triad variants has not been previously reported. To test for changes in O2 activation due to the D201X substitutions, steady-state kinetics with O2 as the varied substrate were measured using fixed concentrations of CTAD (80 μM), saturating concentrations of FeSO4 (25 μM), αKG (100 μM), and ascorbate (2 mM) in 50 mM HEPES, 100 mM NaCl, pH 7.00 (Figure 3). The conservative substitution in the D201E variant led to depressed kinetic parameters (kcat/KM(O2) < 0.02 μM−1min−1), whereas the D201G variant exhibited kinetics that were similar to those of WT-FIH (kcat/KM(O2) ~ 0.02 μM−1min−1); the D201A variant failed to hydroxylate CTAD at both high and low O2 concentrations (Table 2). Although these results suggested a very complex relationship between the identity of residue 201 and O2-activation, it is important to consider that reaction rates were obtained by measuring CTAD hydroxylation. As defects in the coupling of the active oxidant to CTAD hydroxylation could have led to low yields of hydroxylated CTAD, we measured the succinate formation in these variants to account for all consumed αKG.
Figure 3.

Steady-state kinetics varying O2 for WT (■) and D201G (●) (Insert) Steady-state kinetics varying O2 for D201E (▼). All assays contained ascorbic acid (2mM), αKG (100 μM), FeSO4 (25 μM) and CTAD (80 μM).
Table 2.
O2 Activation Kinetic parameters for FIH and Asp201 variantsa.
| kcatapp (min−1) | kcat/KM(O2)app (μM−1 min−1) | KM(O2)app (μM) | |
|---|---|---|---|
| WT FIHb | 33 ± 3 | 0.17 ± 0.03 | 200 ± 40 |
| D201E | 0.81 ± 0.02 | 0.014 ± 0.002 | 70 ± 15 |
| D201G | 20 ± 1.0 | 0.18 ± 0.02 | 115 ± 20 |
Reactions contained ascorbate (2 mM), αKG (100 μM), FeSO4 (25 μM), CTAD (80 μM) and O2 (0–950 μM) in 50 mM HEPES containing 100 mM NaCl pH 7.00, 37.0 °C.
From Ref. ([25])
Succinate forms stoichiometrically with the active oxidant (Eq. 1a), making succinate quantitation a direct measurement of O2 activation regardless of the fate of the putative [Fe(IV)=O]2+ oxidant identified in related enzymes [5, 14, 44] (Eq. 1b, c). As the putative ferryl oxidant can hydroxylate CTAD to form the normal product (Eq. 1b, R = CTAD-Asn803), inactivate by autohydroxylating (Eq. 1b, R = FIH-Trp296) [37, 38], or potentially react with an exogenous reagent as seen for related enzymes AtsK and CS2 (Eq. 1c) [45, 46], there is the very real potential for the D201X substitutions to lead to normal O2 activation chemistry that is simply poorly coupled to CTAD hydroxylation.
| (1a) |
| (1b) |
| (1c) |
Although each of the facial triad variants was reported to produce significant levels of succinate, the level of coupling between O2 activation and CTAD hydroxylation was not reported [13]. We measured the ratio of succinate to hydroxylated CTAD, in order to identify the coupling ratio (C = [succinate]/[CTADOH]) for each variant. As seen previously [10], WT-FIH produced 1.05 equivalents of succinate for every equivalent of CTADOH formed (C = 1.05) and exhibited tight coupling between O2 activation and the subsequent hydroxylation step. In contrast, the other variants produced excess succinate (C ≫ 1), with appreciable uncoupling of O2 activation to CTAD hydroxylation (Table 3). The conservative D201E variant exhibited C = 20 and D201G exhibited C = 2.0; the D201A variant exhibited C ≫ 80, however the CTADOH level was undetectable such that C could be significantly larger than reported.
Table 3.
Coupling ratios for facial triad variants of FIH.
| Variant | C x kcat/KM(O2)app (μM−1 min−1) | kcat/KM(O2)app (μM−1 min−1) | C |
|---|---|---|---|
| WT (Asp201) | 0.18 ± 0.03 | 0.17 ± 0.03 | 1.05 ± 0.05 |
| D201G | 0.36 ± 0.05 | 0.18 ± 0.02 | 2.0 ± 0.20 |
| D201A | ---- | ---- | ≫ 80a |
| D201E | 0.28 ± 0.06 | 0.014 ± 0.002 | 20 ± 3 |
C = (moles succinate)/(moles CTAD-OH)
Estimated based on moles of succinate produced and the limit of detection for hydroxylation.
The product of C x kcat/KM(O2)app yields the rate constant for total O2 activation independent of whether or not the active oxidant succeeds in hydroxylating CTAD. Intriguingly, this product for both D201G and D201E (0.36 ± 0.05 μM−1 min−1 and 0.28 ± 0.06 μM−1 min−1, respectively) is greater than that of WT-FIH (0.18 ± 0.03 μM−1 min−1), indicating that O2 activation to was not impaired by either of these variants (Table 3). Furthermore, although the hydroxylated CTAD levels could only be estimated for the D201A variant, even this variant produced succinate at nearly the same rate as WT-FIH. As the trend in Lewis acidity for the Fe(II) cofactor (D201G ~ D201A > WT-FIH ~ D201E) did not track with the O2 activation trend (D201G > D201E > WT-FIH > D201A), it is unlikely that the increased O2 activation rates for the D201G and D201E variants arose from an intrinsically more reactive Fe(II). Further, it is apparent that the Asp201 in WT-FIH was not essential to support O2 activation but rather played a decisive role in coupling O2 activation to the subsequent hydroxylation step.
Autohydroxylation
The rapid kinetic parameters for O2 activation suggested some of these variants were constitutively triggered, which would allow for increased O2 activation even in the absence of CTAD. As WT-FIH was previously shown to autohydroxylate slowly in the absence of CTAD [37, 38], the D201X variants were tested for increased autohydroxylation kinetics. Samples containing a D201X variant, FeSO4 and αKG were prepared anaerobically and then exposed to air allowing for the slow auto-hydroxylation of the D201X variant. UV-Vis spectroscopy was used to monitor the changes in the (Fe+αKG)FIH absorption spectra as a function of time.
The slow oxidation of (Fe+αKG)WT-FIH resulted in the formation of the previously identified indigo-coloured species with a characteristic visible absorption spectrum (λmax = 583 nm) at a rate of 0.35 μM min−1. This species results from the auto-hydroxylation of FIH at Trp296, forming the Fe(III)-O-Trp296 chromophore [37, 38] which can be thought of as the basal rate for O2 activation in when WT-FIH is not triggered. Autohydroxylation of the D201E variant formed the same chromophore as observed for WT-FIH (λmax = 583 nm) but at significantly faster initial rate (2.2 μM s−1) (Figure 4). This faster auto-hydroxylation by D201E was consistent with an enzyme that was constitutively triggered to react with O2.
Figure 4.

Autohydroxylation following exposure to air of FIH variants (100 μM) mixed anaerobically with FeSO4 (100 μM) and αKG (500 μM) in 50 mM HEPES pH 7.00 containing 100 mM NaCl. (A) Timecourse for WT-FIH and the D201E variant; inset, D201E spectra. (B) Timecourse for the D201G variant; inset, D201G spectra.
When analogous samples containing D201G or D201A mixed with FeSO4 and αKG were exposed to air, pink chromophores formed with λmax = 515 nm (ε515 = 2500 M−1 cm−1) and λmax = 510 nm (ε510 = 2000 M−1 cm−1) respectively (Figure 4B and S3). The formation of these chromophores was O2-dependent, as they did not form in the absence of oxygen. The rate of formation was significantly higher (1.4, 2.2 μM min−1) when compared to that of WT FIH, and within a factor of two of the rate observed for the D201E variant, suggesting that ~ 2 μM−1 s−1 may be the upper limit for O2 activation by FIH variants. The new chromophores observed for D201G and D201A indicated an altered electronic environment of the Fe site, which could include coordination by a catecholate formed from autohydroxylation of a nearby Tyr residue instead of the Trp296, or the binding of an exogenous anion.
When TauD autohydroxylates to form an Fe(III)-catecholate species, that chromophore absorbs visible light (λmax = 720 nm) that can blue-shift to 550 nm upon the addition of excess bicarbonate [47]. Inspection of the structure of FIH shows that the aromatic residue closest to the Fe center is Trp296 (4 Å), with Tyr102 (8 Å) significantly further away, consistent with Trp296 as the site of autohydroxylation in WT-FIH and suggesting that Tyr102 would be an unlikely site for modification. Similarly, comparing the electronic spectra of the autohydroxylation products from the FIH D201(G/A) variants (λmax = 515 nm/510 nm) with that of TauD (λmax = 720 nm) suggests that catecholate formation is unlikely in these D201(G/A) variants. As the crystal structure of the D201A variant exhibited electron density near the Fe that was refined as CO32−, it is more likely that both D201G and D201A autohydroxylate at Trp296 but bind a bicarbonate or carbonate anion, leading to the blue –shifted UV-Vis absorption for autohydroxylated D201(G/A).
Structure of Asp201 Variants
In the consensus mechanism, substrate binding leads to aquo release from the resting enzyme, thereby creating the site to bind O2 (Scheme 1). While there is strong evidence that the aquo ligand is released from FIH upon binding CTAD, structural information explaining why this happens is limited. In part this is because the initially reported X-ray crystal structures of (Fe+NOG)FIH and (Fe+αKG)FIH did not contain a resolved aquo ligand to the metal center [32, 33], although a hydrogen bond between the backbone amide of CTAD-Asn803 and Asp201 was noted to link the Fe(II) binding site and the CTAD binding site [33]; a more recent structure for (Fe+αKG)FIH included a resolved aquo ligand with an Fe-OH2 bond length of 2.00 Å [34], however it is worth noting that precise identification of non-protein ligands near metal centers via crystallography is quite challenging. MCD spectroscopy showed that the Fe(II) converts from a 6-coordinate geometry in the (Fe+αKG)FIH state to a mixed 5- and 6-coordinate geometry in the (Fe+αKG)FIH/CTAD structure [20], consistent with aquo release to ensure that O2 activation only occurs once CTAD is present. Asp201 of the His2Asp facial triad is oriented to permit a polar contact between the distal O-atom and the Fe(II)-bound water molecule or to the CTAD-Asn803 backbone, suggesting that shifted polar contacts are central to this coupling – but sterics with residues near this aquo ligand site have been implicated to play a significant role in O2 binding/reactivity with WT-FIH [34]. A comparison of the structures and reactivity of the D201X variants suggests that both polar contacts and sterics involving the D201X residue impact communication between the O2 and CTAD binding sites.
Although the structures of the D201A and D201G variants have been reported [13], there were very few structural differences from WT-FIH even near the Fe(II) site. The most notable difference was the loss of the facial triad carboxylate and its accompanying polar contacts, although a large planar molecule in the D201A structure, refined as CO32−, occupying the place of the missing carboxylate sidechain from the D201A variant. As both the D201G and D201A variants uncoupled, the authors proposed that the loss of polar contacts from Asp201 weakened the interaction with the aquo ligand and led to a more facile O2 reaction in the absence of CTAD [13]. Our results agree with this prior report, supporting a strong role for polar contacts near the diffusible ligand site in defining the O2 access.
The great mystery was that the conservative substitution in the D201E variant retained the polar contacts from the facial triad, and yet this variant uncoupled to a large extent. This observation pointed to some factor in addition to polar contacts as determining the ordered addition of O2 to the enzyme. We hoped that structural data would help to resolve this puzzling observation and provide insight into how this variant could react so poorly with CTAD but so efficiently with O2. Crystals of the D201E variant containing Fe(II) were grown anaerobically while crystals containing Zn(II) were grown aerobically using the hanging drop method. The structure of (Fe+αKG)D201E was refined to 2.3 A (Rwork = 18.6% and Rfree = 22.7% and the (Zn+αKG)D201E structure was refined to 2.4 Å (Rwork = 18.0% and Rfree = 22.7%. The protein backbone of (Fe+αKG)D201E and (Zn+αKG)D201E were essentially identical to each other, and very similar to that of wild-type (Fe+αKG)FIH (PDB:4Z2W 2.4 Å)[34] (Figure 5), indicating that the D201E substitution did not significantly alter the structure relative to WT-FIH (rmsd = 0.178 Å over residues 9-349).
Figure 5.

(A) Active site overlays of (Fe+αKG)FIH (PDB: 4Z2W[34], white), (Fe+αKG)D201E (PDBID: to be assigned, green) and (Zn+αKG)D201E (PDBID: to be assigned, cyan). Fe is shown in brown and Zn in grey. (B) Active site overlays of (Fe+αKG)FIH/CTAD (PDB: 1H2L[33], white) and (Zn+αKG)D201E/CTAD (cyan). Fe is shown in brown and Zn in grey. In the D201E variant, the Glu residue crowds the diffusible ligand site.
Each of the four facial triad variants exhibited nearly identical protein structures [13, 32–34], indicating that the reactivity changes did not arise from gross conformational changes. The active site metal retained pseudo-octahedral geometry in the D201X variants [13], and was coordinated by His199, His279, αKG with the C-1 carboxylate bound trans to His199, and a variable ligand for each variant: Glu201 (D201E), Asp201 (WT), H2O (D201G), CO32− (D201A). That loss of polar contacts to the diffusible ligand site with the Gly and Ala substitutions led to uncoupling, and rapid autohydroxylation, pointed to an active site that easily releases bound H2O from the Fe(II), allowing O2 to access to the metal center without regard to the presence of CTAD as previously proposed [13].
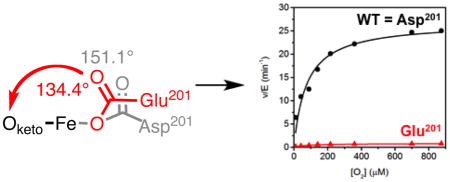
The D201E variant structure showed a slight expansion of the beta-barrel near residues 199 – 202 to accommodate the longer Glu201 sidechain in a suitable bonding configuration with the metal ion. WT-FIH exhibited fairly typical M-O distances for a monodentate carboxylate from Asp201: Fe-O1 (1.9 Å); Fe-O2 (3.1 Å). Both Fe-O distances were changed by less than 0.1 Å in the D201E variant (Table 5), showing that D201E retained the potential to form adequate polar contacts to the putative aquo ligand. Although bonded contacts involving the facial triad carboxylate were only slightly altered by the D201E substitution, the slight rotation of the carboxylate in this variant led to the distal O-atom impinging on the sterics of the diffusible ligand site (Figure 5A). The O2–Oketo distance was decreased by 0.3 Å for the D201E variant from that of WT-FIH (Table 5), which narrowed the available space for the exchangeable ligand site. This is well demonstrated as the exchangeable site angle (Oketo – Fe – O2) decreased from 151° in WT-FIH to 134° by the D201E substitution, indicating an increased steric ‘push’ in the D201E variant (Figure 6). Although polar contacts from Glu201 to a bound H2O ligand would be possible, sterics appear to be sufficient to disfavor this ligand based on the rapid inactivation of the D201E variant.
Table 5.
Selected bond lengths (Å) and angles for (Fe+αKG)WT-FIH and (Fe+αKG)D201E.
Figure 6.

Comparison of the carboxylate location and exchangeable site angle (Oketo-Fe-O2) in (Fe+αKG)FIH variants; WT-FIH (Asp201) (PDBID 1H2N [33]); D201E (Glu201) (PDBID to be assigned).
Further support for steric imposition upon the diffusible ligand site by the distal O-atom in the D201E variant was found by collecting the X-ray crystal structure of the CTAD-bound form of enzyme. The structure of (Zn+αKG)D201E/CTAD was refined to 2.1 A resolution with an Rwork = 17.0% and Rfree = 21.0%. The (Zn+αKG)D201E/CTAD structure is similar to the published structure of (Fe+αKG)FIH/CTAD (PDB:1H2L, rmsd = 0.164 A) [33]. There were few changes in contacts to the facial triad carboxylate in the D201E variant when compared to WT-FIH (Figure 5B). In the presence of CTAD, both WT-FIH and D201E possessed a 5-coordinate metal center but the distal O-atom of Glu201 was shifted closer to the metal by ~0.3 Å relative to WT, leading to a 2.9 Å Fe-O distance (Table 5). There were two significant differences caused by CTAD binding for both WT-FIH and the D201E variant: he distal O2 formed a polar contact with the backbone amide of CTAD-Asn803, and the exchangeable site angle (Figure 6) decreased by roughly 10°. While the Asp→Glu substitution did not alter polar contacts related to substrate binding, it did add steric hindrance to the exchangeable ligand site.
We propose that Asp201 links the O2 binding site to CTAD binding from both polar contacts and sterics within the active site. The Asp201 residue of WT-FIH is well positioned to stabilize an H2O ligand [32–34] prior to CTAD binding, whereas CTAD binding leads to new polar contacts between this residue and the CTAD-Asn803 as noted previously [33]. Substituting this facial triad carboxylate ligand in D201X variants maintained the same capacity for O2 activation, even when eliminating the carboxylate group as a ligand (X = Gly, Ala), however led to poor coupling and increased autohydroxylation. As the conservative D201E substitution retained this polar carboxylate ligand in a similar orientation as found for WT-FIH in the presence and absence of CTAD but uncoupled significantly, these polar contacts were insufficient to explain the timing of O2 reactivity in FIH. The narrowed approach angle to the diffusible ligand site for D201E appears to disfavor the bulkier H2O from binding while also adding greater steric push that would orient bound O2 toward a reactive orientation with αKG – we note that reorientation of NO was observed upon FIH/CTAD binding, and attributed to sterics from the CTAD-Asn803 sidechain [34]. Similarly, the poor coupling in the D201G and D201A variants could simply reflect the absence of stabilizing polar contacts to the aquo ligand, leading to variants that bind H2O poorly and are constitutively primed for O2 activation.
Conclusion
The αKG oxygenases utilize a sequential chemical mechanism in which O2 activation precedes the chemical reaction with substrate. The kinetic and structural data from D201X variants of FIH suggest a model for coupling of the two half reactions in which the native carboxylate residue of the His2Asp facial triad forms essential polar and steric contacts linkage between substrate binding to enzyme and O2 binding to Fe(II). As the distal O-atom of the facial triad carboxylate is a polar contact for an aquo ligand in the (Fe+αKG)FIH state, properly orienting this residue may be crucial for holding that H2O onto the Fe(II) site until CTAD binds. Once primary substrate binds, the distal O-atom is slightly pulled away from the aquo ligand[33] which allows O2 to reach the Fe leading to decarboxylation of αKG. Altering the facial triad ligand in the D201X variants did not impede O2 reactivity, but rather perturbed the structural linkage thereby disrupting the timing of O2 activation relative to CTAD binding. This relied on the contacts between the D201X residue, the Fe(II)-OH2 moiety, and incoming CTAD substrate; with any perturbation from the WT residue leading to uncoupled turnover in FIH. These results suggest that the sequential chemical mechanism found in the broad family of Fe(II)/αKG-dependent oxygenases may be a result of second-sphere contacts to the facial triad carboxylate residue.
Supplementary Material
Table 4.
Data collection and refinement statistics for the FIH D201E variant. Values in parentheses represent the highest resolution shell.
| Data set | (Zn2++αKG)D201E | (Fe2++αKG)D201E | (Zn2++αKG)D201E/CTAD |
|---|---|---|---|
| PDB ID | 5JWL | 5JWK | 5JWP |
| λ (Å) | 1.08 | 1.08 | 1.08 |
| Space group | P41212 | P41212 | P41212 |
| Unit cell (Å, °) | a = b = 86.17, c = 149.06 | a = b = 86.13, c = 149.21 | a = b = 86.22, c = 14.90 |
| α = β = γ = 90 | α = β = γ = 90 | α = β = γ = 90 | |
| Resolution (Å) | 50–2.4 (2.44 – 2.40) | 50 – 2.3 (2.34 – 2.30) | 34 – 2.1 (2.14 – 2.1) |
| Rsym (%) | 13.0 (100) | 12.4 (88) | 7.0 (100) |
| Redundancy | 9.0 (9.5) | 8.2 (7.9) | 5.2 (5.3) |
| Mean I/σ (I) | 27 (2.7) | 23.4 (2.4) | 24.8 (2.3) |
| Completeness | 98.2 (100) | 99.4(100) | 99.5 (99.8) |
| Refinement | |||
| Resolution range (Å) | 37 – 2.4 | 37 – 2.3 | 34 – 2.1 |
| Number of Reflections | 22314 | 25594 | 28327 |
| Average B factor (Å2) | 67 | 66 | 52 |
| Rwork/Rfree (%) | 18.0/22.6 | 18.6/22.7 | 17.0/21.0 |
| RMS deviations | |||
| Bond lengths (Å) | 0.016 | 0.016 | 0.019 |
| Bond angles (°) | 1.755 | 1.866 | 1.904 |
| Ramachandran plot, residues in (%) | |||
| Preferred region | 92.9 | 94.4 | 96.0 |
| Allowed region | 7.1 | 5.6 | 4.0 |
| Disallowed region | 0 | 0 | 0 |
Highlights.
The enzyme factor-inhibiting hypoxia inducible factor-1α is known as FIH
A His2-Asp facial triad is a common motif in α-ketoglutarate-dependent oxygenases
The Asp structurally links O2 and substrate binding sites
Replacing the Asp ligand leads to uncoupled O2 activation
The Asp ligand is essential for tight coupling in FIH
Acknowledgments
Funding Sources
This research was supported by the U.S. National Institutes of Health (1R01-GM077413 to M.J.K). J.A.H and C.Y.T. were supported in part by the NIH Chemistry-Biology Interface Predoctoral Training Grant T32-GM008515. These crystallographic data were collected at the Advanced Photon Source on the Northeastern Collaborative Access Team beamline 24-ID-C, which is supported by a grant from the National Institute of General Medical Sciences (P41 GM103403) from the National Institutes of Health.
We thank Jeanne A. Hardy for support of the crystallography on this project, and Scott Garman for helpful comments. We also thank Kay Perry at beamline 24- ID-C at the Advanced Photon Source at Argonne National Laboratories for assisting us with remote collection. This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357.
Abbreviations
- αKG
α-ketoglutarate
- CTAD
C-terminal transactivation domain
- FIH
factor-inhibiting HIF
- HAT
hydrogen atom transfer
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- HIF
hypoxia inducible factor-1α
- MALDI-TOF-MS
Matrix assisted laser desorption ionization-time of flight-mass spectrometry
- MCD
magnetic circular dichroism
- MLCT
metal-to-ligand charge-transfer
- MPH
MES-PIPES-HEPES buffer
- NOG
N-oxalylglycine
- P4H
prolyl-4-hydroxylase
- TauD
taurine dioxygenase
- TFA
trifluoroacetic acid
- VTVH
variable temperature variable field
Footnotes
The authors declare no competing financial interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hausinger RP. Crit Rev Biochem Mol Biol. 2004;39:21–68. doi: 10.1080/10409230490440541. [DOI] [PubMed] [Google Scholar]
- 2.Kovaleva EG, Lipscomb JD. Nat Chem Biol. 2008;4:186–193. doi: 10.1038/nchembio.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hangasky JA, Taabazuing CY, Valliere MA, Knapp MJ. Metallomics. 2013;5:287–301. doi: 10.1039/c3mt20153h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hangasky JA, Saban E, Knapp MJ. Biochemistry. 2013;52:1594–1602. doi: 10.1021/bi3015482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Matthews ML, Krest CM, Barr EW, Vaillancourt FH, Walsh CT, Green MT, Krebs C, Bollinger JM. Biochemistry. 2009;48:4331–4343. doi: 10.1021/bi900109z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li M, Miiller TA, Fraser BA, Hausinger RP. Arch Biochem Biophys. 2008;470:44–53. doi: 10.1016/j.abb.2007.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Phelan RM, Townsend CA. J Am Chem Soc. 2013;135:7496–7502. doi: 10.1021/ja311078s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hotopp JCD, Hausinger RP. Biochemistry. 2002;41:9787–9794. doi: 10.1021/bi026057a. [DOI] [PubMed] [Google Scholar]
- 9.McCusker KP, Klinman JP. J Am Chem Soc. 2010;132:5114–5120. doi: 10.1021/ja909416z. [DOI] [PubMed] [Google Scholar]
- 10.Saban E, Chen YH, Hangasky JA, Taabazuing CY, Holmes BE, Knapp MJ. Biochemistry. 2011;50:4733–4740. doi: 10.1021/bi102042t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hangasky JA, Ivison GT, Knapp MJ. Biochemistry. 2014;53:5750–5758. doi: 10.1021/bi500703s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pektas S, Taabazuing CY, Knapp MJ. Biochemistry. 2015;54:2851–2857. doi: 10.1021/bi501540c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hewitson KS, Holmes SL, Ehrismann D, Hardy AP, Chowdhury R, Schofield CJ, McDonough MA. J Biol Chem. 2008;283:25971–25978. doi: 10.1074/jbc.M804999200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grzyska PK, Ryle MJ, Monterosso GR, Liu J, Ballou DP, Hausinger RP. Biochemistry. 2005;44:3845–3855. doi: 10.1021/bi048746n. [DOI] [PubMed] [Google Scholar]
- 15.Bollinger JM, Price JC, Hoffart LM, Barr EW, Krebs C. Eur J Inorg Chem. 2005:4245–4254. [Google Scholar]
- 16.Aik W, McDonough MA, Thalhammer A, Chowdhury R, Schofield CJ. Curr Opin Struc Biol. 2012;22:691–700. doi: 10.1016/j.sbi.2012.10.001. [DOI] [PubMed] [Google Scholar]
- 17.Blomberg MRA, Borowski T, Himo F, Liao RZ, Siegbahn PEM. Chemical Reviews. 2014;114:3601–3658. doi: 10.1021/cr400388t. [DOI] [PubMed] [Google Scholar]
- 18.Solomon EI, Light KM, Liu LV, Srnec M, Wong SD. Acc Chem Res. 2013;46:2725–2739. doi: 10.1021/ar400149m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhou J, Kelly WL, Bachmann BO, Gunsior M, Townsend CA, Solomon EI. J Am Chem Soc. 2001;123:7388–7398. doi: 10.1021/ja004025+. [DOI] [PubMed] [Google Scholar]
- 20.Light KM, Hangasky JA, Knapp MJ, Solomon EI. J Am Chem Soc. 2013;135:9665–9674. doi: 10.1021/ja312571m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Neidig ML, Brown CD, Light KM, Fujimori DG, Nolan EM, Price JC, Barr EW, Bollinger JM, Krebs C, Walsh CT, Solomon EI. J Am Chem Soc. 2007;129:14224–14231. doi: 10.1021/ja074557r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hoffart LM, Barr EW, Guyer RB, Bollinger JM, Jr, Krebs C. Proc Natl Acad Sci USA. 2006;103:14738–14743. doi: 10.1073/pnas.0604005103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hegg EL, Whiting AK, Saari RE, McCracken J, Hausinger RP, Que L. Biochemistry. 1999;38:16714–16726. doi: 10.1021/bi991796l. [DOI] [PubMed] [Google Scholar]
- 24.Mirica LM, McCusker KP, Munos JW, Liu HW, Klinman JP. J Am Chem Soc. 2008;130:8122. doi: 10.1021/ja800265s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hangasky JA, Gandhi H, Valliere MA, Ostrom NE, Knapp MJ. Biochemistry. 2014;53:8077–8084. doi: 10.1021/bi501246v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Grzyska PK, Appelman EH, Hausinger RP, Proshlyakov DA. Proc Natl Acad Sci USA. 2010;107:3982–3987. doi: 10.1073/pnas.0911565107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wong SD, Srnec M, Matthews ML, Liu LV, Kwak Y, Park K, Bell CB, Alp EE, Zhao JY, Yoda Y, Kitao S, Seto M, Krebs C, Bollinger JM, Solomon EI. Nature. 2013;499:320. doi: 10.1038/nature12304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fujimori DG, Barr EW, Matthews ML, Koch GM, Yonce JR, Walsh CT, Bollinger JM, Krebs C, Riggs-Gelasco PJ. J Am Chem Soc. 2007;129:13408. doi: 10.1021/ja076454e. [DOI] [PubMed] [Google Scholar]
- 29.Proshlyakov DA, Henshaw TF, Monterosso GR, Ryle MJ, Hausinger RP. J Am Chem Soc. 2004;126:1022–1023. doi: 10.1021/ja039113j. [DOI] [PubMed] [Google Scholar]
- 30.Riggs-Gelasco PJ, Price JC, Guyer RB, Brehm JH, Barr EW, Bollinger JM, Krebs C. J Am Chem Soc. 2004;126:8108–8109. doi: 10.1021/ja048255q. [DOI] [PubMed] [Google Scholar]
- 31.Price JC, Barr EW, Hoffart LM, Krebs C, Bollinger JM. Biochemistry. 2005;44:8138–8147. doi: 10.1021/bi050227c. [DOI] [PubMed] [Google Scholar]
- 32.Dann CE, Bruick RK, Deisenhofer J. Proc Natl Acad Sci USA. 2002;99:15351–15356. doi: 10.1073/pnas.202614999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Elkins JM, Hewitson KS, McNeill LA, Seibel JF, Schlemminger I, Pugh CW, Ratcliffe PJ, Schofield CJ. J Biol Chem. 2003;278:1802–1806. doi: 10.1074/jbc.C200644200. [DOI] [PubMed] [Google Scholar]
- 34.Taabazuing CY, Fermann J, Garman S, Knapp MJ. Biochemistry. 2016;55:277–286. doi: 10.1021/acs.biochem.5b01003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Taabazuing CY, Hangasky JA, Knapp MJ. J Inorg Biochem. 2014;133:63–72. doi: 10.1016/j.jinorgbio.2013.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Coleman ML, McDonough MA, Hewitson KS, Coles C, Mecinovic J, Edelmann M, Cook KM, Cockman ME, Lancaster DE, Kessler BM, Oldham NJ, Ratcliffe PJ, Schofield CJ. J Biol Chem. 2007;282:24027–24038. doi: 10.1074/jbc.M704102200. [DOI] [PubMed] [Google Scholar]
- 37.Chen YH, Comeaux LM, Eyles SJ, Knapp MJ. Chem Commun. 2008:4768–4770. doi: 10.1039/b809099h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen YH, Comeaux LM, Herbst RW, Saban E, Kennedy DC, Maroney MJ, Knapp MJ. J Inorg Biochem. 2008;102:2120–2129. doi: 10.1016/j.jinorgbio.2008.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Otwinowski Z, Minor W. Method Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 40.Mccoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Emsley P, Cowtan K. Acta Crystallogr D. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 42.Murshudov GN, Vagin AA, Dodson EJ. Acta Crystallogr D. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 43.Bailey S. Acta Crystallogr D. 1994;50:760–763. [Google Scholar]
- 44.Price JC, Barr EW, Tirupati B, Bollinger JM, Jr, Krebs C. Biochemistry. 2003;42:7497–7508. doi: 10.1021/bi030011f. [DOI] [PubMed] [Google Scholar]
- 45.Saban E, Flagg SC, Knapp MJ. J Inorg Biochem. 2011;105:630–636. doi: 10.1016/j.jinorgbio.2011.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Salowe SP, Marsh EN, Townsend CA. Biochemistry. 1990;29:6499–6508. doi: 10.1021/bi00479a023. [DOI] [PubMed] [Google Scholar]
- 47.Ryle MJ, Koehntop KD, Liu AM, Que L, Hausinger RP. Proc Natl Acad Sci USA. 2003;100:3790–3795. doi: 10.1073/pnas.0636740100. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.