

Summary
BinAB is a naturally occurring paracrystalline larvicide distributed worldwide to combat the devastating diseases borne by mosquitoes. These crystals are composed of homologous molecules, BinA and BinB, which play distinct roles in the multi-step intoxication process, transforming from harmless, robust crystals, to soluble protoxin heterodimers, to internalized mature toxin, and finally toxic oligomeric pores. The small size of the crystals, 50 unit cells per edge, on average, has impeded structural characterization by conventional means. Here, we report the structure of BinAB solved de novo by serial-femtosecond crystallography at an X-ray free-electron laser (XFEL). The structure reveals tyrosine and carboxylate-mediated contacts acting as pH switches to release soluble protoxin in the alkaline larval midgut. An enormous heterodimeric interface appears responsible for anchoring BinA to receptor-bound BinB for co-internalization. Remarkably, this interface is largely composed of propeptides, suggesting that proteolytic maturation would trigger dissociation of the heterodimer and progression to pore formation.
Keywords: BinAB mosquito-larvicidal toxin, pore forming toxin, XFEL, serial femtosecond crystallography, de novo MIRAS phasing, in vivo crystals
Mosquitoes continue to be the insects most injurious to human health, spreading devastating diseases such as malaria, filariasis, Dengue fever, West Nile encephalitis [WHO 2012], and more recently, Zika virus. Synthetic chemical insecticides are a cost effective means for reducing mosquito vector populations, but their intensive application results in resistance to them. Most recently, resistance to pyrethroid insecticides threatens the control of malaria. To deal with the problems of resistance and develop more environmentally sound and sustainable approaches to vector control, various biological and genetic-based control strategies are under development. These include the use of microorganisms such as wolbachial endosymbionts that interfere with pathogen development and transmission1, genetically engineered female mosquitoes incapable of flight or pathogen transmission2, and pathogens such as bacteria or fungi engineered for higher efficacy against larvae or adults3,4.
Currently, one of the environmentally safest and most efficient means of controlling mosquitos is the distribution of naturally occurring protein crystals from Bacillus thuringiensis subsp. israelensis (Bti) and Lysinibacillus sphaericus (formerly Bacillus sphaericus) as practiced in the United States, Germany, China, Thailand and Africa5. These proteins are toxic to their targets, but harmless to humans and other animals. The exquisite specificity and efficiency of the L. sphaericus binary toxin, BinAB, presumably arise from its structural complexity which allows it to navigate through a series of barriers en route and recognize only its intended target. Originating as protoxins inside L. sphaericus, BinA and BinB, in a 1:1 ratio, pack into crystals, themselves stored inside spores. The spores are distributed in aquatic environments where mosquitos breed. Larvae ingest the crystals, whereupon they dissolve in the alkaline midgut juices (pH 8 – 11), releasing heterodimers. These are activated by cleavage of four terminal propeptides. BinB recognizes a glycolipid-anchored maltase receptor located at the microvillar surface of midgut cells6 and assists the internalization of BinA, which carries the toxic function and encodes the host range7. Finally, the homologs re-associate into a toxic pore. Recently, the structure of BinB from L. sphaericus was determined8. Here, we elucidate the structure of BinAB crystals, revealing features which endow BinA and BinB with their respective functions, and we suggest how alkalinity and proteolytic activation trigger a series of structural rearrangements that navigate BinAB past barriers to reach its target while maintaining the resilient 1:1 association throughout the life cycle9.
Structure determination of the BinAB toxin
We collected data from the native BinAB complex and three heavy atom derivatives using serial femtosecond crystallography (SFX) methods at the Linear Coherent Light Source (LCLS) Coherent X-ray Imaging (CXI) instrument10, with the aim of phasing by multiple isomorphous replacement with anomalous scattering (MIRAS). We produced nanocrystals of L. sphaericus BinAB (Fig. 1a, b) by recombinant expression in B. thuringiensis cells and jetted them across the pulsed XFEL beam using either a gas dynamic virtual nozzle (pH 7 crystals)11 or electrospin injector (pH 5 and pH 10 crystals)12 (Extended Data Fig. 1a). Initial structure factor amplitudes were obtained with cctbx.xfel13, and then corrected for partiality with cctbx.prime14 (Extended Data Table 1). This procedure was essential to the success of phasing (Fig. 1c). Heavy-atom sites were successfully located for all three derivatives (Extended Data Fig. 1b-d), and their phasing information combined (Extended Data Fig. 1e and Extended Data Fig. 2a) producing a map of sufficient quality that 60% of the BinAB complex could be traced automatically (Extended Data Fig. 2a-c). Subsequent manual building led to a model with Rwork/Rfree of 0.164/0.200 at 2.25 Å resolution. (Extended Data Fig. 2b, c and Extended Data Table 1).
Figure 1. De novo phasing of SFX data collected by from nanocrystals of L. sphaericus BinAB.
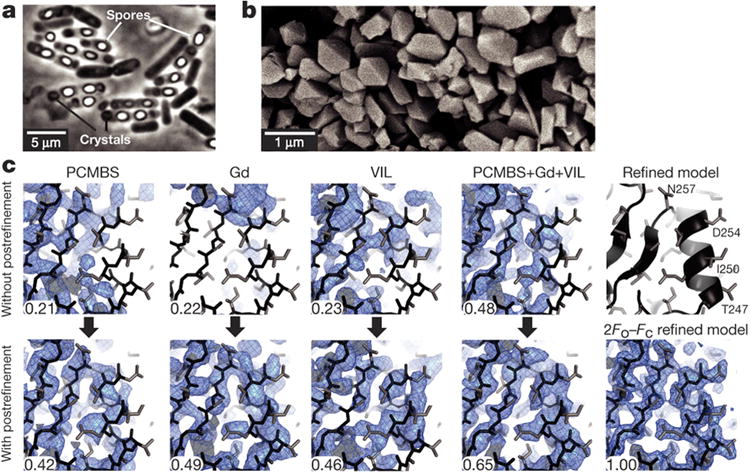
a-b, Phase contrast micrograph of B. thuringiensis sporulated cells engineered to produce BinAB nanocrystals (a) and scanning electron micrograph of isolated BinAB nanocrystals (a). c, The contribution of partiality refinement to the accuracy of the experimental phases is illustrated by maps calculated without (top row) and with (bottom row) benefit of the post-refinement procedure14. The left three columns show SIRAS-phased maps from PCMBS, Gd, and VIL derivatives. The fourth column shows MIRAS phased maps from all three derivatives. The right column shows the refined model and 2Fo-Fc map. The improvement from post-refinement is quantified by the correlation coefficient (CC) between each experimental map and the map phased by the final refined coordinates. Main chain and side chain atoms are shown in black and grey sticks, respectively.
The maps revealed two rod-shaped molecules, BinA and BinB, each approximately 100 Å long and 25-30 Å in diameter (Fig. 2a). They resemble each other closely (1.7 Å RMSD for 329 pairs of α-carbons) (Extended Data Fig. 2d), reflecting the 28% sequence identity (46% similarity) covering nearly their full lengths (Extended Data Fig. 2e). Each subunit comprises two domains: an N-terminal β-trefoil domain (BinA residues 1-155, BinB residues 1-198) and a C-terminal pore-forming domain (PFD) (BinA residues 156-370, BinB residues 199-448).
Figure 2. BinA and BinB folds and carbohydrate binding modules.
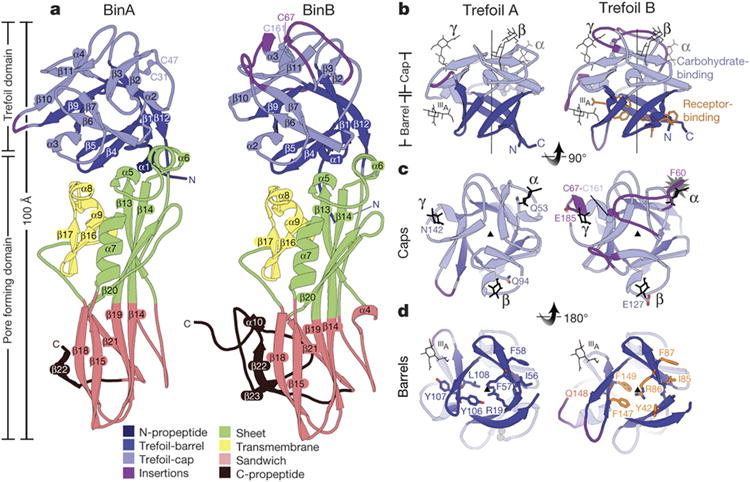
a, BinA and BinB are structurally similar to each other, each composed of trefoil and pore forming domains. The most noticeable differences correspond to insertions in surface loops on the trefoil domains (purple). b, The trefoil domains are composed of barrel and cap subdomains. In the cap subdomain, there are three canonical carbohydrate modules (α, β, γ). Carbohydrate molecules superimposed from structures of hemagglutinin (3AH1)29 and hemolytic lectin (1W3G)30 are shown in black sticks. These occupy the α, β, and γ binding modules. A fourth carbohydrate binding site, marked IIIA, is a minor site observed in hemagglutinin29. c, View of the cap subdomains along the pseudo-three fold symmetry axis; loop insertions (purple) break the symmetry in BinB. The starburst indicates a steric overlap between the modelled carbohydrate and the α module of BinB. The conflict arises from the 9-residue insertion in this loop, tethered by a disulphide bond, C67-C161. d, View of the barrel subdomains along the pseudo three-fold symmetry axis. BinB residues implicated in receptor binding are shown in sticks (orange). Structurally analogous residues are shown on BinA.
The most notable structural differences between BinA and BinB are located on the carbohydrate binding modules of the trefoil domains, suggesting that these may contribute to the distinct roles of BinA and BinB in intoxication. All three modules (α, β, γ) of BinA appear structurally capable of binding carbohydrate; however BinB contains four loop insertions relative to BinA (residues 62-70, 111-117, 139-143, and 179-185), which distort the trefoil's pseudo threefold-symmetry (Fig. 2c, Extended Data Figs. 2e and 3a). One insertion, 62-70, completely obstructs the α-module through a disulphide-linked tether, Cys67-Cys161 (Fig. 2c and Extended Data Fig. 3b). These differences suggest a lesser role for BinB in carbohydrate binding, or perhaps an adaptation to diverse carbohydrates. Carbohydrate binding has been linked with toxicity. Numerous mutations in these modules reduce toxicity (Supplementary Discussion; Supplementary Tables 1 and 2) and sugars such as chitobiose are potent antagonists of the BinA toxin15. The outward facing orientation of the modules in the BinAB dimer (Fig. 3a) indicates accessibility to cell surface glycoproteins and glycolipids, which may aid in concentrating BinAB at the cell surface, before receptor binding (Fig. 3b).
Figure 3. BinAB dimer assembly is weakened by proteolysis.
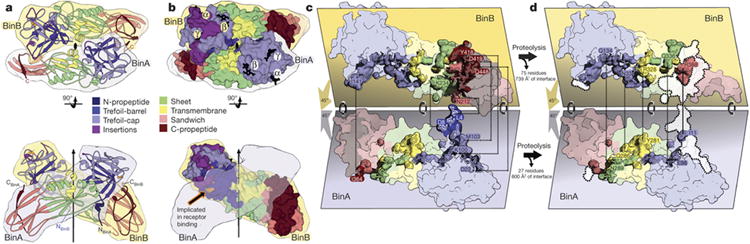
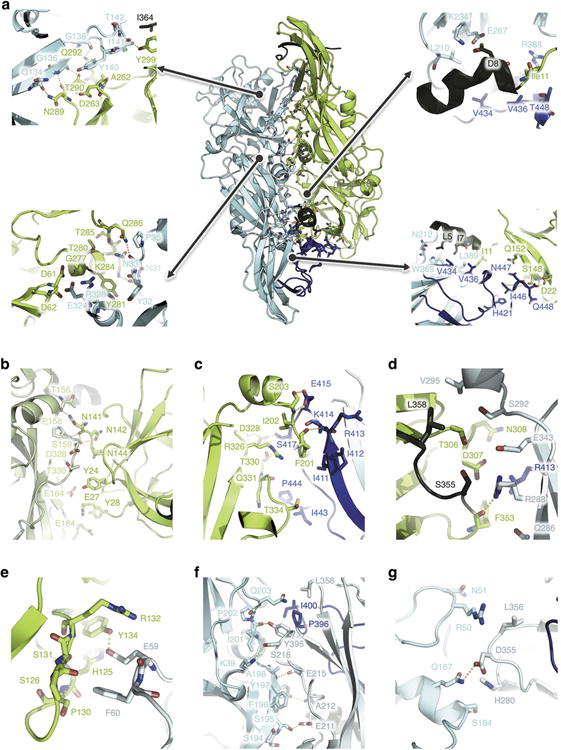
a, The BinAB dimer shown here manifests the largest intermolecular interface in the crystal. The vertical arrow and lens shaped symbol (black) indicate the position of the pseudo two-fold rotation axis that relates BinA and BinB. The interface extends over all four domains in both molecules. Propeptides (dark blue and dark red) play a substantial role in the interface. b, Sphere representation of the BinAB dimer shows that the canonical carbohydrate modules are accessible, but the receptor binding epitope is not, implying that carbohydrate binding occurs first, and then a conformational change exposes the receptor binding epitope. c, d, e Transformation of the BinAB interface accompanying proteolysis. Panel (c) illustrates the BinAB dimer split apart to reveal the interface. (d), All four subdomains and three propeptides contribute to the BinAB interface. Dashed lines connect select propeptide residues in contact across the dimer interface. (e) Dissociation of the propeptides following proteolysis eliminates 42% of the interface. Dotted lines encompassing white patches mark the interface lost after dissociation of the propeptides. Dashed lines connect select residues remaining in contact across the dimer interface following propeptide dissociation. The TM subdomain is the only subdomain that does not lose contacts after proteolysis.
Relatively few structural features distinguish BinA from BinB in the PFD, suggesting pore assembly may be heteromeric16. The PFD topology is characteristic of the aerolysin family of pore-forming toxins, comprising a 60 Å long antiparallel ß-sheet, folded into a sandwich at one end (sandwich subdomain), and adherent to a putative membrane-spanning segment (TM subdomain)17 at the other end (sheet subdomain) (Fig. 2a and Extended Data Fig. 4). This structural homology suggests that BinAB may form pores by a mechanism similar to aerolysin18, in which the TM subdomain refolds into a hairpin, oligomerizes into a β-barrel, and inserts into the membrane. However, no experiment has revealed whether these pores would be homo- or hetero-oligomeric. We find that the degree of structural similarity between the PFDs of BinA and BinB (1.4 Å RMSD over 203 α-carbons) (Extended Data Fig. 2d) is even closer than that observed between LukF and Hlg2 (1.5 Å RMSD over 241 α-carbons) which adopt alternate positions around the ring-shaped heterooctameric pore of γ-hemolysin19. A heteromeric assembly analogous to γ-hemolysin might explain why BinA and BinB form pores more efficiently in combination than either component separately16,20.
Propeptides at the BinAB dimer interface
The crystal packing reveals a unique and exceptionally large heterodimer interface, specifying a resilient 1:1 association between the two components. In this interface, the 100 Å-long BinA and BinB molecules cross each other at a 30° angle, forming an “X” shape (Fig. 3a, b). This interface is so large (burying 1855 Å2 on BinA and 1800 Å2 on BinB) that it accounts for nearly half of all the intermolecular interface area in the crystal. It exceeds by far the threshold value (856 Å2 per monomer) estimated to discriminate between biological and artificial dimers21. It is three times larger than the next largest interface in the crystal, and therefore it is likely to be the only one of the total seven intermolecular interfaces preserved after dissolution of the crystals (Extended Data Figs. 5 and 6, and Supplementary Tables 3-9). Its shape complementarity (0.65) is similar to that observed in antibody-antigen interfaces22 and electrostatic complementarity is evident (Extended Data Fig. 7a). The fit and extent of this heterodimer association implies stability, which likely contributes to attaining the 1:1 molar ratio shown to be optimal for receptor binding and toxicity (Supplementary Tables 1 and 2)
Further examination of the heterodimer interface reveals that its size would be severely reduced by proteolytic activation and this loss would appear to threaten access of BinA into the cell. Remarkably, 42% of the heterodimer interface involves propeptide segments, i.e. 1539 Å2 combined interface surface area (Fig. 3c, d, Extended Data Fig. 5c, Extended Data Fig. 6a, c, d, f, and Supplementary Tables 3 and 10). Therefore, removal of these cleavable segments before internalization would dismantle what may be the essential interface tethering BinA to its chaperone, the cell surface receptor-bound BinB. Indeed, deletion of the largest of the four propeptides (BinB residues 396-448) reduces the ability of this truncated mutant to direct the regional binding of BinA to target cells23. However, we note that due to the large surface area it buries in BinAB (3790 Å2), this 53-residue propeptide would be slow to release. Slow release may delay heterodimer dissociation until after internalization, when at the targeted location, it signals transformation into a pore.
Alkaline triggered release of BinAB dimers
Of all the transformations undergone by BinAB over its life cycle, our data bears most directly on the pH-signalled transformation from crystal to soluble dimers. Using crystallography as a means of structure elucidation, we can see in detail the crystal contacts which are the focus of this transformation and monitor their perturbation with elevation of pH from 7 to 10. The most fascinating attribute of this transformation is the seemingly contradictory combination of both stability and sensitivity that evolved to adapt the crystal to different stages of the life cycle. Crystal stability preserves and stores potency in harsh environments before ingestion, yet alkalinity readily dissolves the crystal in the larval midgut (pH 8-11) after ingestion, releasing BinAB to access cell surface receptors and activating proteases. The mechanism by which alkalinity lowers the high barrier to crystal dissolution is evident at three levels: amino acid composition, local, and global structural changes.
Alkalinity may facilitate crystal dissolution through the concerted deprotonation of BinAB's 49 tyrosine hydroxyl groups, occurring around pH 10 (pKaTyr = 10.1) (Extended Data Fig. 7b). The frequency of tyrosine residues (6%) is almost twice greater than the average for proteins (3.5%)24. We calculate that pH elevation from 7 to 10 would change the net charge on BinAB from -13.9 to -73.5 e, thereby destabilizing the crystals through negative electrostatic repulsion (Extended Data Fig. 7c, d). An analogous mechanism was proposed for the dissolution of the ultra-stable viral spindle crystals, which also contain an unusually high frequency of tyrosines (8.6%), some located strategically at crystal contacts25.
Alkalinity observably perturbs four regions in BinAB crystals, identified by peaks in a difference Fourier map (Fo-Fo) computed between datasets collected at pH 7 and pH 10 (Fig. 4, Extended Data Fig. 8 and Supplementary Tables 11 and 12). These regions involve contacts between molecules or subdomains. Two of these perturbations increase accessibility of propeptide segments to proteases. The N-terminal propeptide of BinA (residues 1-10) unravels from a helix to an extended conformation, following the loss of a hydrogen bond between Gly15(O) and Tyr213(OH) of BinA (Fig. 4c and Extended Data Fig. 9). Similarly, a pair of hydrogen bonds is broken in the BinAB dimer interface between the C-terminal carboxylate of the BinB propeptide (Gln448) and BinA Asp22 side-chain carboxylate (Fig. 4d). The third region is a lattice contact outside the dimer interface, between BinA Tyr134(OH) and BinB Glu59(OE2) which breaks at elevated pH (Fig. 4e and Supplementary Table 7). The fourth region involves loss of a hydrogen-bond between BinA Asp342(OD1) and Glu240(OE1). Located at the junction between the TM and sandwich subdomains, this break might be an early step toward pore formation (Fig. 4f and Extended Data Fig. 8). In each of these four regions, deprotonation is presumably the cause of hydrogen bond disruption, involving either a tyrosine hydroxyl or a carboxylate group paired with an obligate hydrogen bond acceptor (Fig. 4 and Supplementary Discussion).
Figure 4. pH sensing in BinAB crystals.
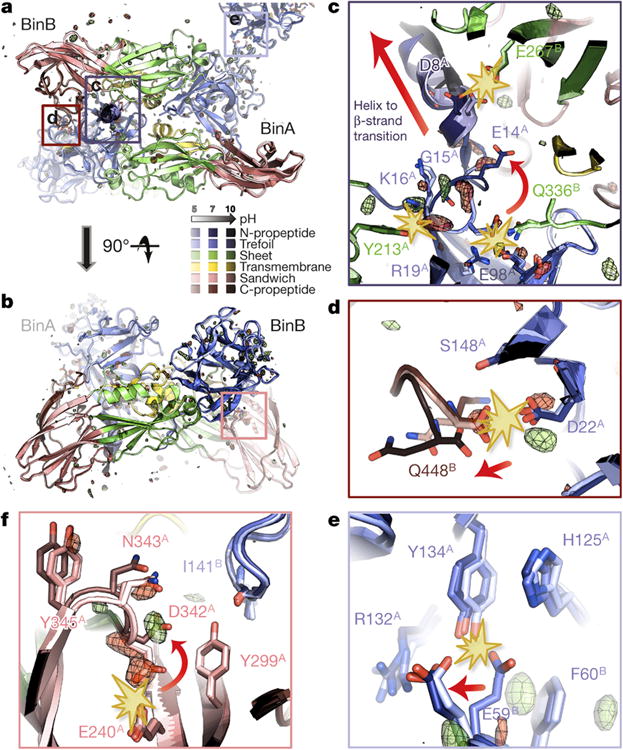
a-f, BinA and BinB cartoons are coloured by subdomain. The pH 5, pH 7 and pH 10 structures are increasingly shaded. Symmetry-related molecules are coloured similarly. The Fo[pH10] - Fo[pH7] map is superimposed at ± 3.5 σ, with positive and negative peaks shown in green and red, respectively. Panels (a) and (b) show orthogonal views of the BinAB dimer. The Fo-Fo map reveals four regions (c, d, e, f) of the BinAB crystal that are perturbed by elevated pH, probably reflecting early events in crystal dissolution. Rupture of H-bonds and conformational changes are highlighted by starbursts and arrows, respectively. (c); Deprotonation of Asp8, Glu14 and Tyr213 triggers a helix to extended β-strand conformational change in BinA N-term propeptide, increasing its accessibility to proteolytic cleavage. (d); Deprotonation of BinB Gln448 terminal carboxylate breaks its H-bond with BinA Asp22, weakening the BinA-BinB dimer interface and making the C-terminal propeptide of BinB available for proteolysis. (e); Deprotonation of BinA Tyr134 and His125 results in the rupture of their H-bonds with BinB Glu59. Loss of these contacts directly weakens the lattice. (f); Negative electrostatic repulsion pushes Asp342 away from Glu240 in BinA. This rearrangement could be an early step in the transformation into a pore owing to Asp342's location at the junction between the three PFD subdomains.
Alkalinity also induces global hinge motions, potentially straining the crystal lattice and thereby contributing to its dissolution. The trefoils of BinA and BinB move about 0.5 Å closer to their respective PFDs (Extended Data Fig. 9 and Supplementary Discussion). These motions might also foreshadow a rearrangement of the dimer interface, to expose the receptor binding motif, which is otherwise buried in the dimer interface (Fig. 3a, b).
To validate our model for alkaline-induced crystal dissolution and address concerns that the structural changes we attribute to pH elevation may have instead originated from unintended radiation damage (Supplementary Discussion), we mutated one of the four pH sensitive switches that we identified, BinA Asp22. Recall that upon pH elevation, withdrawal of donor hydrogen atoms breaks the bifurcated hydrogen bond between carboxylates of Asp22 and the C-terminus of BinB (Fig. 4d). Mutation from Asp to Asn should reduce toxicity by decreasing sensitivity of this interaction to alkaline-induced rupture and thereby delay release of the C-terminus for proteolytic activation. Affirmatively, this mutation did not change the appearance of the crystals; however, it reduced crystal solubility by 30% in the period 30-90 minutes after pH elevation (Extended Data Fig. 7e) and increased LC50 and LC95 by 11.6-fold and 24-fold, respectively (Supplementary Table 13). The striking effect of this single conservative substitution in a 93 kDa complex provides a convincing validation of our model.
Our structure of BinAB illuminates several important molecular events in the life cycle of the toxin. These include discovery of (1) four pH sensitive switches that facilitate crystal dissolution in the larval midgut, (2) a large heterodimer interface explaining how heterodimers persist after dissolution, (3) three competent carbohydrate binding modules in BinA that may assist in directing heterodimers to the cell surface, and (4) the potential to disrupt the heterodimer interface by proteolytic activation, thereby signalling remodelling. Our success in de novo MIRAS phasing of a crystallographic asymmetric unit nearly three times larger than any previously phased de novo by SFX26–28, and from crystals approximately 50 unit cells per edge, suggests this approach could be applied again in other cases where crystal size is limiting.
Methods
In vivo production of Lysinibacillus sphaericus BinAB crystals
L. sphaericus BinAB crystals were grown in a Bacillus thuringiensis strain engineered to improve crystal expression, which allowed growth of much larger crystals than occur naturally in L. sphaericus. There is no evidence that growth of the L. sphaericus BinAB crystals in B. thuringiensis in anyway affected the structure of BinAB; the toxicity per unit mass of the Bin crystals produced in B. thuringiensis is the same as that produced in L. sphaericus. Assuming that the cytosolic composition of L. sphaericus and B. thuringiensis cells do not strongly differ, crystal packing interactions are expected to persist and crystals formed by BinAB in the two types of cells should be identical in terms of space group and unit cell constants. Five hundred millilitres of glucose-yeast-salts (GYS) growth medium [0.1% glucose, 0.2% yeast extract, 0.05% K2HPO4, 0.2% (NH4)2SO4, 0.002% MgSO4, 0.005% MnSO4, and 0.008% CaCl2] supplemented with 25 μg/mL erythromycin was sterilized in a 2 L baffled flask and inoculated with spores from a lyophilized 5-day lysate of B. thuringiensis subsp. israelensis strain 4Q7 containing plasmid pPHSP-1 (Bti4Q7/pPHSP-1), which encodes BinA and BinB from L. sphaericus strain 2362 (Park et al., 1998)31. Of all known L. sphaericus strains, this strain produces the most potent toxin. Cultures were harvested after growth for 5 days at 30° C with shaking at 250 rpm, monitored by phase contrast light microscopy until sporulation and cell lysis were observed.
We attempted to obtain crystals of BinAB toxin incorporating the unnatural amino acid 3-iodo-tyrosine (3iTyr) at tyrosine positions in BinA and BinB. By exposing early sporulating cells to excess 3iTyr, phenylalanine, and tryptophan, we aimed to block aromatic amino acid biosynthesis and force the utilization of the iodine-labeled amino acid. Bti4Q7/pPHSP-1 cells were grown to late log phase and exposed to 3iTyr, phenylalanine, and tryptophan at concentrations of 100 mM each. Two further additions of the three amino acids, each increasing the concentration by 65 mM, were made at 24-hours intervals. Cultures were harvested after 4 days. However, SDS-PAGE analysis showed partial or no incorporation of 3iTyr in the BinAB crystals. Furthermore, there is no evidence of incorporation in the final structure of BinAB. Following culture lysis, spores, crystals, cells, and cell debris were pelleted by centrifugation at 6,000 × g for 30 min, then resuspended in 50 mL sterile water. The concentrated lysate was sonicated for 3 min on ice [1 s on, 1 s off (6 min elapsed time); 60% intensity] to lyse remaining cells. The sonicated lysate was pelleted at 6,000 × g for 30 min at 4° C. All subsequent steps were performed at 4° C. The pellet was washed in 50 mL cold water to remove soluble material and some of the spores, then re-pelleted, and resuspended in 15 mL water. Crystals were isolated from the suspension by sucrose gradient centrifugation as described in Sawaya et al., 2014 32. Before injecting the sample into the beam, large particles were removed by filtering the material with a 10 μm stainless steel frit.
Construction of pPHSP1 D22N and pBUSP-1 and pBUSP-1 D22N clones
The recombinant plasmid, pPHSP-1, containing BinA and BinB open reading frames under control of the pSTAB promoter 33 was used as template to amplify two DNA fragments each containing the D22N mutation in the BinA ORF, using Q5 polymerase (New England Biolabs, Beverly, MA). The first fragment began at the MluI site near the 3′ end of the BinB ORF and went through the D22 region of the BinA ORF, and contained the BinA D22N mutation; this fragment was amplified using primer pairs [1]. The second fragment began at the D22 region and continued to the end of the Bin operon terminator, and was amplified with primer pair [2]. Both fragments had approximately 15 bp overlapping homology regions with the vector on one end and the second fragment on the other. The two fragments were assembled along with pPHSP-1 linearized with MluI and PstI by recombineering using the Choo-Choo Cloning Kit (MCLAB, S. San Francisco, CA). The entire Bin operon of the resulting clone was sequenced to confirm the presence of the D22N mutation. Full-length fragments of the entire Bin operon of either wild-type pPHSP-1 or pPHSP-1-D22N, including approximately 15 bp vector homology regions on both ends, were amplified using primers [3], and assembled by recombineering in pBU434 linearized with KpnI and SalI. The resulting plasmids, pBUSP-1 and pBUSP-1 D22N, which also contained the 20-kDa helper protein gene as previously described35,36 were sequenced and used to transform Bacillus thuringiensis strain 4Q7 by electroporation33.
Primer pairs:
TTCACACTAAAACGCGTTAATGGTGAAATTG (BinA D22N For 1) and CTCGCTATTATAAAAATTCATAACGCGAATGTACTTTCCTTCTG (BinA D22N Rev 1)
CGTTATGAATTTTTATAATAGCGAGTATCCTTTCTGTATACATGCACC (Bin D22N Frag 2 F2 New) and CCAAGCTTGCATGCCTGCAGC (BinA D22N Rev 2)
GTGAATTCGAGCTCGGTACCGAATTCTATTTTCGATTTCAAATTTTCCAAAC (HSP Promoter For) and GGGTGTTAACGTCGACAAACAACAACAGTTTACATTCGA (HSP Bin Term. Rev)
Bioassay for mosquito larvicide activity
Lyophilized cultures containing spores and parasporal bodies of the L. sphaericus BinAB and BinAD22NB strains were resuspended in ddH2O. Suspensions were diluted to 6 to 7 different concentrations, ranging from 0.5 ng/ml to 1 μg/ml, in 6 oz cups in a final volume of 100 ml. Bioassays were replicated three times using 30 fourth-instars of S-Lab (Bin-sensitive) strains of Cx. quinquefasciatus. After 24 h of exposure at 28°C, dead larvae were counted and the 50% and 95% lethal concentrations, respectively, LC50 and LC95, were calculated by Probit analysis (POLO-PC; LeOra Software, Berkeley, CA).
Assay for alkaline induced crystal dissolution for wild type BinA-BinB and BinA(D22N)-BinB
The strains producing BinAB (4Q7/pPHSP-1) and BinAB(D22N) (4Q7/pBU-D22N) were grown in Nutrient Broth + Glucose (NBG) supplemented with, respectively, erythromycin (25 mg/ml) and tetracycline (3 mg/ml), for 5 days until > 95% of cells had sporulated and lysed. To isolate crystalline inclusions, spore/crystal mixtures collected from 50 ml cultures were resuspended in 15 ml ddH2O and sonicated twice at 50% duty cycle for 15 s using the Ultrasonic Homogenizer 4710 (Cole-Parmer Instrument Co.). Five-milliliter samples were loaded onto a sucrose gradient cushion (30-65% w/v), which was then centrifuged at 20,000 g for 45 min at 20°C in a Beckman L7-55 ultracentrifuge using the SW28 rotor. Bands containing inclusions were collected and washed three times in ddH2O, followed by centrifugation at 6,500 g for 15 min at 4°C after each wash, then lyophilized for storage at -20 °C until use. For solubilization assays, 200 μg or 800 μg of each crystal preparation was resuspended in, respectively, 100 μl of 10 mM Tris-Cl (pH 7; baseline control) or 400 μl of 10 mM Tris-Cl (pH 10; solubilization buffer). Assays were performed in triplicate. For each sample, three-25 μl aliquots were collected and spun at 16,300 × g for 2 min to pellet undissolved crystals, and supernatants were transferred to fresh microfuge tubes containing 75 μl of 10 mM Tris-Cl (pH 10). Absorbance at A280 nm was recorded at various time intervals.
Preparation of the mercury derivative
We produced the mercury derivative by soaking the BinAB crystals in 1 mM p-chloromercuribenzene sulfonate (PCMBS) for five months. Specifically, we added 200 μL of 0.1M PCMBS to 15 mL of crystal slurry at room temperature, and then we passed the mixture through a stainless steel frit with 10 μm pore size. We estimated the concentration of crystals in the slurry by pelleting the crystals in a centrifuge, then noting the crystals occupied approximately 25 μL of the 1 mL sample volume. The five months incubation was not intentional. We intended to use the derivative on the day it was prepared, which was during a previous data collection trip; however, this sample immediately clogged the gas dynamic virtual nozzle (GDVN) injector upon its first use. So, virtually none of this large volume of sample passed through the injector. In retrospect, we think the clogging was the result of mercuric iodide precipitation induced by residual iodide ion in the delivery lines; a derivative containing 0.5 M potassium iodide had indeed been run just previously to injecting the PCMBS derivative. To reduce the chance of this same PCMBS sample clogging again, we removed the non-covalently bound PCMBS by washing the crystals twice in deionized water a week before the LCLS experiment. The crystals used for derivatization in this experiment have the same origin as those we used to produce our effectively “native” data set. They were prepared under conditions intended to substitute 3iTyr in place of tyrosine, but the substitution was not efficient and so were effectively native before derivatizing with PCMBS.
Preparation of the gadolinium derivative
We produced the gadolinium derivative by soaking the BinAB crystals in 5 mM GdCl3. More specifically, we added 50 μL of 0.1M GdCl3 to 1 mL of crystal slurry at room temperature one day before the diffraction experiment. We estimated the concentration of crystals in the slurry by pelleting the crystals in a centrifuge, then noting the crystals occupied approximately 25 μL of the 1 mL sample volume. In order to conserve the amount of crystals used in screening for a heavy atom derivative, we recycled the crystal sample that had been used in previous diffraction experiments performed in helium vapour atmosphere (that is, not vacuum). For example, the crystals used in this experiment had been soaking for five months in KI and CsCl since the time they cycled through a GDVN nozzle for data collection in a previous XFEL experiment. The sample could be recycled without fear that the previous experiment damaged the crystals since over 99.9% of the crystals never intercepted the XFEL beam during the previous XFEL experiment. We washed the KI and CsCl from the crystals by pelleting the crystals and replacing the supernatant with water three times before adding GdCl3.
Preparation of the VIL derivative
We produced an iodinated tyrosine derivative by adapting the “vaporizing iodine labelling” (VIL) methods described by Miyatake et al. 37. The method uses gaseous iodine vapour to derivatize the ortho positions of accessible tyrosine residues. In our case, rather than derivatizing a few crystals in a standard 1-5 μL drop, we derivatized 1 mL of crystal slurry. To improve accessibility of the iodine vapour to this relatively larger volume of crystals, we distributed the 1 mL of BinAB crystal slurry evenly over nine wells of a glass depression dish (i.e. 111 μL per well). Four 10 μL aliquots of a KI/I2 mixture (0.67 M / 0.47 M concentration) were placed on the dish, adjacent, but not contacting the crystal slurry in the wells. The plate was sealed inside a sandwich box with 5 mL of water to act as a reservoir. The three components (drops of crystal slurry, the iodine, and the water reservoir) were separated in space but in vapour contact. The sandwich plate was sealed with tape at room temperature. The drops of crystal slurry turned yellow after 3.5 h. The crystals were harvested after 21 h. We estimated the concentration of crystals in the slurry by pelleting the crystals in a centrifuge, then noting the crystals occupied approximately 25 μL of the 1 mL sample volume. The crystals used in this experiment had the same history of origin as those used for the gadolinium derivative. They had been soaking for five months in KI and CsCl for a previous XFEL experiment, then jetted across the beam in helium vapour atmosphere, and washed three times to remove the non-covalently bound heavy atoms before derivatizing with iodine.
Sample injection using GDVN (pH 7 crystals)
BinAB nanocrystals of the native and derivatives (average volume is 0.75*0.5*0.25 μm3) were injected using a gas-focused dynamic virtual nozzle (GDVN) liquid microinjection system38 at the Coherent X-ray Imaging (CXI) instrument of LCLS10. Data were collected on two occasions, one for native data and one for heavy atom data, for a combined time of 245.9 minutes, with the detector framing at 120 Hz.
Preparation of pH 5 BinAB crystals
Crystals of native BinAB at pH 7 were pelleted by centrifugation at 10,000 × g for five minutes, and then resuspended in pH 5 buffer (0.1 M N-cyclohexyl-3-aminopropanesulfonate (CAPS) pH 10.0, 30% glycerol (v/v), 10% PEG 2000 MME, and 0.1 M NaCl). This preparation was originally intended to be at pH 10; however, it was later discovered that one of the components, polyethylene glycol monomethyl ether 2000 (PEG 2000 MME) was acidic (probably due to age), overpowering the pH 10 CAPS buffer, and bringing the final pH to 5. The pH of the crystal slurry was indicated by colour change on pH paper. The crystals were soaked at pH 5 for 10 minutes before the diffraction experiment.
Preparation of pH 10 BinAB crystals
Crystals of native BinAB at pH 7 were pelleted by centrifugation at 10,000 × g for five minutes, and then resuspended in pH 10 buffer (0.11 M CAPS pH 10.0, 33% glycerol (v/v), 11% methylpentanediol, and 0.11 M NaCl). The pH of the crystal suspension was verified by colour change on pH paper. The crystals were soaked at pH 10.0 for 5 hours before the diffraction experiment.
Sample Injection via MESH-on-a-stick (pH 5 and pH 10 crystals)
Crystals of native BinAB in pH 5 and pH 10 buffers were delivered in the microfluidic electrokinetic sample holder (MESH) method, described more fully in Sierra et al. 201212. The MESH injection system was modified (Extended Data Fig. 1a) to interface more readily with the CXI nozzle rods that mount interchangeably into the liquid sample delivery system. The crystal slurries were prepared as described above. A continuous 1.5 m long fused silica, polyamide coated capillary of 100 μm inner diameter and 360 μm outer diameter was used to deliver the sample into the CXI vacuum chamber. Approximately 800 μl of sample slurry with glycerol additive was placed in a microcentrifuge tube and placed in a small pressurized sample holder. The capillary and platinum wire were fed through the pressure cell and immersed in the slurry. A small backing pressure of 5 psig nitrogen gas was applied to aid the injection. The voltage was applied by a Stanford Research Systems PS350 (Sunnyvale, CA) high voltage source and was held between 4300-4500 V (currents < 1 μA) while the counter electrode was grounded. The flow rate was not directly measured, but we estimate that the sample consumption was approximately 2 μl/min, as judged from crude measurements of left-over sample volume. The pH 10 structure presented here was collected in less than one hour of continuous beamtime and consumed less than a millilitre of sample. The sample injection compared favourably to prior attempts at data collection by giving comparable resolution with only hundreds of microliters of sample consumed.
Data collection, indexing, merging and post-refinement
The sample chamber was at room temperature, under vacuum. Native and derivatives pH 7 datasets were collected with a XFEL beam focused to a 1 μm FWHM spot, and characterized by a wavelength of 1.21 Å (native crystals; 1.9 ×1011 photons per pulse) or 1.41 Å (heavy atom soaked crystals; 7.2 × 1011 photons per pulse). We chose a single wavelength for all three derivatives as a compromise of beam stability, time efficiency, and significant f″ for the elements used (Extended Data Fig. 1b). The pH 5 and pH 10 dataset were collected with a XFEL beam focused to a 100 nm FWHM spot (6.4×1011 photons per pulse), and characterized by a wavelength of 1.46 Å. Thus, the pH 5 and pH 10 datasets were collected with a ∼500 fold higher dose than the pH 7 dataset (Extended Data Table 1).
Images were reduced using cctbx.xfel13,39. No thresholding was applied to filter out blank shots; we attempted to index every frame. Further, for the heavy atom soaked data, if a frame indexed successfully, we removed the primary lattice from the list of candidate reflections and attempted to index again, searching for a secondary lattice. 8% of the final number of indexed images from the heavy atom data was from secondary lattices. In total, 24.5% of all recorded frames from the heavy atom data contained indexable patterns (13.2% for the native data), for a final total of 398,971 indexed patterns across the three derivatives and the native data set collected at pH 7. The dataset collected at pH 5 and pH 10 respectively consisted of 17,099 and 27,792 indexed patterns. First we merged and post-refined our data without negative intensities included using cctbx.prime14, which determines relative scale factors for each image, along with partiality estimates for each structure factor measurement. Post-refinement in this manner was critical for multiple isomorphous replacement with anomalous scattering (MIRAS) phasing as described below. Figure 2 illustrates, for the three derivatives and their combination thereof, the extent to which cctbx.prime improved the quality of the intensities, leading to improved phases, and thereby rendering the path of the peptidic chain visible in electron density map, where only broken density was visible before. Resolution cut-offs were determined based on completeness (> 90%), redundancy (> 4) and CC1/2 (> 0.14) ; in all datasets, <I/sigI> in the highest resolution shells are greater than 6. For all data sets, postrefinement corrected for the inherent partiality of SFX data and thus improved the σ-weighted (i.e. variance weighted) average estimate of structure factor amplitudes. Handling of negative intensities was then implemented in cctbx.xfel, allowing the production of meliorated post-refined datasets that are not plagued apparent twinning. We used the same criteria for the resolution cut-off, based on completeness on (> 90%), redundancy (> 4) and CC1/2 (> 0.14); in all datasets, the resulting <I/sigI> in the highest resolution shells are greater than 0.5. While inclusion of negative intensities dramatically affects <I/sigI>, it results in a stabilization of the refinement for the three native structures (pH 5, pH 7 and pH 10), requiring to impose a lower weight on geometry in phenix.refine to obtain a structure with a plateauing Rfree. Inclusion of negative intensities in the datasets also resulted in a decrease of noise levels in q-weighted structure factor amplitude Fourier difference maps (see below). At the atomic level, the only noticeable change between structures refined with and without negative intensities included is an increase in the B-factors, reflecting the increase of the Wilson-B – and possibly giving a more realistic description of the BinAB structures at room-temperature.
Structure determination by MIRAS
Heavy-atom sites were successfully located for all three derivatives using ShelxD40 (Extended Data Fig. 1c, d). The individual derivatives demonstrated limited phasing power, and the maps they produced were not interpretable (Extended Data Figs. 1d and 2a). Fortunately, the three derivatives were isomorphous (Extended Data Fig. 1e), and so could be combined to obtain a map of sufficient quality to automatically trace a partial BinAB model using the Phenix suite41 (Fig. 1c and Extended Data Fig. 2a, b), and extend using Arp/Warp42. The remaining residues were built manually, leading to a model with Rwork/Rfree of 0.164/0.200 at a resolution of 2.25 Å (Extended Data Table 1). After each cycle of model rebuilding, reciprocal space refinement (including refinement of coordinates, atomic displacement parameters, TLS parameters and occupancy) was carried out using phenix.refine. The final MIRAS model features residues 1-357 of BinA and 28-448 of BinB. Figure 2 illustrates the benefit of post-refinement to the accuracy of experimental phasing. For this figure, all SIRAS and MIRAS maps were calculated using MLPhaRe, followed by ten cycles of density modification with DM43 enforcing 58% solvent content, and displayed at 2.8 Å resolution. The CC quantifies the improvement gained from post-refinement. It reports on the whole asymmetric unit, not just the region illustrated in the figure.
Details of MIRAS phasing
We attempted to determine heavy-atom substructures for each of the three derivatives by supplying both isomorphous and anomalous difference signals (SIRAS), or only anomalous difference signal (SAD) to the program ShelxD 40 in super-sharpening mode (‘PSMF=-4′). Success was evidenced by the clear separation between populations of false and true solutions ranked by goodness-of-fit. Super-sharpening enhanced this separation. We found that SAD substructure determination was successful for the PCBMS (mercury) and Gd (gadolinium) derivatives, but not for the VIL (iodide) derivative (Extended Data Fig. 1d). SIRAS substructure determination were however successful for all three derivatives (Extended Data Fig. 1d). SIRAS phases of the mercury and iodide derivatives are of better quality than their SAD counterparts, as evidenced by a sharper separation between populations of false and true solutions ranked by goodness-of-fit (Extended Data Fig. 1d). In contrast, the SAD phases of the gadolinium derivative are better quality than its SIRAS phases.
Yet, for each individual derivative, the correct choice of hand remained ambiguous after initial density modification of SIRAS phased maps and chain tracing with ShelxE, as judged by a lack of separation in map quality statistics between the two hands, such as contrast, connectivity, and correlation coefficient for auto-traced chains (Extended Data Fig. 2a). We sought to maximize the distinction between hands by systematically computing a series of maps at different estimated solvent contents, from 55 to 73% and comparing the map quality statistics from opposing hands (Extended Data Fig. 2a). The true solvent content of BinAB crystals is 59.7%. We found that for each value of solvent content tested between 55% and 65%, a consistent choice of hand was indicated by the correlation coefficient of the automatically traced chain. However, at higher solvent content values tested, the opposite hand was indicated. Correlation coefficient of traces (CCs) were below 10% in all cases, and the differences between the CCs of the two hands in the 0.5-2.5 % range. So, the distinction between hands appeared to remain ambiguous at this point. For each derivative, we chose the hand having the most consistently (repeatedly) better correlation coefficient over the range of solvent content values tested. The refined heavy atom sites were input to Phenix.autosol from the Phenix 41 software suite for phase refinement, density modification and initial model building.
Attempts to solve the structure based on any of the single derivative (including single isomorphous replacement, single anomalous dispersion and single isomorphous replacement with anomalous scattering) were all unsuccessful. The combination of phase information from the three derivatives nonetheless produced usable maps, allowing us to obtain an experimental BinAB model. Our previous inability to distinguish the correct hand for the heavy atom substructure for the individual derivatives was overcome in Phenix through use of cross difference Fourier techniques, an option that becomes available with multiple derivatives (Extended Data Fig. 2a). Briefly, phenix.autosol (using Solve for phase combination) 44 was used to test various phase combinations schemes and to refine phases. While quality maps were obtained by combining anomalous and isomorphous signals from all three derivatives (MIRAS), the best map was obtained combining the anomalous signal of the gadolinium derivative with the combined anomalous and isomorphous differences of the mercury and iodide derivatives. Initial figures of merits (FOMs) were 0.12, 0.14 and 0.19 for the PCMBS, Gd and VIL substructures, respectively. The FOM of the best phase-combined map was 0.19 before density modification in Resolve 44, and 0.26 after density modification. The phase-and-build routine of Phenix was able to trace 296 residues from this map, resulting in a model displaying Rfree and Rfactor of 0.41 and 0.39, respectively (Extended Data Fig. 2b). Of importance, we had to optimize the parameters of phenix.autosol to obtain this model; in particular, we specified that the data were weak, that we needed to perform a thorough search, and that we needed to use “extreme-dm.”. The FOM of the density-modified map obtained after running this script was 0.72. From this map, phenix.autobuild was in 30 cycles able to reconstruct 501 residues, of which 183 were placed, corresponding to Rfree and Rfactor of 0.35 and 0.32, respectively.
This model and the corresponding phases were then input to a first round of Arp/Warp 42, fitting 62 additional residues into the BinA and BinB models (518 residues, 245 placed) (Extended Data Fig. 2b). This experimental yet incomplete BinAB model displayed Rfree and Rfactor of 0.32 and 0.29. The MIRAS model then underwent another cycle of model building in phenix.autobuild and then a second round of Arp/Warp, resulting in a model with 630 residues, of which 450 were putatively in sequence. This automatically fitted model was however unsatisfactory in terms Rfree, Rfactor and goodness of fit of the placed residues in the experimental map. Manual rebuilding was necessary, in order to correct wrongly assigned residues and to fit the complete BinAB sequence into the electron density maps. In one round of manual fitting, an additional 92 residues were built, leading to a total of 722 residues (299 and 423 residues in BinA and BinB, respectively) (Fig. 1c and Extended Data Fig. 2c). A cycle of reciprocal space refinement and yet another cycle of manual rebuilding produced a map which allowed fitting the 59 missing residues of BinA. The final model displays Rfree and Rfactor of 0.200 and 0.164, respectively (Extended Data Table 1), with 96.5% of residues in favoured region of the Ramachandran plot (0.5% Ramachandran outliers, 1.2% rotamer outliers). Of note, we could also phase and build the structure using MIRAS phases of all derivatives, i.e. without excluding the isomorphous differences from Gd, but the initial model turned out to be less complete, further highlighting the higher quality of the SAD phases of the gadolinium derivative, as compared to SIRAS (Extended Data Fig. 2b).
Determination of pH 7 structure by molecular replacement
Molecular replacement for BinAB (strain 2362) was performed with Phaser 45 using as a starting model the activated BinB structure 8 (PDB ID: 3WA1) from L. sphaericus strain 2297. BinB from the two strains differ in only five amino acids (A104S, R267K, L314Y, F317L, L389M). This search model became available only after we had begun our search for heavy atom derivatives. BinB shares only 28% identity with BinA. Two copies of BinB were found (corresponding to a solvent content of 68 %), one of which indeed corresponded to BinB, but the other to BinA. Corresponding residues were mutated and fit manually into the electron density maps using Coot 46. After each cycle of model rebuilding, reciprocal space refinement (including refinement of coordinates and atomic displacement parameters) was carried out using phenix.refine 41. The last cycle of refinement was performed using Buster 47 and included TLS refinement. Rwork and Rfree of the final model are 15.8 and 20.3%, respectively, at a resolution of 2.25 Å. Note that residues 1-11 of BinA and residues 1-34 of BinB were disordered in the electron density map and therefore not included in the model. This model is nearly identical to the independently built and refined MIRAS model (0.102 Å RMSD over 773 aligned α-carbons; see Supplementary Table 14). The BinAB model obtained by molecular replacement was never used in building the BinAB model to the experimental map. In fact, the two models were built independently by different authors. The similarity between the independently obtained models provided additional assurance of model accuracy.
Structure determination of BinAB at pH 5 and 10
We obtained experimental insights into pH induced conformational changes by calculating structure factor amplitude Fourier difference maps (Fo-Fo) between the pH 5, pH 7 and pH 10 datasets. To improve the estimate of structure factor amplitude differences, Fo-Fo maps were q-weighted as described48 and produced using a CNS49 custom-written script50. Application of the Q-weighting scheme to the diffraction data sets was essential to eliminate noise and amplify the difference signal. Figure 4 shows the FopH10-FopH7 map (Riso=0.28) phased with the pH 7 model (φpH7) and highlights 4 four regions highly sensitive to pH elevation (Fig. 4c-f). Extended Data Figure 8 shows, for each of these regions (Extended Data Fig. 8a-d) and for the two BinAB disulfides (Extended Data Fig. 8e-f), the six possible FopHj-FopHi, φpHi maps calculated from the pH 5, pH 7 and pH 10 datasets and structures. Supplementary Tables 11 and 12 respectively list all BinA and BinB residues that feature peaks higher than ± 3.5 σ and larger than 2 voxels in the FopH10-FopH7, φpH7 map. Sequence-wise integrations of the FopH10-FopH7, φpH7 and FopH5-FopH7, φpH7 map around BinA and BinB are given in Extended Data Figs. 8g and 8h, respectively.
We phased the pH 5 and pH 10 dataset by molecular replacement with Phaser45 using as a starting model the refined MIRAS (pH 7) structure. Conformational changes with respect to the latter were modelled manually. Reciprocal space refinement was performed using phenix.refine and included positional, B-factor, TLS and occupancy refinements. Rwork and Rfree of the final pH 5 model are 0.211 and 0.262, respectively, at a resolution of 2.5 Å, with 97.6% of residues in favoured region of the Ramachandran plot (0.4% Ramachandran outliers, 1.5% rotamer outliers). Rwork and Rfree of the final pH10 model are 0.165 and 0.211, respectively, at a resolution of 2.4 Å, with 98.9% of residues in favoured region of the Ramachandran plot (0.0% Ramachandran outliers, 0.0% rotamer outliers). Difference distance matrices (DDMs) are presented in Extended Data Fig. 9. Fo-Fo maps integration and DDM calculations were performed using custom-written scripts.
Electrostatic potential maps
pKa values were assigned using PROPKA51 and structures protonated in the AMBER force field at pH 7.5 and pH 10.5 using PDB2PQR52. Figures were produced using PyMOL53.
Extended Data
Extended Data Table 1. Data collection, phasing and refinement statistics.
| Native, pH 7 5F0Ya | PCMBSb | Gdc | VILd | Native, pH 10 5F0Ze | Native, pH 5 5G37f | |
|---|---|---|---|---|---|---|
| Data collection | ||||||
| Space group | P212121 | P212121 | P212121 | P212121 | P212121 | P212121 |
| Cell dimensions | ||||||
| a, b, c (Å) | 86.9, 97.4, 128.3 | 87.1, 97.8, 128.4 | 87.0, 97.5, 128.1 | 86.7, 97.3, 127.7 | 86.7, 97.3, 127.7 | 86.8, 97.0, 127.0 |
| α, β, γ (°) | 90, 90, 90 | 90, 90, 90 | 90, 90, 90 | 90, 90, 90 | 90, 90, 90 | 90, 90, 90 |
| Wavelength (Å) | 1.212 | 1.408 | 1.408 | 1.408 | 1.459 | 1.459 |
| X-ray beam focus (μm) | 1.3 | 1.3 | 1.3 | 1.3 | 0.25 | 0.25 |
| Photons/pulse (× 1011) | 1.9 | 7.2 | 7.2 | 7.2 | 6.4 | 6.4 |
| Pulse duration (fs) | 31.9 | 45.1 | 45.1 | 45.1 | 40.8 | 40.8 |
| Absorbed dose (GGy)g | 0.03 | 0.15 | 0.15 | 0.15 | 14.4 | 14.4 |
| Number of collected frames | 312,659 | 761,052 | 431,986 | 264,770 | 371,386 | 493,979 |
| Number of indexed patterns | 41,206 | 184,091 | 131,137 | 42,537 | 27,792 | 17099 |
| Number of indexed images accepted by cctbx.prime | 40,794 | 170,616 | 123,484 | 39,789 | 26,022 | 16224 |
| Resolution (Å)h | 43.5-2.25 (2.29-2.25) | 43.5-2.40 (2.44-2.40) | 43.5-2.35 (2.39-2.35) | 43.5-2.60 (2.64-2.60) | 43.5-2.37 (2.44-2.37) | 43.5-2.50 (2.54-2.50) |
| Number of observationsi | 12,854,588 | 53,361,546 | 39,314,393 | 11,249,082 | 12,069,895 | 5,495,157 |
| I/σI | 2.4 (0.5) | 2.8 (0.7) | 3.1 (0.6) | 3.7 (0.9) | 2.7 (0.9) | 6.34 (2.46) |
| CCl/2 | 96.8 (26.2) | 98.6 (16.2) | 98.4 (3.5) | 96.3 (17.5) | 97.3 (34.1) | 86.8 (28.0) |
| Completeness (%) | 99.7 (99.7) | 99.8 (96.9) | 99.8 (97.0) | 99.4 (96.2) | 99.9 (100.0) | 99.9 (89.4) |
| Multiplicity | 65.9 (4.8) | 372 (4.3) | 260.1 (4.4) | 100.4 (4.7) | 88.8 (19.2) | 76.62 (8.61) |
| Refinement | ||||||
| Refinement target function | Phased maximum-likelihood | Maximum-likelihood | Maximum-likelihood | |||
| Resolution (Å) | 2.25 (2.30 – 2.25) | 2.40 (2.46 – 2.40) | 2.50 (2.56 – 2.50) | |||
| Number of reflections | 52379 (3523) | 42817 (2923) | 37785(2557) | |||
| Rwork / Rfreeg | 0.164(0.288)/ 0.200 (0.327) | 0.165 (0.264)/ 0.211(0.311) | 0.211(0.357)/ 0.262 (0.427) | |||
| Number of atoms | ||||||
| Protein | 6479 | 6350 | 6415 | |||
| Water | 571 | 730 | 720 | |||
| B-factors (Å2) | ||||||
| Protein (BinA/BinB) | 47.5 / 40.7 | 52.8 / 47.3 | 39.2/36.8 | |||
| Water | 50.8 | 62.4 | 41.01 | |||
| R.m.s. deviations | ||||||
| Bond lengths (Å) | 0.006 | 0.007 | 0.002 | |||
| Bond angles (°) | 1.359 | 0.8 | 0.5 |
41,206 crystals were used to produce the native, pH 7 dataset.
184,091 crystals were used to produce the PCMBS dataset.
131,137 crystals were used to produce the Gd dataset.
42,537 crystals were used to produce the VIL dataset.
27,792 crystals were used to produce the native, pH 10 dataset.
17099 crystals where used to produce the native, pH 5 dataset.
As calculated by Raddose-3D70. Note that these dose calculations do not yet take into account the escape of photoelectrons from the diffracting volume. The track length in protein and water of photoelectrons has been estimated to 3 μm, for 1 Å wavelength X-rays71, suggesting that most photoelectrons travel outside of the diffracting volume without losing all their energy within it (crystals are 0.25 * 0.35 * 0.75 μm3 on average). Although these are not the only electrons to contribute to the damage, a preliminary photoelectron escape model incorporated into an unreleased version of RADDOSE-3D suggests that the absorbed dose is overestimated by a factor of ∼100 (J. Brooks-Bartlett and E. Garman, personal communication) for these small crystals.
Values in parentheses are for highest-resolution shell.
Including negative intensity observations.
Extended Data Figure 1. Data collection and heavy atom substructure determination.
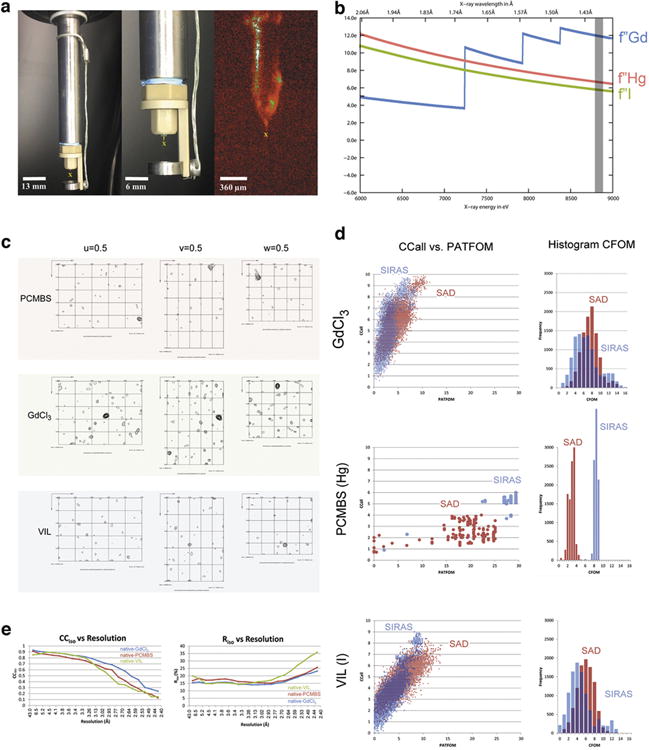
a, The “MESH-on-a-stick” sample injector configuration (see Methods). In the three panels, the yellow “X” below the capillary indicates the X-ray path into the page. The middle panel shows a closer view of the injector tip; the right panel shows an on-axis view of the sample injecting during the experiment. b, Our choice of X-ray wavelength for diffraction and MIRAS phasing was a compromise between maximizing the heavy atom anomalous signals, f″, as indicated by the curves for each element, and maximizing the number of data sets collected in the time allotted for the experiment. The grey bar corresponds to the wavelength we used, 1.41 Å. c, Difference Patterson maps calculated at 2.8 Å resolution. Sharpening (-5.0Å2) was applied to Hg and VIL maps. Coefficients for the PCMBS and VIL maps were obtained from both isomorphous and anomalous differences. The Gd difference Patterson map was calculated from anomalous differences only. Contours start at the 1.5 σ level and continue at 0.5 σ intervals. Peaks corresponding to vectors between heavy atoms stand out as high peaks, up to 7.5 σ. d, Heavy atom sites were located successfully for each of the three derivatives using the program, ShelxD. We compared the quality of potential heavy atom substructure solutions obtained from two sources of heavy atom signal: single wavelength anomalous dispersion (SAD) and a combination of anomalous dispersion and isomorphous differences (SIRAS). Ten thousand independent trials were performed for each derivative and signal source. Each dot in the scatter plots indicates the quality of an individual substructure solution. The vertical axis, labeled CCall, indicates the consistency between the potential solution and the diffraction data as the correlation coefficient between normalized structure factors, Ecalc and Eobs. The horizontal axis, labeled PATFOM (Patterson figure of merit), indicates the consistency between observed difference Patterson map and that predicted by the potential solution. Successful substructure determination is suggested by the appearance of a sharp separation between two populations of potential solutions: a cluster with lower values of CCall and PATFOM (incorrect solutions) and a cluster with higher values (correct solutions). Such is the case for all the trials performed, except for VIL using the SAD signal, where only a single population of solutions is observed. Evidently, the SAD signal was insufficient for accurate location of iodine sites. For VIL, we relied on the accuracy of sites obtained from the SIRAS signal. In most cases, the SIRAS (blue) signal is stronger than the SAD signal (red), indicating good isomorphism between native and derivative data sets. Only in the case of GdCl3, does the SAD signal appear better than the SIRAS signal. The histograms in the right column indicate the number of potential substructure solutions with given values of CFOM (combined figure of merit). The histograms recapitulate the trends observed in the scatter plots. e, The correlation coefficient (CCiso) measures the agreement and Riso measures discrepancy between the native structure factors and those of each of the derivatives. Each of our three derivative data sets shows isomorphism with the native data set up to 2.8 Å resolution.
Extended Data Figure 2. Structure solution and model building.
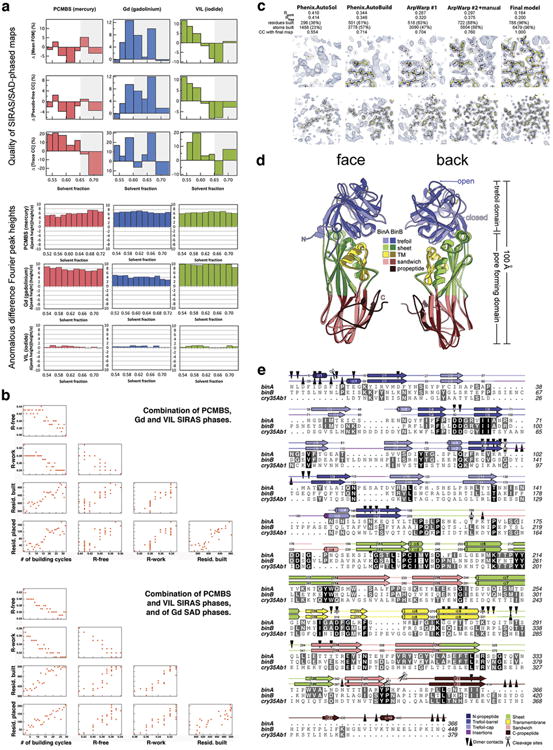
a, Evidence for choosing the correct hand of heavy atom substructures. We illustrate here two types of comparisons we used for choosing the correct hand of the heavy atom substructures: (1) comparisons of the quality of the SIRAS/SAD phased maps (upper three panels) and (2) comparisons of the heights of anomalous difference Fourier peaks (lower three panels). These comparisons are made between maps calculated in opposite hands; the correct hand is indicated by the individual with higher values. The disparity in values (Δ) is indicated on the vertical axes of the graphs. Greater |Δ| values indicate a stronger phasing power and more reliable choice of hand. There are six comparisons shown for each of the three heavy derivatives: PCMBS (mercury) in red, GdCl3 (gadolinium) in blue, and VIL (iodide) in green. The top three panels illustrate the percent difference between hands in the mean figure of merit (Mean-FOM), the pseudo-free correlation coefficient (Pseudo-free CC) of the density modified map, and the correlation coefficient of the trace (Trace-CC) as reported by ShelxE. The sites and phases were obtained from SIRAS signal for mercury and iodide, and from SAD signal for Gd. The most probable solvent content is ∼59 %, corresponding to one BinAB complex per asymmetric unit. However, we note conflicting choices of hand indicated by fluctuations in the sign of Δ accompanying small variations in the solvent content used in the density modification step (horizontal axes). We found that the difference Fourier maps (lower three panels) offered a stronger and more consistent indication of the choice of hand even when the statistics from SIRAS/SAD phased maps themselves differed little between hands. In these panels, SIRAS phases from the each heavy atom were used to compute three anomalous difference Fourier maps, using as coefficients, the anomalous differences from each derivative (rows 4, 5, 6). The graphs show that for all three derivatives, the original hand choice was correct (indicated by positive Δ), consistent across choices of solvent content (all Δ have the same sign within a graph), and consistent across sources of anomalous differences (all Δ have the same sign within a column). b, Automated tracing and model-building. Phases from the three derivatives were combined using SOLVE (see Methods). The hands which we decided on during the phasing step (a) were specified as ‘known’ to phenix.autosol. We then used RESOLVE (see Methods) to trace the density and build a model. Upper and lower panels show the progress of model building, depending on whether anomalous and isomorphous differences were combined from all derivatives (upper panel) or mixed phases (i.e. anomalous and isomorphous differences from PCMBS and VIL, and anomalous differences from the Gd derivative) (lower panel). Each panel shows scatter plots of R-free, R-factor, number of residues built and number of residues placed in sequence as a function of the number of cycle. The use of mixed phases allowed obtaining a better model, faster (lower panel). c, Electron density maps at various stages of model building. From left to right, the panels illustrate progressive improvement in map and model at two representative regions of BinAB (upper vs. lower panels). The number of residues built (including residues without side chains) is noted at each stage, as well as the number of protein atoms built. The quality of the maps at each stage is reported as a correlation coefficient with the map obtained from the final model. Approximately 60% of the total atoms in BinAB were built automatically. d, Comparison of BinA and BinB structures. Superposition of BinA (lighter colours) and BinB (darker colours) shows similarity between molecules, which superimpose with an RMSD of 1.7 Å over 329 pairs of α-carbons. The “face” view displays the surface involved in the BinA-BinB dimer interface, and the barrel subdomain of the trefoil is oriented toward the viewer. The back view displays the outward faces of the molecules, with the putative carbohydrate binding modules, in the cap subdomain, oriented toward the viewer. One of the largest structural differences is located in a surface loop on the back face, in the trefoil domain (blue). In BinB, a disulphide bond (Cys67-Cys161, yellow sticks) pins a surface loop (residues 60-74) away from the opening in the trefoil domain (open), whereas in BinA, the analogous loop (residues 34-46) is stabilized by a different disulphide bond (Cys31-Cys47, yellow sticks) to take a conformation that covers the opening in the trefoil domain (closed). e, Structure-based sequence alignment of BinA, BinB, and cry35Ab1. The secondary structure of BinA and BinB are shown above the sequences. Heterodimer contacts and cleavage sites are noted.
Extended Data Figure 3. Trefoil domains of BinA and BinB.
a, Structural relationships among trefoil domains illustrated by a phylogenetic tree plot. Four of the structures used for comparison were identified from a structural similarity search through the Protein Data Bank conducted by the Dali server (using BinA residues 6-156 as the probe). The top 4 hits occupy the top half of the plot (3AH1, 3VT2, 2E4M, and 4JP0). The remaining structures chosen for comparison (1W3G and 3ZXG) were selected based on their membership in the aerolysin family of toxins, of which BinA and BinB are members. That is, these are trefoils covalently linked to aerolysin type pore-forming domains. These are highlighted in blue text and include another insecticidal protein from B. thuringiensis, Cry35Ab1 (4JP0). Note that BinA and BinB are nearly as distant from each other as they are from the closest homologs, hemagglutinin, ricin, and Cry35Ab1. Carbohydrate molecules are shown in sticks where coordinates are available. Notable loop insertions in BinA and BinB are coloured in orange and magenta, respectively. b, Carbohydrate binding modules of BinA and BinB display different levels of structural integrity. No carbohydrates were included or observed in the crystals structure of BinAB. To investigate the structural integrity of the putative carbohydrate binding pockets of BinAB we superimposed coordinates of lectin (1W3G) and hemagglutinin (3AH1). These crystal structures illustrated in the left column are carbohydrate complexes chosen for their structural similarity to BinAB. Some modules appear competent for carbohydrate binding, such as the β and γ modules of BinA and the β module of BinB. Others show steric clash (yellow starburst) such as the α module of BinA and β module of BinB, which could be overcome by allowing adjustments in torsion angles. Notably, the α module of BinB is completely occluded by the insertion in its sequence (magenta) and stapled shut by a disulphide bond. In addition to the canonical α, β, and γ binding modules, 3AH1 displays another weakly bound carbohydrate marked site IIIA (bottom panel). This site is illustrated here because its superimposed coordinates lie adjacent to Y150 in BinB. The Y150A mutation causes complete loss of receptor binding 54.
Extended Data Figure 4. Pore-forming domains (PFD) of BinA and BinB.
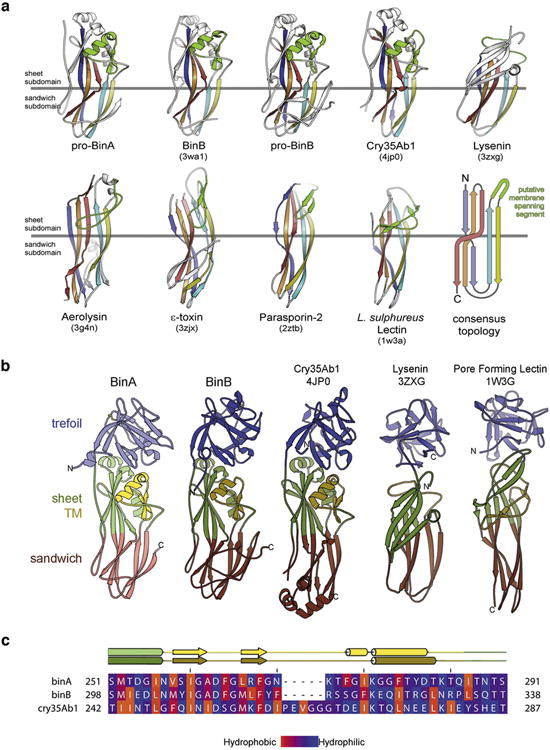
a, Topology of the aerolysin family of pore forming toxins. These share a core topology composed of five antiparallel β-strands and a putative membrane-spanning segment (green). PDB ID codes are included in parentheses. For clarity, we exclude from this illustration any accessory domains outside the pore forming module (PFM) of these toxins. The PFM is divided into two subdomains: a β-sheet subdomain at one end (above the horizontal grey line) and a β-sandwich subdomain at the opposite end (below the horizontal grey line). The length, twist, and number of strands vary between toxins. Also, the putative membrane-spanning segment (green) varies widely in secondary structure. However, in all cases this putative membrane-spanning segment is located between the second and third strands, suggesting these toxins might share a common mechanism of pore formation. b, Members of the aerolysin family that also contain a β-trefoil domain like BinAB. These are: Cry35Ab1 toxin from B. thuringiensis (4jp0) 55, lysenin, a haemolytic toxin from the earthworm Eisenia fetida (3zxg) 56, and a pore-forming lectin from the mushroom Laetiporus suphureus (1w3g) 30. c, Amphipathicity is evident in the sequence of the putative transmembrane spanning subdomain (TM) of BinA and BinB. The observed secondary structures of BinA and BinB are shown above the sequence alignment. The range of the TM subdomain is coloured yellow. Amino acids are coloured by hydrophobicity according to the scale given at the bottom. Note the alternating hydrophobic-hydrophilic pattern is especially prominent in the N-terminal half of the TM subdomain. This pattern is consistent with the proposal of an oligomeric membrane-spanning β-barrel. The figure was made using the program Jalview 57.
Extended Data Figure 5. Overview and analysis of molecular interfaces in the BinAB crystal.
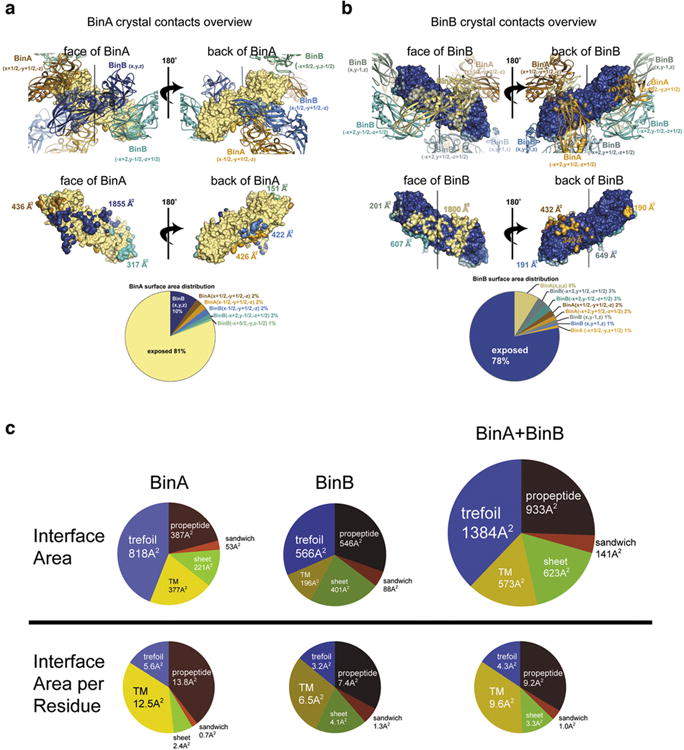
a, Overview of the six molecular interfaces involving BinA in the BinAB crystal. The reference copy of the BinA molecule is depicted as a beige coloured molecular surface and its six neighbouring molecules are shown as cartoon ribbons (upper panels). “Face” and “back” views (left and right panels), reveal opposite surfaces of the BinA molecule. The largest interface is with BinB (x,y,z) which is shown most clearly in the “face” view (left panels) in dark blue colour. It is the only interface of the six which is large enough to stretch over most of the length of the molecule. In all views, the pseudo two-fold axis relating BinA and BinB is in a vertical orientation (black line in upper panels). The areas of contact are illustrated on the BinA molecular surface (middle panels) in colours corresponding to the cartoon ribbons (upper panels). BinA molecules and surfaces are shown in amber colours; BinB molecules and surfaces are shown in blue-green colours. The pie chart shows the relative amounts of total BinA surface area buried by each of the six crystal contacts and the remainder that is solvent exposed. b, Overview of the eight molecular interfaces involving BinB in the BinAB crystal. The reference copy of the BinB molecule is depicted as a dark blue coloured molecular surface and its eight neighbouring molecules are shown as cartoon ribbons (upper panels). “Face” and “back” views (left and right panels), reveal opposite surfaces of the BinB molecule. The largest interface is with BinA (x,y,z) which is shown most clearly in the “face” view (left panels) in beige colour. It is the only interface of the eight which is large enough to stretch over most of the length of the molecule. In all views, the pseudo two-fold axis relating BinA and BinB is in a vertical orientation (black line in upper panels). The areas of contact are illustrated on the BinA molecular surface (middle panels) in colours corresponding to the cartoon ribbons (upper panels). BinA molecules and surfaces are shown in amber colours; BinB molecules and surfaces are shown in blue-green colours. The pie chart shows the relative amounts of total BinB surface area buried by each of the eight crystal contacts and the remainder that is solvent exposed. c, Distribution of BinA-BinB interface area over its subdomains. The pie charts in the upper half shows the area contributions to the principal BinA-BinB interface from each of the five named regions: trefoil domain, TM subdomain, sheet subdomain, sandwich subdomain, and combined N and C-terminal propeptides. The lower charts show analogous contributions on a per residue basis. That is, the area contributed by each region is divided by the total number of residues comprising that region. These pie charts emphasize the role of the TM domain in the dimer interface, perhaps to restrain this domain from inserting into a membrane until after the BinAB dimer dissociates. Interestingly, the higher efficiency of pore formation of BinA compared to BinB 20 correlates with the greater protection of its TM domain (12.5 Å2 buried per residue versus 6.5 Å2 buried per residue) in the dimer.
Extended Data Figure 6. Detailed views of the molecular interfaces in the BinAB crystal.
a-g, BinA and BinB are shown as green and cyan ribbon diagrams, respectively. The C-terminal pro-peptide of BinB (residues 396-448) is highlighted in blue, while the N- terminal (residues 1-10) and C-terminal (residues 354-367) propeptides of BinA are shown in dark green. Contacting residues are shown as sticks. Polar interactions within a 3.6 Å cut-off are highlighted by yellow dashes. The contacts illustrated in panels a, b, c, d, e, f and g are detailed in Supplementary Tables 3-9, respectively. (a); Molecular contacts between BinA (x,y,z) (green) and BinB (x,y,z) (cyan), i.e. within the biological dimer. A large part of this interface involves the C-terminal pro-peptide of BinB. (b); Molecular contacts between BinA (x,y,z) (green) and BinA (x+1/2,-y+1/2,-z) (lime green). (c); Molecular contacts between BinA (x,y,z) (green) and BinB (x-1/2,-y+1/2,-z) (cyan). This interface involves the pro-peptide of BinB (residues 396-448). (d); Molecular contacts between BinA (x,y,z) (green) and BinB (-x+2, y-1/2,-z+1/2) (cyan). This interface involves the pro-peptide of BinB (residues 396-448). (e); Molecular contacts between BinA (x,y,z) (green) and BinB (-x+5/2, -y,-z+1/2) (cyan). (f); Molecular contacts between BinB (x,y,z) (cyan) and BinB (-x+2, y-1/2,-z+1/2) (teal). A small part of this interface involves the pro-peptide of BinB (residues 396-448). (g); Molecular contacts between BinB (x,y,z) (cyan) and BinB (x, y-1, z) (teal).
Extended Data Figure 7. Electrostatic complementarity, tyrosine distribution, predicted electrostatic changes upon pH elevation and crystal solubilisation assays.
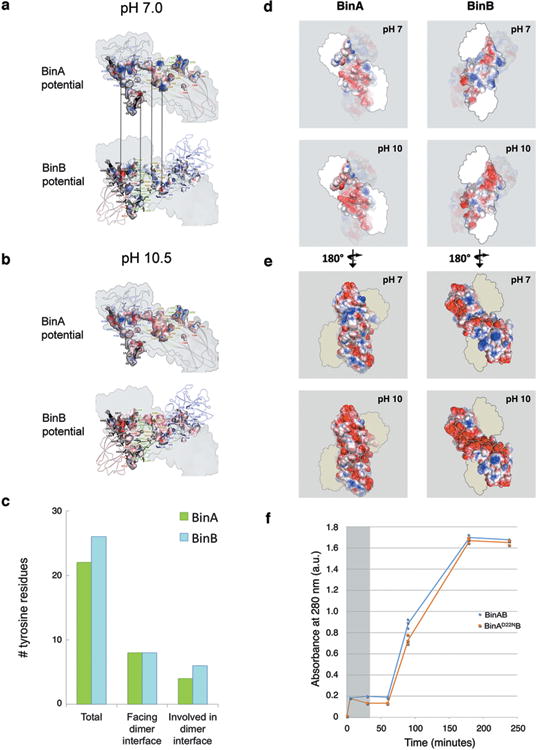
a, Electrostatic surface complementarity of the BinA-BinB interface. At pH 7.0 (left column), complementary charges are notable between the BinA electrostatic surface potential (top) and the BinB electrostatic surface potential (bottom). The complementarity in potential is highlighted by the vertical arrows connecting adjacent patches on opposing surfaces of the interface. At pH 10.5 (right column), deprotonation of tyrosine and increased negative charge on acid residues causes a reduction in electrostatic complementarity from 0.37 to 0.29 58. All panels depict the BinA surface of the BinAB dimer interface. In the upper panels, this surface is coloured by electrostatic surface potential of BinA; in the lower panel, this surface is coloured by electrostatic surface potential of BinB. Residues lining the interface (sticks) are labelled with colour corresponding to the domain to which it belongs. The colour scheme is as described in Extended Data Fig. 2a. BinA residues are labelled in the upper panel. BinB residues are labelled in the lower panel. In all panels, the pseudo two-fold axis relating BinA and BinB is in a vertical orientation (black line in Fig. 3a, lower panel).b, Distribution of tyrosine residues in the BinAB dimer. Of the total 49 tyrosine residues, 48 are ordered in the crystal structure. Of these, 20% are located in the dimer interface, which itself accounts for only 10% of the total molecular surface. Thus, the distribution of tyrosine residues is slightly more concentrated on the dimer interface compared to the remainder of the BinAB surface. Tyrosines outside the dimer interface are probably more prone to deprotonation than those within the dimer interface, due to differences in solvent accessibility. c-d, Electrostatic potential map of BinA and BinB. The surface of the BinAB dimer is depicted, coloured by electrostatic surface potential of BinA, on the left, and by that of BinB, on the right. In (e), the regions of BinA (left) and BinB (right) that participate in the dimer interface are highlighted. In (f), the external surface of the dimer is highlighted. In both (e) and (f), the upper and lower panels show the electrostatic surface potentials of BinA (left) and BinB (right) at pH 7 and pH 10.5, respectively. e, Alkaline induced crystal dissolution is delayed for BinA(D22N) mutant compared to wild type. Our structural data suggested that BinA Asp22 was an important pH sensor for triggering crystal dissolution at the high pH characteristic of the mosquito midgut. We reasoned that a D22N mutation in BinA would render the crystal less sensitive to pH by stabilizing a hydrogen bond with the BinB C-terminal carboxylate. We constructed a BinA D22N mutant and measured solubility of BinA(D22N)BinB crystals at pH 10, colleting three data points for each time point. We found its solubility in vitro decreased by 30% between 30 and 90 minutes at pH 10 compared to wild type crystals, but not at pH 7. After 90 minutes, crystals of wild type BinAB and BinA(D22N)BinB mutant are completely dissolved. This delay in crystal dissolution up to the 90 minutes time point is an important difference because in the fourth-instar Culex larvae, the larval feeding rate from the time particles are ingested until they are digested and exit the hindgut is 30 minutes (indicated by gray shading). Hence, the 60 minute delay that we see in our experiments with D22N is long enough to contribute to the striking loss of toxicity of more than 20-fold at the LC95 level (Supplementary Table 13). These results are consistent with the model of Asp22 serving as a pH sensor for crystallization.
Extended Data Figure 8. Comparison of FopHi-FopHj maps obtained from crystals receiving different X-ray doses suggests that the structural changes observed are due to pH change and not radiation damage.
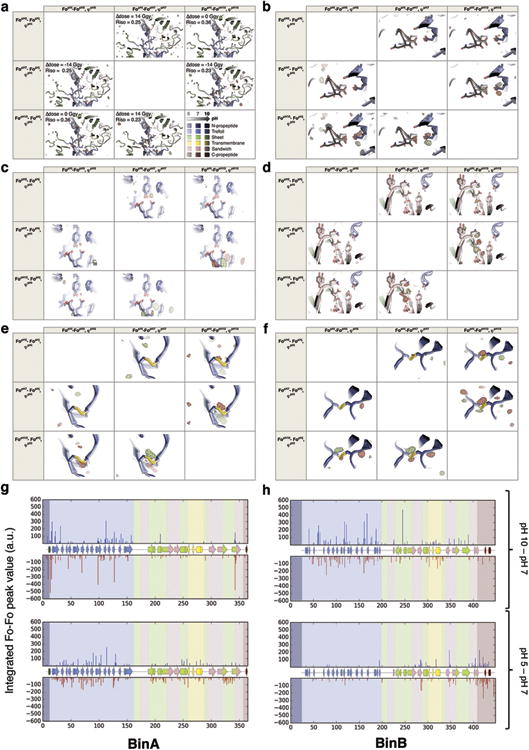
a-f, The pH 5 and pH 10 datasets were collected with a ∼500-fold higher dose than the pH 7 dataset, raising the concern that some of the peaks in the FopH10-FopH7, φpH7 map result from radiation damage, most notably those observed on disulphides. Panels a-d show, for each of the four regions identified as highly sensitive to pH elevation in Figure 4 (Fig. 4c-f), the six possible FopHj-FopHi, φpHi maps calculated from the pH 5, pH 7 and pH 10 datasets and structures. Panels e-f show these maps around the disulphides of BinA (e) and BinB (f). BinA and BinB are shown as cartoons, coloured by subdomain, as in Fig. 4. The cartoons range in colour from pale to medium, to dark, signifying the pH values 5, 7, and 10. Consistent with the hypothesis that a 500-fold difference in dose causes no significant structural change, we see a lack of peaks in the FopH5-FopH7, φpH7 map (Riso=0.26) around disulphides (e, f) and other pH sensitive residues (a-d). Consistent with the hypothesis that the peaks observed in the FopH10-FopH7, φpH7 map (Riso=0.23) are caused by pH change, these peaks are reproduced in the FopH10-FopH5, φpH5 and FopH5- FopH10, φpH10 maps (Riso =0.35). We interpret this pattern of peaks as implying movement of the disulphide bonds rather than their disruption. This movement accompanies pH-sensitive rigid-body motion of the trefoil domains (Extended Data Fig. 9). g-h, Peaks stronger than ± 3.5 σ were integrated contiguously in the FopH10- FopH7, φpH7 (upper panels) and FopH5-FopH7, φpH7 map (lower panels) and then assigned to the closest residue. The secondary structures of BinA (g) and BinB (h) are shown as cartoons, coloured by subdomain as in Fig. 4. The background of the sequence is also coloured by subdomain. The sequence-wise integration of the FopH10-FopH7, φpH7 map reveals that BinB is more affected by the pH elevation than BinA, and in both chains, the trefoil is more affected than the pore-forming domain. The propeptide and TM regions of both proteins are also sensitive to pH elevation. Peaks in the FopH5-FopH7, φpH7 map (lower panels) are smaller in magnitude and concentrated in the trefoil domain of BinA and the C-terminal propeptide of BinB. They correspond to side chains reorientation rather than increased dynamics or domain motion (Extended Data Fig. 9). The marked difference in pattern between the FopH10-FopH7, φpH7 and FopH5-FopH7, φpH7 map integrations is consistent with the hypothesis that the peaks observed in these maps are not due to radiation damage, but rather to pH induced conformational changes.
Extended Data Figure 9. Conformational changes in the BinAB dimer upon pH elevation from 7 to 10.
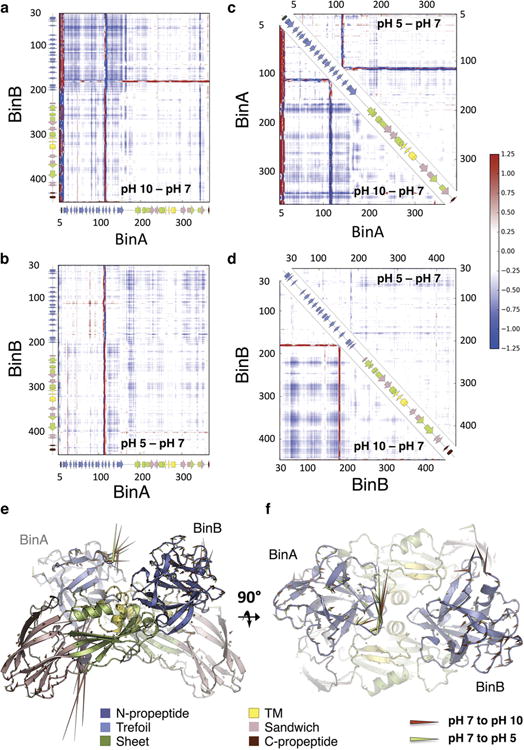
a-d, Distance difference matrices (DDMs) calculated between the pH 7 (reference) structure and either the pH 10 or the pH 5 structure. Blue and red indicate decreases and increases in Cα-Cα distances in the pH 10 or pH 5 structures as compared to the pH 7 structure, respectively. The secondary structures of BinA (a-c) and BinB (a, b, d) are recapitulated by cartoons on the side or the diagonal of the DDMs. These cartoons are coloured by subdomain as in Fig. 4. (a) Intermolecular (BinA vs. BinB) DDM between the pH 10 and the pH 7 structures. This DDM illustrates that the BinAB dimer contracts upon pH elevation, with the two trefoil domains coming closer to one another. This might be due to electrostatic repulsion at crystal contact zone 5 (Fig. 4e, Extended Data Fig. 6e and Supplementary Table 7), which involves the trefoils of BinA and BinB from two symmetry related dimers. (b); Intermolecular (BinA vs. BinB) DDM between the pH 5 and the pH 7 structures. The pH5 structure is overall slightly more compact than the pH 7 structures but shows no major conformational changes. (c, d); Intramolecular DDMs of BinA (c) and BinB (d). Changes in Cα-Cα distances between the pH 10 and the pH 7 structures are reported below the diagonal, while those between the pH 7 and the pH 5 structures are shown above the diagonal. The pH 5 and pH 7 structures of BinA. (c) and BinB (d) are overall similar, with only the BinA loop Ile110-Arg120 and BinB loop Lys175-Ser184 showing a noticeable change of conformation. In contrast, the pH 10 structures of BinA (c) and BinB (d) appear more compact. On the local level, striking conformational changes are observed upon pH elevation in the N-terminal propeptide of BinA, in loops Ile110-Thr120 (trefoil) and Asn341-Tyr345 (Pore forming domain: PFD) of BinA, and in loop Lys175-Ser184 (trefoil) of BinB. The increase in compactness is due to the trefoil domain coming closer to the PFD, in both BinA and BinB. BinA loop Ile110-Thr120 appears sensitive to both pH elevation and decrease. e-f, Porcupine plots depicting differences between structures of BinAB for pH 7 vs. 5 (green arrows) and pH 7 vs. 10 (red arrows). The pH7 structure of BinAB is shown, coloured by subdomain as in Fig. 4. The movement of Cα atoms is indicated by arrows on the ribbon representation, with the magnitude of motions illustrated by length of arrows exaggerated by 2.5 Å to increase visibility (for all atoms that move by more than 0.1 Å). (e); View of the BinAB dimer, in a orientation similar to Fig. 4b. As compared to Fig. 3a, b, this view is rotated by 180° around the vertical axis. (f); View from the top of the trefoil domains; this face of the BinAB dimer is presumably that interacting with the apical membrane of larvae midgut cells. The view in (f) is 90° apart from that in (e).
Supplementary Material
Acknowledgments
We acknowledge the help of the following people during data collection: Sangho Lee, Jake Koralek, Robert Shoeman, Sabine Botha, Bruce Doak, Oliver Zeldin. We thank Anne Volveda for advice regarding sequence-wise Fourier difference map integration, Jonathan Brooks-Bartlett and Elspeth Garman for help with dose calculations, and Martin Weik for stimulating discussions and continuing support. We thank the HCIA program of HHMI, the W.M. Keck Foundation (Grant 2843398), NIH (Grant AG-029430), National Science Foundation (Grant MCB 0958111) and DOE (DE-FC02-02ER63421) (to D.S.E.), the France Alzheimer Foundation (FA-AAP-2013-65-101349) and the Agence Nationale de la Recherche (ANR-12-BS07-0008-03) (to J.-P.C.), NIH Grants GM095887 and GM102520 for data-processing methods (to N.K.S.), and NIH Grant AI45817 (to B.A.F.). Support by the CNRS (PEPS-SASLELX-2013, PEPS-SASLELX-2014) funded travel to LCLS. Use of the Linac Coherent Light Source (LCLS), SLAC National Accelerator Laboratory, is supported by the U.S. Department of Energy, Office of Science, and Office of Basic Energy Sciences under Contract no. DE-AC02-76SF00515.The CXI instrument was funded by the Linac Coherent Light Source Ultrafast Science Instruments project funded by the DOE Office of Basic Energy Sciences. Parts of the sample injector used at LCLS for this research was funded by the National Institutes of Health, P41GM103393, formerly P41RR001209.
Footnotes
Supplementary Information is linked to the online version of the paper at www.nature.com/nature
Author Contributions: J.-P.C., M.R.S., M.G., J.A.R., D.C., B.A.F. and D.S.E designed and coordinated the project. M.G. carried out the in vivo production of BinAB nano-crystals. H.W.P., D.K.B., and B.A.F., engineered bacterial strain to produce large BinAB crystals. R.L.R and B.A.F. performed toxicity assays. R.H.H. designed the mutagenesis protocol. M.R.S. performed heavy atom derivatizations. J.-P.C., M.R.S., J.A.R., D.C., A.S.B., T.M.-C., S.B., G.J.W., M.M., D.P.D., R.G.S., H.L., J.E.K., M.S.H., N.C., M.U. and N.K.S. acquired and processed data. J.-P.C. and M.R.S, carried out the MIRAS and MR phasing, and built and refined the atomic models. R.G.S. and H.L. developed the MESH-on-a-stick injector. J.-P.C., M.R.S., and D.S.E prepared the manuscript with input from M.G., J.A.R., D.C., A.S.B., T.M.-C., R.G.S., M.S.H., A.T.B., B.A.F., and N.K.S.
Atomic coordinates and structure factors have been deposited in the Protein Data Bank under accession codes 5FOY (pH 7 structure), 5FOZ (pH 10 structure) and 5G37 (pH 5 structure).
The authors declare no competing financial interests.
Readers are welcome to comment on the online version of the paper.
References
- 1.Moreira LA, et al. A Wolbachia symbiont in Aedes aegypti limits infection with dengue, Chikungunya, and Plasmodium. Cell. 2009;139:1268–1278. doi: 10.1016/j.cell.2009.11.042. [DOI] [PubMed] [Google Scholar]
- 2.Alphey N, Bonsall MB, Alphey L. Combining pest control and resistance management: synergy of engineered insects with Bt crops. J Econ Entomol. 2009;102:717–732. doi: 10.1603/029.102.0233. [DOI] [PubMed] [Google Scholar]
- 3.Federici BA, Park HW, Bideshi DK. Overview of the basic biology of Bacillus thuringiensis with emphasis on genetic engineering of bacterial larvicides for mosquito control. Open Toxinol J. 2010;3:83–100. [Google Scholar]
- 4.Fang W, et al. Development of transgenic fungi that kill human malaria parasites in mosquitoes. Science. 2011;331:1074–1077. doi: 10.1126/science.1199115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Worrall E, Fillinger U. Large-scale use of mosquito larval source management for malaria control in Africa: a cost analysis. Malar J. 2011;10:338. doi: 10.1186/1475-2875-10-338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Darboux I, Nielsen-LeRoux C, Charles JF, Pauron D. The receptor of Bacillus sphaericus binary toxin in Culex pipiens (Diptera: Culicidae) midgut: molecular cloning and expression. Insect Biochem Mol Biol. 2001;31:981–990. doi: 10.1016/s0965-1748(01)00046-7. [DOI] [PubMed] [Google Scholar]
- 7.Berry C, et al. Genetic determinants of host ranges of Bacillus sphaericus mosquito larvicidal toxins. J Bacteriol. 1993;175:510–8. doi: 10.1128/jb.175.2.510-518.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Srisucharitpanit K, et al. Crystal structure of BinB: a receptor binding component of the binary toxin from Lysinibacillus sphaericus. Proteins. 2014;82:2703–2712. doi: 10.1002/prot.24636. [DOI] [PubMed] [Google Scholar]
- 9.Opota O, et al. Bacillus sphaericus binary toxin elicits host cell autophagy as a response to intoxication. PloS One. 2011;6:e14682. doi: 10.1371/journal.pone.0014682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liang M, et al. The Coherent X-ray Imaging instrument at the Linac Coherent Light Source. J Synchrotron Radiat. 2015;22:514–519. doi: 10.1107/S160057751500449X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.DePonte DP, et al. SEM imaging of liquid jets. Micron. 2009;40:507–509. doi: 10.1016/j.micron.2008.12.009. [DOI] [PubMed] [Google Scholar]
- 12.Sierra RG, et al. Nanoflow electrospinning serial femtosecond crystallography. Acta Crystallogr D Biol Crystallogr. 2012;68:1584–1587. doi: 10.1107/S0907444912038152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hattne J, et al. Accurate macromolecular structures using minimal measurements from X-ray free-electron lasers. Nat Methods. 2014;11:545–548. doi: 10.1038/nmeth.2887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Uervirojnangkoorn M, et al. Enabling X-ray free electron laser crystallography for challenging biological systems from a limited number of crystals. eLife. 2015;4 doi: 10.7554/eLife.05421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Broadwell AH, Baumann P. Proteolysis in the gut of mosquito larvae results in further activation of the Bacillus sphaericus toxin. Appl Environ Microbiol. 1987;53:1333–1337. doi: 10.1128/aem.53.6.1333-1337.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cokmus C, Davidson EW, Cooper K. Electrophysiological effects of Bacillus sphaericus binary toxin on cultured mosquito cells. J Invertebr Pathol. 1997;69:197–204. doi: 10.1006/jipa.1997.4660. [DOI] [PubMed] [Google Scholar]
- 17.Melton JA, Parker MW, Rossjohn J, Buckley JT, Tweten RK. The identification and structure of the membrane-spanning domain of the Clostridium septicum alpha toxin. J Biol Chem. 2004;279:14315–14322. doi: 10.1074/jbc.M313758200. [DOI] [PubMed] [Google Scholar]
- 18.Degiacomi MT, et al. Molecular assembly of the aerolysin pore reveals a swirling membrane-insertion mechanism. Nat Chem Biol. 2013;9:623–629. doi: 10.1038/nchembio.1312. [DOI] [PubMed] [Google Scholar]
- 19.Yamashita K, et al. Crystal structure of the octameric pore of staphylococcal γ-hemolysin reveals the β-barrel pore formation mechanism by two components. Proc Natl Acad Sci U S A. 2011;108:17314–17319. doi: 10.1073/pnas.1110402108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schwartz JL, et al. Permeabilization of model lipid membranes by Bacillus sphaericus mosquitocidal binary toxin and its individual components. J Membr Biol. 2001;184:171–183. doi: 10.1007/s00232-001-0086-1. [DOI] [PubMed] [Google Scholar]
- 21.Ponstingl H, Henrick K, Thornton JM. Discriminating between homodimeric and monomeric proteins in the crystalline state. Proteins. 2000;41:47–57. doi: 10.1002/1097-0134(20001001)41:1<47::aid-prot80>3.3.co;2-#. [DOI] [PubMed] [Google Scholar]
- 22.Lawrence MC, Colman PM. Shape complementarity at protein/protein interfaces. J Mol Biol. 1993;234:946–950. doi: 10.1006/jmbi.1993.1648. [DOI] [PubMed] [Google Scholar]
- 23.Oei C, Hindley J, Berry C. Binding of purified Bacillus sphaericus binary toxin and its deletion derivatives to Culex quinquefasciatus gut: elucidation of functional binding domains. J Gen Microbiol. 1992;138:1515–1526. doi: 10.1099/00221287-138-7-1515. [DOI] [PubMed] [Google Scholar]
- 24.Klapper MH. The independent distribution of amino acid near neighbor pairs into polypeptides. Biochem Biophys Res Commun. 1977;78:1018–1024. doi: 10.1016/0006-291x(77)90523-x. [DOI] [PubMed] [Google Scholar]
- 25.Chiu E, et al. Structural basis for the enhancement of virulence by viral spindles and their in vivo crystallization. Proc Natl Acad Sci U S A. 2015 doi: 10.1073/pnas.1418798112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Barends TRM, et al. De novo protein crystal structure determination from X-ray free-electron laser data. Nature. 2014;505:244–247. doi: 10.1038/nature12773. [DOI] [PubMed] [Google Scholar]
- 27.Yamashita K, et al. An isomorphous replacement method for efficient de novo phasing for serial femtosecond crystallography. Sci Rep. 2015;5:14017. doi: 10.1038/srep14017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fukuda Y, et al. Redox-coupled proton transfer mechanism in nitrite reductase revealed by femtosecond crystallography. Proc Natl Acad Sci. 2016;201517770 doi: 10.1073/pnas.1517770113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nakamura T, et al. Sugar-binding sites of the HA1 subcomponent of Clostridium botulinum type C progenitor toxin. J Mol Biol. 2008;376:854–867. doi: 10.1016/j.jmb.2007.12.031. [DOI] [PubMed] [Google Scholar]
- 30.Mancheño JM, Tateno H, Goldstein IJ, Martínez-Ripoll M, Hermoso JA. Structural analysis of the Laetiporus sulphureus hemolytic pore-forming lectin in complex with sugars. J Biol Chem. 2005;280:17251–17259. doi: 10.1074/jbc.M413933200. [DOI] [PubMed] [Google Scholar]
Additional references for Methods, Supplementary Discussion and Supplementary Tables
- 31.Park HW, Ge B, Bauer LS, Federici BA. Optimization of Cry3A yields in Bacillus thuringiensis by use of sporulation-dependent promoters in combination with the STAB-SD mRNA sequence. Appl Environ Microbiol. 1998;64:3932–3938. doi: 10.1128/aem.64.10.3932-3938.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sawaya MR, et al. Protein crystal structure obtained at 2.9 Å resolution from injecting bacterial cells into an X-ray free-electron laser beam. Proc Natl Acad Sci U S A. 2014;111:12769–12774. doi: 10.1073/pnas.1413456111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Park HW, et al. Recombinant larvicidal bacteria with markedly improved efficacy against culex vectors of west nile virus. Am J Trop Med Hyg. 2005;72:732–738. [PubMed] [Google Scholar]
- 34.Bourgouin C, Delécluse A, de la Torre F, Szulmajster J. Transfer of the toxin protein genes of Bacillus sphaericus into Bacillus thuringiensis subsp israelensis and their expression. Appl Environ Microbiol. 1990;56:340–344. doi: 10.1128/aem.56.2.340-344.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Park HW, Hice RH, Federici BA. Effect of Promoters and Plasmid Copy Number on Cyt1A Synthesis and Crystal Assembly in Bacillus thuringiensis. Curr Microbiol. 2016;72:33–40. doi: 10.1007/s00284-015-0911-x. [DOI] [PubMed] [Google Scholar]
- 36.Wu D, Federici BA. A 20-kilodalton protein preserves cell viability and promotes CytA crystal formation during sporulation in Bacillus thuringiensis. J Bacteriol. 1993;175:5276–5280. doi: 10.1128/jb.175.16.5276-5280.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Miyatake H, Hasegawa T, Yamano A. New methods to prepare iodinated derivatives by vaporizing iodine labelling (VIL) and hydrogen peroxide VIL (HYPER-VIL) Acta Crystallogr D Biol Crystallogr. 2006;62:280–289. doi: 10.1107/S0907444905041909. [DOI] [PubMed] [Google Scholar]
- 38.Weierstall U, Spence JCH, Doak RB. Injector for scattering measurements on fully solvated biospecies. Rev Sci Instrum. 2012;83:035108. doi: 10.1063/1.3693040. [DOI] [PubMed] [Google Scholar]
- 39.Sauter NK, Hattne J, Grosse-Kunstleve RW, Echols N. New Python-based methods for data processing. Acta Crystallogr D Biol Crystallogr. 2013;69:1274–1282. doi: 10.1107/S0907444913000863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sheldrick GM. A short history of SHELX. Acta Crystallogr A. 2008;64:112–122. doi: 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- 41.Adams PD, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Langer G, Cohen SX, Lamzin VS, Perrakis A. Automated macromolecular model building for X-ray crystallography using ARP/wARP version 7. Nat Protoc. 2008;3:1171–1179. doi: 10.1038/nprot.2008.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Winn MD, et al. Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr. 2011;67:235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Terwilliger TC. SOLVE and RESOLVE: automated structure solution and density modification. Methods Enzymol. 2003;374:22–37. doi: 10.1016/S0076-6879(03)74002-6. [DOI] [PubMed] [Google Scholar]
- 45.McCoy AJ, et al. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 47.Blanc E, et al. Refinement of severely incomplete structures with maximum likelihood in BUSTER-TNT. Acta Crystallogr D Biol Crystallogr. 2004;60:2210–2221. doi: 10.1107/S0907444904016427. [DOI] [PubMed] [Google Scholar]
- 48.Ursby T, Bourgeois D. Improved Estimation of Structure-Factor Difference Amplitudes from Poorly Accurate Data. Acta Crystallogr A. 1997;53:564–575. [Google Scholar]
- 49.Brunger AT. Version 1.2 of the Crystallography and NMR system. Nat Protoc. 2007;2:2728–2733. doi: 10.1038/nprot.2007.406. [DOI] [PubMed] [Google Scholar]
- 50.Colletier JP, et al. Shoot-and-Trap: Use of specific x-ray damage to study structural protein dynamics by temperature-controlled cryo-crystallography. Proc Natl Acad Sci. 2008;105:11742–11747. doi: 10.1073/pnas.0804828105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Olsson MHM, Søndergaard CR, Rostkowski M, Jensen JH. PROPKA3: Consistent Treatment of Internal and Surface Residues in Empirical pKa Predictions. J Chem Theory Comput. 2011;7:525–537. doi: 10.1021/ct100578z. [DOI] [PubMed] [Google Scholar]
- 52.Dolinsky TJ, Nielsen JE, McCammon JA, Baker NA. PDB2PQR: an automated pipeline for the setup of Poisson–Boltzmann electrostatics calculations. Nucleic Acids Res. 2004;32:W665–W667. doi: 10.1093/nar/gkh381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.The PyMOL Molecular Graphics System. Schrödinger, LLC; [Google Scholar]
- 54.Singkhamanan K, Promdonkoy B, Chaisri U, Boonserm P. Identification of amino acids required for receptor binding and toxicity of the Bacillus sphaericus binary toxin. FEMS Microbiol Lett. 2010;303:84–91. doi: 10.1111/j.1574-6968.2009.01865.x. [DOI] [PubMed] [Google Scholar]
- 55.Kelker MS, et al. Structural and biophysical characterization of Bacillus thuringiensis insecticidal proteins Cry34Ab1 and Cry35Ab1. PloS One. 2014;9:e112555. doi: 10.1371/journal.pone.0112555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.De Colibus L, et al. Structures of Lysenin Reveal a Shared Evolutionary Origin for Pore-Forming Proteins And Its Mode of Sphingomyelin Recognition. Structure. 2012;20:1498–1507. doi: 10.1016/j.str.2012.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Waterhouse AM, Procter JB, Martin DMA, Clamp M, Barton GJ. Jalview Version 2—a multiple sequence alignment editor and analysis workbench. Bioinformatics. 2009;25:1189–1191. doi: 10.1093/bioinformatics/btp033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.McCoy AJ, Chandana Epa V, Colman PM. Electrostatic complementarity at protein/protein interfaces. J Mol Biol. 1997;268:570–584. doi: 10.1006/jmbi.1997.0987. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.