

Abstract
Cutaneous exposure to solar ultraviolet (UV) radiation is a major causative factor in skin carcinogenesis, and improved molecular strategies for efficacious chemoprevention of non-melanoma skin cancer (NMSC) are urgently needed. Toll-like receptor 4 (TLR4) signaling has been shown to drive skin inflammation, photoimmunosuppression and chemical carcinogenesis. Here we have examined the feasibility of genetic and pharmacological antagonism targeting cutaneous TLR4 for the suppression of UV-induced NF-κB and AP-1 signaling in keratinocytes and mouse skin. Using immunohistochemical and proteomic microarray analysis of human skin, we demonstrate for the first time that a significant increase in expression of TLR4 occurs in keratinocytes during the progression from normal skin to actinic keratosis (AK), also detectible during further progression to squamous cell carcinoma. Next, we demonstrate that siRNA-based genetic TLR4 inhibition blocks UV-induced stress signaling in cultured keratinocytes. Importantly, we observed that resatorvid (TAK-242), a molecularly-targeted clinical TLR4 antagonist, blocks UV-induced NF-κB and MAP kinase/AP-1 activity and cytokine expression (Il-6, Il-8, and Il-10) in cultured keratinocytes and in topically treated murine skin. Taken together, our data reveal that pharmacological TLR4 antagonism can suppress UV-induced cutaneous signaling, and future experiments will explore the potential of TLR4-directed strategies for prevention of NMSC.
Graphical Abstract
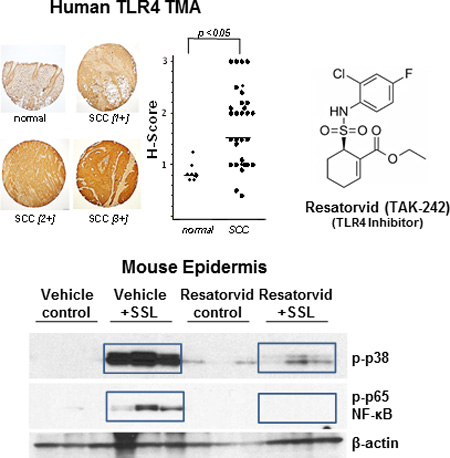
Toll-like Receptor 4 (TLR4) expression is elevated during tumorigenic progression of normal human skin through actinic keratosis (AK) to squamous cell carcinoma (SCC; tissue microarray analysis, top left panel). Topical use of the pharmacological TLR4 antagonist resatorvid (TAK-242, chemical formula, top right panel) suppresses UV-induced stress signaling mediated through NF-κB and AP-1/MAP kinases in SKH-1 mouse epidermis (immunoblot analysis, bottom panel).
INTRODUCTION
Skin cancer is the most commonly diagnosed cancer worldwide. Keratinocyte-derived non-melanoma skin cancers (NMSCs) represent by far the largest proportion of these diagnoses. Recent analysis of cancer prevalence rates indicate that annual diagnoses of skin cancers have increased by 1.5 million between the years of 2002–2006 and 2007–2011, to 4.9 million cases per year in the United States. Meanwhile, the cost for treating these cancers has increased by 126.2% (1). Although preventive measures such as limiting exposure by seeking shade, covering up, and proper application of sunscreen can provide significant protection from ultraviolet (UV) light, novel topical preventive and treatment regimes targeting NMSC are clearly needed (2–5).
Dysregulation of inflammatory signaling is a known hallmark of tumorigenic progression for many malignancies including NMSC (6, 7). The innate immune response receptor Toll-Like Receptor 4 (TLR4) has recently been identified as a major driver of cutaneous inflammation. Elegant work has described the contribution of TLR4 to skin anti-microbial defense, wound healing, regulation of DNA repair, and inflammatory cell-related skin responses (8–11). Importantly, TLR4 overexpression has now been documented in human cutaneous SCC and melanoma, and genetic antagonism targeting TLR4 has been shown to block metastasis of malignant melanoma in mice (12–14). Moreover, a causative role of TLR4-dependent signaling in UV-induced systemic photoimmunosuppression and chemical skin carcinogenesis has been substantiated in murine models (10, 13).
TLR4 is now recognized as a major mediator of innate immune defenses, serving as a receptor for PAMPs [pathogen-associated molecular patterns, including lipopolysaccharide (LPS)] and DAMPs [damage-associated molecular patterns such as HMGB1 (14), HSP70 and HSP90 (15), SAA1 (16) and NAMPT/PBEF (17)]. The canonical TLR4 agonist LPS induces activation of TLR4 resulting in stimulation of MyD88-dependent and MyD88-independent pathways, both of which contribute to activation of inflammatory and stress-response signaling (18). Both the MyD88-dependent and –independent pathways regulate activity of the NF-κB transcription factor. TLR4 also regulates the activity of MAP kinases (p38, ERKs, JNKs, for example) as well as the PI3 kinase/Akt signaling pathway (reviewed in (19) and (18)). Both NF-κB and AP-1/CREB (downstream of MAP kinases) stimulate innate immune response genes as part of the stress signaling network of early response genes. Notably, all of these pathways are also well known to be stimulated in keratinocytes by UV treatment (20, 21).
In skin, TLR4 expression has been documented in myeloid-derived inflammatory cells, dendritic cells and keratinocytes (22). While TLR4 activation is documented in keratinocytes in response to LPS stimulation (23), it remains unknown whether UV can contribute to TLR4-dependent activation of NF-κB, MAP kinases and AP-1 signaling in these cells. Activation of these pathways in response to acute and chronic UV exposure is known to contribute to photocarcinogenesis, and upstream receptors of these pathways represent valid targets for photochemoprevention (e.g. EGFR) (20, 24). We therefore examined TLR4 expression in human skin as a function of tumorigenic progression towards NMSC and tested the hypothesis that pharmacological antagonism of TLR4 may suppress UV-induced AP-1/NF-κB signaling. Here we provide novel evidence that a significant increase in expression of TLR4 occurs in keratinocytes during the progression from normal human skin to actinic keratosis (AK), also detectable during further progression to SCC. We also demonstrate for the first time that topical application of the TLR4-specific pharmacological antagonist resatorvid (TAK-242, CLI-095), a molecularly-targeted TLR4 antagonist that has reached the stage of clinical testing (25), blocks UV-induced NF-κB and MAP kinase/AP-1 activity in cultured keratinocytes and murine skin.
MATERIALS AND METHODS
Materials
Resatorvid (TAK-242) and Ibudilast were purchased from MedChem Express (Monmouth Junction, NJ). ST-2825 was purchased from Apexbio Technology (Houston, TX). Carbenoxolone disodium was purchased from LKT Laboratories (St. Paul, MN). Most antibodies were purchased from Cell Signaling including phospho-NF-κB p65 (3033), phospho-p38 (9215), phospho-Akt (4060), phospho-JNK (9255), and beta tubulin (5666). The antibody for TLR4 western blots (sc-293072) was purchased from Santa Cruz Biotechnology (Dallas, TX), and the antibody for TLR4 IHC (ab22048) was purchased from Abcam (Cambridge, MA).
Immunohistochemistry and tissue microarray
All human skin samples were obtained through the Skin Cancer Institute Patient Registry and Tissue Bank as approved by the University of Arizona Institutional Review Board (Protocol number 11-0212-04). De-identified biopsies (n=10) obtained from matched normal sun-protected buttock skin, sun-damaged (SD) forearm skin and actinic keratosis (AK) were used to examine TLR4 expression as skin progresses to a severely damaged state. Staining for TLR4 was performed on the Discovery XT automated stainer from Ventana Medical Systems (VMS), a member of the Roche family. Briefly, the TLR4 mouse monoclonal antibody (Abcam) diluted 1:6000 in PSS diluent from VMS was added to 3 µm cut formalin-fixed, paraffin-embedded (FFPE) biopsy slides (100 µL/slide). Staining was performed using the DAB Map Kit together with the streptavidin-biotin peroxidase detection system (VMS, Tucson, Arizona). Slides underwent antigen retrieval using a borate buffer for 60 minutes, followed by primary antibody incubation for 32 minutes. Detection was performed using DAB and counter stained with hematoxylin and Bluing agent. Measurement of TLR4 staining thickness was performed using Image J. Briefly, a micrometer picture was used to calibrate Image J, and the thickness of TLR4 staining from a representative image taken of each slide (200×) were each measured at 8 sites per image. The average from all measurements/slide were used to generate the data shown in Figure 1b. Additional samples were evaluated using a tissue microarray containing normal human skin (n = 9) and human NMSC specimens [including cutaneous SCCs (n = 31)] purchased from Novus (NBP-2-30229), and interrogated with the same TLR4 antibody and same methodology.
Figure 1. TLR4 is expressed by basal epidermal keratinocytes in normal human skin and is up-regulated in actinic keratosis and SCC.

(A) Matched biopsies from sun-protected normal skin, sun-damaged (SD) skin and actinic keratoses (AK) were stained for TLR4 expression. TLR4 strongly stains in the basal layer of the epidermis in normal samples and increases in cell layer involvement during the progression to AK. (B) The thickness of TLR4 stained cell layers in (A) was quantified using Image J and statistically compared using student’s t test. (C) A tissue microarray (NBP-2-30229, Novus) containing 9 normal specimens and 31 cutaneous SCCs was stained for TLR4 (left and middle). H-scores for the samples were statistically compared using the Mann-Whitney test (right). (D) Reverse phase protein microarray (RPPA) was used to quantify expression of TLR4 and key related inflammatory proteins in SD epidermis, AKs and SCCs. The horizontal line in the box is the median of the dataset. Expression of each analyte is significantly increased during progression according to the Kruskal-Wallis test (p < 0.05).
For staining of mouse skin for cleaved caspase-3, FFPE slides were processed and stained as described previously (26). For quantification, the average positive staining in 5 fields/slide were counted for each mouse (400× magnification) and the average staining of the three mice per group was calculated. Two independent, blinded reviewers scored the staining.
Reverse-Phase Protein Microarray (RPPA)
Epidermal protein lysates were generated from frozen slides from human biopsies of sun damaged (SD) forearm skin, AK and cutaneous SCC and processed according to standard protocols (27, 3). Slides were stained with H&E and laser-capture microdissection (LCM) was applied to separate the area of interest (epidermis) from the surrounding stroma. Prior to LCM, a pathologist confirmed the grading of the sample and marked the area of interest for harvest. Analysis of the RPPA outputs was used to generate the box-and-whisker plots shown in Figure 1d. Non-parametric Kruskal-Wallis analysis was used to compare each group. Patient sample numbers (n) for individual analyte proteins differed as follows: For MyD88, NF-κB p65, IL-10: sun-damaged skin (SD) group, n=18; AK group, n=35; SCC group n=16. For TLR4: SD group, n=17; AK group, n=34; SCC group, n=15.
Ultraviolet light treatment
All UV exposures were performed as previously published (28, 3). For cell culture, both UVB bulbs (FS20 bulbs, National Biological Corp. Beachwood, OH) and solar-simulated light (SSL) bulbs (UVA340 bulbs, Q-Lab Corporation, Westlake, OH) were utilized within a biosafety cabinet. Control cells were mock exposed in an identical cabinet. For in vivo exposures, a bank of 6 UVA340 bulbs was utilized within an approved ventilated animal rack. Fluence intensity for both types of bulbs was measured as described previously (3).
Keratinocyte cell culture
All cell culture was maintained in a 37°C, 5% CO2 incubator and checked routinely for mycoplasma contamination. The human immortalized keratinocyte HaCaT cell line was previously stably transfected with a luciferase reporter vector driven by an AP-1-responsive TPA-response element (TRE). This “HCl14” cell line has been used historically in our laboratory and is well characterized as a tool for studying the regulation of the AP-1 transcription factor and cellular stress (29–31). These cells were grown in DMEM with 10% FBS and pen/strep. Similarly, the JB6 P+ mouse epidermal keratinocyte cell line stably transfected with an NF-κB-driven luciferase reporter (a kind gift of Dr. Zigong Dong) was cultured in Minimal Essential Media (MEM, Corning, Corning, NY) with 5% FBS and pen/strep (32). All cells were serum starved at least 12 to 16 hours prior to LPS or UV treatment. Adult human primary epidermal keratinocytes were purchased from ThermoFisher Scientific, grown as described previously and used within three passages (3).
siRNA Transfection
For genetic knockdown of TLR4 in keratinocytes, siRNA against either mouse TLR4 (two separate constructs), human TLR4 (three pooled constructs) or control non-targeting pooled siRNA was purchased from GE Dharmacon and reconstituted in 1× siRNA buffer (GE Dharmacon, La Fayette, CO). Cells were transfected using Lipofectamine RNAi MAX (Life Technologies, Grand Island, NY) according to manufacturer’s protocols. Cells were transfected one day after seeding and incubated for 48 hr (HCl-14 cells) or 72 hr (NF-κB cells) prior to either harvest for mRNA analysis or treatment with UV. Results representative of n=3.
AP-1 and NF-κB Luciferase reporter assays
Serum-starved HCl-14 (HaCaT) or NF-κB (JB6+) cells were treated with indicated doses of LPS or UVB and harvested 6hr (for NF-κB luciferase) or 12hr (for AP-1 luciferase) later. Cells were lysed in Cell Culture Lysis Buffer (Promega) and proteins were quantified using the BCA assay (BioRad, Hercules, CA). Experimental triplicates were assayed for luciferase activity according to the manufacturer’s instructions using the Luciferase Assay System (Promega, Madison, WI) as described (29). The means for each experiment (results representative of n ≥ 3) were averaged and analyzed by Student’s t test for statistical significance.
Western blot analysis
Cultured keratinocytes were lysed in RIPA buffer containing 1× HALT phosphatase/protease inhibitors (Invitrogen, Waltham, MA) and PMSF (Sigma-Aldrich, St. Louis, MO) and briefly sonicated on ice as described (29). For epidermal lysates, frozen mouse skin was scraped to remove the epidermal layer, which was then ground using a frozen mortar and pestle, placed in skin lysis buffer containing 1× HALT and PMSF and processed as described (26). Protein concentrations for epidermal samples were determined using the Dc protein assay (BioRad, Hercules, CA). Samples were separated on SDS-PAGE, transferred to PVDF membranes and processed using established protocols (26). Protein bands were visualized using Amersham ECL reagents (GE Healthcare Life Sciences, Pittsburgh, PA). Results are representative of n≥3 for cell culture studies; 3 mice/group for mouse epidermis studies which were repeated at least two times.
Real-Time Quantitative RT-PCR
Total RNA was extracted from either scraped mouse skin or cultured keratinocytes using an RNeasy Mini Kit (Qiagen, Frederick, MD) according to the manufacturer’s protocol. Human TLR4 (Hs00152939_m1), IRF3 (Hs01547283_m1), IL-6 (Hs00985639_m1), IL-8 (Hs00174103_m1), GAPDH (Hs99999905_m1) and mouse IL-10 (Mm01288386_m1), IRF3 (Mm00516784_m1), and GAPDH (Mm99999915_m1) primer/probes were obtained from ABI (Applied Biosystems, Branchburg, NJ). cDNAs from three individual samples for each control/treatment experiment were synthesized from 500 ng of total RNA in a 50 ul reaction with master mix containing 10×RT buffer, 5.5 mM MgCl2, 2 mM dNTPs, 2.5 µM random hexamers, 2 Units of RNase Inhibitor and 62.5 Units of Multi Scribe Reverse Transcriptase. All MasterMix reagents were purchased from ABI (Applied Biosystems, Branchburg, NJ). Reactions were performed in a MJ Thermocycler PTC-200 (MJ Research, Inc., Watertown, MA) under the following conditions: 25 °C for 10 min, 48 °C for 30 min and 95 °C for 5 min. 10 ng of cDNA was then used to amplify the mouse or human sequences. The conditions for quantitative PCR reactions were: 10 min at 95 °C followed by 15 s at 95 °C, 1 min at 60 °C for 40 cycles by using ABI7000 Real-Time PCR System (Applied Biosystems, Foster City, CA). PCR amplification of the human or mouse GAPDH was used to control quality of the cDNA. Non-template controls were included on each PCR plate. Amplification plots were generated and the Ct values (cycle number at which fluorescence reaches threshold) recorded. Target gene levels were normalized to the GAPDH control [ΔCt=Ct(gene of interest)−Ct(housekeeping gene)]. Results are representative of n=3 for cell culture studies, 3 mice/group for in vivo studies.
In vivo studies
Female SKH-1 mice (SKH1-Hrhr) were utilized for analysis of the effect of resatorvid on epidermal stress responses in vivo. Six to eight-week old mice were purchased from Charles River laboratories and housed in accordance with The University of Arizona Animal Care and Use Committee standards under an approved protocol. Solar-simulated light (SSL) exposure was performed as described above. Mice were divided into four groups (n = 3) and either held as controls or exposed to a single dose of SSL (105 kJ/m2 UVA/6.4 kJ/m2 UVB). After SSL (or mock) exposure, all mice were immediately post-treated with vehicle (acetone) or resatorvid (200 µL/back, 0.5%). Mice were sacrificed 24hr later and their back skins were divided for snap freezing or formalin fixation for IHC. Epidermal lysates were derived from scraped frozen epidermis as described above.
Flow cytometric cell death assay
HaCaT keratinocytes were serum starved for 12 hr and pretreated with either vehicle (DMSO) or resatorvid (10 µM) 1 hr prior to exposure to SSL (40 kJ/m2 UVA/2.6 kJ/m2 UVB). Cells were also post-treated with the same agents. 16 hr later, cells were harvested and processed for Annexin V/propidium iodide uptake as described previously (29). Results representative of n=3.
Statistical methods
Statistical analysis was performed as described in the respective methodology section. Student’s t-test was performed if no other analysis method is specified.
RESULTS
TLR4 expression increases during tumorigenic progression from normal human skin to actinic keratosis and SCC
First, TLR4 expression was profiled, comparing matched human samples of normal (sun-protected) buttock skin, sun-damaged (SD) forearm skin and AK from ten individuals employing immunohistochemistry (IHC, Fig. 1a,b). This analysis revealed that TLR4 expression in normal (sun-protected) skin is primarily confined to the basal layer of epidermal keratinocytes. In contrast, in biopsies taken from SD skin, TLR4 expression is detectible in multiple layers of the basal epidermis, and the thickness of TLR4 staining is significantly enhanced in matched biopsies of actinic keratosis (AK) compared to the normal controls and SD skin (Fig. 1a,b). Moreover, TLR4 expression analysis comparing normal human skin biopsies and cutaneous SCC samples employing a commercial tissue microarray (TMA) indicates that expression of this receptor is significantly increased in skin tumors (p < 0.05) (Fig. 1c). As an independent confirmation, proteomic analysis of patient skin samples (epidermis only) was performed using the reverse-phase protein microarray (RPPA) format. This analysis confirms a significant increase in expression of TLR4 and related signaling/inflammatory proteins [MyD88, p-p65 (NF-κB) and IL-10], observable during progression from SD epidermis to SCC.
Genetic or pharmacological (resatorvid-based) inhibition of TLR4 blocks UV-induced stress signaling in cultured murine and human immortalized keratinocytes
To test the involvement of TLR4 in UV-induced keratinocyte stress signaling, we first employed genetic antagonism of TLR4 expression using an siRNA-based methodology. To this end, both JB6+ murine and HaCaT human keratinocytes stably expressing UV-inducible luciferase reporters for NF-κB or AP-1, respectively, were employed as previously published (29, 32). In these cells, significant and sustained knockdown of TLR4 mRNA was achieved 72hr post transfection when employing TLR4-directed siRNA, an effect not observed with non-targeted siRNA controls [(Fig. 2a, data obtained in JB6+ cells; identical results were obtained using HaCaT cells (data not shown)]. Strikingly, TLR4 knockdown resulted in significant inhibition of UV-induced NF-κB- and AP-1-controlled luciferase activity compared to UV-exposed siRNA controls (Fig. 2b,c). We then confirmed the occurrence of TLR4 signaling in cultured keratinocytes exposed to the canonical TLR4 agonist LPS, a treatment that caused significant induction of NF-κB luciferase activity (Fig. 2d). Furthermore, treatment with the TLR4-specific small molecule inhibitor resatorvid (TAK-242, CLI-095) significantly suppressed LPS-induced activation of NF-κB-luciferase signaling, suggesting the feasibility of blocking TLR4-dependent signaling using resatorvid in keratinocytes (Fig. 2d).
Figure 2. Genetic and pharmacological TLR4 antagonism attenuates UV-induced NF-κB and AP-1 stress signaling in immortalized keratinocytes.

(A) Mouse JB6+ cultured keratinocytes were transfected with two types of TLR4 siRNA or non-targeted control siRNA for 72hr and the level of TLR4 mRNA was quantified using quantitative real-time RT-PCR. (B) JB6+ keratinocytes which stably express an NF-κB reporter (“NF-κB keratinocytes”) were transfected as in (A) and treated with UVB (250 kJ/m2). TLR4 siRNA transfection resulted in significant inhibition of NF-κB luciferase signaling. (C) Human HaCaT keratinocytes which stably express an AP-1 luciferase reporter (“AP-1 keratinocytes”) were transfected with a mix of three TLR4-specific siRNAs or non-targeted control siRNA 72 hr prior to UVB treatment (250 kJ/m2). TLR4 siRNA significantly inhibited UV-induced AP-1 signaling in these cells. (D) NF-κB keratinocytes were stimulated with the classical TLR4 agonist LPS (0.6 µg/mL) in the presence of resatorvid (10 µM, pre and post) or vehicle (DMSO) and harvested 6hr later. Resatorvid significantly inhibited LPS-induced NF-κB-driven luciferase signaling in these cells. (E) Similarly, NF-κB keratinocytes were treated with UVB +/− 10 µM resatorvid (or vehicle) and harvested 6 hr later. Resatorvid significantly inhibited UV-induced NF-κB luciferase signaling in keratinocytes. (F) AP-1 keratinocytes also showed significant inhibition of UVB-induced AP-1 luciferase signaling due to pre and post-treatment with 5 µM or 10 µM of resatorvid. (G and H) Likewise, additional pharmacological inhibitors of the TLR4 signaling pathway are shown to significantly inhibit UVB-induced signaling in NF-κB or AP-1 luciferase reporter assays, respectively. ST2825 (10 µM) is an inhibitor of the TLR4 signaling cofactor MyD88. Ibudilast (25 µM) is a direct inhibitor of TLR4 activity. Carbenoxolone (40 µM) is an inhibitor of HMGB1 activity. (I) HaCaT keratinocytes were pre- and post-treated with resatorvid (10 µM), exposed to SSL (40 kJ/m2 UVA/2.6 kJ/m2 UVB) and harvested for Western blot analysis at the indicated timepoints. (J) JB6+ mouse keratinocytes were pre- and post-treated with 10 µM resatorvid or vehicle and SSL (same dose as in I). Cells were harvested 24 hr later for analysis of IRF3 gene expression using quantitative RT-PCR.
Next, we tested the hypothesis that UV exposure can elicit TLR4 signaling in keratinocytes which is amenable to resatorvid-based pharmacological intervention. As observed in the context of LPS-induced TLR4 signaling (Fig. 2d), resatorvid significantly suppressed UV-induced NF-κB and AP-1 activation (Fig. 2e,f). Likewise, similar blockade of UV-induced signaling in both cell types was observed when employing the MyD88 inhibitor ST2825 (33), the alternative TLR4 inhibitor Ibudilast (34, 35), and the HMBG1 release inhibitor carbenoxolone (36). These findings suggest that blocking TLR4 signaling from several pharmacological vantage points effectively reduces UV-induced responses in keratinocytes (Fig. 2g,h). For data presented in Figure 2, panels b-h, UVB was used as the irradiation source in order to assure consistency with our prior published luciferase assays interrogating UV-driven AP-1 and NF-κB activation and its pharmacological modulation (30, 32). We then examined the effect of pharmacological inhibition of TLR4 on UV-induced MAP kinase signaling employing solar simulated light (SSL). SSL uses a light source which replicates the relative contributions of UVA and UVB found in natural sunlight, as previously published (37, 3). We observed that stimulation of p38 MAP kinase phosphorylation in response to SSL was attenuated in the presence of resatorvid (Fig. 2i). SSL-induced transcription of interferon regulatory factor-3 (IRF-3), a MyD88-independent TLR4 downstream target, was also significantly inhibited by this compound (Fig. 2j).
Resatorvid-based pharmacological inhibition of TLR4 blocks UV-induced stress signaling in human primary keratinocytes
Next, we examined if pharmacological modulation of UV-induced TLR4 signaling as observed in immortalized keratinocytes (Fig. 2) may also occur in primary human epidermal keratinocytes (HEKs). To this end, HEKs were exposed to SSL (40 kJ/m2 UVA/2.6 kJ/m2 UVB) and post-treated with either vehicle (DMSO) or resatorvid. Western blot analysis revealed that resatorvid-based inhibition of TLR4 was able to block UV-induced phosphorylation of MAP kinases (p38 and JNK) and Akt (Fig. 3a). Likewise, UV-induced transcription of the inflammatory mediators Il-6 and Il-8 was significantly inhibited by resatorvid in these cells (Fig. 3b,c).
Figure 3. Resatorvid inhibits UV-induced stress signaling in human primary keratinocytes.

(A) Primary human keratinocytes were treated with SSL (40 kJ/m2 UVA/ 2.7kJ/m2 UVB), post-treated with resatorvid and harvested for Western blot 1 hr later. (B, C) Primary human keratinocytes were treated as in (A) and harvested at 18 hr for qRT-PCR analysis of IL-6 and IL-8 mRNA, respectively (p < 0.05).
Resatorvid blocks TLR4 signaling and potentiates UV-induced cell death in mouse epidermis
Next, the feasibility of cutaneous TLR4 modulation by topical application of resatorvid was examined in vivo, using SKH-1 mouse skin exposed to acute UV. To this end, adult female SKH-1 mice were exposed to an acute dose of SSL and immediately post-treated with topical resatorvid [14 mM; 0.5% in acetone (200uL)] or vehicle alone and harvested 24hr later. Subsequent Western blot analysis of epidermal lysates revealed that SSL treatment causes the expected phosphorylation of both p38 MAP kinase and the p65 subunit of NF-κB, and that post-treatment with resatorvid dramatically inhibited these phosphorylation events (Fig. 4a). Consistent with this finding, UV-induced expression of the inflammatory marker Il-10 was also significantly inhibited by topical resatorvid (Fig. 4b). Next, we performed IHC staining of skin specimens for cleaved caspase 3 in order to detect UV-induced keratinocyte cell death. As expected, SSL exposure caused an increase in epidermal keratinocytes positive for cleaved caspase 3. Interestingly, a two-fold increase in cleaved caspase-3 positive keratinocytes was observed upon combined exposure to resatorvid and UV, whereas resatorvid alone did not display any epidermal cytotoxic effects (Fig. 4c,d). Likewise, while resatorvid treatment in cell culture did not display toxicity at doses relevant to our studies, the compound significantly sensitized HaCaT keratinocytes to UV-induced cell death as compared to vehicle controls observable by flow cytometric analysis of Annexin V/PI-labeled cells (Fig. 4e). Taken together, these data indicate that topical application of a pharmacological TLR4 antagonist can block UV-induced epidermal stress signaling while potentially enhancing apoptotic clearance of damaged keratinocytes.
Figure 4. Resatorvid inhibits UV-induced acute inflammatory signaling and potentiates cell death in mouse epidermis.

SKH-1 mice were treated with an acute dose of SSL (105 kJ/m2 UVA/6.4 kJ/m2 UVB) and post-treated topically with 0.5% resatorvid or acetone (vehicle). 24 hr later, mice were sacrificed and back skins were harvested. (A) Epidermal protein lysates were analyzed by immunoblot analysis for UV-induced phosphorylation of p38 and p65/NF-κB. (B) qRT-PCR of epidermal RNA shows induction of IL-10 mRNA which is significantly inhibited by resatorvid. (C) Skins were sectioned for IHC and stained for cleaved caspase 3, 600× magnification is shown. (D) Quantification of stained cells per field in 400× images from skins in C (p < 0.05). (E) HaCaT keratinocytes were pretreated with 10 µM resatorvid for 1 hr prior to SSL treatment (40 kJ/m2 UVA/ 2.7kJ/m2 UVB) and post-treated with the same dose of the compound. Cells were harvested 18 hr later for analysis of cell death using annexin V/PI staining via flow cytometry.
DISCUSSION
Cumulative experimental evidence indicates that TLR4 plays an important role in cutaneous inflammatory dysregulation and may also be involved in tumorigenesis (14, 36, 9, 10, 13). Here we show for the first time that pharmacological antagonism of TLR4 can block UV-induced inflammatory signaling in cultured keratinocytes and mouse skin. We also confirm a strong upregulation of TLR4 expression during the progression from normal skin to cutaneous SCC using several methodologies. RPPA has the advantage of allowing for simultaneous quantitative analysis of additional proteins in the TLR4-related pathway. Through this methodology, a strong correlation was found between MyD88, NF-κB and IL-10 expression levels in samples representing the progression from sun-damaged skin to AK to SCC. We note that while expression of TLR4 in normal (sun-protected) skin was limited to one or two layers of the basal epidermis, the actual staining intensity of these cells was variable between individuals. This finding indicates an inter-subject variability of expression potentially driven by differences such as genetic polymorphisms, which will need to be investigated.
Using keratinocytes in culture, we also present genetic and pharmacological evidence supporting an active role of TLR4 in these cells, contributing to UV-induced inflammatory/stress signaling as summarized in Figure 5. Resatorvid, a cyclohexene derivative which covalently binds to TLR4 at Cys747 (38), proves itself to be quite effective at inhibiting both LPS- and UV-induced NF-κB and AP-1 luciferase activity in mouse and human keratinocytes. Beyond luciferase assays, resatorvid potently inhibits UV-induced p38 MAP kinase phosphorylation as well as the MyD88-independent pathway as evidenced by IRF-3 transcriptional inhibition. Primary human keratinocytes also confirmed the ability of resatorvid to significantly block UV-induced inflammatory signaling as evidenced by IL-6 and IL-8 inhibition. Additional studies revealed that resatorvid, at the concentration range used in culture, does not display relevant UVB absorptivity and that results similar to those detailed in Figures 2 and 3 could be obtained irrespective of whether the compound was used in a pre-only or post-only UV regimen (data not shown).
Figure 5.

TLR4 signaling and TLR4-directed modulatory strategies in keratinocytes. Apart from ligand-dependent activation (exogenous: LPS; endogenous: HMGB1), UV-induced TLR4 activation in keratinocytes plays a role in the control of inflammation, proliferation, cell survival and differentiation, partly though modulation of stress signaling pathways (NF-κB, AP-1). Small molecule antagonism of TLR4-dependent signaling employing resatorvid or other pharmacological modulators may represent a novel approach for the suppression of UV-induced inflammatory signaling and tumorigenesis.
The dramatic inhibition of UV-induced stress/inflammatory signaling by post-treatment of SKH-1 mouse skin with resatorvid provides evidence that this compound is readily absorbed into the skin and provides a robust response, even upon single application. The potentiation of apoptosis also noted in the epidermis of these samples (and in cultured keratinocytes) is consistent with the survival functions of NF-κB and AP-1 signaling during UV exposure (24). Interestingly, TLR4 has been shown to antagonize UV-induced PARP cleavage in APCs (9). Therefore, it may also be speculated that resatorvid-based enhancement of UV-induced cell death may occur as a result of increased PARP-dependent NAD+ pool depletion and energy crisis, a hypothesis to be substantiated by future experiments.
Our findings suggest a heretofore underappreciated role of TLR4 in UV-induced stress signaling in keratinocytes, with mechanistic implications for skin tumorigenesis. Therefore, pharmacological interventions that suppress TLR4-dependent signaling may represent a novel approach for skin photochemoprevention (Figure 5). The prototype TLR4 antagonist resatorvid, as used in our studies, may therefore represent an emerging class of novel chemopreventive agents. Consistent with this scenario, TLR4-directed chemoprevention may also be relevant to other target organ systems including liver, breast, and colon (reviewed in (19)). Moreover, the role of TLR4 in chemotherapy-driven resistance and metastasis has recently been described, and it may therefore be speculated that TLR4-directed antagonism may provide therapeutic benefit in advanced stages of tumorigenesis (39, 14, 38).
Here we have shown good cutaneous bioavailability and robust activity of this compound in an acute exposure model (Fig. 4), and our ongoing studies are therefore focusing on chronic exposure regimens targeting photocarcinogenesis. Based upon a low toxicity profile observed upon topical and systemic administration, and its prior use in human clinical trials for the treatment of septic shock (25), it is anticipated that resatorvid-based inhibition of TLR4 may prove useful for both topical inhibition of UV-induced skin inflammation and potentially tumorigenesis. Future experiments will test the feasibility of resatorvid, possibly in combination with other agents, for skin cancer photochemoprevention of NMSC.
Acknowledgments
Immunohistochemical data generated by the Tissue Acquisition and Cellular/Molecular Analysis Shared Resource (TACMASR) shown in Figure 1 was supported by the University of Arizona Cancer Center Support Grant, NIH CA023074. All other data was generated with the support of the following NIH grants: NCI P01 CA027502, R03 CA167580, and pilot funding from the University of Arizona Skin Cancer Institute (SCI).
REFERENCES
- 1.Guy GP, Jr, Machlin SR, Ekwueme DU, Yabroff KR. Prevalence and costs of skin cancer treatment in the U.S., 2002–2006 and 2007–2011. Am. J. Prev. Med. 2015;48:183–187. doi: 10.1016/j.amepre.2014.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tao S, Park SL, Rojo de la Vega M, Zhang DD, Wondrak GT. Systemic administration of the apocarotenoid bixin protects skin against solar UV-induced damage through activation of NRF2. Free Rad. Biol. & Med. 2015;89:690–700. doi: 10.1016/j.freeradbiomed.2015.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dickinson SE, Janda J, Criswell J, Blohm-Mangone K, Olson ER, Liu Z, Barber C, Petricoin EF, 3rd, Calvert VS, Einspahr J, Dickinson JE, Stratton SP, Curiel-Lewandrowski C, Saboda K, Hu C, Bode AM, Dong Z, Alberts DS, Timothy Bowden G. Inhibition of Akt Enhances the Chemopreventive Effects of Topical Rapamycin in Mouse Skin. Cancer Prev. Res. 2016;9:215–224. doi: 10.1158/1940-6207.CAPR-15-0419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dickinson SE, Melton TF, Olson ER, Zhang J, Saboda K, Bowden GT. Inhibition of activator protein-1 by sulforaphane involves interaction with cysteine in the cFos DNA-binding domain: implications for chemoprevention of UVB-induced skin cancer. Cancer Res. 2009;69:7103–7110. doi: 10.1158/0008-5472.CAN-09-0770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wondrak GT. Sunscreen-Based Skin Protection Against Solar Insult: Molecular Mechanisms and Opportunities. In: Alberts D, editor. Fundamentals of Cancer Prevention. Springer Science & Business Media; 2014. pp. 301–320. [Google Scholar]
- 6.Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature. 2008;454:436–444. doi: 10.1038/nature07205. [DOI] [PubMed] [Google Scholar]
- 7.Anuja K, Roy S, Ghosh C, Gupta P, Bhattacharjee S, Banerjee B. Prolonged inflammatory microenvironment is crucial for pro-neoplastic growth and genome instability: a detailed review. Inflamm. Res. 2016 doi: 10.1007/s00011-016-0985-3. E-pub ahead of print. [DOI] [PubMed] [Google Scholar]
- 8.Chen L, Guo S, Ranzer MJ, DiPietro LA. Toll-like receptor 4 has an essential role in early skin wound healing. J Invest. Derm. 2013;133:258–267. doi: 10.1038/jid.2012.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Harberts E, Zhou H, Fishelevich R, Liu J, Gaspari AA. Ultraviolet Radiation Signaling through TLR4/MyD88 Constrains DNA Repair and Plays a Role in Cutaneous Immunosuppression. J. Imm. 2015;194:3127–3135. doi: 10.4049/jimmunol.1402583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lewis W, Simanyi E, Li H, Thompson CA, Nasti TH, Jaleel T, Xu H, Yusuf N. Regulation of ultraviolet radiation induced cutaneous photoimmunosuppression by toll-like receptor-4. Arch. Biochem. Biophys. 2011;508:171–177. doi: 10.1016/j.abb.2011.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang X, Bi Z, Wang Y, Wang Y. Increased MAPK and NF-kappaB expression of Langerhans cells is dependent on TLR2 and TLR4, and increased IRF-3 expression is partially dependent on TLR4 following UV exposure. Mol. Med. Rep. 2011;4:541–546. doi: 10.3892/mmr.2011.450. [DOI] [PubMed] [Google Scholar]
- 12.Weng H, Deng Y, Xie Y, Liu H, Gong F. Expression and significance of HMGB1, TLR4 and NF-kappaB p65 in human epidermal tumors. BMC Cancer. 2013;13:311. doi: 10.1186/1471-2407-13-311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mittal D, Saccheri F, Venereau E, Pusterla T, Bianchi ME, Rescigno M. TLR4-mediated skin carcinogenesis is dependent on immune and radioresistant cells. EMBO J. 2010;29:2242–2252. doi: 10.1038/emboj.2010.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bald T, Quast T, Landsberg J, Rogava M, Glodde N, Lopez-Ramos D, Kohlmeyer J, Riesenberg S, van den Boorn-Konijnenberg D, Homig-Holzel C, Reuten R, Schadow B, Weighardt H, Wenzel D, Helfrich I, Schadendorf D, Bloch W, Bianchi ME, Lugassy C, Barnhill RL, Koch M, Fleischmann BK, Forster I, Kastenmuller W, Kolanus W, Holzel M, Gaffal E, Tuting T. Ultraviolet-radiation-induced inflammation promotes angiotropism and metastasis in melanoma. Nature. 2014;507:109–113. doi: 10.1038/nature13111. [DOI] [PubMed] [Google Scholar]
- 15.Triantafilou M, Triantafilou K. Heat-shock protein 70 and heat-shock protein 90 associate with Toll-like receptor 4 in response to bacterial lipopolysaccharide. Biochem. Soc. Trans. 2004;32:636–639. doi: 10.1042/BST0320636. [DOI] [PubMed] [Google Scholar]
- 16.Han S, Jin SP, Oh JH, Seo EY, Park CH, Yoon HS, Lee DH, Chung JH. Serum amyloid A1 secreted from UV-irradiated keratinocytes induces matrix metalloproteinase-1 in fibroblasts through toll-like receptor 4. Exper. Derm. 2016;25:526–531. doi: 10.1111/exd.12979. [DOI] [PubMed] [Google Scholar]
- 17.Camp SM, Ceco E, Evenoski CL, Danilov SM, Zhou T, Chiang ET, Moreno-Vinasco L, Mapes B, Zhao J, Gursoy G, Brown ME, Adyshev DM, Siddiqui SS, Quijada H, Sammani S, Letsiou E, Saadat L, Yousef M, Wang T, Liang J, Garcia JG. Unique Toll-Like Receptor 4 Activation by NAMPT/PBEF Induces NFkappaB Signaling and Inflammatory Lung Injury. Scientif. Rep. 2015;5:13135. doi: 10.1038/srep13135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Akira S, Takeda S. Toll-like receptor signalling. Nature Rev. Immunol. 2004;4:499–511. doi: 10.1038/nri1391. [DOI] [PubMed] [Google Scholar]
- 19.Mai CW, Kang YB, Pichika MR. Should a Toll-like receptor 4 (TLR-4) agonist or antagonist be designed to treat cancer? TLR-4: its expression and effects in the ten most common cancers. OncoTargets Ther. 2013;6:1573–1587. doi: 10.2147/OTT.S50838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bowden GT. Prevention of non-melanoma skin cancer by targeting ultraviolet-B-light signalling. Nature Rev. Cancer. 2004;4:23–35. doi: 10.1038/nrc1253. [DOI] [PubMed] [Google Scholar]
- 21.Bode AM, Dong Z. Signal transduction pathways: targets for chemoprevention of skin cancer. Lancet. Oncol. 2000;1:181–188. doi: 10.1016/s1470-2045(00)00029-2. [DOI] [PubMed] [Google Scholar]
- 22.Burns EM, Yusuf N. Toll-like receptors and skin cancer. Front. Immunol. 2014;5:135. doi: 10.3389/fimmu.2014.00135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Min W, Ahmad I, Chang ME, Burns EM, Qian Q, Yusuf N. Baicalin Protects Keratinocytes From Toll-Like Receptor-4 Mediated DNA Damage and Inflammation Following Ultraviolet Irradiation. Photochem. Photobiol. 2015:1435–1443. doi: 10.1111/php.12505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cooper SJ, Bowden GT. Ultraviolet B regulation of transcription factor families: roles of nuclear factor-kappa B (NF-kappaB) and activator protein-1 (AP-1) in UVB-induced skin carcinogenesis. Curr. Cancer Drug Target. 2007;7:325–334. doi: 10.2174/156800907780809714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rice TW, Wheeler AP, Bernard GR, Vincent JL, Angus DC, Aikawa N, Demeyer I, Sainati S, Amlot N, Cao C, Ii M, Matsuda H, Mouri K, Cohen J. A randomized, double-blind, placebo-controlled trial of TAK-242 for the treatment of severe sepsis. Crit. Care Med. 2010;38:1685–1694. doi: 10.1097/CCM.0b013e3181e7c5c9. [DOI] [PubMed] [Google Scholar]
- 26.Dickinson SE, Olson ER, Zhang J, Cooper SJ, Melton T, Criswell PJ, Casanova A, Dong Z, Hu C, Saboda K, Jacobs ET, Alberts DS, Bowden GT. p38 MAP kinase plays a functional role in UVB-induced mouse skin carcinogenesis. Mol. Carcinogen. 2011;50:469–478. doi: 10.1002/mc.20734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Einspahr JG, Calvert V, Alberts DS, Curiel-Lewandrowski C, Warneke J, Krouse R, Stratton SP, Liotta L, Longo C, Pellacani G, Prasad A, Sagerman P, Bermudez Y, Deng J, Bowden GT, Petricoin EF., 3rd Functional protein pathway activation mapping of the progression of normal skin to squamous cell carcinoma. Cancer Prev. Res. 2012;5:403–413. doi: 10.1158/1940-6207.CAPR-11-0427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Olson ER, Melton T, Dickinson SE, Dong Z, Alberts DS, Bowden GT. Quercetin potentiates UVB-Induced c-Fos expression: implications for its use as a chemopreventive agent. Cancer Prev. Res. 2010;3:876–884. doi: 10.1158/1940-6207.CAPR-09-0220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dickinson SE, Olson ER, Levenson C, Janda J, Rusche JJ, Alberts DS, Bowden GT. A novel chemopreventive mechanism for a traditional medicine: East Indian sandalwood oil induces autophagy and cell death in proliferating keratinocytes. Arch. Biochem. Biophys. 2014;558:143–152. doi: 10.1016/j.abb.2014.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhu M, Zhang Y, Cooper S, Sikorski E, Rohwer J, Bowden GT. Phase II enzyme inducer, sulforaphane, inhibits UVB-induced AP-1 activation in human keratinocytes by a novel mechanism. Mol. Carcinogen. 2004;41:179–186. doi: 10.1002/mc.20052. [DOI] [PubMed] [Google Scholar]
- 31.Barthelman M, Chen W, Gensler HL, Huang C, Dong Z, Bowden GT. Inhibitory effects of perillyl alcohol on UVB-induced murine skin cancer and AP-1 transactivation. Cancer Res. 1998;58:711–716. [PubMed] [Google Scholar]
- 32.Jung SK, Lee KW, Byun S, Kang NJ, Lim SH, Heo YS, Bode AM, Bowden GT, Lee HJ, Dong Z. Myricetin suppresses UVB-induced skin cancer by targeting Fyn. Cancer Res. 2008;68:6021–6029. doi: 10.1158/0008-5472.CAN-08-0899. [DOI] [PubMed] [Google Scholar]
- 33.Loiarro M, Capolunghi F, Fanto N, Gallo G, Campo S, Arseni B, Carsetti R, Carminati P, De Santis R, Ruggiero V, Sette C. Pivotal Advance: Inhibition of MyD88 dimerization and recruitment of IRAK1 and IRAK4 by a novel peptidomimetic compound. J. Leuk. Biol. 2007;82:801–810. doi: 10.1189/jlb.1206746. [DOI] [PubMed] [Google Scholar]
- 34.Awasthi S. Toll-like receptor-4 modulation for cancer immunotherapy. Front. Immunol. 2014;5:328. doi: 10.3389/fimmu.2014.00328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ruiz-Perez D, Benito J, Polo G, Largo C, Aguado D, Sanz L, Gomez de Segura IA. The Effects of the Toll-Like Receptor 4 Antagonist, Ibudilast, on Sevoflurane's Minimum Alveolar Concentration and the Delayed Remifentanil-Induced Increase in the Minimum Alveolar Concentration in Rats. Anesth. Analges. 2016;122:1370–1376. doi: 10.1213/ANE.0000000000001171. [DOI] [PubMed] [Google Scholar]
- 36.Li W, Li J, Sama AE, Wang H. Carbenoxolone blocks endotoxin-induced protein kinase R (PKR) activation and high mobility group box 1 (HMGB1) release. Mol. Med. 2013;19:203–211. doi: 10.2119/molmed.2013.00064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lee MH, Lim DY, Kim MO, Lee SY, Shin SH, Kim JY, Kim SH, Kim DJ, Jung SK, Yao K, Kundu JK, Lee HS, Lee CJ, Dickinson SE, Alberts D, Bowden GT, Stratton S, Curiel C, Einspahr J, Bode AM, Surh YJ, Cho YY, Dong Z. Genetic ablation of caspase-7 promotes solar-simulated light-induced mouse skin carcinogenesis: the involvement of keratin-17. Carcinogenesis. 2015;36:1372–1380. doi: 10.1093/carcin/bgv110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Takashima K, Matsunaga N, Yoshimatsu M, Hazeki K, Kaisho T, Uekata M, Hazeki O, Akira S, Iizawa Y, Ii M. Analysis of binding site for the novel small-molecule TLR4 signal transduction inhibitor TAK-242 and its therapeutic effect on mouse sepsis model. Brit. J. Pharm. 2009;157:1250–1262. doi: 10.1111/j.1476-5381.2009.00297.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ran S. The Role of TLR4 in Chemotherapy-Driven Metastasis. Cancer Res. 2015;75:2405–2410. doi: 10.1158/0008-5472.CAN-14-3525. [DOI] [PMC free article] [PubMed] [Google Scholar]