

Abstract
Chagas disease, leishmaniasis, and sleeping sickness affect 20 million people worldwide and lead to more than 50,000 deaths annually1. The diseases are caused by infection with the kinetoplastid parasites Trypanosoma cruzi, Leishmania spp. and Trypanosoma brucei spp., respectively. These parasites have similar biology and genomic sequence, suggesting that all three diseases could be cured with drug(s) modulating the activity of a conserved parasite target2. However, no such molecular targets or broad spectrum drugs have been identified to date. Here we describe a selective inhibitor of the kinetoplastid proteasome (GNF6702) with unprecedented in vivo efficacy, which cleared parasites from mice in all three models of infection. GNF6702 inhibits the kinetoplastid proteasome through a non-competitive mechanism, does not inhibit the mammalian proteasome or growth of mammalian cells, and is well-tolerated in mice. Our data provide genetic and chemical validation of the parasite proteasome as a promising therapeutic target for treatment of kinetoplastid infections, and underscore the possibility of developing a single class of drugs for these neglected diseases.
Kinetoplastid infections affect predominantly poor communities in Latin America, Asia and Africa. Available therapies suffer from multiple shortcomings, and new drug discovery for these diseases is limited by insufficient investment3. We sought low molecular weight compounds with a growth inhibitory effect on Leishmania donovani (L. donovani)4,5, Trypanosoma cruzi (T. cruzi)6,7 and Trypanosoma brucei (T. brucei)5,8. Our approach was to test 3 million compounds in proliferation assays on all three parasites (Supplementary Information Tables 1–3), followed by triaging of active compounds (half-maximum inhibitory concentration value EC50<10 μM) to select those with a clear window of selectivity (>5-fold) with respect to growth inhibition of mammalian cells. An azabenzoxazole, GNF5343, was identified as a hit in the L. donovani and T. brucei screens. Although GNF5343 was not identified in the T. cruzi screen, we noted potent anti-T. cruzi activity of this compound in secondary assays.
Optimization of GNF5343 involved the design and synthesis of ~3,000 compounds, and focused on improving bioavailability and potency on inhibition of L. donovani growth within macrophages (Fig. 1). A critical modification involved replacement of the azabenzoxazole center with C6-substituted imidazo- and triazolopyrimidine cores, which yielded compounds up to 20-fold more potent on intra-macrophage L. donovani (e.g. GNF2636). Replacement of the furan group with a dimethyloxazole ring reduced the risk of toxicity associated with the furan moiety, and replacement of the chlorophenyl group with a fluorophenyl improved selectivity over mammalian cell growth inhibition (e.g. GNF3849). These changes also resulted in low clearance and acceptable bioavailability. Further substitutions at the core C6 position led to GNF6702 and a 400-fold increase in intra-macrophage L. donovani potency compared to GNF5343.
Figure 1. Chemical evolution of GNF6702 from the phenotypic hit GNF5343.

L. donovani: amastigotes proliferating within primary mouse macrophages; T. brucei: the bloodstream form trypomastigotes; T. cruzi: amastigotes proliferating in 3T3 fibroblast cells; macrophage: mouse primary peritoneal macrophages; EC50 and CC50: half-maximum growth inhibition concentration; F: oral bioavailability in mouse after administering single compound dose (20 mg/kg) as a suspension; CL: plasma clearance in mouse after single iv bolus dose (5 mg/kg); N.D.: not determined; all EC50 and CC50 values correspond to means ± s.e.m. (n=4 technical replicates).
L. donovani parasites cause a majority of visceral leishmaniasis (VL) cases in East Africa and India9. In mice infected with L. donovani10, oral dosing with GNF6702 effected a more pronounced reduction in liver parasite burden than miltefosine, the only oral anti-leishmanial drug available in clinical practice5 (Fig. 2a). The miltefosine regimen for VL efficacy studies was chosen to approximate the drug plasma concentration of the clinical regimen11. We noted a greater than three log reduction in parasite load after eight day treatment with 10 mg/kg of GNF6702 twice-daily with the free concentration of GNF6702 (fraction unbound in plasma=0.063) staying above the L. donovani EC99 value (the concentration inhibiting 99% of intra-macrophage parasite growth in vitro) for the duration of the dosing period (Extended Data Fig. 1a). Characterization of efficacy of ten analogues in the series at various doses revealed a significant correlation (r2=0.89, p<0.01) between i) the ratio of mean free plasma compound concentration to the L. donovani EC90 value and ii) reduction of the liver parasite burden. We found that 90% parasite burden reduction in the mouse model was achieved when the mean free compound plasma concentration during treatment equaled a 0.94-fold multiple of the L. donovani EC90 value (Fig. 2b). Cutaneous leishmaniasis (CL) affects about a million people per year, causing skin lesions that can resolve into scar tissue12. In parts of the Middle East, CL has reached epidemic proportions13. After footpad infection of BALB/c mice with the dermatotropic L. major strain14,15, treatment with GNF6702 at 10 mg/kg twice-daily caused a 5-fold decrease in footpad parasite burden and a reduction in footpad swelling (Fig. 2c). Both 3 mg/kg and 10 mg/kg twice-daily regimens of GNF6702 were superior to 30 mg/kg once-daily miltefosine regimen (p<0.01), which translates into ~2-fold higher miltefosine plasma concentration in mice than observed in clinical dosing11.
Figure 2. GNF6702 clears parasites in mouse models of kinetoplastid infections.

a, Post-treatment L. donovani liver burdens in mouse model of VL as assessed by qPCR (n=5 mice). b, PK/PD relationship for ten GNF6702 analogues, each administered at several doses; circles: mean liver burdens associated with individual compound regimens (30 regimens in total; n=5 mice per regimen) relative to vehicle; horizontal dotted line: 90% reduction in the liver L. donovani burden; vertical dotted line: 0.94-fold multiple of the mean free compound plasma concentration/the L. donovani EC90 value ratio. c, Post-treatment L. major footpad burdens in the BALB/c mouse model of CL as assessed by qPCR (n= 6 mice); the p values (two-tailed distribution) relate parasite burdens in compound-treated mice with those from vehicle-treated mice; left inset picture: a representative mouse footpad after treatment with vehicle; right inset picture: a representative mouse footpad after treatment with GNF6702 10 mg/kg twice-daily regimen. d, T. cruzi burden in mouse blood (circles), colon (triangles) and heart (diamonds) as assessed by qPCR after 20 days of treatment and four weeks of immunosuppression (n=8 mice). e, Whole body in vivo imaging of bioluminescent T. brucei before and after treatment; Trypanosoma brucei–infected mice were treated by a single intraperitoneal injection of diminazene aceturate (n=3 mice) or by oral administration of GNF6702 once-daily for 7 days (n=6 mice); filled symbols show whole body bioluminescence values for individual mice; several mice from the untreated and diminazene aceturate-treated groups were euthanized between days 28 and 56 due to CNS infection symptoms; background bioluminescence values shown for uninfected mice (grey-filled squares; n=4) were collected independently from mice aged-matched for day 0 using the same acquisition settings. Red dotted lines in a, c and d plots show limit of parasite detection by qPCR; plot symbols below the red dotted line: mice with no detectable parasites; data points below the limit of detection are ‘jittered’ to show number of animals in a group; thick horizontal lines: means of the treatment groups; RU: relative units (parasite burden relative to the mean burden of the vehicle-treated group).
We further tested if GNF6702 can cure additional kinetoplastid parasite infections. An estimated 25% of the 8 million people infected with T. cruzi will develop chronic Chagas disease, manifesting as cardiac or intestinal dysfunction16,17. Benznidazole is broadly used for treatment of acute and indeterminate stages of Chagas disease in Latin America18,19. However, benznidazole has side-effects that frequently lead to treatment interruption18,20–22 and a better tolerated drug is needed. To model treatment in the indeterminate disease stage, we infected mice with T. cruzi parasites and began treatment 35 days after infection, when the immune system of the mice had controlled parasite burden23. We increased the parasite detection sensitivity by immunosuppressing the mice after 20 days of treatment23,24. In this model, GNF6702 dosed twice-daily at 10 mg/kg matched the efficacy of benznidazole at 100 mg/kg once-daily; all but one treated mice had no detectable parasites in blood, colon or heart tissue, even after 4 weeks of immunosuppression (Fig. 2d).
Finally, we tested GNF6702 in a mouse model of stage II sleeping sickness (human African trypanosomiasis - HAT)25. Mortality of stage II HAT is caused by infection of the CNS and, in this mouse model, luciferase-expressing T. brucei parasites establish a CNS infection by day 21 post-infection. GNF6702 was administered at 100 mg/kg once-daily to account for low exposure in the brain relative to the plasma (~10%, Extended Data Fig. 1b). Diminazene aceturate, a stage I drug that poorly crosses the blood-brain barrier, effected apparent clearance of parasites from the blood after a single dose, but did not prevent parasite recrudescence 21 days later. By contrast, treatment with GNF6702 for seven days caused a sustained clearance of parasites (days 42 and 92 post-infection in Fig. 2e, Extended Data Fig. 2a, Supplementary Information Tables 4 and 5). Significantly, mice treated with GNF6702 had no detectable parasites in the brain at termination of the experiment, though parasites were clearly detected in the brains of mice treated with diminazene aceturate (Extended Data Fig. 2b, Supplementary Information Table 6).
As GNF6702 showed compelling efficacy in four mouse models of kinetoplastid infections: VL, CL, Chagas disease and stage II HAT, we reasoned that mechanistic studies of GNF6702 might identify a pan-kinetoplastid drug target that could inform target-based drug discovery efforts. We attempted to evolve L. donovani strains resistant to GNF3943 and GNF8000 (early analogues from the series, Extended Data Fig. 3) through 12 months of parasite culture under drug pressure without success. However, we were able to select two drug-resistant T. cruzi epimastigote isolates, one resistant to GNF3943, and another to GNF8000. Both T. cruzi lines exhibited at least 40-fold lower susceptibility to GNF6702 than wild type T. cruzi (Extended Data Fig. 4a and 4b). Using whole genome sequencing, we found that the GNF3943-resistant line had a homozygous mutation encoding a substitution of isoleucine for methionine at amino acid 29 in the proteasome beta 4 subunit (PSMB4I29M/I29M) and a heterozygous mutation P82L in dynein heavy chain gene. The GNF8000-resistant line had a heterozygous F24L mutation in PSMB4, and four other heterozygous mutations (Extended Data Table 1). We focused our attention on the proteasome as a likely target for the compound series because we found two independent mutations in the PSMB4 gene, and because the proteasome is an essential enzyme in eukaryotic cells. We also note that the Plasmodium falciparum proteasome has recently been the target of promising drug discovery efforts for malaria26.
We first asked whether two prototypic inhibitors of mammalian proteasome, bortezomib and MG132, could also block T. cruzi growth. Indeed, both compounds inhibited T. cruzi epimastigote proliferation with sub-micromolar potency. However, in contrast to GNF6702, bortezomib and MG132 inhibited proliferation of the two resistant lines (PSMB4I29M/I29M, PSMB4wt/F24L) with comparable potency to the wild type parasites. Additionally, the PSMB4 mutant lines were not resistant to nifurtimox, an anti-kinetoplastid drug with an unrelated mechanism of action (Extended Data Fig. 4a and 4b). To determine whether the F24L mutation was sufficient to confer resistance to GNF6702, we engineered T. cruzi epimastigote lines that ectopically expressed either wild type or F24L-mutated PSMB4. Overexpression of PSMB4WT had little effect on the EC50 value for GNF6702, whereas overexpression of PSMB4F24L caused a greater than 10-fold reduction in GNF6702 potency, but not in that of bortezomib (Fig. 3a, Extended Data Fig. 4c). Previously, bortezomib was also shown to inhibit the growth of T. brucei, suggesting that proteasome activity is essential for growth in this parasite as well27. To test whether PSMB4F24L can rescue growth inhibition by GNF6702 in T. brucei, we engineered two parasite strains that ectopically expressed wild type and F24L-mutated PSMB4, respectively. Similar to T. cruzi, overexpression of PSMB4F24L in T. brucei conferred a high level of resistance to GNF6702 (~70-fold shift in EC50 value), while having no effect on parasite susceptibility to bortezomib (Fig. 3b, Extended Data Fig. 4c).
Figure 3. F24L mutation in proteasome beta 4 subunit confers selective resistance to GNF6702.

a, growth inhibition of T. cruzi epimastigote strains ectopically expressing PSMB4WT or PSMB4F24L protein by GNF6702 and bortezomib; non-induced/induced: culture medium without/with tetracycline to modulate expression of tetracycline-inducible PSMB4 genes. b, growth inhibition of T. brucei bloodstream form trypomastigotes constitutively overexpressing PSMB4WT or PSMB4F24L protein by GNF6702 and bortezomib. EC50 values for each strain/compound pair are listed inside a and b plot panels next to corresponding strain/compound symbol (defined in plot legends); means from n=3 technical replicates are shown; error bars represent s.e.m. values; for data points lacking error bars, s.e.m. values are smaller than circles representing means; due to limited aqueous solubility, the highest tested GNF6702 concentration was 10 μM. RU (relative units) in a and b corresponds to parasite growth relative to the DMSO control (%).
We next asked whether GNF6702 could inhibit any of three T. cruzi proteasome proteolytic activities in biochemical assays. As predicted from the T. cruzi genome28, mass spectrometry analysis of purified T. cruzi proteasome identified seven alpha and seven beta proteasome subunits, including PSMB4 (Supplementary Tables 7 and 8). Using substrates that are specific for each of the chymotrypsin-like, trypsin-like and caspase-like proteolytic activities, we found that only the chymotrypsin-like activity of the T. cruzi proteasome was inhibited by GNF6702 (IC50=35 nM), while the other two activities were not affected (IC50>10 μM). In contrast, bortezomib inhibited the chymotrypsin-like (IC50=91 nM), the caspase-like (IC50=370 nM) and the trypsin-like (IC50=1.7 μM) activities. We further found that the chymotrypsin-like activity of the PSMB4I29M T. cruzi proteasome was at least 300-fold less susceptible to GNF6702 (IC50>10 μM) and ~3-fold less susceptible to bortezomib (IC50=0.26 μM), while susceptibility of the other two mutant proteasome proteolytic activities to the two inhibitors were not affected (Fig. 4a, Extended Data Table 2).
Figure 4. Compounds from GNF6702 series inhibit growth of kinetoplastid parasites by inhibiting parasite proteasome chymotrypsin-like activity.

a, Inhibition of three proteolytic activities of purified wild type (PSMB4WT) and PSMB4I29M T. cruzi proteasomes by GNF6702 and bortezomib; IC50 values for proteasome proteolytic activities are listed inside plots. b, Correlation between inhibition of chymotrypsin-like activity of purified L. donovani proteasome (IC50) and L. donovani axenic amastigote growth inhibition (EC50; data points correspond to means of 2 technical replicates); red circles: IC50>20 μM; blue circles: EC50>25 μM; yellow circles: IC50>20 μM and EC50>25 μM; data for 317 analogues are shown. c, Lineweaver-Burk plot of inhibition of T. cruzi proteasome chymotrypsin-like activity by GNF6702 at increasing concentrations of a peptide substrate. d, Effect of GNF6702 and bortezomib on three proteolytic activities of human constitutive proteasome; IC50 values for proteasome proteolytic activities are listed inside plots. Data shown in a, c and d represent means ± s.e.m. (n=3 technical replicates; for data points lacking error bars, s.e.m. values are smaller than circles representing means). Due to limited aqueous solubility, the highest tested GNF6702 concentration in experiments shown in a and d was 10 μM.
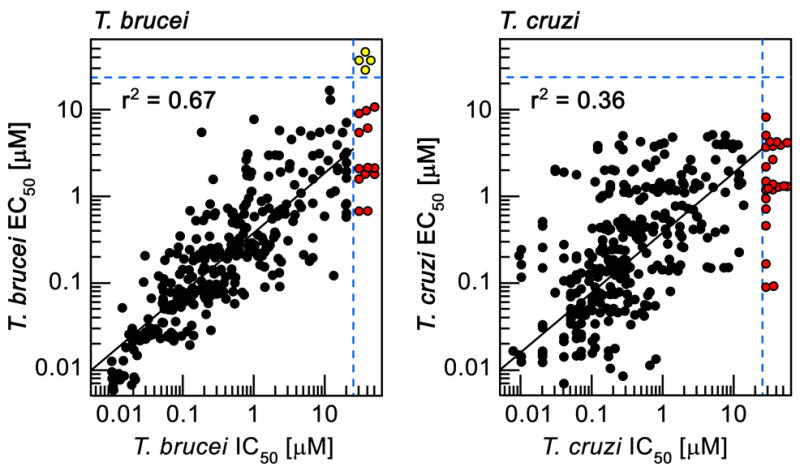
We reasoned that if the primary mechanism of parasite growth inhibition by the compound series was through inhibition of the proteasome chymotrypsin-like activity, then the IC50 values for this proteolytic activity should correlate with EC50 values for parasite proliferation. Indeed, a tight correlation between the two parameters was observed for L. donovani axenic amastigotes and T. brucei bloodstream form trypomastigotes (r2=0.78 and r2=0.67, respectively) over a 2,000-fold potency range for 317 analogues, thus indicating that inhibition of parasite proteasome activity was driving the anti-parasitic activity of these compounds. We observed a weaker correlation between IC50 and EC50 values for intracellular T. cruzi (r2=0.36, p<0.01), perhaps reflecting more complex cellular pharmacokinetics resulting from compounds having to access T. cruzi parasites within the cytosol of mammalian cells (Fig. 4b, Extended Data Fig. 5).
Both resistant T. cruzi lines retained sensitivity to bortezomib, which is a substrate-competitive inhibitor, suggesting that GNF6702 might have an alternative mode of inhibition. A Lineweaver-Burk plot of chymotrypsin-like activity at increasing concentrations of peptide substrate showed that GNF6702 has a non-competitive mode of inhibition clearly distinct from the competitive mechanism described for MG132 and bortezomib29,30. We were also able to extend these observations to proteasome from L. donovani (Fig. 4c, Extended Data Table 3). We further note that GNF6702 had no measurable activity on the human proteasome (Fig. 4d, Extended Data Table 2). Interestingly, human proteasome beta 4 subunit has a methionine at the 29th amino acid position, mirroring the I29M mutation in the GNF3943-resistant T. cruzi line (Extended Data Fig. 6a).
In summary, GNF6702 blocks the chymotrypsin-like activity harbored by the beta 5 subunit without competing with substrate binding, and mutations in the beta 4 subunit, which is in direct physical contact with the beta 5 subunit, confer resistance to this inhibition. Next we used homology modeling of the T. cruzi proteasome to look for evidence of an allosteric inhibitor binding site. In the T. cruzi proteasome model, the F24 and I29 beta 4 residues are positioned at the interface between the beta 4 and beta 5 subunits, on the outer limit of the beta 5 active site. Adjacent to these two beta 4 residues and the beta 5 active site is a plausible binding pocket for GNF6702 (Extended Data Fig. 6b and 6c).
Finally, we tested whether GNF6702 can inhibit proteasome activity in intact T. cruzi cells. Cellular proteins entering the proteasome degradation pathway are first tagged with ubiquitin, and proteasome inhibition results in intracellular accumulation of ubiquitylated proteins. Treatment of T. cruzi epimastigotes with GNF6702 led to significant buildup of ubiquitylated proteins (Extended Data Fig. 7a) with the half-maximal effect (EC50) achieved at 130 nM compound concentration (Extended Data Fig. 7c). This EC50 value correlated well with the half-maximal growth inhibitory concentration of GNF6702 on T. cruzi epimastigotes (EC50=150 nM; Extended Data Fig. 4b). For comparison, similar experiments with bortezomib yielded comparable inhibitor potencies in the two T. cruzi assays (ubiquitylation EC50=62 nM vs growth inhibition EC50=160 nM; Extended Data Fig. 4b and 7c). We did not observe any detectable accumulation of ubiquitylated proteins in mammalian 3T3 cells treated with GNF6702 (Extended Data Fig. 7b and 7c), further confirming high selectivity of this compound.
Validation of the parasite proteasome as the target of GNF6702 is supported through several lines of evidence: i) point mutations in the PSMB4 gene are sufficient to confer resistance to biochemical proteasome inhibition and cellular T. cruzi growth inhibition; ii) GNF6702 is a selective inhibitor of parasite proteasome activity and does not inhibit the human proteasome, mirroring the selective inhibition of parasite growth over mammalian cell growth; and iii) potency of GNF6702 and analogues in parasite proteasome assays predict potency in parasite growth inhibition assays.
In this work we show that in mouse disease models, GNF6702 was able to eradicate parasites from diverse niches that included the cytosol (T. cruzi), phagolysosome (L. donovani, L. major) of infected host cells, and brain (T. brucei). GNF6702 has also good pharmacokinetic properties, and the compound did not show activity in panels of human receptor, enzyme and ion channel assays (Supplementary Tables 9–11). Going forward, GNF6702, or analogues thereof, has potential to yield a new treatment for several kinetoplastid infections and it is currently being evaluated in preclinical toxicity studies. It is unclear if the clinical utility of GNF6702 could extend to the treatment of stage II HAT as GNF6702 was tested in the HAT mouse model only at one high dose (100 mg/kg once-daily). We also note that identification of a broadly active pan-kinetoplastid drug might not be feasible (or desirable) as such a drug would need to reach high concentrations in varied tissues/subcellular compartments, and might carry increased toxicity risk. Instead, alternative analogues from this series with different pharmacological profiles might be needed for treatment of different kinetoplastid infections. Nevertheless, there are only scarce resources for drug development in these diseases, and identification of a common target and chemical scaffold with potential across multiple indications provides new hope for improved treatment options for some of the world’s poorest people.
METHODS
Ethics statement for animal models
All procedures involving mice were performed in accordance with AAALAC standards or under UK Home Office regulations, and were reviewed and approved in accordance with the Novartis Animal Welfare Policy. Sample size was determined on the basis of the minimum number of animals required for good data distribution and statistics. Blinding was not possible in these experiments but animals were selected randomly for each group.
Determination of IC50, EC50, and CC50 values
Reported IC50/EC50/CC50 values were calculated by averaging IC50/EC50/CC50 values obtained from individual technical replicate experiments (n; specified in relevant Figure captions and Methods sub-sections). Each technical replicate experiment was performed on a different day with freshly prepared reagents. Reported standard errors of mean (s.e.m.) were calculated using IC50/EC50/CC50 values determined in individual technical replicate experiments. To calculate IC50/EC50/CC50 values, measured dose response values were fitted with 4-parameter logistic function y=A+(B−A)/(1+(x/C)^D) (model 201, XLfit, IDBS), where x refers to compound concentration and y corresponds to an assay readout value.
Leishmania donovani axenic amastigote growth inhibition assay
RPMI 1640 medium (HyClone) was supplemented with 20% heat-inactivated fetal bovine serum (Omega Scientific), 23 μM folic acid (Sigma-Aldrich), 100 μM adenosine (Sigma-Aldrich), 22 mM D-glucose (Sigma-Aldrich), 4 mM L-glutamine (Hyclone), 25 mM 2-(4-morpholino) ethanesulfonic acid (Sigma-Aldrich) and 100 IU penicillin/100 μg/mL streptomycin (HyClone), and adjusted to pH= 5.5 with 6 M hydrochloric acid (Fisher Scientific) at 37 °C. Leishmania donovani MHOM/SD/62/1S-CL2D axenic amastigotes were cultured in 10 mL of this medium (Axenic Amastigote Medium) in T75 CELL-STAR flasks (Greiner Bio-One) at 37 °C/5% CO2 and passaged once a week.
To determine compound growth inhibitory potency on L. donovani axenic amastigotes, 100 nL of serially diluted compounds in DMSO were transferred to the wells of white, solid bottom 384-well plates (Greiner Bio-One) by Echo 555 acoustic liquid handling system (Labcyte). Then, 1 × 103 of L. donovani axenic amastigotes in 40 μL of Axenic Amastigote Medium were added to each well, and plates were incubated for 48 hours at 37 °C/5% CO2. Parasite numbers in individual plate wells were determined through quantification of intracellular ATP. The CellTiter-Glo luminescent cell viability reagent (Promega) was added to plate wells, and ATP-dependent luminescence signal was measured on an EnVision MultiLabel Plate Reader (Perkin Elmer). Luminescence values in wells with compounds were divided by the average luminescence value of the plate DMSO controls, and used for calculation of compound EC50 values as described above.
Axenic amastigote EC50 values shown in Fig. 4b correspond to means of 2 technical replicates.
Isolation and maintenance of Leishmania donovani splenic amastigotes
Female BALB/cJ mice (Envigo) infected with L. donovani MHOM/ET/67/HU3 (ATCC) for 50–80 days were euthanized, and infected spleens were removed and weighed. The weight of an infected spleen ranged from 300 to 600 mg. For comparison, spleens from non-infected age-matched BALB/cJ mice weighed ~100 mg. Infected spleens were washed in Axenic Amastigote Medium (composition described above) and placed into Falcon 50 mL conical centrifuge tubes (Fisher Scientific) containing ice-cold Axenic Amastigote Medium (15 mL per infected spleen). Spleens were homogenized on ice in a Dounce homogenizer and centrifuged at 200 × g for 15 minutes at 4 °C to remove tissue debris. Leishmania donovani amastigotes present in the supernatant were pelleted by centrifugation at 1,750 × g for 15 min at 4 °C and re-suspended either in Axenic Amastigote Medium (when used for in vitro macrophage infections) or in Hanks’ Balanced Salt Solution (when used for mouse infections; Hyclone). Suspensions of splenic amastigotes were kept on ice and used for in vitro or in vivo infections within 2–3 hours. To propagate L. donovani amastigotes in vivo, 6 to 7 weeks old female BALB/cJ mice were infected with 8 × 107 purified splenic amastigotes in 200 μL of Hanks’ Balanced Salt Solution by tail vein injection.
Leishmania donovani intra-macrophage amastigote growth inhibition assay
In vitro compound potencies on intra-macrophage L. donovani MHOM/ET/67/HU3 were determined using primary murine peritoneal macrophages infected with L. donovani splenic amastigotes. Primary macrophages were elicited in female BALB/c mice for 72 hours following the injection of 500 μL of sterile aqueous 2% starch (J. T. Baker) solution into the mouse peritoneal cavity. The protocol used for isolation of peritoneal macrophages was described in detail previously31. The isolated macrophages were re-suspended in Macrophage Infection Medium (RPMI-1640 medium supplemented with 2 mM L-glutamine, 10% heat-inactivated fetal bovine serum, 10 mM sodium pyruvate (Hyclone), and 100 IU penicillin/100 μg/mL streptomycin), and 50 μL of macrophage suspension (8 × 105 macrophages/mL) were added to microscopy-grade, clear-bottom, black 384-well plates (Greiner Bio-One). Following overnight incubation at 37 °C/5% CO2, plate wells were washed with Macrophage Infection Medium to remove non-adherent cells using ELx405 Select microplate washer (BioTek), and then filled with 40 μL of Macrophage Infection Medium. Leishmania donovani HU3 splenic amastigotes isolated from infected spleens were re-suspended in Macrophage Infection Medium at a concentration of 6 × 107 cells/mL, and 10 μL of the suspension were added to assay plate wells containing adherent macrophages. After a 24-hour infection period at 37 °C/5% CO2, plate wells were washed with Macrophage Infection Medium to remove residual extracellular parasites and re-filled with 50 μL of the medium. Leishmania donovani-infected macrophages were subsequently treated with DMSO-dissolved compounds (0.5% final DMSO concentration in the assay medium) in dose response for 120 hours at 37 °C/5% CO2. Next, treated macrophages were washed with the phosphate-buffered saline buffer (PBS; Sigma-Aldrich) supplemented with 0.5 mM magnesium chloride (Sigma-Aldrich) and 0.5 mM calcium chloride (Sigma-Aldrich), fixed with 0.4% paraformaldehyde (Sigma-Aldrich) in PBS, permeabilized with 0.1% Triton X-100 (Sigma-Aldrich) in PBS, and stained with SYBR Green I nucleic acid stain(Invitrogen, 1:100,000 dilution in PBS) overnight at 4 °C. Image collection and enumeration of macrophage cells and intracellular L. donovani amastigotes was performed using the OPERA QEHS automated confocal microscope system equipped with 20x water immersion objective (Evotec Technologies) and the OPERA Acapella software (Evotec Technologies) as described previously32.
All reported intra-macrophage L. donovani EC50 values were calculated from at least 3 technical replicates (n= 3 or n= 4; specified in relevant Figure captions).
Trypanosoma brucei growth inhibition assay
Bloodstream form Trypanosoma brucei Lister 427 parasites were continuously passaged in HMI-9 medium formulated from IMDM medium (Invitrogen), 10% heat-inactivated fetal bovine serum, 10% Serum Plus medium supplement (SAFC Biosciences), 1 mM hypoxanthine (Sigma-Aldrich), 50 μM bathocuproine disulfonic acid (Sigma-Aldrich), 1.5 mM cysteine (Sigma-Aldrich), 1 mM pyruvic acid (Sigma-Aldrich), 39 μg/mL thymidine (Sigma-Aldrich), and 14 μL/L beta-mercapthoethanol (Sigma-Aldrich); all concentrations of added components refer to those in complete HMI-9 medium. The parasites were cultured in 10 mL of HMI-9 medium in T75 CELL-STAR tissue culture flasks at 37 °C/5% CO2.
To determine compound growth inhibitory potency on T. brucei bloodstream form parasites, 100 nL of serially diluted compounds in DMSO were transferred to the wells of white, solid bottom 384-well plates (Greiner Bio-One) by Echo 555 acoustic liquid handling system. Then, 5 × 103 of T. brucei parasites in 40 μL of HMI-9 medium were added to each well, and the plates were incubated for 48 hours at 37 °C/5% CO2. Parasite numbers in individual plate wells were determined through quantification of intracellular ATP amount. The CellTiter-Glo luminescent cell viability reagent was added to plate wells, and ATP-dependent luminescence signal was measured on an EnVision MultiLabel Plate Reader. Luminescence values in wells with compounds were divided by the average luminescence value of the plate DMSO controls, and used for calculation of compound EC50 values as described above.
Trypanosoma brucei EC50 values shown in Fig. 1 and Extended Data Fig. 3 correspond to means of 4 technical replicates.
Trypanosoma cruzi amastigote growth inhibition assay
NIH 3T3 fibroblast cells (ATCC) were maintained in RPMI 1640 medium (Life Technologies) supplemented with 10% heat-inactivated fetal bovine serum and 100 IU penicillin/100 μg/mL streptomycin at 37 °C/5% CO2. Trypanosoma cruzi Tulahuen parasites constitutively expressing Escherichia coli beta-galactosidase33 were maintained in tissue culture as an infection in NIH 3T3 fibroblast cells. Briefly, 2 × 107 T. cruzi trypomastigotes were used to infect 6 × 105 NIH 3T3 cells growing in T75 CELL-STAR tissue culture flasks and cultured at 37 °C/5% CO2 until proliferating intracellular parasites lysed host 3T3 cells and were released into the culture medium (typically 6–7 days). During the infection, the tissue culture medium was changed every two days. Number of T. cruzi trypomastigotes present in one mL of medium was determined using a hemocytometer.
To determine compound potency on intracellular T. cruzi amastigotes, NIH 3T3 cells were re-suspended in phenol red-free RPMI 1640 medium containing 3% heat-inactivated fetal bovine serum and 100 IU penicillin/100 μg/mL streptomycin, seeded at 1,000 cells/well (40 μL) in white, clear bottom 384-well plates (Greiner Bio-One), and incubated overnight at 37 °C/5% CO2. The following day, 100 nL of each compound in DMSO were transferred to individual plate wells by Echo 555 acoustic liquid handling system. After one hour incubation, 1 × 106 of tissue culture-derived T. cruzi trypomastigotes, in 10 μL of phenol red-free RPMI 1640 medium supplemented with 3% heat-inactivated fetal bovine serum and 100 IU penicillin/100 μg/mL streptomycin were added to each well. Plates were then incubated for 6 days at 37 °C/5% CO2. Intracellular T. cruzi parasites were quantified by measuring the activity of parasite-expressed beta-galactosidase. Ten microliters of a chromogenic beta-galactosidase substrate solution (0.6 mM chlorophenol red-β-D-galactopyranoside/0.6% NP-40 in PBS; both reagents from Calbiochem) were added to each well and incubated for 2 hours at room temperature. After incubation, absorption was measured at 570 nM on SpectraMax M2 plate reader (Molecular Devices). Measured absorbance values in wells with compounds were divided by the average absorbance value of the plate DMSO controls, and used for calculation of compound EC50 values as described above.
Trypanosoma cruzi amastigote EC50 values shown in Fig. 1 and Extended Data Fig. 3 correspond to means of 4 technical replicates.
Trypanosoma cruzi epimastigote proliferation assay
Trypanosoma cruzi CL epimastigotes were continuously passaged in LIT medium containing 9 g/L liver infusion broth (Difco), 5 g/L bacto-tryptose (Difco), 1 g/L sodium chloride, 8 g/L dibasic sodium phosphate (Sigma-Aldrich), 0.4 g/L potassium chloride (Sigma-Aldrich), 1 g/L D-glucose, 10 % heat-inactivated fetal bovine serum and 10 ng/mL of hemin (Sigma-Aldrich). The medium was adjusted to pH= 7.2 with 6 M hydrochloric acid. The parasites were cultured in 10 mL of LIT medium in T75 CELL-STAR tissue culture flasks at 27 °C.
To determine compound growth inhibitory potency on T. cruzi epimastigotes, 100 nL of serially diluted compounds in DMSO were transferred to the wells of white, solid bottom 384-well plates (Greiner Bio-One) by an Echo 555 acoustic liquid handling system. Then, 5 × 103 of T. cruzi epimastigotes in 40 μL of LIT medium were added to each well, and the plates were incubated for 7 days at 27 °C. Parasite numbers in individual plate wells were determined through quantification of intracellular ATP amount. The CellTiter-Glo luminescent cell viability reagent was added to plate wells, and ATP-dependent luminescence signal was measured on an EnVision MultiLabel Plate Reader. Luminescence values in wells with compounds were divided by the average luminescence value of the plate DMSO controls, and used for calculation of compound EC50 values as described above.
Trypanosoma cruzi epimastigote EC50 values shown in Extended Data Fig. 4 correspond to means of 3 technical replicates.
Mouse fibroblast NIH 3T3 growth inhibition assay
NIH 3T3 fibroblast cells were maintained in RPMI medium 1640 with glutamine (Life Technologies) supplemented with 5% heat-inactivated fetal bovine serum and 100 IU penicillin/100 μg/mL streptomycin (3T3 Medium) at 37 °C/5% CO2. NIH 3T3 fibroblast cells were purchased from ATCC. We did not perform cell line authentication and did not test the cells for mycoplasma contamination. This cell line is not listed in the database of commonly misidentified cell lines maintained by ICLAC and NCBI Biosample.
To determine compound potency, NIH 3T3 cells re-suspended in 3T3 medium were seeded at 1,000 cells/well (50 μL) in white 384-well plates (Greiner Bio-One) and incubated overnight at 37 °C/5% CO2. The following day, 100 nL of each compound in DMSO were transferred to individual plate wells by Echo 555 acoustic liquid handling system and plates were incubated for five days at 37 °C/5% CO2. Cell numbers in individual plate wells were determined through quantification of intracellular ATP amount. The CellTiter-Glo luminescent cell viability reagent was added to plate wells, and ATP-dependent luminescence signal was measured on an EnVision MultiLabel Plate Reader. Luminescence values in wells with compounds were divided by the average luminescence value of the plate DMSO controls, and used for calculation of compound CC50 values as described above.
NIH 3T3 CC50 values shown in Fig. 1 and Extended Data Fig. 3 correspond to means of 4 technical replicates.
Primary macrophage cytotoxicity assay
Primary macrophage cell viability was determined on mouse peritoneal macrophages infected with L. donovani and was expressed as the ratio of the number of macrophage cells in wells treated with a compound to those in wells treated with DMSO. The number of macrophage cells in wells was determined by high content microscopy as described previously32.
All reported macrophage CC50 values were calculated from 4 technical replicates (n= 4; also specified in Figure 1 and Extended Data Figure 3 captions).
Selection of GNF3934- and GNF8000-resistant T. cruzi mutants
T. cruzi epimastigotes cultures resistant to GNF3943 and GNF8000 were generated using a methodology described previously32. Briefly, epimastigotes were initially cultured in the presence of compound concentration equivalent to its EC20 value (GNF3943 EC20= 1.5 μM and GNF8000 EC20= 0.2 μM in 0.2% DMSO) or 0.2% DMSO (control). Once a week, parasites were counted and growth rates were determined. If the parasite cultures exhibited a reduced growth rate compared to 0.2% DMSO-treated parasites, epimastigotes were cultured at the same compound concentration. Once the growth rates matched that of the control epimastigote culture (0.2% DMSO), parasites were transferred into medium containing two-fold higher compound concentration. The process was repeated until significant resistance was achieved (~10- to 20-fold increase in corresponding EC50 value). The time required for generation of cultures with such a level of resistance was approximately five months. Resistant clones were isolated via cloning by limiting dilution, and two independent clones were analyzed by whole genome sequencing.
T. cruzi whole genome sequencing
Chromosomal DNA isolation from GNF3943- and GNF8000-resistant T. cruzi clones, whole genome sequencing and sequence analysis were performed as described previously32. Sequencing reads were aligned to the T. cruzi CL Brenner genome34.
Generation of T. cruzi strains ectopically expressing proteasome beta 4 subunit variants
PSMB4 TcCLB503891.100 was amplified from T. cruzi CL Brenner genomic DNA using KOD Hot Start DNA Polymerase (EMD Millipore), and sense (5′-AAAGCGGCCGCATGTCGGAGACAACCATTG-3) and antisense (5-CCATGATCTTGATGTAATATAAGGCATTCAGCCCTGCTG-3) primers. The PSMB4F24L gene was generated from the wild type PSMB4 construct by site-directed mutagenesis using mutagenic sense (5-CAGCAGGGCTGAATGCCTTATATTACATCAAGATCATGG-3′) and antisense (5′-CCATGATCTTGATGTAATATAAGGCATTCAGCCCTGCTG-3′) primers and QuikChange II Site-Directed Mutagenesis Kit (Stratagene). The sequences of the wild type and mutant PSMB4 genes were verified by sequencing and both gene versions were subcloned into the T. cruzi expression vector pTcIndex1 under control of a T7 promoter35. Trypanosoma cruzi CL Brenner epimastigotes were first transfected as described previously36 with the pLEW13 plasmid37 harboring a tetracycline-inducible T7 RNA polymerase gene. Transfected epimastigotes were selected in medium supplemented with neomycin (G418) at 500 μg/ml, and then transfected a second time with either pTcIndex1-PSMB4wt or pTcIndex1-PSMB4F24L plasmid. Double transfected epimastigotes were selected in the presence of 500 μg/mL of G418 (Sigma-Aldrich) and 500 μg/mL of hygromycin (Sigma-Aldrich). Susceptibility of double transfected epimastigote cell lines to compounds was assessed using induced (+5 mg/mL of tetracycline) and non-induced parasite cultures after five days of compound treatment. Parasite viability was determined with AlamarBlue (ThermoFisher Scientific).
Reported EC50 values for T. cruzi epimastigotes ectopically expressing PSMB4 proteins were calculated from 3 technical replicates (n= 3; also specified in the Figure 3a caption).
Generation of T. brucei strains ectopically expressing proteasome beta 4 subunit variants
PSMB4 (Tb927.10.4710) was amplified from T. brucei Lister 427 genomic DNA using PCR SuperMix High Fidelity (Invitrogen), sense (5′-GCAAGCTTATGGCAGAGACGACTATCGG-3) and antisense (5′-GCGGATCCCTAGCTTACAGATTGCACTC-3′) primers. The PSMB4F24L gene was generated from the wild type PSMB4 construct by site-directed mutagenesis using mutagenic sense (5′-gctgcggggttaaatgcgttatactacattaagataacgg-3′), antisense (5′-ccgttatcttaatgtagtataacgcatttaaccccgcagc-3′) primers and QuikChange II Site-Directed Mutagenesis Kit (Stratagene). The sequences of the wild type and mutant PSMB4 genes were verified by sequencing and both gene versions were cloned into the T. brucei expression vector pHD1034 under control of a ribosomal RNA promoter. Transfected T. brucei Lister 427 cells were selected in medium supplemented with puromycin at 1 μg/ml. Susceptibility of transfected T. brucei cell lines to compounds were assessed after 2 days of compound treatment. Parasite viability was determined with CellTiter-Glo.
Reported EC50 values for T. brucei parasites ectopically expressing PSMB4 proteins were calculated from 3 technical replicates (n= 3; also specified in the Figure 3b caption).
Purification of parasite 20S proteasomes
T. cruzi CL epimastigotes, L. donovani MHOM/SD/62/1S-CL2D axenic amastigotes and T. brucei Lister 27 bloodstream form trypomastigotes were grown to log phase and harvested by centrifugation. The corresponding cell pellets were stored at −80 °C until further use. Prior to purification, 10 g of cell pellets were thawed, re-suspended in lysis buffer (50 mM Tris-HCl pH = 7.5, 1 mM TCEP, 5 mM EDTA, and 10 μM E-64), and lysed by passing cell suspension three times through a needle (22 gauge) and by subsequent three freeze/thaw cycles. The lysate was first cleared of cellular debris by two centrifugation steps (15,000 × g at 4 °C for 15 minutes followed by 40,000 × g at 4 °C for 60 minutes) and then fractionated through ammonium sulfate precipitation. The protein fraction precipitated between 45% and 65% of ammonium sulfate saturation was re-suspended in 25 mM Tris-HCl pH = 7.5, 1 mM TCEP buffer, and dialyzed overnight at 4 °C against the same buffer. Proteasomes were further purified by anion exchange chromatography (Resource Q column, GE Healthcare Life Sciences) and size exclusion chromatography (Superose 6 column, GE Healthcare Life Sciences) as described elsewhere38. Active fractions from the latter purification step were pooled and used in proteasome biochemical assays.
Subunit composition analysis of purified T. cruzi 20S proteasome by LC/MS/MS
Purified T. cruzi proteasome sample was buffer-exchanged and concentrated into 100 mM trimethylamine bicarbonate-HCl pH= 8.0, 150 mM NaCl buffer using a 10 kDa molecular weight cut-off micro-concentrator (Milipore Amicon Ultra). The resulting proteasome sample (200 μl, 1 mg/ml) was mixed with 5 μl of a TMTsixplex reagent (Pierce). After 60 second incubation to label primary amines, the reaction was stopped by adding 25 μl of 5% hydroxylamine. The labeled sample was run on 4–20% Bis-Tris PAGE gel (Invitrogen) to separate polypeptides. The gel was stained with eStain 2.0 (GenScript). Stained protein bands were cut out and in-gel digested separately with elastase (Promega) and asparaginase (Roche). Peptides generated by the digestions were resolved by HPLC using a vented column setup with a 2 cm Poros 10 R2 (Life Technologies, Carlsbad, CA) self-packed pre-column, and a PepMap Easy-Spray C18 analytical column (15 cm × 75 μm ID, Thermo Scientific). Resin-bound proteolytic fragments were eluted with 2 to 40% acetonitrile/0.1% formic acid operated at 300 nL/min for 120 min. Spectra of eluted peptide species were determined by a column-coupled Q Exactive hybrid quadrupole orbitrap mass spectrometer (Thermo Scientific). Proteome Discoverer v1.4 software (Thermo Scientific) was used to search the T. cruzi genome28 with identified spectra for presence of 20S proteasome subunits (Supplementary Table 7). Search parameters included fixed carbamidomethyl modification of cysteine, and variable oxidation of methionine, deamidation of asparagine, pyro-glu of N-terminal glutamine, and TMT(6-plex) modification of lysine residues.
Measuring proteasome proteolytic activities
The activity of purified parasite and human 20S proteasomes was monitored by measuring cleavage of various rhodamine-labelled fluorogenic substrates. Purified 20S proteasomes were diluted in proteasome assay buffer (25 mM Tris-HCl pH 7.5, 1 mM dithiothreitol (Sigma-Aldrich), 10 mM sodium chloride, 25 mM potassium chloride, 1 mM magnesium chloride, 0.05% (w/v) CHAPS (Sigma-Aldrich) and 0.9% DMSO) at a final concentration of 162 nM (parasite proteasomes) or 25 nM (human proteasome), and pre-incubated with compound (40 nL; 0.2% final DMSO concentration) for 1 hour. Next, the following substrates (Biosynthan GmbH) were added at 3 μM final concentration to monitor specific proteolytic activities (Suc-LLVY-Rh110-dPro: chymotrypsin-like activity; Ac-RLR-Rh110-dPro: trypsin-like activity; Ac-GPLD-Rh110-dPro: caspase-like activity). The reaction was allowed to proceed for two hours at room temperature and fluorescence as a measure of purified 20S proteasome activity was monitored using the EnVision® plate reader (excitation at 485 nm/emission at 535 nm). Km and Ki values were calculated using GraphPad Prism (GraphPad Software) ‘Non-competitive enzyme inhibition’ function.
Data shown in Fig. 4a, 4c, 4d and Extended Data Table 3 represent means of 3 technical replicates (n= 3). Data shown in Fig. 4b and Extended Data Fig. 5 represent means of 2 technical replicates (n= 2).
Monitoring accumulation of ubiquitylated proteins in intact cells
Growing T. cruzi epimastigotes were seeded into 24-well tissue culture plate (1 × 107 cells/per well) in LIT medium and treated for 2–12 hours with DMSO (0.2%) or various concentrations of bortezomib and GNF6702 at 27 °C. Following the treatment, parasites were collected by centrifugation (3,500 g for 6 minutes) and washed twice with phosphate-buffered saline (PBS). Epimastigotes were lysed by resuspending washed cells in a buffer containing 50 mM Tris-HCl pH= 7.4, 150 mM sodium chloride, 1% CHAPS, 20 μM E-64 (Sigma-Aldrich), 10 mM EDTA(Sigma-Aldrich), 5 mM N-ethylmaleimide(Sigma-Aldrich), 1 mM phenylmethylsulfonyl fluoride (Sigma-Aldrich), 10 μg/mL leupeptin (Sigma-Aldrich), 10 μg/mL aprotinin (Sigma-Aldrich), and incubating the suspension on ice for 20 minutes. Cell lysates were cleared by centrifugation at 21,000 g for 30 min at 4 °C.
For 3T3 cells, 2 × 105 cells/well were seeded into 24-well tissue culture plates in RPMI medium 1640 supplemented with 10% heat-inactivated fetal bovine serum, and incubated overnight at 37 °C to allow cells to attach. Attached cells were treated for 2 hours with DMSO (0.25%) or various concentrations of bortezomib and GNF6702. Treated cells were washed twice with PBS and then lysed by incubating cells in modified RIPA buffer (50 mM Tris-HCl pH= 7.4, 1% Triton X-100, 0.2% sodium dodecylsulfate, 1 mM EDTA, 1 mM phenylmethylsulfonyl fluoride, 5 μg/mL aprotinin, 5 μg/mL leupeptin) for 30 min at 4 °C. Cell lysates were cleared by centrifugation at 21,000 g for 30 min at 4 °C.
Protein concentration in cell extracts was determined with BCA assay (ThermoFisher), and 10 μg of cell extracts were loaded on NuPAGE Novex 4–12% Bis-Tris gel (Invitrogen). After electrophoresis, resolved proteins were transferred to nitrocellulose membrane. Ubiquitylated proteins were detected with polyclonal anti-ubiquitin primary antibody (Proteintech, catalogue number 10201-2-AP) and rabbit anti-mouse IgG-peroxidase antibody (Sigma-Aldrich, catalogue number A0545), and then imaged using ECL Prime Western Blotting Detection Reagent (Amersham) on Chemidoc XR+ imaging system (BioRad). Collected western blot images were quantified using Image Lab software (BioRad). Briefly, rectangles of identical size and shape were drawn around each blot lane to include inside the shape all ubiquitylated protein bands within 17 – 198 kDa molecular mass range. Next, integrated signal intensities within the rectangles (reported by the Image Lab software) were used for calculation of EC50 values. Three technical replicate experiments (n= 3) for each different dose response experiment (GNF6702 on T. cruzi epimastigotes; GNF6702 on 3T3 cells; bortezomib on T. cruzi epimastigotes; bortezomib on 3T3 cells) were performed.
Trypanosoma cruzi proteasome modeling studies
The homology model of T. cruzi 20S proteasome was built using ‘Prime’ protein structure prediction program (Schrödinger) and X-ray structure of bovine 20S proteasome (pdb accession code 1IRU)39 as the template. The model was subjected to restrained minimization to relieve inter-chain clashes. ‘SiteMap’ program (Schrödinger) was used to identify pockets on a protein surface suitable for small molecule binding. Flexible ligand docking was performed using ‘Glide 5.8’ (Schrödinger). The grid box was centered in a middle of the identified pocket and extended by 10 Å, with outer box extending additional 20Å. The ligand was docked using the standard precision (SP) algorithm and scored using ‘GlideScore’ (Schrödinger). The GNF6702 GlideScore is equal to −8.5.
Receptor, enzyme and ion channel assays
GNF6702 profiling was performed at 10 μM concentration in a selectivity panel at Eurofins (www.eurofinspanlabs.com/Catalog/AssayCatalog/AssayCatalog.aspx). Listed values % change in the assay readout relative to the DMSO control. To determine inhibition of a subset of human tyrosine kinases by GNF6702, the inhibitor was profiled on a panel of Ba/F3 cell lines expressing individual Tel-activated kinases as described previously40. All assays were performed as single technical repeats.
Determination of GNF6702 thermodynamic solubility
The solubility of GNF6702 was assessed in a high throughput thermodynamic solubility assay as described previously41. First, 25 μL of GNF6702 DMSO solutions were transferred to individual wells of a 96-well plate. DMSO was evaporated and 250 μL of 67 mM potassium phosphate buffer pH 6.8 were added to yield projected final compound concentrations from 1 μM to 100 μM. The plate was sealed to prevent solvent loss and shaken for 24 hours at room temperature. The plate was then filtered to remove non-dissolved material. Concentration of GNF6702 in individual plate wells was determined by measuring solution UV absorbance with reference to a GNF6702 calibration curve.
Determination of GNF6702 permeability in Caco-2 assay
A 96-Multiwell Insert System (BD Biosciences) was used for the Caco-2 cell culture and permeability assay as described previously42. Caco-2 cells were seeded onto insert wells at a density of 1.48 × 105 cells per ml and allowed to grow for 19–23 days before assays. To measure both absorptive (apical to basolateral [A-B]) and secretory (basolateral to apical [B-A]) compound transport, a solution of GNF6702 at 10 μM concentration in 0.5% DMSO were added to donor wells. The plate was incubated at 37°C for 2 hours, with samples taken at the beginning and end of the incubation from both donor and acceptor wells. The concentration of GNF6702 was determined by LC-MS/MS.
Apparent drug permeability (Papp) was calculated using the following equation:
where dQ/dt is the total amount of a test compound transported to the acceptor chamber per unit of time (nmol/s), A is the surface area of the transport membrane (0.0804 cm2), Cin is the initial compound concentration in the donor chamber (10 μM), and Papp is expressed as cm/s).
Determination of human CYP450 inhibition by GNF6702
Extent of inhibition of major human CYP450 isoforms 2C9, 2D6 and 3A4 by GNF6702 was determined using pooled human liver microsomes and the known specific substrates of various CYP450 isoforms: diclofenac (5 μM), bufuralol (5 μM), midazolam (5 μM), and testosterone (50 μM). Probe substrate concentrations were used at concentrations equal to their reported Km values. The CYP450 inhibition assays with probe substrates diclofenac (2C9) or midazolam (3A4) were incubated at 37 °C for 5 to 10 minutes using a microsomal protein concentration of 0.05 mg/mL. Probe substrates bufuralol (2D6) and testosterone (3A4) were incubated at 37 °C for 20 minutes using microsomal concentration 0.5 mg/mL. The test concentrations of GNF6702 ranged from 0.5 to 25 μM in the presence of 1% DMSO. The reactions were initiated by adding NADPH (1 mM final concentration; Sigma-Aldrich) after a 5-min pre-incubation. Incubations were terminated by the addition of 300 μL of acetonitrile to 100 μL of a sample. No significant cytochrome P450 inhibition was observed. Extent of CYP450 isoform inhibition was determined by quantifying residual concentrations of individual CYP450 substrate probes at the end of reactions by LC/MS/MS.
Determination of GNF6702 in vitro metabolic stability
The intrinsic metabolic stability of GNF6702 was determined in mouse and human liver microsomes using the compound depletion approach and LC/MS/MS quantification. The assay measured the rate and extent of metabolism of GNF6702 by measuring the disappearance of the compound. The assay determined GNF6702 in vitro half-life (T1/2) and hepatic extraction ratios (ER) as described previously43. GNF6702 was incubated for 30 minutes at 1.0 μM concentration in a buffer containing 1.0 mg/mL liver microsomes. Samples (50 μL) were collected at 0, 5, 15 and 30 minutes and immediately quenched by addition of 150 μL of ice-cold acetonitrile/methanol/water mixture (8/1/1). Quantification of GNF6702 in samples was performed by LC/MS/MS, and the in vitro intrinsic clearance was determined using the substrate depletion method. The intrinsic clearance, CLint was calculated using the following equation:
where T1/2 is the in vitro half-life, V (μL) is the reaction volume, and M (mg) is the microsomal protein amount. Finally the hepatic extraction ratio is calculated as:
where CLh = hepatic clearance, Qh = hepatic blood flow.
Clh was calculated using the following equation:
where fu = fraction unbound to protein (assumed to be 1).
Pharmacokinetic studies
An outline of various in vitro and in vivo DMPK assays used in this study for compound profiling was summarized previously44. The pharmacokinetic properties of GNF compounds and calculation of pharmacokinetic parameters was performed as described previously23. Mean compound plasma concentrations were calculated from fitted functions approximating compound plasma profile throughout 8 days of dosing. Blinding was not possible in these experiments.
Bioanalysis of GNF6702 in plasma
Plasma concentration of GNF6702 was quantified using a LC/MS/MS assay. Solution of 20 ng/mL of verapamil hydrochloride (Sigma-Aldrich) in acetonitrile/methanol mixture (3/1 by volume), was used as an internal standard. Twenty microliters of plasma samples were mixed with 200 μl of internal standard solution. The samples were vortexed and then centrifuged in an Eppendorf Centrifuge 5810R (Eppendorf) at 4,000 rpm for 5 minutes at 4 °C to remove precipitated plasma proteins. The supernatants (150 μl) were transferred to a 96-well plate and mixed with 150 μl H2O. The samples (10 μl) were then injected onto a Zorbax SB-C8 analytical column (2.1 × 30 mm, 3.5 μm; Agilent Technologies) and separated using a three step gradient (1st step: 1.5 mL of 0.05% formic acid in 10% acetonitrile; 2nd step: 0.5 mL of 0.05% formic acid in 100% acetonitrile; 3rd step: 0.5 mL of 0.05% formic acid in 10% acetonitrile) at flow rate of 700 μl/min. GNF6702 and verapamil were eluted at retention time 1.19 and 1.17 minutes, respectively. The HPLC system, consisting of Agilent 1260 series binary pump (Agilent Technologies), Agilent 1260 series micro vacuum degasser (Agilent Technologies) and CTC PAL-HTC-xt analytics autosampler (LEAP Technologies) was interfaced to a SCIEX API 4000 triple quadrupole mass spectrometer (Sciex). Mass spectrometry analysis was carried out using atmospheric pressure chemical ionization (APCI) in the positive ion mode. GNF6702 (430.07 > 333.20) and verapamil (455.16 > 164.90) peak integrations were performed using AnalystTM 1.5 software (Sciex). The lower limit of quantification (LLOQ) in plasma was 1.0 ng/mL. Samples were quantified using seven calibration standards (dynamic range 1 – 5,000 ng/mL) prepared in plasma and processed as described above.
Formulation of study drugs for in vivo efficacy experiments
All compounds administered to mice during efficacy experiments were formulated as suspensions in distilled water containing 0.5% methylcellulose (Sigma-Aldrich) and 0.5% Tween 80 (Sigma-Aldrich). During a treatment course, each mouse received 0.2 ml of drug suspension per dose by oral gavage.
Mouse model of visceral leishmaniasis
Female BALB/c mice (Envigo; 6–8 weeks old) were infected by tail vein injection with 4 × 107 L. donovani MHOM/ET/67/HU3 splenic amastigotes (protocol number P11-319). Seven days after infection, animals were orally dosed for eight days with vehicle (0.5% methylcellulose/0.5% Tween 80, miltefosine (12 mg/kg once-daily; Sigma-Aldrich), or a GNF compound (twice-daily). On the first day of dosing, three mice were used for collection of blood for PK determination and euthanized afterwards. On the last day of dosing, PK samples were collected from remaining five mice, which were also used for determination of compound efficacy (n= 5 mice per group). Liver samples were collected from these five mice and L. donovani parasite burdens were quantified by qPCR as follows. Total DNA was extracted from drug-treated mice livers using the DNeasy Blood and Tissue Kit (Qiagen). Two types of DNA were quantified in parallel using the TaqMan assay: L. donovani major surface glycoprotein gp63 (Ldon_GP63) and mouse GAPDH. L. donovani GP63 DNA was quantified with the following set of primers: TGCGGTTTATCCTCTAGCGATAT (forward), AGTCCATGAAGGCGGAGATG (reverse), and TGGCAGTACTTCACGGAC (TaqMan MGB probe, 5′-FAM-labeled reporter dye, non-fluorescent quencher). Mouse GAPDH DNA was quantified with the following set of primers: GCCGCCATGTTGCAAAC (forward primer), CGAGAGGAATGAGGTTAGTCACAA (reverse primer), and ATGAATGAACCGCCGTTAT (TaqMan MGB probe, 5′-FAM-labeled reporter dye, non-fluorescent quencher). Each qPCR reaction (10 μL) included 5 μl of TaqMan Gene Expression Master Mix (Life Technologies), 0.5 μL of a 20X primer/probe mix (Life Technologies), and 4.5 μL (50 ng) of total DNA from liver samples. DNA amount was quantified using the Applied Biosystems 7900HT instrument. L. donovani parasite burden (RU: relative units) was expressed as the abundance of L. donovani GP63 DNA relative to the abundance of mouse GAPDH DNA.
Mouse footpad model of cutaneous leishmaniasis
L. major MHOM/SA/85/JISH118 metacyclic promastigotes were generated and purified by the peanut agglutinin method as described elsewhere45. To establish the L. major footpad infection, female BALB/c mice (Envigo; 6–8 weeks old; protocol number P11-319) were injected with suspension of L. major metacyclic promastigotes (1 × 106 parasites in 50 μL) into each hind footpad. After eight days of infection, animals were dosed with vehicle, miltefosine (30 mg/kg once-daily), or indicated regimens of GNF6702 for seven days (n=6 mice per group). The progress of infection was monitored by measuring the size (length and thickness) of hind footpad swelling using digital calipers. At the end of the study, the mice were euthanized, and the footpad tissues were extracted and used for genomic DNA isolation with the DNeasy Blood and Tissue kit (Qiagen). The L. major footpad burden was determined by qPCR quantification of kinetoplastid minicircle DNA (forward primer: 5′-TTTTACACCTCCCCCCAGTTT-3′; reverse primer: 5′-CCCGTTCATAATTTCCCGAAA-3′; Taqman MGB probe: 5′-AGGCCAAAAATGG-3′, 5′-FAM [6-carboxyfluorescein]-labeled reporter dye, non-fluorescent quencher). The amounts of mouse chromosomal DNA in extracted samples were quantified in parallel qPCR using a glyceraldehyde-3-phosphate dehydrogenase (GAPDH) TaqMan assay as described for mouse VL model above. L. major burden in footpad was expressed as the ratio of kinetoplast minicircle DNA to mouse GAPDH. P values for the between-groups differences in efficacies were calculated with a Student’s paired t test with a two-tailed distribution.
Mouse model of Chagas disease
Compound efficacy in mouse model of Chagas disease was determined as described previously23. Female C57BL/6 mice (Envigo; 6–8 weeks old; protocol number P11-316) were infected by intraperitoneal injection with 103 tissue culture-derived T. cruzi CL trypomastigotes. Starting at 35 days after infection, the animals were dosed orally once-daily with 100 mg/kg benznidazole (Sigma-Aldrich) and indicated doses of GNF6702 (1, 3, and 10 mg/kg twice-daily, n=8 per group) for 20 days. Ten days following the end of drug treatment, the mice underwent four cycles of cyclophosphamide immunosuppression, each cycle lasting one week. During each immunosuppression cycle, mice were dosed by oral gavage once-daily with 200 mg/kg cyclophosphamide (suspension in 0.5% methylcellulose/0.5% Tween80 aqueous solution) on day 1 and day 4 of the cycle. After the fourth immunosuppression cycle, blood samples were collected from the orbital venous sinus of each mouse, mice were euthanized and heart and colon samples were collected. Samples from treated mice were used for extraction of total DNA using the High Pure PCR template preparation kit (Roche). The amounts of T. cruzi satellite DNA (195-bp fragment) in extracted DNA samples were quantified by real-time qPCR TaqMan assay (Life Technologies) with the following set of primers: AATTATGAATGGCGGGAGTCA (forward primer), CCAGTGTGTGAACACGCAAAC (reverse primer), and AGACACTCTCTTTCAATGTA (TaqMan MGB probe, 5′-FAM [6-carboxyfluorescein]-labeled reporter dye, non-fluorescent quencher). The amounts of mouse chromosomal DNA in extracted samples were quantified in parallel qPCR reactions using a GADPH (glyceraldehyde-3-phosphate dehydrogenase) TaqMan assay as described for mouse VL model above. Each qPCR mixture (10 μl) included 5 μl of TaqMan Gene Expression master mix (Life Technologies), 0.5 μl of a 20x primer/probe mix (Life Technologies), and 4.5 μl (50 ng) of total DNA extracted from blood samples. PCRs were run on the Applied Biosystems 7900HT instrument. T. cruzi parasitemia was expressed as the abundance of T. cruzi microsatellite DNA relative to the abundance of mouse GAPDH DNA.
Mouse model of stage II HAT
Female CD1 (Charles River UK; ~8 weeks old; protocol number PPL 60/4442) mice were infected by injection into the peritoneum with 3 × 104 T. brucei (GVR35-VSL2) bloodstream form parasites46. Starting on day 21, mice were dosed by oral gavage once-daily with GNF6702 (n= 6) at 100 mg/kg for 7 days or a single dose of diminazene aceturate (Sigma-Aldrich) at 40 mg/kg in sterile water was administered by ip injection (n= 3). A group of untreated mice (n= 3) was included as controls.
Mice were monitored weekly for parasitemia from day 21 post-infection. T. brucei was quantified in blood samples from the tail vein by microscopy, and in vivo bioluminescence imaging of infected mice was performed before treatment on day 21 post-infection and in weeks following the treatment (day 28, 35, 42, 56, 63, 72, 84, 92 post-infection). Imaging on groups of three mice was performed 10 min after ip injection of 150 mg D-luciferin (Promega)/kg body weight (in PBS) using an IVIS Spectrum (PerkinElmer) as described previously25. A group of uninfected mice (aged-matched for day 0 time point; n= 4) were imaged using the same acquisition settings to show the background bioluminescence (Fig. 2e, grey-filled squares) in the absence of luciferase-expressing T. brucei after day 92 of the experiment. Untreated and diminazene-treated mice were euthanized on days 32 and 35, and day 42, respectively, due to high parasitemia or the development of symptoms related to CNS infection. GNF6702-treated mice were euthanized on day 92. No parasitemia or clinical symptoms were observed at this point. At the specified endpoints mice were sacrificed by cervical dislocation, after which whole brains were removed and imaged ex vivo within 10 minutes after administration of 100 μL of D-luciferin onto the brain surface. Data analysis for bioluminescence imaging was performed using Living Image Software. The same rectangular region of interest (ROI) covering the mouse body was used for each whole body image to show the bioluminescence in total flux (photons per second) within that region. Image panels of whole mouse bodies are composites of the original images with areas outside the ROI cropped out to save space. For ex vivo brain images the same oval shaped ROI was used to display the bioluminescence detected for each mouse brain at the respective endpoints.
Chemical synthesis
The detailed procedures for chemical synthesis are presented in Supplementary Information.
Extended Data
Extended Data Figure 1. Pharmacokinetic profile of GNF6702 in mouse.

a, Time profiles of mean free plasma concentration of GNF6702 in mouse model of visceral leishmaniasis; free GNF6702 concentration values were predicted from measured total plasma concentration values collected on day 1 and day 8 of treatment. Dashed blue lines correspond to intra-macrophage L. donovani EC50 of 18 ± 1.8 nM and EC99 of 42 ± 5.6 nM. Circles: means ± s.d.; n=3 mice for treatment day 1; n=5 mice for treatment day 8; fraction unbound in mouse plasma=0.063. For data points lacking error bars, standard deviations are smaller than circles representing means. b, Time course of total GNF6702 concentration in mouse plasma and brain after single oral dose (20 mg/kg); n=2 mice per time point; circles: measured values; rectangles: means.
Extended Data Figure 2. GNF6702 clears parasites from mice infected with T. brucei.

a, In vivo quantification of bioluminescent T. brucei in infected mice before and after treatment. ip: intraperitoneal; day 21: start of treatment; day 28: 24 hours after last GNF6702 dose; day 42: evaluation of early parasite recrudescence in mice treated with diminazene aceturate (n=3); day 42 and 92: absence of parasite recrudescence in mice treated with GNF6702 (n=6). Images from uninfected mice (3 mice of 4 are shown) aged-matched for day 0 were collected independently using the same acquisition settings. Parasitemia (blue font) and whole mouse total flux (black font) values of each animal are shown above the image; N.D.: not detectable. Within each group the mouse numbers in yellow (top left in each image) refer to the same mouse imaged throughout. Complete sets of parasitemia and whole mouse total flux values collected on individual mice throughout the experiment are listed in Supplementary Tables 4 and 5. b, Brains from mice shown in panel a were soaked in luciferin and imaged for presence of bioluminescent T. brucei at the indicated time points. For three diminazene-treated mice, two images of each brain are shown, one at a lower sensitivity (left) and the other at a high signal intensity scale.
Extended Data Figure 3. Structures and profiles of GNF3943 and GNF8000 used for selection of resistant T. cruzi lines.

L. donovani: amastigotes proliferating within primary mouse macrophages; T. brucei: the bloodstream form trypomastigotes; T. cruzi: amastigotes proliferating in 3T3 fibroblast cells; macrophage: mouse primary peritoneal macrophages; EC50 and CC50 : half-maximum growth inhibition concentration; F: oral bioavailability in mouse after administering single compound dose (20 mg/kg) as a suspension; CL: plasma clearance in mouse after single iv bolus dose (5 mg/kg); all EC50 and CC50 values correspond to means ± s.e.m. (n=4 technical replicates).
Extended Data Figure 4. Mutations in proteasome beta 4 subunit confer resistance to GNF6702 in T. cruzi and T. brucei.

a, growth curves of wild type, GNF3943-resistant and GNF8000-resistant T. cruzi epimastigote strains in the presence of increasing concentrations of GNF6702, nifurtimox, bortezomib and MG132; RU (relative units) corresponds to parasite growth relative to the DMSO control (%); for data points lacking error bars, standard errors are smaller than circles representing means; due to limited aqueous solubility, the highest tested GNF6702 concentration was 10 μM. b, growth inhibition EC50 values of GNF6702, bortezomib, MG132 and nifurtimox on indicated T. cruzi strains. c, growth inhibition EC50 values of GNF6702 and bortezomib on T. cruzi epimastigotes and T. brucei bloodstream form trypomastigotes overexpressing PSMB4WT or PSMB4F24L. Data shown in panels a, b and c correspond to means ± s.e.m. (n=3 technical replicates).
Extended Data Figure 5. Correlation between inhibition of parasite proteasome chymotrypsin-like activity and parasite growth inhibition by the GNF6702 compound series.
IC50: half-maximum inhibition of indicated parasite proteasome; T. brucei EC50: half-maximum growth inhibition on T. brucei bloodstream form trypomastigotes; T. cruzi EC50: half-maximum growth inhibition on T. cruzi amastigotes proliferating inside 3T3 cells; data points correspond to means of 2 technical replicates; red circles: IC50>20 μM; yellow circles: IC50>20 μM and EC50>25 μM; data for 317 analogues are shown.
Extended Data Figure 6. Hypothetical model of GNF6702 binding to T. cruzi proteasome beta 4 subunit.

a, Alignment of amino acid sequences of proteasome beta 4 subunits (PSMB4) from L. donovani, T. cruzi, T. brucei and H. sapiens. Green: amino acid residues conserved between human and kinetoplastid PSMB4 proteins; blue: amino acid residues conserved only among kinetoplastid PSMB4 proteins; black: amino acids mutated in T. cruzi mutants resistant to analogues from the GNF6702 series. b, Surface representation of the modeled T. cruzi 20S proteasome structure showing relative positions of the beta 5 and beta 4 subunits. Beta 4 amino acid residues F24 and I29 (colored yellow) are located at the interface of the two beta subunits. GNF6702 is depicted in a sphere representation bound into a predicted pocket on the beta 4 subunit surface with carbon, nitrogen, oxygen and hydrogen atoms colored magenta, blue, red and grey, respectively. The other T. cruzi 20S proteasome subunits are colored gray. c, Close-up of the beta 5 and beta 4 subunits. The beta 5 subunit active site (pocket 1, chymotrypsin-like activity) is colored pale green. The predicted beta 4 pocket (pocket 2) with bound GNF6702 is colored blue. The inhibitor is shown in a stick representation with atoms colored as described in caption for the b panel. Beta 4 residues F24 and I29 are colored yellow. The proteasome model shown in panels b and c was produced by The PyMol Molecule Graphics System, Version 1.8, Schrodinger, LLC.
Extended Data Figure 7. Effect of GNF6702 on accumulation of ubiquitylated proteins by T. cruzi epimastigotes and 3T3 cells.

a, Western blot analysis of T. cruzi whole cell extracts with anti-ubiquitin antibody after treatment with GNF6702 and bortezomib. b, Western blot analysis of 3T3 whole cell extracts with anti-ubiquitin antibody after treatment with GNF6702 and borteomib. c, Concentrations of GNF6702 and bortezomib effecting half-maximum accumulation of ubiquitylated proteins in T. cruzi and 3T3 cells (means ± s.e.m.; n=3 technical replicates); total ubiquitin signal values in individual blot lanes shown in panels a and b were quantified and used for calculation of the listed EC50 values. In a and b, numbers above the blot lanes indicate compound concentrations and D indicates control, DMSO-treated cells. For western blot source data, see Supplementary Figure 1.
Extended Data Table 1.
Point mutations identified by whole genome sequencing in GNF3943- and GNF8000-resistant T. cruzi epimastigotes
| Gene ID | GNF3943R mutant | GNF80008 mutant | |
|---|---|---|---|
| Number of reads (clone 1/ clone 2) | 78×108/ 63×108 | 48×106/ 68×108 | |
| Mapped reads (clone 1/ clone 2) [%] | 87/ 87 | 90/ 90 | |
| Average genome coverage (clone 1/ clone 2) | 82x/ 66x | 51x/ 66x | |
| Proteasome beta 4 subunit | 3540409 | I29M/ I29M | wt/ F24L |
| Dynein heavy chain | 3548195 | wt/ P82L | wt/ wt |
| Trans-sialidase | 3542504 | wt/ wt | wt/ G90E |
| Trans-sialidase | 3542504 | wt/ wt | wt/ L93P |
| Hypothetical protein TCSYLVIO_005989 | 3547397 | wt/ wt | wt/ L627P |
| Hypothetical protein TCSYLVIO_005986 | 3547401 | wt/ wt | wt/ S55P |
Extended Data Table 2.
Enzyme inhibition IC50 values of bortezomib and GNF6702 on three proteolytic activities of wild type T. cruzi, PSMB4I29M T. cruzi, and H. sapiens proteasomes
| GNF6702 IC50 [μM]* | bortezomib IC50 [μM]* | ||
|---|---|---|---|
| wild type T. cruzi proteasome | chymotrypsin | 0.035 ± 0.0013 | 0.091 ± 0.0075 |
| caspase | >10 | 0.37 ± 0.012 | |
| trypsin | >10 | 1.7 ± 0.088 | |
| PSMB4I29M T. cruzi proteasome | chymotrypsin | >10 | 0.26 ± 0.040 |
| caspase | >10 | 0.54 ± 0.012 | |
| trypsin | >10 | 1.6 ± 0.058 | |
| H. sapiens constitutive proteasome | chymotrypsin | >10 | 0.030 ± 0.0070 |
| caspase | >10 | 0.16 ± 0.007 | |
| trypsin | >10 | 7.9 ± 0.15 |
mean ± s.e.m.; n= 3 technical replicates
Extended Data Table 3.
Inhibition kinetics parameters of GNF6702 on L. donovani and T. cruzi proteasomes
| chymotrypsin-like activity | caspase-like activity | trypsin-like activity | ||||
|---|---|---|---|---|---|---|
| L. donovani | T. cruzi | L. donovani | T. cruzi | L. donovani | T. cruzi | |
| Ki ± s.e.m. [μM] | 0.055 ± 0.006* | 0.079 ± 0.003* | > 10 | > 10 | > 10 | > 10 |
| Km ± s.e.m. [μM] | 3.6 ± 0.60* | 2.6 ± 0.15* | N.A.† | N.A.† | N.A.† | N.A.† |
| Mode of inhibition | non-competitive | non-competitive | N.A.† | N.A.† | N.A.† | N.A.† |
| R2 (goodness of fit) | 0.91 | 0.97 | N.A.† | N.A.† | N.A.† | N.A.† |
mean ± s.e.m.; n= 3 technical replicates
not applicable
Supplementary Material
Acknowledgments
This work was supported in part by grants from the Wellcome Trust (091038/Z/09/Z to R.J.G. and F.S., and 104976/Z/14/Z, 104111/15/Z to J.C.M. and E.M.) and NIH (AI106850 to F.S.B.). We thank Simon Croft, Rob Don, Lars Gredsted, Alan Hudson and John Mendlein for discussions, Rick Tarleton for T. cruzi CL strain, and George Cross for T.b. brucei Lister 427 strain. We thank Andreas Kreusch for help with proteasome purification, and Fabio Luna for help with T. cruzi whole genome sequencing. We acknowledge technical assistance of Omeed Faghih in generating the plasmids for ectopic expression of PSMB4 in T. cruzi, Ryan Ritchie for IVIS in vivo imaging, and Annie Mak, Jason Matzen and Paul Anderson for execution of high throughput screens. We thank John Isbell and Thomas Hollenbeck for profiling GNF6702 in ADME assays.
Footnotes
Supplementary Information can be found at the end of this manuscript.
Contributions
A.B., F.L., C.J.N.M., P.K.M., A.S.N., J.L.T. and V.Y. designed chemical analogues, and performed chemical synthesis and purification of synthesized analogues. F.S.B., J.B., J.R.G., S.K., H.X.Y.K., Y.H.L., S.P.S.R., F.S., and X.L. conducted and analyzed data from in vitro growth inhibition assays. L.C.D., X.L., J.C.M., E.M., I.C.R., S.P.S.R., M.S., F.S., and B.G.W. conducted and analyzed data from in vivo efficacy assays. J.B., M.-Y.G., S.K., and F.S. conducted proteasome purification, proteasome inhibition assays and biochemical data analysis. S.W.B., G.F., S.K., F.S., and J.R.W. designed, conducted and analyzed experiments resulting in identification of proteasome resistance mutations. G.S. and B.B. built the homology model of T. cruzi proteasome structure and performed GNF6702 docking. A.B. and J.D.V. analyzed T. cruzi proteasome by mass spectrometry. A.N., T.G., M.S., F.S., and T.T. designed, conducted, and analyzed PK data. A.N. and V.M. led the chemistry team. F.S. led the biology team. R.J.G. and F.S. supervised and led the overall project, and led the writing of the manuscript. All authors contributed to writing of the manuscript.
Competing financial interests
Patents related to this work has been filed (WO 2015/095477 A1, WO 2014/151784 A1, WO 2014/151729). Several authors own shares of Novartis.
References
- 1.World Health Organization. Research priorities for Chagas disease human African trypanosomiasis leishmaniasis. 2012. pp. 1–100. WHO Technical Report Series 975. [PubMed] [Google Scholar]
- 2.El-Sayed NM, et al. Comparative genomics of trypanosomatid parasitic protozoa. Science. 2005;309:404–409. doi: 10.1126/science.1112181. [DOI] [PubMed] [Google Scholar]
- 3.Bilbe G. Infectious diseases. Overcoming neglect of kinetoplastid diseases. Science. 2015;348:974–976. doi: 10.1126/science.aaa3683. [DOI] [PubMed] [Google Scholar]
- 4.Sundar S, Chakravarty J. An update on pharmacotherapy for leishmaniasis. Expert Opinion on Pharmacotherapy. 2015;16:237–252. doi: 10.1517/14656566.2015.973850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nagle AS, et al. Recent developments in drug discovery for leishmaniasis and human African trypanosomiasis. Chemical Reviews. 2014 doi: 10.1021/cr500365f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bern C. Chagas’ Disease. The New England Journal of Medicine. 2015;373:456–466. doi: 10.1056/NEJMra1410150. [DOI] [PubMed] [Google Scholar]
- 7.Chatelain E. Chagas disease drug discovery: toward a new era. Journal of Biomolecular Screening. 2015;20:22–35. doi: 10.1177/1087057114550585. [DOI] [PubMed] [Google Scholar]
- 8.Kennedy PG. Clinical features, diagnosis, and treatment of human African trypanosomiasis (sleeping sickness) The Lancet. Neurology. 2013;12:186–194. doi: 10.1016/S1474-4422(12)70296-X. [DOI] [PubMed] [Google Scholar]
- 9.World Health Organization. Control of the leishmaniases. 2010. pp. 37–39. WHO Technical Report Series 949. [Google Scholar]
- 10.Yardley V, Croft SL. A comparison of the activities of three amphotericin B lipid formulations against experimental visceral and cutaneous leishmaniasis. International Journal of Antimicrobial Agents. 2000;13:243–248. doi: 10.1016/s0924-8579(99)00133-8. [DOI] [PubMed] [Google Scholar]
- 11.Dorlo TP, et al. Pharmacokinetics of miltefosine in Old World cutaneous leishmaniasis patients. Antimicrobial Agents and Chemotherapy. 2008;52:2855–2860. doi: 10.1128/AAC.00014-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McGwire BS, Satoskar AR. Leishmaniasis: clinical syndromes and treatment. QJM: Monthly journal of the Association of Physicians. 2014;107:7–14. doi: 10.1093/qjmed/hct116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hotez PJ. Combating the next lethal epidemic. Science. 2015;348:296–297. doi: 10.1126/science.348.6232.296-b. [DOI] [PubMed] [Google Scholar]
- 14.Sacks D, Anderson C. Re-examination of the immunosuppressive mechanisms mediating non-cure of Leishmania infection in mice. Immunological Reviews. 2004;201:225–238. doi: 10.1111/j.0105-2896.2004.00185.x. [DOI] [PubMed] [Google Scholar]
- 15.Nelson KG, Bishop JV, Ryan RO, Titus R. Nanodisk-associated amphotericin B clears Leishmania major cutaneous infection in susceptible BALB/c mice. Antimicrobial Agents and Chemotherapy. 2006;50:1238–1244. doi: 10.1128/AAC.50.4.1238-1244.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nunes MC, et al. Chagas disease: an overview of clinical and epidemiological aspects. Journal of the American College of Cardiology. 2013;62:767–776. doi: 10.1016/j.jacc.2013.05.046. [DOI] [PubMed] [Google Scholar]
- 17.Coura JR, Borges-Pereira J. Chagas disease: 100 years after its discovery. A systemic review. Acta Tropica. 2010;115:5–13. doi: 10.1016/j.actatropica.2010.03.008. [DOI] [PubMed] [Google Scholar]
- 18.Bern C. Antitrypanosomal therapy for chronic Chagas’ disease. The New England Journal of Medicine. 2011;364:2527–2534. doi: 10.1056/NEJMct1014204. [DOI] [PubMed] [Google Scholar]
- 19.Viotti R, et al. Towards a paradigm shift in the treatment of chronic Chagas disease. Antimicrobial Agents and Chemotherapy. 2014;58:635–639. doi: 10.1128/AAC.01662-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Molina I, et al. Randomized trial of posaconazole and benznidazole for chronic Chagas’ disease. The New England Journal of Medicine. 2014;370:1899–1908. doi: 10.1056/NEJMoa1313122. [DOI] [PubMed] [Google Scholar]
- 21.Morillo CA, et al. Randomized trial of benznidazole for chronic Chagas’ cardiomyopathy. The New England Journal of Medicine. 2015;373:1295–1306. doi: 10.1056/NEJMoa1507574. [DOI] [PubMed] [Google Scholar]
- 22.Viotti R, et al. Side effects of benznidazole as treatment in chronic Chagas disease: fears and realities. Expert Review of Anti-Infective Therapy. 2009;7:157–163. doi: 10.1586/14787210.7.2.157. [DOI] [PubMed] [Google Scholar]
- 23.Khare S, et al. Antitrypanosomal treatment with benznidazole is superior to posaconazole regimens in mouse models of Chagas disease. Antimicrobial Agents and Chemotherapy. 2015;59:6385–6394. doi: 10.1128/AAC.00689-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bustamante JM, Bixby LM, Tarleton RL. Drug-induced cure drives conversion to a stable and protective CD8+ T central memory response in chronic Chagas disease. Nature Medicine. 2008;14:542–550. doi: 10.1038/nm1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Myburgh E, et al. In vivo imaging of trypanosome-brain interactions and development of a rapid screening test for drugs against CNS stage trypanosomiasis. PLoS Neglected Tropical Diseases. 2013;7:e2384. doi: 10.1371/journal.pntd.0002384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li H, et al. Structure- and function-based design of Plasmodium-selective proteasome inhibitors. Nature. 2016;530:233–236. doi: 10.1038/nature16936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Steverding D, Wang X. Trypanocidal activity of the proteasome inhibitor and anti-cancer drug bortezomib. Parasites & Vectors. 2009;2:29. doi: 10.1186/1756-3305-2-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ivens AC, et al. The genome of the kinetoplastid parasiteLeishmania major. Science. 2005;309:436–442. doi: 10.1126/science.1112680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li X, et al. Effect of noncompetitive proteasome inhibition on bortezomib resistance. Journal of the National Cancer Institute. 2010;102:1069–1082. doi: 10.1093/jnci/djq198. [DOI] [PubMed] [Google Scholar]
- 30.Fernandez Y, et al. Chemical blockage of the proteasome inhibitory function of bortezomib: impact on tumor cell death. The Journal of Biological Chemistry. 2006;281:1107–1118. doi: 10.1074/jbc.M511607200. [DOI] [PubMed] [Google Scholar]
- 31.Zhang X, Goncalves R, Mosser DM. The isolation and characterization of murine macrophages. Curr Protoc Immunol. 2008;Chapter 14(Unit1 4.1) doi: 10.1002/0471142735.im1401s83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Khare S, et al. Utilizing chemical genomics to identify cytochrome b as a novel drug target for Chagas disease. PLoS Pathogens. 2015;11:e1005058. doi: 10.1371/journal.ppat.1005058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Buckner FS, Verlinde CL, La Flamme AC, Van Voorhis WC. Efficient technique for screening drugs for activity against Trypanosoma cruzi using parasites expressing beta-galactosidase. Antimicrobial Agents and Chemotherapy. 1996;40:2592–2597. doi: 10.1128/aac.40.11.2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Logan-Klumpler FJ, et al. GeneDB--an annotation database for pathogens. Nucleic Acids Research. 2012;40:D98–108. doi: 10.1093/nar/gkr1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Taylor MC, Kelly JM. pTcINDEX: a stable tetracycline-regulated expression vector forTrypanosoma cruzi. BMC Biotechnology. 2006;6:32. doi: 10.1186/1472-6750-6-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hariharan S, Ajioka J, Swindle J. Stable transformation of Trypanosoma cruzi: inactivation of the PUB12.5 polyubiquitin gene by targeted gene disruption. Molecular and Biochemical Parasitology. 1993;57:15–30. doi: 10.1016/0166-6851(93)90240-x. [DOI] [PubMed] [Google Scholar]
- 37.Wirtz E, Leal S, Ochatt C, Cross GA. A tightly regulated inducible expression system for conditional gene knock-outs and dominant-negative genetics in Trypanosoma brucei. Molecular and Biochemical Parasitology. 1999;99:89–101. doi: 10.1016/s0166-6851(99)00002-x. [DOI] [PubMed] [Google Scholar]
- 38.Wilk S, Chen W-E. Purification of the eukaryotic 20S proteasome. Curr Protoc Protein Sci. 2001;Chapter 21 doi: 10.1002/0471140864.ps2106s25. [DOI] [PubMed] [Google Scholar]
- 39.Unno M, et al. The structure of the mammalian 20S proteasome at 2.75 A resolution. Structure. 2002;10:609–618. doi: 10.1016/s0969-2126(02)00748-7. [DOI] [PubMed] [Google Scholar]
- 40.Melnick JS, et al. An efficient rapid system for profiling the cellular activities of molecular libraries. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:3153–3158. doi: 10.1073/pnas.0511292103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Waters NJ, Jones R, Williams G, Sohal B. Validation of a rapid equilibrium dialysis approach for the measurement of plasma protein binding. Journal of Pharmaceutical Sciences. 2008;97:4586–4595. doi: 10.1002/jps.21317. [DOI] [PubMed] [Google Scholar]
- 42.Wang J, Skolnik S. Recent advances in physicochemical and ADMET profiling in drug discovery. Chemistry & Biodiversity. 2009;6:1887–1899. doi: 10.1002/cbdv.200900117. [DOI] [PubMed] [Google Scholar]
- 43.Kalvass JC, Tess DA, Giragossian C, Linhares MC, Maurer TS. Influence of microsomal concentration on apparent intrinsic clearance: implications for scaling in vitro data. Drug Metabolism and Disposition: the Biological Fate of Chemicals. 2001;29:1332–1336. [PubMed] [Google Scholar]
- 44.Li C, et al. A modern in vivo pharmacokinetic paradigm: combining snapshot, rapid and full PK approaches to optimize and expedite early drug discovery. Drug Discovery Today. 2013;18:71–78. doi: 10.1016/j.drudis.2012.09.004. [DOI] [PubMed] [Google Scholar]
- 45.Sacks DL, Melby PC. Animal models for the analysis of immune responses to leishmaniasis. Curr Protoc Immunol. 2001;Chapter 19(Unit 19.12) doi: 10.1002/0471142735.im1902s28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.McLatchie AP, et al. Highly sensitive in vivo imaging of Trypanosoma brucei expressing “red-shifted” luciferase. PLoS Neglected Tropical Diseases. 2013;7:e2571. doi: 10.1371/journal.pntd.0002571. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.