

Abstract
Lower serum chloride (Cl) levels are strongly associated with increased long-term mortality after admission for acute heart failure (AHF). However, the therapeutic implications of serum Cl levels during AHF are unknown. We sought to determine the short-term clinical response and post-discharge outcomes associated with serum Cl levels in AHF. Serum Cl was measured at randomization (N=358) and during hospitalization from patients with AHF in the in the Renal Optimization Strategies Evaluation in Acute Heart Failure (ROSE-AHF) trial. Outcomes included diuretic response and renal function at 72 hours and death and rehospitalization at 60 and 180-days. Baseline Cl tertiles were: 84–98 meq/L; 99–102 meq/L; and 103–117 meq/L. Baseline Cl level was associated with diuretic efficiency (P<.001), but not change in cystatin C (P=0.30) at 72 hours; and was associated with 60-day death (HR 0.86, P=0.029), 60-day death and rehospitalization (HR 0.90, P=0.01), and 180-day death (HR 0.91, P=0.049). These associations were attenuated with additional adjustment for loop diuretic dose (P>0.05). Chloride change correlated with weight change (rho 0.18, P=0.001), cystatin C change (rho −0.35, P<.001), and cumulative sodium excretion (rho −0.21, P<.001), but was not associated with any clinical outcomes (P>0.05 for all). In conclusion, serum Cl levels in AHF were inversely associated with loop diuretic response and were prognostic. However, changes in Cl levels were associated with parameters of decongestion, but not with clinical outcomes.
Keywords: heart failure, electrolytes, and diuretics
INTRODUCTION
Serum chloride (Cl) levels are commonly checked as a part of routine chemistry panels during hospitalization for acute heart failure (AHF). While serum sodium (Na) levels in AHF have received much attention, serum Cl levels are more closely related to prognosis in AHF.1 The exact reasons for this are unclear, though are likely multi-factorial. In addition to arginine vasopressin-mediated Cl dilution,2,3 Cl levels may also become depleted with loop or thiazide diuretic use, 4–6 and play a prominent role in the acid-base homeostasis.7 Despite growing knowledge about the importance of serum Cl in heart failure, its prognostic impact may vary in different clinical settings. Since loop diuretics can deplete Cl,8 the propensity for acute depletion of Cl may have consequences on prognosis and diuretic efficacy. The short-term clinical implications of Cl levels during decongestive therapy and the variability of serum Cl levels in AHF are unknown. The Renal Optimization Strategies Evaluation in Acute Heart Failure (ROSE-AHF) clinical trial provides a unique opportunity to address these questions.9 We additionally sought to verify prior evidence of the association of Cl and post-discharge outcomes.1
METHODS
We included patients who were randomly assigned within the ROSE-AHF trial, a study conducted within the National Heart, Lungs, and Blood Institute-sponsored Heart Failure Clinical Trials Network. The protocol was approved by the Institutional Review Boards at each site, and written informed consent was obtained from every patient prior to randomization. The trial was conducted in the United States and Canada.
The ROSE-AHF trial (conducted from 2010–2013 at 26 clinical sites) sought to determine the effectiveness of decongestive therapies and renal consequences of placebo compared to low-dose dopamine (2μg kg−1 min−1) or low-dose nesiritide (0.005 μg kg−1 min−1) in hospitalized patients with AHF and renal dysfunction.9 These patients received 2.5 times their total daily oral outpatient furosemide dose up to 600 mg/day. All patients were placed on both Na and fluid restriction consistent with the Heart Failure Society of America 2010 heart failure practice guidelines.10 Daily history and physical examinations, fluid and volume status, medication reconciliation, and blood work (including Cl and other electrolytes) were collected by study personnel.
The primary endpoints of ROSE-AHF were assessed 72 hours after randomization with additional chemistries and biomarkers measured upon discharge or day 7. Worsening or persisting heart failure and freedom from congestion were determined by 72 hours and have been previously defined.9 Diuretic efficiency was defined as net-fluid output per 40 mg of intravenous furosemide equivalents.11,12 For all analyses whereby diuretic efficiency was an outcome at 72 hours, one extreme outlier was excluded (13,793 ml/40 mg furosemide equivalents) as this was 3 times the interquartile range above the third quartile and physiologically anomalous. Cardiorenal biomarkers included serum cystatin C and aminoterminus pro-B-type natriuretic peptide (NT-proBNP) and were analyzed at a biomarker core laboratory at the University of Vermont, Burlington, VT. Daily 24-hour urine volume and Na concentrations were collected for 72 hours. Urinary Na concentrations were normalized to 24 hours. In order to parallel these endpoints, 72 hour changes in serum Cl were used for comparison. Post-hospitalization clinical outcomes included death and rehospitalization at 60 and 180-days.
Continuous variables were presented as median (25th, 75th percentile) and categorical variables as number (%). Baseline characteristics were stratified by tertiles of baseline Cl levels with comparisons by the Kruskall-Wallis test for continuous and Pearson chi-square for categorical variables. Linear or logistic regression was used to determine the association of continuous markers of diuretic response or clinical outcomes where appropriate. Change in Cl was calculated by subtracting the 72-hour Cl level from the baseline Cl level and changes in other continuous variables were calculated similarly. Non-parametric correlations were presented as Spearman correlations coefficients. The association between baseline treatment (dopamine, nesiritide, or placebo and diuretic dose in furosemide equivalents) and serial Cl levels was analyzed by a random intercept, random slope model. The Kaplan-Meier method and Log-Rank test were used to demonstrate differences in cumulative death rates across Cl tertiles. Cox-proportional hazards models determined the association of baseline Cl levels or changing Cl levels with post-discharge outcomes. Two models were constructed for risk-adjustment: 1) Na, bicarbonate, cystatin C, white race; and 2) these previous variables with pre-hospitalization loop diuretic dose in furosemide equivalents. These variables were selected a priori for their property to confound the Cl-risk relationship. Cystatin C was selected given the association of renal function and diuretic response.13 Models with changing Cl levels were additionally adjusted for baseline Cl level with outcomes assessed from 72 hours onward. The validity of the proportional hazards assumption was determined by plotting the Schoenfeld residuals by time and then testing for a non-zero slope. All analyses were done for complete cases. Two-sided P-values <0.05 were considered statistically significant. Statistical analyses were completed using SAS software, version 9.3 (SAS Institute Inc., Cary, North Carolina).
RESULTS
Of the 360 patients randomly assigned, 358 had Cl levels measured on admission, 344 at 24 hours, 347 at 48 hours, 334 at 72 hours, and 274 at discharge or day 7. Baseline Cl levels were normally distributed (100 ± 5 meq/L, Supplement) and tertiles were: 84–98 meq/L (N=115); 99–102 meq/L (N=125); and 103–117 meq/L (N=118). Baseline Cl levels were correlated to both baseline Na and bicarbonate levels (rho=0.57, P<.001 and rho=−0.47, P<.001; respectively). Table 1 illustrates the baseline characteristics of the cohort stratified by Cl tertile, which are representative of a contemporary cohort of patients hospitalized with acute heart failure. Chloride levels were not correlated with age (rho 0.061, P=0.25), body mass index (rho −0.006, P=0.94), or LVEF (rho −0.004, P=0.94). Importantly, Cl was correlated with systolic blood pressure (rho 0.30, P<.001), blood urea nitrogen (rho −0.21, P<.001), and Na (rho 0.57, P<.001). Additionally, there was a modest negative correlation with admission Cl levels and the pre-hospitalization diuretic dose in furosemide equivalents (rho=−0.28, P<.001).
Table 1.
Baseline Characteristics According to Admission Chloride Tertile
| Admission Serum Chloride (meq/L) | |||||
|---|---|---|---|---|---|
| Characteristic* | Overall (N=358) | 84–98 (N=115) | 99–102 (N=125) | 103–117 (N=118) | P-value† |
| Age (years) | 70 (62– 79) | 69 (61– 77) | 71 (62– 81) | 70 (62– 81) | 0.31 |
| Men | 264 (73.7%) | 87 (75.7%) | 96 (76.8%) | 81 (68.6%) | 0.30 |
| Black | 74 (20.7%) | 10 (8.7%) | 26 (20.8%) | 38 (32.2%) | <.001 |
| White | 270 (75.4%) | 102 (88.7%) | 94 (75.2%) | 74 (62.7%) | |
| Other | 14 (3.9%) | 3 (2.6%) | 5 (4.0%) | 6 (5.1%) | |
| Body Mass Index (kg/m2) | 30.7 (26.5– 36.9) | 30.6 (26.6– 36.7) | 30.7 (25.7– 37.3) | 31.2 (27.1– 36.9) | 0.90 |
| Ejection Fraction (%) (N=356) | 34.0 (21.5– 53.0) | 34.0 (20.0– 54.0) | 35.0 (23.0– 54.5) | 32.0 (23.0– 50.0) | 0.73 |
| Systolic Blood Pressure (mm Hg) | 115 (103– 127) | 108 (100– 119) | 114 (103– 129) | 122 (112– 135) | <.001 |
| Jugular Venous Pressure (cm H2O) (N=341) | 0.049 | ||||
| <8 | 16 (4.7%) | 5 (4.6%) | 3 (2.5%) | 8 (7.1%) | |
| 8–12 | 74 (21.7%) | 18 (16.7%) | 23 (19.2%) | 33 (29.2%) | |
| 13–16 | 121 (35.5%) | 35 (32.4%) | 51 (42.5%) | 35 (31.0%) | |
| >16 | 130 (38.1%) | 50 (46.3%) | 43 (35.8%) | 37 (32.7%) | |
| Diabetes Mellitus | 200 (55.9%) | 63 (54.8%) | 70 (56.0%) | 67 (56.8%) | 0.95 |
| Implantable Cardioverter Defibrillator | 156 (43.6%) | 56 (48.7%) | 59 (47.2%) | 41 (34.7%) | 0.060 |
| Chronic Obstructive Pulmonary Disease | 95 (26.5%) | 30 (26.1%) | 39 (31.2%) | 26 (22.0%) | 0.27 |
| New York Heart Association Class (N=342) | 0.16 | ||||
| II | 16 (4.7%) | 2 (1.8%) | 7 (5.9%) | 7 (6.1%) | |
| III | 229 (67.0%) | 77 (70.6%) | 84 (70.6%) | 68 (59.6%) | |
| IV | 97 (28.4%) | 30 (27.5%) | 28 (23.5%) | 39 (34.2%) | |
| Sodium (meq/L) | 138.5 (136.0– 141.0) | 136.0 (133.0– 138.0) | 138.0 (137.0– 141.0) | 141.0 (139.0– 142.0) | <.001 |
| Bicarbonate (meq/L) (N=343) | 27.0 (24.0– 30.0) | 29.0 (27.0– 32.0) | 28.0 (25.0– 30.0) | 25.0 (23.0– 28.0) | <.001 |
| Blood urea nitrogen (mg/dL) (N=356) | 37.0 (27.5– 51.0) | 44.0 (30.0– 61.0) | 37.0 (26.8– 50.8) | 32.0 (26.0– 44.0) | <.001 |
| Creatinine (mg/dL) | 1.7 (1.4– 2.1) | 1.7 (1.4– 2.2) | 1.7 (1.4– 2.0) | 1.7 (1.4– 2.0) | 0.92 |
| Cystatin C (mg/L) (N=334) | 1.7 (1.4– 2.2) | 107, 1.8 (1.5– 2.2) | 123, 1.7 (1.4– 2.2) | 113, 1.7 (1.4– 2.1) | 0.31 |
| NT-proBNP (pg/mL) (N=343) | 4972 (2330– 10280) | 4123 (1999– 9511) | 4339 (2194– 9879) | 6196 (3286– 10937) | 0.11 |
| ACEI or ARB | 179 (50.0%) | 46 (40.0%) | 68 (54.4%) | 65 (55.1%) | 0.034 |
| Beta-Blocker | 298 (83.2%) | 93 (80.9%) | 110 (88.0%) | 95 (80.5%) | 0.21 |
| Aldosterone Antagonist | 108 (30.2%) | 48 (41.7%) | 33 (26.4%) | 27 (22.9%) | 0.004 |
| Furosemide equivalent dose, mg/d (N=338) | 80.0 (60.0– 160.0) | 130.0 (80.0– 200.0) | 80.0 (60.0– 120.0) | 80.0 (40.0– 100.0) | <.001 |
Values are presented as median (interquartile range) for continuous and n(%) for categorical variables.
P-values for continuous variables by the Kruskal-Wallis test and for categorical variables by the Pearson chi-square test or Fisher’s exact test.
Baseline Cl level was not associated with incident worsening or persistent heart failure or freedom from congestion at 72 hours (n/N=20/325, OR 0.98 95% CI 0.83–1.15, P=0.78; and n/N=41/297, OR 1.09, 95% CI 0.97–1.22, P=0.14; respectively). Components of diuretic response by 72 hours are shown in Table 2. Lower admission Cl levels were associated with a relatively lower loop diuretic responsiveness. Although baseline Cl level was not associated with cumulative urine volume (P=0.698), or cumulative urine Na excretion (P=0.09); it was strongly associated with cumulative loop diuretic dose (P<.001) with a strong inverse association with diuretic efficiency (P<.001).
Table 2.
Association of Baseline Chloride Levels and Components of Diuretic Response at 72 Hours
| Clinical parameters from randomization to 72 hours | Models | Estimate: per 1 mEq/L increase in Chloride | Standard Error | P-value |
|---|---|---|---|---|
| Furosemide equivalent dosing (mg) (N=308) | Unadjusted | −28.29 | 4.68 | <.001 |
| Adjusted** | −32.66 | 8.13 | <.001 | |
| Diuretic efficiency (ml/40 mg furosemide equivalents) (N=298) | Unadjusted | 17.04 | 4.77 | <.001 |
| Adjusted** | 27.98 | 8.33 | <.001 | |
| Cumulative urine volume (ml) (N=259) | Unadjusted | −45.65 | 36.23 | 0.21 |
| Adjusted** | −23.97 | 61.69 | 0.70 | |
| Change in cystatin C (mg/L) (N=286) | Unadjusted | 0.01 | 0.004 | <.001 |
| Adjusted** | 0.01 | 0.006 | 0.30 | |
| Cumulative sodium excretion* (mmol) (N=251) | Unadjusted | 6.37 | 3.60 | 0.078 |
| Adjusted** | 10.43 | 6.19 | 0.093 | |
| Weight change (lbs) (N=295) | Unadjusted | −0.16 | 0.08 | 0.052 |
| Adjusted** | −0.38 | 0.15 | 0.010 | |
| Change in NT-proBNP (pg/mL) (N=286) | Unadjusted | −98.78 | 50.38 | 0.051 |
| Adjusted** | −63.18 | 85.40 | 0.46 |
Abbreviation: NT-proBNP is amino terminus pro B-type natriuretic peptide.
Normalized to 24 hours
Multivariable adjustment for sodium, bicarbonate, and cystatin C
With the exception of an inverse correlation between Cl and blood urea nitrogen (rho −0.21, P<.001), baseline Cl level was not correlated with other cardiorenal biomarkers: cystatin C (rho=−0.075, P=0.17) or NT-proBNP (rho=0.10, P=0.054). After adjustment, baseline Cl level was not associated with either change in cystatin C level (P=0.09) or change in NT-proBNP (P-0.46).
Chloride levels from baseline until discharge or day 7 were highly correlated (rho=0.75, P<.001) and decreased during the hospitalization as shown in Figure 1. From the random intercept, random slope models (N=340), the estimated change in Cl/day for placebo was −0.89 meq/L/day (P<.001) and this trend was not associated with either dopamine (0.15 meq/L/day, P=0.34) or nesiritide (0.17 meq/L/day, P=0.28). Although small in magnitude, higher in-hospital loop diuretic dosing was associated with lower serial Cl levels (−0.041 meq/L/day/25 mg furosemide IV equivalent, P=0.008).
Figure 1.
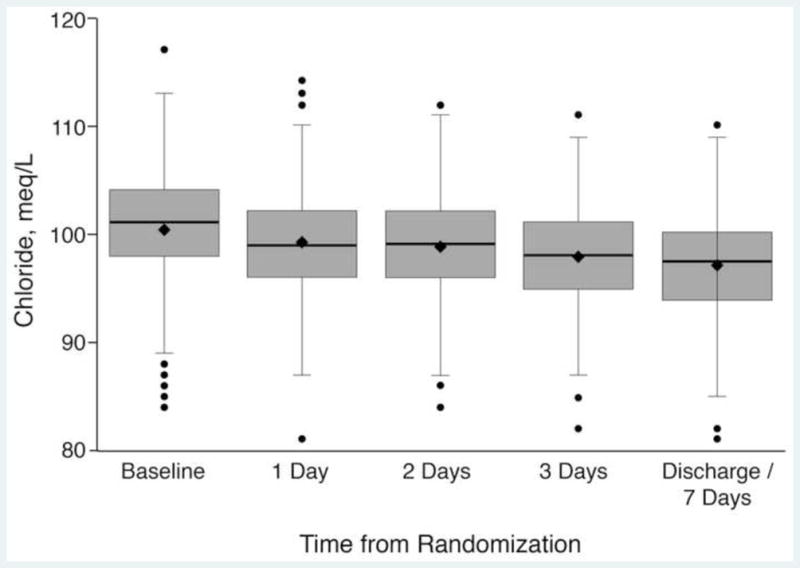
Chloride Levels Over Time
The median (25th, 75th) change in Cl levels from baseline to 72 hours was −3 (−5, 0) meq/L. Correlations of mediators and markers of diuretic response with change in Cl level from baseline to 72 hours are shown in Table 3. Change in Cl was correlated with diuretic efficiency (P=0.004), cumulative urine volume (diuresis, P=0.039), cumulative urine Na (natriuresis, P<.001), and change in NT-proBNP (P=0.037). Interestingly, change in Cl levels demonstrated the strongest (negative) correlation with change in cystatin C (P<.001).
Table 3.
Components of Diuretic Response and Changing Chloride Levels
| Change in chloride from randomization to 72 hours | ||
|---|---|---|
| Clinical parameters from randomization to 72 hours | Spearman Rho | P-value |
| Furosemide equivalent dosing (mg) (N=332) | 0.04 | 0.53 |
| Diuretic efficiency (ml/40 mg furosemide equivalents) (N=327) | −0.16 | 0.004 |
| Cumulative urine volume (mL) (N=280) | −0.12 | 0.039 |
| Change in cystatin C (mg/L) (N=302) | −0.35 | <.001 |
| Cumulative sodium excretion* (mmol) (N=272) | −0.21 | <.001 |
| Weight change (lbs) (N=323) | 0.18 | 0.001 |
| Change in NT-proBNP (pg/mL) (N=302) | 0.12 | 0.037 |
Abbreviation: NT-proBNP is amino terminus pro B-type natriuretic peptide.
Normalized to 24 hours
Mortality was 9.5% at 60-days (N=31/358) and 20% at 180-days (N=71/358). There was a 24% (N=85/352) event rate for death or rehospitalization by 60-days. Kaplan-Meier estimates for 180-day death for admission Cl level were 27%, 21%, and 12%, for tertiles 1, 2, and 3, respectively (Figure 2, Log-rank P=0.031). Baseline Cl levels were inversely associated with all 3 post-discharge outcomes (Table 4): 180-day death (P<.001), 60-day death (P<.001), and 60-day death or rehospitalization (P=0.010) with comparable findings after adjustment for Na, bicarbonate, white race, and cystatin C (P=0.049, P=0.029, and P=0.010) but not after the addition of pre-hospital loop diuretic dose (P=0.39, P=0.082, and P=0.094). In contrast, change in Cl was not associated with any of these outcomes in either unadjusted or adjusted analyses (P>0.5 for all). There was no interaction between Cl levels and white race for any of these outcomes (P>0.1 for all).
Figure 2.
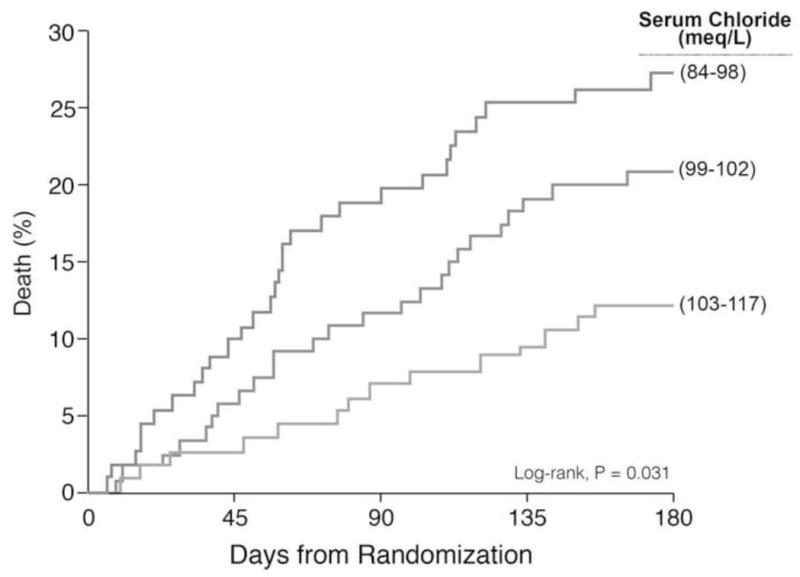
Kaplan-Meier Estimates of 180-Day Mortality Across Chloride Tertiles
Chloride tertiles: Tertile 1, 84–98 meq/L; Tertile 2, 99–102 meq/L; and Tertile
Table 4.
The Association of Baseline Chloride Level and Change in Chloride Level to 72 Hours with 60- and 180-day Outcomes
| Outcome | Events | Model | Hazard Ratio† | Hazard Ratio‡ | 95% Lower CL | 95% Upper CL | P-value |
|---|---|---|---|---|---|---|---|
| 180 Day Mortality | 60/319 | Unadjusted | 0.92 | -- | 0.88 | 0.97 | <.001 |
| 60/319 | Adjusted* | 0.91 | -- | 0.83 | 1.00 | 0.049 | |
| 59/300 | Adjusted** | 0.96 | -- | 0.87 | 1.06 | 0.39 | |
| 180 Day Mortality from 72 hours | 55/297 | Unadjusted | -- | 0.99 | 0.92 | 1.07 | 0.75 |
| 55/297 | Adjusted* | -- | 0.99 | 0.92 | 1.07 | 0.88 | |
| 60 Day Mortality | 27/319 | Unadjusted | 0.89 | -- | 0.82 | 0.95 | <.001 |
| 27/319 | Adjusted* | 0.86 | -- | 0.75 | 0.99 | 0.029 | |
| 27/300 | Adjusted** | 0.88 | -- | 0.76 | 1.02 | 0.082 | |
| 60 Day Mortality from 72 hours | 23/297 | Unadjusted | -- | 0.96 | 0.86 | 1.08 | 0.53 |
| 23/297 | Adjusted* | -- | 0.97 | 0.86 | 1.09 | 0.60 | |
| 60 Day Death or HF Rehospitalization | 76/314 | Unadjusted | 0.94 | -- | 0.90 | 0.99 | 0.010 |
| 76/314 | Adjusted* | 0.90 | -- | 0.83 | 0.98 | 0.010 | |
| 73/295 | Adjusted** | 0.93 | -- | 0.85 | 1.01 | 0.094 | |
| 60 Day Death or HF Rehospitalization from 72 hours | 71/297 | Unadjusted | -- | 1.02 | 0.96 | 1.10 | 0.51 |
| 71/297 | Adjusted* | -- | 1.02 | 0.95 | 1.09 | 0.57 |
Multivariable adjustment for sodium, bicarbonate, white race, and cystatin C
Multivariable adjustment for sodium, bicarbonate, white race, cystatin C, and pre-hospitalization loop diuretic dose
Hazard ratios represent a 1meq/L increase of baseline chloride
Hazard ratios represent a 1 meq/L increase in change of chloride from baseline to 72 hours and are additionally adjusted for baseline chloride levels
DISCUSSION
This post-hoc analysis from ROSE-AHF has several findings incremental to our understanding of Cl levels in AHF. First, lower Cl levels were associated with lower loop diuretic responsiveness. Second, Cl levels decreased over time during decongestive treatments, and this was augmented by loop diuretic dose paralleling decongestion. Third, like prior observations, this analysis demonstrated a link between Cl and adverse long-term events. This risk was attenuated, however, with adjustment for loop diuretic dosage. Further, acute changes in Cl level during treatment did not show a long-term impact. Taken together, these findings suggest that chronically perturbed Cl homeostasis is associated with lower diuretic efficacy and may impact long-term clinical outcomes above and beyond acute depletion of Cl levels by loop diuretics in AHF.
Serum Cl homeostasis is tightly regulated by two major mechanisms: 1) maintaining electroneutrality between the intra- and extracellular spaces and 2) the ability of the kidney to maintain serum Cl balance. In heart failure, this balance can be disturbed shifting more Cl into the intracellular space where, normally, it is virtually absent.14 Additionally, renal regulation of serum Cl is disrupted by salt restriction and loop diuretic which limit tubular delivery and resorption, and higher arginine vasopressin secretion which may contribute to dilution.15–18
A poor diuretic response is recognized as an adverse prognostic factor in the setting of AHF.11,13 Our finding that lower baseline serum Cl was associated with lower diuretic response (lower diuretic efficiency and higher total in-hospital IV furosemide dose with comparable Na excretion and weight change/urine volume) is consistent with prior findings suggesting that lower Na was inversely associated with lower diuretic responsiveness.13 In our cohort, baseline Cl was correlated to baseline Na (rho 0.57, P<.001). In contrast to Na depletion, however, Cl depletion may be a more specific electrolyte marker for impaired diuretic response because this association was independent of both Na, bicarbonate, and cystatin C. Murine models have suggested that systemic Cl depletion is essential for mediating a contraction alkalosis and can effect tubuloglomerular feedback, independent of volume expansion.19,20 As a result, Cl depletion may lead to diuretic resistance by fueling adaptive renal mechanisms to maintain volume status.
To the best of our knowledge, this is the first analysis showing that serum Cl levels may decrease during treatment (Figure 1) and correlate with both diuresis and natriuresis. This trend was affected by loop diuretic dose, but not vasoactive therapy (nesiritide) or by attempting to augment renal vasodilation (dopamine), supporting the iatrogenicity of Cl depletion by loop diuretic therapy. In fact, human models of Cl depletion have been generated by dietary Na restriction with loop diuretic usage.8 Importantly, these changes were not associated with adverse events, but they suggest that long-term loop diuretic use may contribute to chronic Cl depletion. In the loop diuretic-mediated acutely lowered Cl state, there may be transient volume redistribution from the intracellular and extracellular spaces into the vasculature that may resolve with the transition to lower-dose loop diuretics when congestion resolves. Chronically, however, escalating loop diuretic dosage over time may impede this homeostatic resolution.
The inverse correlation between changing Cl levels and changing cystatin C highlights the pathobiology of Cl losses and renal function changes. As Cl depletion evolves, lower levels of Cl signal a reduction in glomerular filtration rate, which is the normal homeostatic response to lower delivery of bicarbonate to the distal nephron, thus attenuating further Na and fluid losses.20 Therefore, changing renal function during decongestive therapies may be mediated, at least in part, by Cl imbalance underscoring the complex interplay between hemodynamic and non-hemodynamic determinants of renal function during AHF.21
In addition to supporting prior findings, this analysis extends the prognostic role of Cl to heart failure rehospitalizations.1 The precise reasons why hypochloremia identifies higher risk are unclear, but this association between Cl levels and adverse outcomes has been seen in other diseases.22,23 In heart failure, however, serum Cl levels may be more disease-specific given its role in the cardiorenal axis. Loop diuretic usage may lead to chronic Cl depletion. Chloride, not Na, suppresses plasma renin activity – which is supported in this analysis by the attenuation of risk by adjustment for loop diuretic dose.24 Their use may contribute to higher renin levels, more neurohormonal activation, and thus a worse prognosis.25 In aggregate, these findings support the hypothesis that strategies to preserve Cl homeostasis might impact outcomes in heart failure, especially in the setting of chronic diuretic use.
These results must be interpreted within the context of several limitations. First, Cl levels were measured by clinically collected blood specimens as part of routine care of patients with AHF as opposed to the core lab. Second, urine Cl levels were not measured, which would have clarified Cl depletion or dilution. Third, it was unknown if therapies that increase serum Cl levels were administered to any participants. However, this seems unlikely since their use is not commonplace, and would have likely biased these findings to the null. Fourth, there was a low prevalence of thiazide diuretic usage (10.3% at baseline, 10.9% on day 1, 15.1% on day 2, 14.0% on day 3) and it was therefore not added in the multivariable models. Despite these limitations, these data were collected within a clinical trial with detailed daily clinical data and clearly defined outcomes supporting the validity of these results.
CONCLUSIONS
Baseline serum Cl levels were associated with impaired diuretic efficiency during treatment for AHF and post-discharge adverse outcomes. Change in serum Cl levels was correlated with changing markers of decongestion and inversely correlated with cystatin C change, although acute decreases in Cl levels were not associated with adverse post-discharge outcomes. These findings support the hypothesis that lower baseline serum Cl levels may identify a high-risk phenotype with relatively lower diuretic response, but that short term decreases in Cl levels mediated by loop diuretics may not lead to adverse outcomes. They also support the interrelation between serum Cl levels and loop diuretics in heart failure.
Supplementary Material
Acknowledgments
Financial Support: The Heart Failure Clinical Research Network is supported by the NHLBI, National Institutes of Health (U10HL084904 for coordinating center; and U10HL084861, U10HL084875, U10HL084877, U10HL084889, U10HL084890, U10HL084891, U10HL084899, U10HL084907, and U10HL084931 for clinical centers). Dr. Testani is funded by NIH Grants, K23HL114868, L30HL115790 and R01HL128973.
All authors have no conflicts of interest. The ROSE-AHF study and this ancillary study is sponsored by the National Institutes of Health, which has provided input in the design, conduct, and reporting of this paper.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Grodin JL, Simon J, Hachamovitch R, Wu Y, Jackson G, Halkar M, Starling RC, Testani JM, Tang WH. Prognostic Role of Serum Chloride Levels in Acute Decompensated Heart Failure. J Am Coll Cardiol. 2015;66:659–666. doi: 10.1016/j.jacc.2015.06.007. [DOI] [PubMed] [Google Scholar]
- 2.Sica DA. Sodium and water retention in heart failure and diuretic therapy: basic mechanisms. Cleve Clin J Med. 2006;73(Suppl 2):S2–7. doi: 10.3949/ccjm.73.suppl_2.s2. discussion S30–33. [DOI] [PubMed] [Google Scholar]
- 3.Sica DA. Hyponatremia and heart failure--treatment considerations. Congest Heart Fail. 2006;12:55–60. doi: 10.1111/j.1527-5299.2006.04844.x. [DOI] [PubMed] [Google Scholar]
- 4.Ellison DH. The physiologic basis of diuretic synergism: its role in treating diuretic resistance. Ann Intern Med. 1991;114:886–894. doi: 10.7326/0003-4819-114-10-886. [DOI] [PubMed] [Google Scholar]
- 5.Hropot M, Fowler N, Karlmark B, Giebisch G. Tubular action of diuretics: distal effects on electrolyte transport and acidification. Kidney int. 1985;28:477–489. doi: 10.1038/ki.1985.154. [DOI] [PubMed] [Google Scholar]
- 6.Tannen RL. Effect of potassium on renal acidification and acid-base homeostasis. Semin Nephrol. 1987;7:263–273. [PubMed] [Google Scholar]
- 7.Luke RG, Galla JH. It is chloride depletion alkalosis, not contraction alkalosis. J Am Soc Nephrol. 2012;23:204–207. doi: 10.1681/ASN.2011070720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rosen RA, Julian BA, Dubovsky EV, Galla JH, Luke RG. On the mechanism by which chloride corrects metabolic alkalosis in man. Am J Med. 1988;84:449–458. doi: 10.1016/0002-9343(88)90265-3. [DOI] [PubMed] [Google Scholar]
- 9.Chen HH, Anstrom KJ, Givertz MM, Stevenson LW, Semigran MJ, Goldsmith SR, Bart BA, Bull DA, Stehlik J, LeWinter MM, Konstam MA, Huggins GS, Rouleau JL, O’Meara E, Tang WH, Starling RC, Butler J, Deswal A, Felker GM, O’Connor CM, Bonita RE, Margulies KB, Cappola TP, Ofili EO, Mann DL, Davila-Roman VG, McNulty SE, Borlaug BA, Velazquez EJ, Lee KL, Shah MR, Hernandez AF, Braunwald E, Redfield MM Network NHFCR. Low-dose dopamine or low-dose nesiritide in acute heart failure with renal dysfunction: the ROSE acute heart failure randomized trial. JAMA. 2013;310:2533–2543. doi: 10.1001/jama.2013.282190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Heart Failure Society of A. Lindenfeld J, Albert NM, Boehmer JP, Collins SP, Ezekowitz JA, Givertz MM, Katz SD, Klapholz M, Moser DK, Rogers JG, Starling RC, Stevenson WG, Tang WH, Teerlink JR, Walsh MN. HFSA 2010 Comprehensive Heart Failure Practice Guideline. J Card Fail. 2010;16:e1–194. doi: 10.1016/j.cardfail.2010.04.004. [DOI] [PubMed] [Google Scholar]
- 11.Testani JM, Brisco MA, Turner JM, Spatz ES, Bellumkonda L, Parikh CR, Tang WH. Loop diuretic efficiency: a metric of diuretic responsiveness with prognostic importance in acute decompensated heart failure. Circ Heart Fail. 2014;7:261–270. doi: 10.1161/CIRCHEARTFAILURE.113.000895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.ter Maaten JM, Valente MA, Damman K, Hillege HL, Navis G, Voors AA. Diuretic response in acute heart failure-pathophysiology, evaluation, and therapy. Nat Rev Cardiol. 2015;12:184–192. doi: 10.1038/nrcardio.2014.215. [DOI] [PubMed] [Google Scholar]
- 13.ter Maaten JM, Dunning AM, Valente MA, Damman K, Ezekowitz JA, Califf RM, Starling RC, van der Meer P, O’Connor CM, Schulte PJ, Testani JM, Hernandez AF, Tang WH, Voors AA. Diuretic response in acute heart failure-an analysis from ASCEND-HF. Am Heart J. 2015;170:313–321. doi: 10.1016/j.ahj.2015.05.003. [DOI] [PubMed] [Google Scholar]
- 14.Flear CT, Crampton RF. Evidence for the entry of chloride into cells in congestive cardiac failure. Clin Sci. 1960;19:495–504. [PubMed] [Google Scholar]
- 15.Greger R, Schlatter E. Properties of the basolateral membrane of the cortical thick ascending limb of Henle’s loop of rabbit kidney. A model for secondary active chloride transport. Pflugers Arch. 1983;396:325–334. doi: 10.1007/BF01063938. [DOI] [PubMed] [Google Scholar]
- 16.Adrogue HJ, Madias NE. Hyponatremia. N Engl J Med. 2000;342:1581–1589. doi: 10.1056/NEJM200005253422107. [DOI] [PubMed] [Google Scholar]
- 17.Goldsmith SR, Francis GS, Cowley AW, Jr, Levine TB, Cohn JN. Increased plasma arginine vasopressin levels in patients with congestive heart failure. J Am Coll Cardiol. 1983;1:1385–1390. doi: 10.1016/s0735-1097(83)80040-0. [DOI] [PubMed] [Google Scholar]
- 18.Francis GS, Benedict C, Johnstone DE, Kirlin PC, Nicklas J, Liang CS, Kubo SH, Rudin-Toretsky E, Yusuf S. Comparison of neuroendocrine activation in patients with left ventricular dysfunction with and without congestive heart failure. A substudy of the Studies of Left Ventricular Dysfunction (SOLVD) Circulation. 1990;82:1724–1729. doi: 10.1161/01.cir.82.5.1724. [DOI] [PubMed] [Google Scholar]
- 19.Galla JH, Bonduris DN, Dumbauld SL, Luke RG. Segmental chloride and fluid handling during correction of chloride-depletion alkalosis without volume expansion in the rat. J Clin Invest. 1984;73:96–106. doi: 10.1172/JCI111211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Galla JH, Bonduris DN, Sanders PW, Luke RG. Volume-independent reductions in glomerular filtration rate in acute chloride-depletion alkalosis in the rat. Evidence for mediation by tubuloglomerular feedback. J Clin Invest. 1984;74:2002–2008. doi: 10.1172/JCI111622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Damman K, Testani JM. The kidney in heart failure: an update. Eur Heart J. 2015;36:1437–1444. doi: 10.1093/eurheartj/ehv010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.De Bacquer D, De Backer G, De Buyzere M, Kornitzer M. Is low serum chloride level a risk factor for cardiovascular mortality? J Cardiovasc Risk. 1998;5:177–184. [PubMed] [Google Scholar]
- 23.McCallum L, Jeemon P, Hastie CE, Patel RK, Williamson C, Redzuan AM, Dawson J, Sloan W, Muir S, Morrison D, McInnes GT, Freel EM, Walters M, Dominiczak AF, Sattar N, Padmanabhan S. Serum chloride is an independent predictor of mortality in hypertensive patients. Hypertension. 2013;62:836–843. doi: 10.1161/HYPERTENSIONAHA.113.01793. [DOI] [PubMed] [Google Scholar]
- 24.Kotchen TA, Luke RG, Ott CE, Galla JH, Whitescarver S. Effect of chloride on renin and blood pressure responses to sodium chloride. Ann Intern Med. 1983;98:817–822. doi: 10.7326/0003-4819-98-5-817. [DOI] [PubMed] [Google Scholar]
- 25.Packer M, Lee WH, Kessler PD, Gottlieb SS, Bernstein JL, Kukin ML. Role of neurohormonal mechanisms in determining survival in patients with severe chronic heart failure. Circulation. 1987;75:IV80–92. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.