

Abstract
Single particle cryo-Electron Microscopy (cryo-EM) is a technique that allows the structural characterization of macromolecules without the need for crystallization. For certain type of samples that are ideally suited for cryo-EM studies it has been possible to reach high-resolution structures following relatively standard procedures. Other biological systems remain highly challenging, even for cryo-EM. Challenges may involve the scarcity of the sample, poor stability of the complexes, and most often, the intrinsic flexibility of biological molecules. Among these challenging samples are large eukaryotic transcription complexes, which suffer from all such shortcomings. Here we report how we have recently tried to overcome those challenges in order to improve our structural understanding of the human transcription pre-initiation complex assembly and the transcription initiation process. Parallel efforts have also been carried out for budding yeast transcription initiation complexes, allowing comparisons that establish both the overall conservation and the specific structural differences between the two systems.
Single particle cryo-electron microscopy (cryo-EM) is a structural technique that overcomes the need for crystallization, requires very small amounts of sample, and is ideally suited to study large macromolecular assemblies. These properties have made it an ideal complement to X-ray crystallography. But while in use now for several decades, only recently has cryo-EM become a mainstream structural biology method capable of producing high-resolution structures at a competitive pace, in great part due to both new detector technology and new software tools for image processing of easy use [1,2]. These technical advances are resulting in an ever increasing number of atomic structures for molecules or assemblies that were considered structurally unreachable or highly challenging due to the difficulty to obtain crystals. However, certain samples still remain challenging, even for cryo-EM. Here, we use our studies of human transcription initiation as an example of the type of challenges and limitation that cryo-EM still needs to face.
Challenge number 1 - Scarcity
Gene transcription requires the assembly at the core promoter of a large pre-initiation complex (PIC) that includes RNA Polymerase II (Pol II) and the general transcription factors (GTFs) TFIID, TFIIA, TFIIB, TFIIF, TFIIE and TFIIH. With ten or more subunits each, human TFIID, TFIIH and Pol II have proven refractory to efficient reconstitution and are generally obtained by immuno-purification from HeLa cells. Given their scarcity, especially for TFIID, the amounts and concentrations obtained are extremely limiting for structural characterization. The problem is exacerbated when trying to assemble a full PIC on DNA. The final human PIC concentrations we use for cryo-EM studies are in the low hundreds of nanomolar, and volumes are realistically limited to 5-10 μl. At these concentrations, mild crosslinking (e.g. with 0.01-0.05% glutaraldehyde) is useful for keeping the supracomplexes together, and has the added benefit of making samples more resilient during the harsh process of cryo-EM sample preparation. Furthermore, the experimental amount of purified PIC requires concentrating the sample on the EM grid. Conventionally in cryo-EM, the sample is deposited on a carbon film with holes that, after most of the sample is blotted off and plunged into liquid ethane, will be spanned by a suspended layer of vitrified buffer containing the macromolecule of interest. The number of human TFIID or PIC “particles” in a cryo-EM image can be increased, to the point of making data collection practical, with the use of a thin carbon film over the holes that has been properly treated to be hydrophilic. A drop of the sample is then deposited on this surface and is incuvated for a few minutes so that complexes have the chance to touch and adhere to it, resulting in the concentration of the sample by adsorption. Unfortunately, this carbon layer contributes to image background and makes the blotting of the sample into a thin layer less homogeneous and less reproducible.
Challenge number 2 – Conformational complexity
The lack of a crystallization requirement is a great advantage in cryo-EM studies. However, because the macromolecule is not trapped in a crystal lattice, which most often constrains molecular motions, conformational flexibility is likely to be observed in images of the complex. This problem can present itself at different scales. If a small region of a large complex is flexibly attached to the rest, it will appear blurred out or even be invisible after image averaging. This loss is akin to what is seen (or not seen) in crystallographic studies of proteins containing flexible loops. A more difficult challenge involves large movements of significant masses within a structure. The ratcheting of the ribosome is a classical example. This type of heterogeneity needs to be identified early in the analysis so that the appropriate sorting can be done computationally. The ribosome ratchet is an ideal example, as the conformational landscape is made of two well-defined and discrete states (to a first approximation), and it has served as a test case for a number of software solutions to the heterogeneity problem [3,4]. However, macromolecular complexes often display a continuum of states, making sorting a challenge and hampering resolution.
The existence of multiple conformations can be initially detected in a cryo-EM reconstruction when the resolution is clearly below what would be expected given the amount of images being combined. We were able to detect such shortcoming in our initial cryo-EM studies of human TFIID [5]. TFIID is a ∼1 MDa complex of TATA-binding protein (TBP) and 13 TBP-associated factors (TAFs) that are involved in recognition of core promoter motifs and interaction with cofactors and gene specific transcription factors. The use of a bootstrap method developed by Pawel Penczeck to estimate the 3D variance of a cryo-EM reconstruction [6] confirmed that human TFIID suffers from significant heterogeneity. But only more recent analysis allowed us to describe the nature of this flexibility and its biological relevance.
A number of EM studies [7-9], showed human TFIID to have a horseshoe shape made of three main lobes, named A, B and C. A similar overall architecture has also been observed for budding yeast TFIID [10,11]. Careful 2D image classification of the human TFIID images demonstrated that while the B and C lobes form a fairly stable core, lobe A moves dramatically across the complex [12], from interacting with lobe C, to interacting with lobe B, a displacement of ∼100 Å (Figure 1a). TFIID appears to partition between two extreme conformations that we refer to as canonical (lobe A proximal to lobe C) and rearranged (lobe A proximal to lobe B), but the motion is better described as a continuum. Most interestingly, when core promoter DNA and TFIIA were added, the TFIID conformational landscape shifted towards the rearranged state, and 3D reconstructions showed that DNA-bound TFIID was in the rearranged state [12] (Figure 1b). These findings point to a model in which modulation of the conformational landscape of TFIID by regulatory factors would affect DNA binding and thus transcriptional output [13].
Figure 1. Describing the conformational heterogeneity of TFIID.
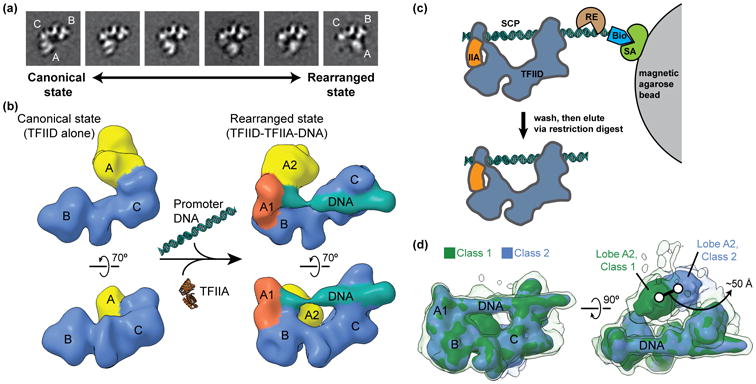
(a) Two-dimensional classification of cryo-EM images of TFIID revealed the conformational rearrangement of TFIID, in which lobe A translocates ∼100 Å across the rigid TFIID core comprised of lobes B and C. Quantification of the occupancy of lobe A in different positions along the BC-core showed that TFIID partitions between the canonical (left) and rearranged (right) states. (b) A multi-model refinement strategy led to 3D reconstructions of TFIID for the both the canonical (left) and rearranged (right) states and showed that TFIID binds to promoter DNA in the rearranged state in the presence of TFIIA (right). (c) Schematic of the strategy used for the purification of promoter-bound TFIID. TFIID was incubated with TFIIA (IIA) and super core promoter (SCP) DNA that was biotinylated (Bio) on one end. The DNA and associated proteins was then immobilized on magnetic streptavidin (SA) coated beads, and excess proteins were washed away. The DNA-bound complexes were then eluted by restriction endonuclease (RE) digest and were subsequently used for the preparation of cryo-EM samples. (d) Characterization of the flexibility of lobe A2 within promoter-bound TFIID. Following alignment to the more rigid promoter-bound BC-core of TFIID (including lobe A1), cryo-EM images of the TFIID-TFIIA-promoter complex were sorted by 3D classification within a mask around the entire range of lobe A. The two major 3D classes are shown in green (class 1) and blue (class 2), with the position of lobe A differing by about 50 Å. Surfaces are displayed at two different intensity thresholds, so that the lower intensity surface in transparency show more flexible elements.
The studies just described had an obvious biological value, but they were very limited in resolution, reflecting the fact that conformational mixing was hampering accurate alignment, and that a two-state partitioning fell short of describing the conformational complexity of TFIID.
Progress dealing with conformational heterogeneity: TFIID binding to core promoter DNA
While heterogeneity is a challenge for cryo-EM studies, it also represents a unique opportunity to describe the conformational behavior of macromolecules. There is a continuous effort in the community to devise both experimental and computational strategies to detect and characterize this heterogeneity and its functional relevance, and to overcome the limitations it imposes to achieving high-resolution structures. Classification via maximum likelihood approaches has been, so far, the most successful one [14-16], but other approaches are also being explored (as examples see [17,18]).
Conformational changes in TFIID are so large that ab initio methods were required to produce alternative and unbiased 3D reconstructions. We used the Orthogonal Tilt Reconstruction (OTR) approach [19,20] to extract the canonical and rearranged states from a data set of negatively stained holo-TFIID. The resulting low-resolution structures were then used as references for cryo-EM studies of TFIID under different conditions (e.g. with and without DNA/TFIIA) in supervised or unsupervised approaches.
Given our findings that DNA-bound TFIID is in the rearranged state, we reduced our biochemical and conformational heterogeneity using a purification strategy that selected DNA-bound complexes. The DNA, which included the super core promoter sequence (to maximize binding affinity) [21], was biotinylated and immobilized on streptavidin-coated beads (Figure 1c). Purified human TFIID and TFIIA were incubated with this DNA, excess protein was washed away, and the DNA-bound complexes were recovered by endonuclease cleavage off the beads. For data collection we used a Gatan K2 direct electron detector, which renders higher contrast images and allows for a movie-mode of data collection and the subsequent alignment of individual frames to correct for beam-induced movement [22,23]. Images were analyzed using RELION [15], which utilizes Bayesian principles for image classification and alignment. After an initial round of 2D and 3D classification in RELION was used to remove a significant number of particle images that likely corresponded to damaged complexes, the reconstruction obtained showed, once again, that while there was a well defined core of lobes B and C, lobe A was blurred out due to motion [24] (Figure 1d shows both the blurring of lobe A (transparent surface) and the sorting into two distinct rearranged states that are part of a continuum (green and blue solid surfaces).
It was possible to significantly improve the resolution of the DNA-bound core, comprising lobes B, C, and a segment of lobe A (named lobe A1), by carrying out further classification and alignment after masking out the large, mobile segment of lobe A (named lobe A2) (Figure 2a). The resulting improved structure[EN1] allowed the unequivocal localization of the TBP-TFIIA-TATA DNA crystal structure at one end of the complex (the lobe A1) (Figure 2b-c). Even within the BC core, lobe C and the downstream DNA appeared less flexible (Figure 2a, inset), so a second focused classification and alignment cycle was carried out that led to 7-10 Å resolution for lobe C (Figure 2a-b). As a result, it was possible to identify the position of a TAF1-TAF7 complex by docking of its crystal structure [25] in the region of contact with downstream DNA. Furthermore, a large segment of TAF2 that shares homology with M1 aminopeptidases [26] could be fitted into the lobe C map adjacent to TAF1-TAF7 using crystal structures [26,27]. Within the remaining unassigned region in lobe C, we identified a HEAT-repeat present in two copies, forming an apparent homodimer that could be readily accommodated by fitting two copies of the TAF6 HEAT-repeat crystal structure from a microsporidium fungi [28] (see Figure 2d for final protein and DNA elements assignment). The resolution in lobes A2 and B was insufficient to make any further subunit assignment. However, this study, which combines cryo-EM maps in the 7-10 Å resolution with crystal structures or homology models of subunits that could be docked with high precision into those maps, has already identified, with close to single amino acid precision, the TFIID structural motifs involved in recognition of most human core promoter elements.
Figure 2. Improving the resolution of the promoter-bound TFIID core.
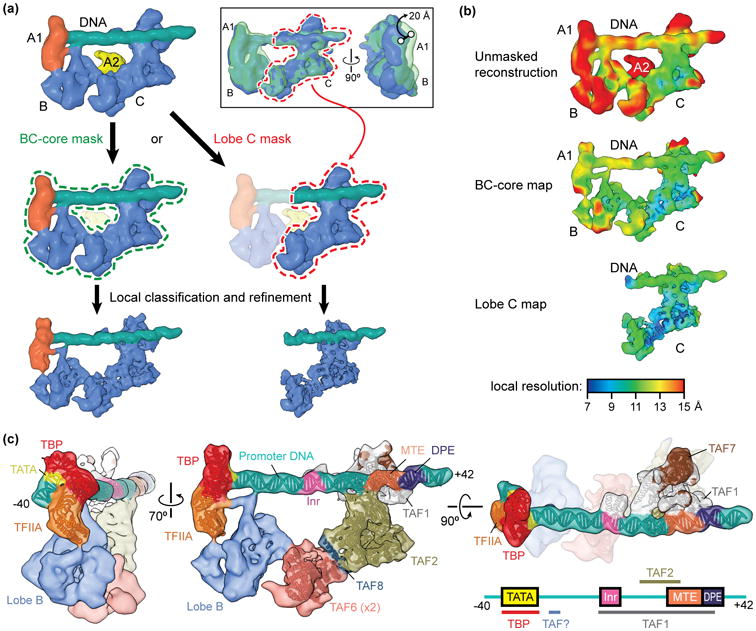
(a) Schematic of focused classification and refinement used to improve the resolution of the promoter-bound BC-core (left) and lobe C (right) of TFIID. Following alignment to the unmasked structure, cryo-EM images of the TFIID-TFIIA-promoter complex were aligned to references with a mask around the entire BC-core or around lobe C only. Images were then sorted by 3D classification within similar masks to extract the best quality particle images within that region. Final refinements of the structures within the masks resulted in improved reconstructions. The inset at the top right shows the flexibility of the lobe B/A1 region of the complex relative to lobe C, as revealed by 3D classification within this region following alignment of the cryo-EM images to lobe C. (b) Local resolution estimation of the unmasked structure (top) versus the BC-core (middle) or lobe C (bottom) reconstructions shows the improvement in resolution obtained by following the focused classification and refinement strategy outlined in (a). (c) Assignments and models of protein subunits and DNA elements within the promoter-bound TFIID structure [24]. The DNA-interacting elements are highlighted in the structure shown on the right, and a diagram of the super core promoter DNA construct used in this study, which contained the TATA, Inr, MTE, and DPE consensus sequences, is shown in the bottom right with the footprints of TFIID subunits indicated, (the identity of one of the DNA-binding TAFs, here labeled “TAF?”, is as of yet unknown).
Notably, Berger and colleagues have succeeded in recombinantly producing a partial human TFIID complex comprising TAF4, TAF5, TAF6, TAF9 and TAF12 [29]. This complex, which appeared to be dimeric, was characterized by cryo-EM and a model of the subunits arrangement was proposed. Those subunits, except for part of TAF6, have not yet been identified within the full human TFIID architecture, and likely correspond to the regions yet unassigned.
Challenge number 3 – Identification of subunits during map interpretation and compositional heterogeneity
The full PIC contains about 50 polypeptide chains. A more amenable complex for cryo-EM is one in which the 14-subunit TFIID complex is substituted by a single protein, TBP in the study of basal transcription from a TATA-based promoter. We reconstituted this TBP-based PIC using the streptavidin bead method described above. Based on the sequential PIC assembly model [30], we used a bootstrap approach that allowed us to identify the position of each PIC component (Figure 3a) [31]. The rational was to generate first a PIC that included DNA-TBP-IIA-IIB-Pol II, for which there were crystallographic structures for all the components or homologues (PDB ID: 1NVP, 1C9B, 3K1F, 3K7A). This step was followed by purification and reconstruction of complexes with one additional component at a time, for which additional density in the EM map would most likely correspond to the newly added component. The cryo-EM density map of the first structure could be fully accounted for by the structures of TBP-TFIIA-TATA and the yeast Pol II-TFIIB, although some adjustments were needed for the relative position of the cyclin folds within TFIIB. The core promoter DNA contained both a TATA box and BREu and BREd (upstream and downstream TFIIB recognition elements) sequences. Only this DNA was visible, indicating that other than the sequence-specific interactions by TBP and TFIIB, there were no other apparent contacts between the DNA and the complex. When the PIC was assembled including those factors plus TFIIF (a heterodimer of Rap30 and Rap74), the reconstruction showed additional density that could be accounted for by the crystal structures of the WH domain of Rap30 and the dimerization domain of Rap30-Rap74. This structure also showed clear density for the full length of the DNA, which was engaged by Rap30, as well as the Rpb1 clamp head and Rpb5 components of Pol II, and conformational changes that included the opening of the clamp. Purification and reconstruction of a complex that included also TFIIE showed extra density interacting with the Rap30 WH domain, the clamp head and the stalk regions of Pol II, which topologically trapped the DNA on the surface of Pol II. Finally, addition of TFIIH showed a large extra density. Because this structure was obtained only for a negative stained sample, the DNA was not visible. However, the DNA position was extrapolated from the previous state (this turned up to be a good approximation). TFIIH was seen to contact the expected DNA, right downstream of the Rpb5 contact, as well as the region of TFIIE proximal to the stalk (Figure 3a).
Figure 3. Structural determination of human TBP-based PIC.
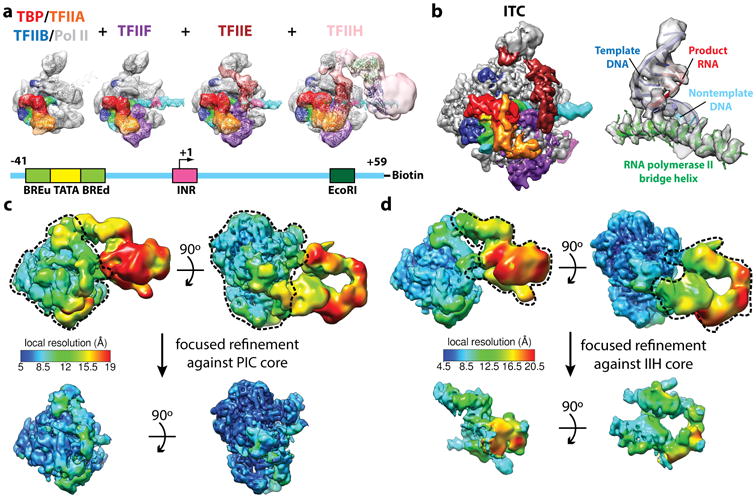
(a) Reconstitution strategy for human PIC by sequential assembly. Color scheme for the components of the PIC is shown at the bottom. Cryo-EM reconstructions of PIC assembly intermediates for (from left to right): TBP–TFIIA–TFIIB–DNA–Pol II, plus TFIIF, plus TFIIE, and negative stain reconstruction of the holo PIC including TFIIH. Schematic of the DNA highlights the relative positions of the core promoter elements and restriction site (bottom). (b) Cryo-EM reconstruction of the core PIC in the ITC state (left) and near-atomic resolution details around the Pol II active site (right). (c,d) Refinement strategies for the “core” PIC in the CC (c) and the TFIIH core complex in the open promoter states (d). The local resolution estimation shows flexibility for TFIIH. Focused refinements (using the masks marked by dashed lines) improved alignment accuracy and thereby the resolution for targeted regions.
The strategy just described built up confidence in the reconstruction procedure, as the sequential structures had to be consistent with each other. It also allowed, as the biochemical complexity built up, to separate and identify coexisting structures that corresponded to partial complexes (where a factor may have fallen off), as corresponding to a previously observed smaller complex. Self-consistency through the entire process gave extra confidence on the experimental design, the cryo-EM reconstructions, and the molecular interpretation. Ultimately, this stepwise assembly and structural characterization allowed the unequivocal assignment of all the relative positions of proteins and DNA within the TBP-based human PIC.
High-resolution PIC structures in different stages of the initiation process
The cryo-EM structure of the TBP-based PIC, in three distinct states throughout the transcription initiation process, has now been obtained at significantly improved resolution using new direct electron detection technology [32]. In addition to the initial close complex (CC), where the PIC engages duplex DNA, we have described an open complex (OC), where the DNA contains an opened transcription bubble, as well as an initial transcribing complex (ITC), containing 6 RNA nucleotides. While these three structures were obtained in the presence of TFIIS, a fourth cryo-EM reconstruction described the ITC in the absence of TFIIS (ITC-(IIS)). Figure 3b (right) shows the active site in Pol II in this ITC-(IIS) structure, an example of higher resolution features within the cryo-EM maps. Resolution improvement was also achieved using the same focused alignment strategy mentioned above, for the core PIC region in CC (Figure 3c) and for the TFIIH complex in the three open promoter states (Figure 3d).
The resolution of the cryo-EM maps and their redundancy has allowed the generation of molecular models for all the components of the “core” PIC (without TFIIH, which is more flexibly attached). Pol II and most of TFIIB are at a resolution of better than 4 Å, thus allowing refinement of the human sequences into the density using the yeast crystal structure as starting point [33] (such as in Fig. 3b (right)). Other regions of the core PIC are between 4.5 and 7 Å resolution, enough to define secondary structure (Fig. 4). We chose to use MDFF [34] as the method to generate atomic models using homology models for individual domains that were then fitted into the cryo-EM map and subsequently refined using Phenix [35]. A number of regions that had been poorly or not characterized previously are now visualized. Examples include the three WH, one HTH and a Zinc-ribbon domains of TFIIE, which were modeled by homology and flexibly fitted into the ∼7Å-resolution density (Figure 4a). As another example, we were able to visualize and model the linker between the Rap30 WH and dimerization domains, which interacts with the protrusion domain, TFIIB and promoter DNA (Figure 4b). Of particular interest is the structure of the TFIIB linker region, which is disordered in the CC state, but becomes ordered upon interaction with the non-transcribed strand of the DNA, playing what appears to be an essential role in the stabilization of the transcription bubble in the OC and ITC states (Figure 4c).
Figure 4. Visualization of new structural elements within the human PIC obtained by resolution improvement.
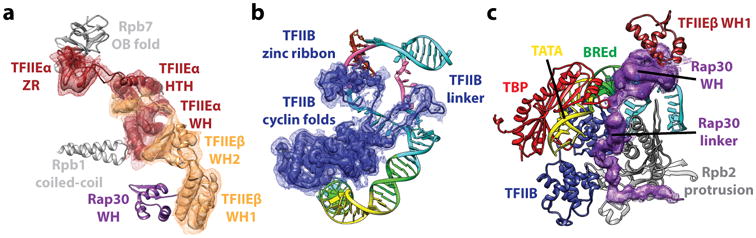
(a) Key domains of TFIIE and their interaction partners within the PIC. (b) Full-length path of TFIIB in the ITC state reveals the TFIIB linker region to be critical for the stabilization of single stranded non-template DNA. (c) RAP30 WH domain and linker within TFIIF. Elements of the PIC in close proximity are indicated.
Cryo-EM studies of the yeast PIC
In addition to the human TBP-based PIC studies described above, there have been parallel efforts using the yeast system [36,37]. These studies show the overall similarity of the arrangements of the different GTFs, confirming the expected conservation of the transcription initiation process across eukaryotes. However, small but significant differences are present between the two systems. Such structural differences are likely to reflect the differences in the detailed mechanisms of promoter clearance and transcription start site (TSS) selection between humans and yeast. Among the structural differences observed is the fact that in the CC state, yeast Pol II does not appear to contact the duplex DNA and its clamp head remains in a closed state. The studies of Cramer and colleagues also showed that in the yeast PIC, the presence of TFIIE was sufficient to generate an OC state in which TFIIE interacts directly with the upstream end of the bubble.
Conclusions and perspectives
The recent revolution in cryo-EM is having a significant effect on our capacity to structurally characterize challenging molecular assemblies that had previously defied analysis, and where only crystal structures of some fragments were accessible. Structures of human and yeast PICs, in different functional states, are among the examples in the recent literature, of how overcoming the requirement of crystallization of large assemblies can lead to powerful strides toward the mechanistic dissection of very complex molecular processes. But these structures are only the beginning. Transcription initiation is subjected to tight regulation that involves layers of molecular complexity where activators, cofactors, chromatin states and other regulatory signals interact with the general machinery to regulate gene expression. Building up on the present structures towards larger and ever more functionally relevant studies is now on the horizon.
Highlights.
Cryo-EM description of human TFIID conformational range and its link to DNA binding
Atomic modeling of TFIID DNA-binding subunits following masked refinement
Visualizing the stepwise assembly of human PIC led to robust subunit localization
High-resolution PIC structures in different states shed light on promoter opening
Acknowledgments
This work was funded by NIGMS (GM63072 to EN). EN is a Howard Hughes Medical Institute Investigator.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Nogales E. The development of cryo-EM into a mainstream structural biology technique. Nat Methods. 2016;13:24–27. doi: 10.1038/nmeth.3694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nogales E, Scheres SH. Cryo-EM: A Unique Tool for the Visualization of Macromolecular Complexity. Mol Cell. 2015;58:677–689. doi: 10.1016/j.molcel.2015.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Penczek PA, Frank J, Spahn CM. A method of focused classification, based on the bootstrap 3D variance analysis, and its application to EF-G-dependent translocation. J Struct Biol. 2006;154:184–194. doi: 10.1016/j.jsb.2005.12.013. [DOI] [PubMed] [Google Scholar]
- 4.Scheres SH, Gao H, Valle M, Herman GT, Eggermont PP, Frank J, Carazo JM. Disentangling conformational states of macromolecules in 3D-EM through likelihood optimization. Nat Methods. 2007;4:27–29. doi: 10.1038/nmeth992. [DOI] [PubMed] [Google Scholar]
- 5.Grob P, Cruse MJ, Inouye C, Peris M, Penczek PA, Tjian R, Nogales E. Cryo- electron microscopy studies of human TFIID: conformational breathing in the integration of gene regulatory cues. Structure. 2006;14:511–520. doi: 10.1016/j.str.2005.11.020. [DOI] [PubMed] [Google Scholar]
- 6.Penczek PA, Yang C, Frank J, Spahn CM. Estimation of variance in single- particle reconstruction using the bootstrap technique. J Struct Biol. 2006 doi: 10.1016/j.jsb.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 7.Andel F, Ladurner AG, Inouye C, Tjian R, Nogales E. Three-dimensional structure of the human TFIID-IIA-IIB complex. Science. 1999;286:2153–2156. doi: 10.1126/science.286.5447.2153. [DOI] [PubMed] [Google Scholar]
- 8.Liu WL, Coleman RA, Grob P, King DS, Florens L, Washburn MP, Geles KG, Yang JL, Ramey V, Nogales E, et al. Structural changes in TAF4b-TFIID correlate with promoter selectivity. Mol Cell. 2008;29:81–91. doi: 10.1016/j.molcel.2007.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu WL, Coleman RA, Ma E, Grob P, Yang JL, Zhang Y, Dailey G, Nogales E, Tjian R. Structures of three distinct activator-TFIID complexes. Genes Dev. 2009;23:1510–1521. doi: 10.1101/gad.1790709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brand M, Leurent C, Mallouh V, Tora L, Schultz P. Three-dimensional structures of the TAFII-containing complexes TFIID and TFTC. Science. 1999;286:2151–2153. doi: 10.1126/science.286.5447.2151. [DOI] [PubMed] [Google Scholar]
- 11.Papai G, Tripathi MK, Ruhlmann C, Layer JH, Weil PA, Schultz P. TFIIA and the transactivator Rap1 cooperate to commit TFIID for transcription initiation. Nature. 2010;465:956–960. doi: 10.1038/nature09080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cianfrocco MA, Kassavetis GA, Grob P, Fang J, Juven-Gershon T, Kadonaga JT, Nogales E. Human TFIID binds to core promoter DNA in a reorganized structural state. Cell. 2013;152:120–131. doi: 10.1016/j.cell.2012.12.005. 2D classification of cryo-EM images revealed that TFIID partitions between two major conformations, the canonical and rearranged states, defined by the translocation of TFIID's lobe A across the more rigid core of lobes B and C. The conformational equilibrium of TFIID was modulated by TFIIA and promoter DNA. A multi-model refinement strategy led to 3D reconstructions of TFIID in both the canonical and rearranged states, and showed that TFIID binds to promoter DNA in the rearranged state. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cianfrocco MA, Nogales E. Regulatory interplay between TFIID's conformational transitions and its modular interaction with core promoter DNA. Transcription. 2013;4 doi: 10.4161/trns.25291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.de la Rosa-Trevin JM, Oton J, Marabini R, Zaldivar A, Vargas J, Carazo JM, Sorzano CO. Xmipp 3.0: an improved software suite for image processing in electron microscopy. J Struct Biol. 2013;184:321–328. doi: 10.1016/j.jsb.2013.09.015. [DOI] [PubMed] [Google Scholar]
- 15.Scheres SH. RELION: implementation of a Bayesian approach to cryo-EM structure determination. J Struct Biol. 2012;180:519–530. doi: 10.1016/j.jsb.2012.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lyumkis D, Brilot AF, Theobald DL, Grigorieff N. Likelihood-based classification of cryo-EM images using FREALIGN. J Struct Biol. 2013;183:377–388. doi: 10.1016/j.jsb.2013.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Frank J, Ourmazd A. Continuous changes in structure mapped by manifold embedding of single-particle data in cryo-EM. Methods. 2016;100:61–67. doi: 10.1016/j.ymeth.2016.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Elmlund D, Elmlund H. SIMPLE: Software for ab initio reconstruction of heterogeneous single-particles. J Struct Biol. 2012;180:420–427. doi: 10.1016/j.jsb.2012.07.010. [DOI] [PubMed] [Google Scholar]
- 19.Leschziner AE, Nogales E. The orthogonal tilt reconstruction method: An approach to generating single-class volumes with no missing cone for ab initio reconstruction of asymmetric particles. J Struct Biol. 2006;153:284–299. doi: 10.1016/j.jsb.2005.10.012. [DOI] [PubMed] [Google Scholar]
- 20.Leschziner A. The orthogonal tilt reconstruction method. Methods Enzymol. 2010;482:237–262. doi: 10.1016/S0076-6879(10)82010-5. [DOI] [PubMed] [Google Scholar]
- 21.Juven-Gershon T, Cheng S, Kadonaga JT. Rational design of a super core promoter that enhances gene expression. Nat Methods. 2006;3:917–922. doi: 10.1038/nmeth937. [DOI] [PubMed] [Google Scholar]
- 22.Campbell MG, Cheng A, Brilot AF, Moeller A, Lyumkis D, Veesler D, Pan J, Harrison SC, Potter CS, Carragher B, et al. Movies of ice-embedded particles enhance resolution in electron cryo-microscopy. Structure. 2012;20:1823–1828. doi: 10.1016/j.str.2012.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li X, Mooney P, Zheng S, Booth CR, Braunfeld MB, Gubbens S, Agard DA, Cheng Y. Electron counting and beam-induced motion correction enable near-atomic-resolution single-particle cryo-EM. Nat Methods. 2013;10:584–590. doi: 10.1038/nmeth.2472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **24.Louder RK, He Y, Lopez-Blanco JR, Fang J, Chacon P, Nogales E. Structure of promoter-bound TFIID and model of human pre-initiation complex assembly. Nature. 2016;531:604–609. doi: 10.1038/nature17394. An improved cryo-EM reconstruction of the promoter-bound core of TFIID, including lobes B, C, and a part of lobe A (lobe A1), allowed modeling of the TBP-TFIIA-TATA subcomplex and the positioning of all the DNA elements within the supercore promoter used. The subnanometer-resolution reconstruction of TFIID's lobe C allowed for an atomic model to be generated for this region, and revealed that TAF1 and TAF2 mediate TFIID's interaction with downstream promoter DNA. A model of the TFIID-based PIC was generated by superposition of the TFIID-TFIIA-promoter structure with the cryo-EM reconstruction of the TBP-based PIC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang H, Curran EC, Hinds TR, Wang EH, Zheng N. Crystal structure of a TAF1-TAF7 complex in human transcription factor IID reveals a promoter binding module. Cell Res. 2014;24:1433–1444. doi: 10.1038/cr.2014.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Malkowska M, Kokoszynska K, Rychlewski L, Wyrwicz L. Structural bioinformatics of the general transcription factor TFIID. Biochimie. 2013;95:680–691. doi: 10.1016/j.biochi.2012.10.024. [DOI] [PubMed] [Google Scholar]
- 27.Kochan G, Krojer T, Harvey D, Fischer R, Chen L, Vollmar M, von Delft F, Kavanagh KL, Brown MA, Bowness P, et al. Crystal structures of the endoplasmic reticulum aminopeptidase-1 (ERAP1) reveal the molecular basis for N-terminal peptide trimming. Proc Natl Acad Sci U S A. 2011;108:7745–7750. doi: 10.1073/pnas.1101262108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Scheer E, Delbac F, Tora L, Moras D, Romier C. TFIID TAF6-TAF9 complex formation involves the HEAT repeat-containing C-terminal domain of TAF6 and is modulated by TAF5 protein. J Biol Chem. 2012;287:27580–27592. doi: 10.1074/jbc.M112.379206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bieniossek C, Papai G, Schaffitzel C, Garzoni F, Chaillet M, Scheer E, Papadopoulos P, Tora L, Schultz P, Berger I. The architecture of human general transcription factor TFIID core complex. Nature. 2013;493:699–702. doi: 10.1038/nature11791. [DOI] [PubMed] [Google Scholar]
- 30.Buratowski S, Hahn S, Guarente L, Sharp PA. Five intermediate complexes in transcription initiation by RNA polymerase II. Cell. 1989;56:549–561. doi: 10.1016/0092-8674(89)90578-3. [DOI] [PubMed] [Google Scholar]
- **31.He Y, Fang J, Taatjes DJ, Nogales E. Structural visualization of key steps in human transcription initiation. Nature. 2013;495:481–486. doi: 10.1038/nature11991. An in vitro reconstituted system was developed for the direct visualization of the stepwise assembly of human TBP-based PIC, ultimate resulting in a comprehensive map of protein-protein and protein-DNA interaction. The localization of the XPB helicase within TFIIH strongly supported a DNA translocation model for promoter opening. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **32.He Y, Yan C, Fang J, Inouye C, Tjian R, Ivanov I, Nogales E. Near-atomic visualization of human transcription promoter opening. Nature. 2016 doi: 10.1038/nature17970. epub ahead of print. Near-atomic resolution models of the human PIC determined by cryo-EM for three major states during transcription initiation. It includes structures for previously uncharacterized components of the PIC, such as TFIIE, TFIIH, and segments of TFIIA, TFIIB, and TFIIF. Comparison of the different structures reveals the sequential conformational changes that accompany the transition from each state to the next throughout the initiation process. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sainsbury S, Niesser J, Cramer P. Structure and function of the initially transcribing RNA polymerase II-TFIIB complex. Nature. 2013;493:437–440. doi: 10.1038/nature11715. [DOI] [PubMed] [Google Scholar]
- 34.Trabuco LG, Villa E, Mitra K, Frank J, Schulten K. Flexible fitting of atomic structures into electron microscopy maps using molecular dynamics. Structure. 2008;16:673–683. doi: 10.1016/j.str.2008.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Murakami K, Tsai KL, Kalisman N, Bushnell DA, Asturias FJ, Kornberg RD. Structure of an RNA polymerase II preinitiation complex. Proc Natl Acad Sci U S A. 2015;112:13543–13548. doi: 10.1073/pnas.1518255112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **37.Plaskchka C, Hantsche M, Dienemann C, Burzinski C, Plitzko J, Cramer P. Transcription initiation complex structures elucidate DNA opening. Nature. 2016 doi: 10.1038/nature17990. epub ahead of print. Near-atomic cryo-EM structures of the yeast “core” PIC, in both the closed and open states, reveals a TFIIH-independent mechanism of transcription bubble opening directly involving TFIIE. Coordinated changes of the protein-DNA interaction network suggests the trapping of an open DNA configuration as a unified model for transcription initiation. [DOI] [PubMed] [Google Scholar]