

Abstract
A variety of human neurologic diseases are caused by inherited defects in DNA repair. In many cases, these syndromes almost exclusively impact the nervous system, underscoring the critical requirement for genome stability in this tissue. A striking example of this is defective enzymatic activity of polynucleotide kinase-phosphatase (PNKP), leading to microcephaly or neurodegeneration. Notably, the broad neural impact of mutations in PNKP can result in markedly different disease entities, even when the inherited mutation is the same. For example microcephaly with seizures (MCSZ) results from various hypomorphic PNKP mutations, as does ataxia with oculomotor apraxia 4 (AOA4). Thus, other contributing factors influence the neural phenotype when PNKP is disabled. Here we consider the role for PNKP in maintaining brain function and how perturbation in its activity can account for the varied pathology of neurodegeneration or microcephaly present in MCSZ and AOA4 respectively.
Keywords: Neurodegeneration, DNA repair, Polynucleotide kinase phosphatase, Single strand break repair, Microcephaly, DNA damage signaling, Neurologic disease, Base excision repair
1. Introduction
Genome stability is essential for normal development and function of an organism. Unrepaired DNA lesions can lead to various pathologic states, including developmental defects, degenerative disease, cancer and aging (Jackson and Bartek, 2009; Madabhushi et al., 2014; McKinnon, 2009; Roy et al., 2012; Vijg and Suh, 2013). DNA lesions are a constant cellular threat and can occur spontaneously via DNA replication or transcription, or can be generated endogenously by reactive oxygen species (ROS) produced during oxidative metabolism (Dizdaroglu and Jaruga, 2012; van Loon et al., 2010). Exogenous agents, such as radiation and chemotherapy can also produce DNA damage. To correct the many types of potential DNA lesions, distinct repair pathways are available that function independently or in concert. The operative pathway depends not only on the particular type of DNA lesion, but also on the cell type replicative status of the cell. Detailed recent reviews of the five major DNA repair pathways and the types of damage that utilizes these pathways are available: Mismatch repair (Kunkel and Erie, 2015; Pena-Diaz and Jiricny, 2012), single strand break repair (SSBR)/base excision repair (BER) (Caldecott, 2008; Krokan and Bjoras, 2013), nucleotide excision repair (Marteijn et al., 2014), crosslink repair (Deans and West, 2011; Kim and D’Andrea, 2012) and double strand break repair (DSBR), which occurs via two independent pathways, homologous recombination (HR) (Heyer et al., 2010; Jasin and Rothstein, 2013) that functions in replicating cells, or non-homologous end-joining (NHEJ), which operates in either dividing or non-dividing cells (Chapman et al., 2012; Lieber, 2010; Ochi et al., 2014; Symington and Gautier, 2011; Williams et al., 2014).
The most frequent DNA damage in cells arises from ROS and mostly results in DNA single strand breaks (SSBs), a lesion that is repaired by the XRCC1-based SSBR/BER pathway (Almeida and Sobol, 2007; Caldecott, 2008; De Bont and van Larebeke, 2004). Inherited defects in SSBR almost exclusively affect the nervous system, leading to neurodegeneration (Caldecott, 2008; McKinnon, 2013; Reynolds and Stewart, 2013). This may relate to the high oxygen consumption of this tissue and the substantial energy requirement and metabolism of neural cells (Magistretti and Pellerin, 1996; Raichle and Gusnard, 2002). Neurons have high transcription rates and ROS and DNA damage accumulation can result in R-loops to block transcription and promote genome instability, which can lead to direct neuronal dysfunction (Aguilera and Garcia-Muse, 2012; Mischo et al., 2011; Sollier and Cimprich, 2015; Yuce and West, 2013). The long life of neural cells and their high transcriptional activity means it is imperative to maintain their genome free from detrimental DNA lesions throughout their lives. Despite the frequency and potential consequences of SSBs, other types of DNA damage also impact the nervous system. For example, unrepaired DNA double strand breaks (DSBs), particularly during neurogenesis can result in neurodevelopmental defects including microcephaly (a condition in which the head circumference is reduced three or more standard deviations below the norm). DSBs can be repaired by either HR, which requires a sister chromatid as a template for repair, and is therefore active in S/G2 phase in replicating cells, or by NHEJ, which promotes direct DNA end ligation and is active throughout the cell cycle (Chapman et al., 2012). While both pathways are important during neurogenesis, NHEJ is critical for DSBR in mature (non-cycling) neurons (Shull et al., 2009). However, the broad spectrum of neurologic disease that can occur from DNA repair deficiency in each of the DNA repair pathways indicates that a variety of DNA lesions can impact the nervous system (Table 1).
Table 1.
Inherited syndromes characterized by DNA damage signaling defects.
| DNA repair defect1 | Gene2 | Hereditary syndrome | Neurological phenotype3 | Extra-neurological features4 | References |
|---|---|---|---|---|---|
|
| |||||
| Replication stress/HR/Fanconi anemia | ATR | Growth retardation | |||
| PCNT | Seckel syndrome | Microcephaly | Immunodeficiency | O’Driscoll et al, 2003 | |
| ATRIP | Skeletal defects | Griffith et al, 2008 | |||
| Blood malignancies | Ogi et al, 2012 | ||||
|
| |||||
| BRCA2 | Growth retardation | Alter et al, 2007 | |||
| RAD51 | Fanconi anemia | Microcephaly | Skeletal defects | Ameziane et al, 2015 | |
|
| |||||
| DSB signaling | Growth retardation | Varon et al, 1998 | |||
| NBS1 | Nijmegen syndrome | Microcephaly | Immunodeficiency | Matsumoto et al, 2011 | |
| MRE11 | Nijmegen-like syndrome | Cancer | |||
|
| |||||
| ATM | Ataxia telangiectasia (A-T), A-T-like syndrome | Neurodegeneration | Immunodeficiency | McKinnon, 2012 | |
| MRE11 | Oculomotor apraxia | Cancer, Elevated AFP5 | |||
|
| |||||
| NHEJ (DSBR) | Growth retardation | ||||
| LIG4 | LIG4 syndrome | Microcephaly | Immunodeficiency | O’Driscoll et al, 2001 | |
| Cancer | |||||
|
| |||||
| XLF | Human immunodeficiency with microcephaly | Growth retardation | |||
| Microcephaly | Immunodeficiency | Buck et al, 2006 | |||
| Skin abnormalities | |||||
|
| |||||
| PNKP6 | MCSZ | Microcephaly | Shen et al, 2010 | ||
| Seizures | |||||
|
| |||||
| Microcephaly with neurodegeneration and polyneuropathy7 | Microcephaly | Poulton at al, 2013 | |||
| Seizures | |||||
| Neurodegeneration | |||||
|
| |||||
| SSBR | Neurodegeneration | Low albumin, Elevated cholesterol | Bras et al, 2015 | ||
| PNKP6 | AOA4 | Oculomotor apraxia | |||
| Dystonia | Elevated AFP5 | ||||
|
| |||||
| TDP1 | SCAN1 | Neurodegeneration | Low albumin, Elevated cholesterol | Takashima et al, 2002 | |
|
| |||||
| APTX | AOA1 | Neurodegeneration | Low albumin, Elevated cholesterol | Barbot et al, 2001 | |
| Oculomotor apraxia | Moreira et al, 2001 | ||||
| Dystonia | |||||
This is a representative list and is not exhaustive. For instance, nucleotide excision repair defects are not listed.
In many cases, the mutations in DSBR factors are hypomorphic, as a complete loss of function is not compatible with embryonic viability.
Only the most prominent neurologic feature is listed.
Only a sample of extra-neurologic features is listed. Many of the DSBR syndromes have multiple additional systemic phenotypes.
AFP is alpha-fetoprotein.
PNKP participates in both DSBR and SSBR, although the exact pathways affected that results in the clinical features is not certain.
This syndrome represents only two siblings from a single family.
Recently, multiple distinct human syndromes have been linked to inherited mutations in the DNA repair factor polynucleotide kinase phosphatase (PNKP) (Table 2). These include, microcephaly with seizures (MCSZ), which is characterized by microcephaly, early-onset, intractable seizures and developmental delay (Shen et al., 2010), progressive cerebellar atrophy and polyneuropathy (Poulton et al., 2013), and ataxia with oculomotor apraxia type 4 (AOA4), characterized by neurodegeneration (Bras et al., 2015). PNKP is a multifunctional DNA repair enzyme important for both the SSBR/BER and NHEJ pathways (Weinfeld et al., 2011). The dual functionality of PNKP in SSBR and DSBR potentially explains the presence of both microcephaly and neurodegeneration in patients with certain inherited PNKP mutations. However, substantial variation exists amongst affected individuals with PNKP mutations, as individuals diagnosed with MCSZ show no neurodegeneration, while individuals with AOA4 show pronounced neurodegeneration without microcephaly (Bras et al., 2015; Shen et al., 2010). More puzzling in understanding these different clinical entities is that individuals with either syndrome can harbor identical PNKP mutations (Table 2). Although some cases involve consanguinity, this does not appear to clearly explain the phenotypic range, although this is suggestive of genetic modifiers impacting PNKP function. However, the underlying reason for phenotypic variability amongst syndromes with PNKP mutations remains uncertain. In this review we will consider the role for PNKP in DNA repair in the brain, and how PNKP mutations impact brain function. Understanding how PNKP prevents human neuropathology will delineate the close relationship between DNA repair and neural homeostasis. This knowledge might be used in the clinic to alleviate neuropathology present in various DNA repair deficiency syndromes.
Table 2.
PNKP mutations present in human syndromes.
| Family | PNKP Mutation | Clinical phenotype | Additional neurologic Findings | |
|---|---|---|---|---|
|
| ||||
| MCSZ | 7x families (mixed-consanguineous) | |||
| Shen et. al., 2012 | 3x Palestinian | E326K | ||
| 1x Turkish | T424GfsX48 | Microcephaly Seizures (range of severity) | White matter defects | |
| 1x Saudi Arabia | T424GfsX48 | |||
| 2x European1 | T424GfsX4/ L176F or 17bp (splice) 1 | |||
|
| ||||
| Poulton et. al., 2013 | Microcephaly | White matter defects | ||
| 1x Dutch family (consanguineous) | T424GfsX48 | Seizures | ||
| Ataxia | ||||
| Neurodegeneration | ||||
|
| ||||
| Nakashima et. al., 2014 | 1x Japanese family (non-consanguineous)2 | A55S & G292R | Microcephaly | White matter defects (hearing loss)2 |
| Seizures | ||||
|
| ||||
| Carvill et al., 2013 | Single individual | P20S | Seizures | Encephalopathy with seizures |
|
| ||||
| AOA4 | G375W | |||
| T408del | Dystonia (common) | |||
| T424GfsX48 (1x) | Ataxia | |||
| Bras et. al., 2015 | 8x Portuguese families3 (mixed-consanguineous) | R439GfsX51 | Oculomotor apraxia | |
| G442AfsX27 | Neurodegeneration | |||
| Q517LfsX24 | ||||
This family in the MCSZ cohort contains compound heterozygous PNKP mutations.
The affected individual contained an additional mutation in PCDH15 that accounted for the hearing deficit.
Four of the eight families were compound heterozygotes for the PNKP mutations, and mutations were all located in the kinase domain of PNKP.
2. PNKP function during DNA repair
PNKP is a DNA processing enzyme in which the C-terminal catalytic domain contains a fused bimodal phosphatase and kinase domain, with a forkhead-associated (FHA) domain at its N-terminus (Weinfeld et al., 2011). Accordingly, PNKP has dual biochemical functionality during DNA repair to provide a 3’-phosphatase and a 5’-kinase activity for modifying the ends of a DNA break (Jilani et al., 1999). Multiple types of damage generate 3’-P and 5’-OH termini at a DNA break that require PNKP to produce ends that are compatible for ligation (i.e. those containing 3’-OH and 5’-P). This enzymatic activity of PNKP is utilized for both SSBR and DSBR (Chappell et al., 2002; Karimi-Busheri et al., 2007; Koch et al., 2004; Shimada et al., 2015; Whitehouse et al., 2001; Zolner et al., 2011). The FHA domain of PNKP is important for interaction with either the XRCC1 or XRCC4 scaffold proteins, which are required for assembling SSBR or DSBR (NHEJ) components respectively (Ali et al., 2009; Bernstein et al., 2005; Koch et al., 2004; Li et al., 2013; Loizou et al., 2004; Lu et al., 2010). SSBs are the most common type of endogenous DNA damage, and PNKP is required for processing of the bulk of these SSBs as most contain 3’-P termini. DNA damage resulting form abortive topoisomerase-1 activity and intermediates formed during the repair of oxidative damage also require PNKP to process 3’-P and 5’-OH termini (Plo et al., 2003; Weinfeld et al., 2011; Wiederhold et al., 2004).
More generally, SSBR involves multiple components that are assembled by XRCC1, a key scaffold factor in this pathway (Almeida and Sobol, 2007; Caldecott, 2008). Amongst these, poly(ADP-ribose) polymerase (PARP) is an enzyme that activates signaling by multiple ADP-ribosylation events (Caldecott, 2008; D’Amours et al., 1999; Kim et al., 2005). SSBs are detected by poly(ADP-ribose) polymerase (PARP), which recruits XRCC1 and other DNA-processing enzymes necessary for SSBR, such as PNKP, Aprataxin (APTX), tyrosyl DNA phosphodiesterase 1 (TDP1) or DNA polymerase beta, amongst others that process the DNA break. APTX and TDP1 participate in the modification of specific DNA lesions, such as adenylation intermediates or trapped topoisomerase-1 complexes (Ahel et al., 2006; Pommier et al., 2006). Other central components include apurinic/apyrimidinic endonuclease 1 (APE1) to initiate repair of oxidative DNA lesions, and PNKP for processing DNA ends prior to DNA ligation (Ahel et al., 2006; Iyama and Wilson, 2013; Weinfeld et al., 2011). Human inherited disorders that disrupt SSBR lead to pathology that is mostly restricted to the nervous system (Fig.1). Examples include; ataxia with oculomotor apraxia-1 (AOA1) caused by mutations in APTX, spinocerebellar ataxia with axonal neuropathy-1 (SCAN1) caused by mutated TDP1 and recently, progressive cerebellar atrophy and AOA4, caused by specific mutations in PNKP (see later for details) (Bras et al., 2015; Date et al., 2001; Poulton et al., 2013; Shen et al., 2010; Takashima et al., 2002). These syndromes underscore the importance of SSBR for normal neural function.
Figure 1. Disease-causing mutations in PNKP.
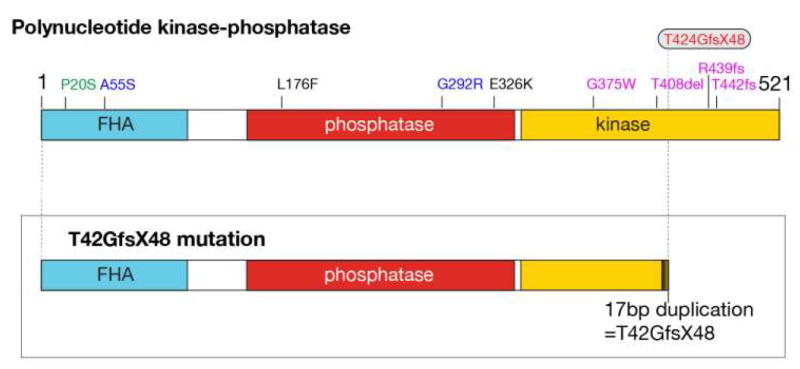
The upper diagram shows the PNKP protein with the forkhead-associated domain (FHA), and the phosphatase and kinase domains indicated. The numbering indicates specific amino acid residues, and also the germline mutation changes found in PNKP in human syndromes. MCSZ mutations are listed in black, AOA4 mutations are purple and blue are mutations found in a compound heterozygote that showed an MCSZ phenotype. The P20S mutation in green is from an individual who manifest only seizures, but not neurodegeneration or microcephaly. The T424GfsX48 common mutation is present in homozygous individuals with MCSZ and also in siblings with both MCSZ, and in a compound heterozygous form in AOA4. The lower illustration indicates the effect of the T424GfsX48 change in the PNKP protein, showing premature termination in the kinase domain.
PNKP is also required for DSBR and loss of PNKP directly impacts this pathway via disruption of NHEJ (Chappell et al., 2002; Karimi-Busheri et al., 2007; Koch et al., 2004; Shimada et al., 2015). Similar to SSBR, there are core NHEJ factors required for recognition, processing and repair of DSBs. The Ku70/80 heterodimers bind to DNA ends and recruits the DNA-PKcs kinases, XLF and other processing factors such as Artemis, with ligation to repair the break completed by the Xrcc4/DNA ligase IV complex (Lieber, 2010).
3. PNKP-associated neurological syndromes
Defective PNKP compromises repair of endogenous DNA lesions leading to increased genomic damage. This is a particular issue in the nervous system as a variety of neuropathology can arise from PNKP dysfunction (Table 2). Unexpectedly, similar mutations of this repair enzyme can result in dissimilar pathology, although the basis for varied clinical presentation remains unclear. In the following, we will examine and discuss the diverse disease-related phenotypes arising from PNKP dysfunction.
3.1. Microcephaly with seizures (MCSZ)
Microcephaly is a common feature of several human syndromes caused by mutations in DNA DSB repair factors (McKinnon, 2013; O’Driscoll and Jeggo, 2006). Note that while microcephaly can be caused by defects in DSB signaling, the etiology of this disease is varied and can involve defective neural progenitor cell cycle regulation or as a result of more diverse agents such as viral infection (Kaindl, 2014; Mahmood et al., 2011; Manicklal et al., 2013; Morris-Rosendahl and Kaindl, 2015; Noyola et al., 2001; Tang et al., 2016; Woods et al., 2005). For example, hereditary mutations in the NHEJ factors LIG4 and XLF or hypomorphic mutations in Nijmegen breakage syndrome 1 (NBS1) and certain mutations of MRE11, both components of the MRN complex that is essential for HR and DSBR, also results in microcephaly (Frappart and McKinnon, 2006; Matsumoto et al., 2011; O’Driscoll and Jeggo, 2006; Stracker and Petrini, 2011). Additionally, hypomorphic mutation of ATR (ATM and Rad3 related), a DNA-damage response kinase that plays a central role in replication-stress signaling leads to Seckel syndrome, a neurodevelopmental disorder characterized by microcephaly (McKinnon, 2012; O’Driscoll and Jeggo, 2006; O’Driscoll et al., 2003; Zeman and Cimprich, 2014).
MCSZ is an autosomal recessive disorder due to defective PNKP and is characterized by microcephaly associated with seizures, hyperactivity and developmental delay (Shen et al., 2010). An OMIM link to MCSZ is available: http://www.omim.org/entry/613402. While PNKP participates in both DSBR and SSBR, the microcephaly in MCSZ, by analogy with other syndromes, implicates defective DSBR. The absence of neurodegenerative changes also suggests defective SSBR does not account for neuropathology. MCSZ was described in seven families that have either homozygous (five families) or compound heterozygous PNKP mutations (two families) (Shen et al., 2010). Three Palestinian families were consanguineous and shared the same homozygous PNKP mutation (exon 11; 975G>A), which result in a non-conservative amino acid change (E326K) in the phosphatase domain. A homozygous mutation caused by a 17bp duplication in exon 14, which results in frame-shift T424GfsX48 in the kinase domain was found in members of two separate non-consanguineous families from Saudi Arabia and Turkey respectively, also with a shared common haplotype, potentially indicating a founder mutation. The remaining two European families were compound heterozygotes containing the same T424GfsX48 mutation together with other PNKP mutations; a point mutation resulting in L176F in the phosphatase domain, or a 17bp deletion in intron 15 that disrupts mRNA splicing. Analysis of PNKP protein levels showed all mutations resulted in reduced protein, indicating these MCSZ mutations are hypomorphic. Thus, a range of mutations in PNKP located in both the phosphates and kinase domains lead to MCSZ (Figure 1).
Ensuing biochemical studies using both recombinant PNKP engineered with MCSZ mutations, and also lymphoid cells derived from the MCSZ individuals revealed that the mutant PNKP protein is unstable at physiologic temperature (Reynolds et al., 2012). Recombinant PNKP protein from the T424GfsX48 and exon15Δfs4X kinase region mutations lacked kinase activity towards an oligomer substrate, as did the L176F mutation but not the E326K mutation despite these being in the phosphatase domain (Reynolds et al., 2012). Conversely, only the E326K mutations had a significant impact on PNKP phosphatase activity. Despite the data using recombinant protein, analysis of all MCSZ cellular extracts resulted in compromised phosphatase activity towards both single and double-stranded DNA oligomers. Importantly, in all tested MCSZ-derived lymphoid lines PNKP protein expression was very low, which compromises available enzymatic activity, and all lines showed a DNA repair defect (Reynolds et al., 2012). The efficient repair of DNA lesions resulting from oxidative damage is dependent upon the 3’-phosphatase function of PNKP, via its interaction with XRCC1 (Breslin and Caldecott, 2009). Thus, rather than specific individual kinase or phosphatase defects, the contributing effect of PNKP mutations to MCSZ is the generation of an unstable protein resulting in marked reduction of the needed enzymatic activity.
3.2 Microcephaly associated with neurodegeneration and polyneuropathy
In a subsequent report (Poulton et al., 2013), the same homozygous PNKP mutation in MCSZ, T424GfsX48, was identified in two affected Dutch siblings with microcephaly, but as distinct to MCSZ, these individuals also developed ataxia and progressive cerebellar degeneration (Figure 1 and Table 2). Why identical PNKP mutations give rise to two different phenotypes isn’t clear, although the most likely explanation points to genetic modifiers. These two affected individuals are siblings from an isolated community in the Netherlands and were born to consanguineous parents. Therefore, its likely there are one or more other genetic or epigenetic modifiers (beside the homozygous T424GfsX48 PNKP mutation) that the two brothers share. This may lead to a shift of the phenotype from classical MCSZ to microcephaly associated with neurodegeneration and polyneuropathy. Thus, PNKP function may be susceptible to genetic modifiers, either as a mechanism to modulate PNKP function, or as an inadvertent consequence of additional mutations arising from consanguinity.s
An additional individual with PNKP mutations presenting with microcephaly and seizures born to non-consanguineous parents was identified in Japan (Nakashima et al., 2014). The particular PNKP mutations present were an A55S (163g>T), and a G292R (874G>A), which are distinct to those found in MCSZ, thus expanding the range of PNKP mutations leading to this syndrome. However, this case was more complex as the individual additionally harbored a PCDH15 mutation known to result in Usher syndrome, which is characterized by hearing loss, a clinical feature also found in this patient.
3.2. AOA4 (Ataxia with Oculomotor Apraxia type 4)
The phenotypic spectrum resulting from PNKP dysfunction was expanded further by the recent identification of an additional clinical syndrome, ataxia with oculomotor apraxia 4 (AOA4), which is characterized by neurodegeneration but not microcephaly. An OMIM link to AOA4 is available: http://www.omim.org/entry/616267. The T424GfsX48 (and other) PNKP mutations were again identified in this cohort of patients (Figure 1 and Table 2). Thus, in quite striking contrast to MCSZ, this syndrome is characterized by neurodegeneration, and microcephaly does not occur.
Ataxia with Oculomotor Apraxia (AOA) comprises a group of at least four separate diseases (for an overview of this group of syndromes see: http://ghr.nlm.nih.gov/condition/ataxia-with-oculomotor-apraxia). These syndromes impact cerebellar function and result in a profound loss of motor control and are characterized by cerebellar degeneration, oculomotor apraxia (abnormal saccadic eye movement) and often dystonia and peripheral neuropathy (Anheim et al., 2012; Barbot et al., 2001). AOA4 is an autosomal recessive ataxia recently identified in 11 Portuguese patients (from 8 different families), caused by PNKP mutation and represents the most common AOA in Portugal (Bras et al., 2015). Amongst the other AOA subgroups, AOA1 is also associated with DNA repair defects, as mutations in the DNA-end processing enzyme APTX underpin this disease (Moreira et al., 2001). Both diseases have early onset, with AOA4 being the earliest (mean age onset 4.3 vs. 6.9 years for AOA1). Extra-neurologic features are also similar as half of the AOA4 cases have low albumin and high cholesterol, features found consistently in older AOA1 patients (Barbot et al., 2001; Bras et al., 2015; Moreira et al., 2001; Yokoseki et al., 2011). A clinically related syndrome AOA2, is caused by mutations in Senataxin (Anheim et al., 2009; Hammer et al., 2012; Le Ber et al., 2004; Moreira et al., 2004), a helicase, which is associated with DNA damage responses, transcriptional control and possibly a SSBR defect (Bennett and La Spada, 2015; Hamperl and Cimprich, 2014; Richard et al., 2013; Suraweera et al., 2007). AOA2 has an older age onset (mean 14.6 years) and affected individuals rarely manifest dystonia and have normal albumin and cholesterol values, but consistently have increased alpha-fetoprotein (Anheim et al., 2012; Schieving et al., 2014). In contrast, AOA3 is reported to result from mutations in the phosphoinositide-3-kinase regulatory subunit 5 (PIK3R5) gene, and cell lines from these patients have been linked to defective DNA damage signaling (Al Tassan et al., 2012; Gueven et al., 2007; Kobayashi et al., 2015). Like AOA2, AOA3 also occurs in the second decade of life and is characterized by normal serum cholesterol and albumin, but elevated alpha-fetoprotein (Al Tassan et al., 2012; Anheim et al., 2012).
In addition to the syndromes above, other neurodegenerative diseases also occur after defective single strand break repair, notably defective TDP1 results in spinocerebellar ataxia with axonal neuropathy (SCAN1). TDP1 is required for the repair of DNA strand breaks arising from abortive topoisomerase-1 activity, and also other damage such as oxidative stress that produces 3’ termini containing phosphate or phosphoglycolate ends (El-Khamisy et al., 2005; Interthal et al., 2005; Katyal et al., 2007; Yang et al., 1996). Similar to AOA1 and AOA4, SCAN1 patients have ataxia with cerebellar atrophy, low or normal albumin levels and high cholesterol; however, the onset of the disease is later (around 14 years old) and oculomotor apraxia and cognitive defects are not reported (Takashima et al., 2002). The presence of a common SSBR defect signature is a unifying theme in the etiology of these neurodegenerative syndromes (McKinnon, 2013).
MCSZ and AOA4 are characterized by profound affects towards the nervous system with a notable lack of defects in other tissue/organs. In general terms, loss of DSBR integrity results in microcephaly, while neurodegeneration is the outcome when SSBR is compromised (Table 1). Given that PNKP functions during both DSBR and SSBR, one would expect that compromised PNKP function lead to both microcephaly and neurodegeneration. While this is the situation in one cohort of patients with PNKP mutation (Poulton et al., 2013), MCSZ and AOA4 reflect different neuropathology, with MCSZ being similar to the phenotype arising from DSBR deficiency and AOA4 from SSBR defects. Although the occurrence of different phenotypes after PNKP mutations is surprising, each disease phenotype is nonetheless consistent with a role of PNKP during NHEJ and SSBR/BER. For instance, during development there may be an increased abundance of specific types of DSBs (e.g. with specific DNA termini) that require PNKP during NHEJ, which could result from replication-related damage, oxidative load or transcriptional stress. Accumulated damage would lead to cell death in neural progenitors resulting in microcephaly. Similarly, since PNKP mutations also compromise SSBR, a higher level of unrepaired SSBs associated with oxidative and transcriptional damage would lead to cell loss in the brain appearing as neurodegeneration. Because of the high mitochondrial activity in the nervous system, excess SSBs will be generated during oxidative metabolism relative to other organs, emphasizing the brain-specific nature of PNKP loss?
4. Modeling PNKP mutations in the mouse
The mouse can be a useful adjunct for analysis of the importance of DNA damage response pathways in the nervous system. The consequences of PNKP loss in the developing murine nervous system was recently reported, revealing that PNKP is an essential enzyme for mouse embryonic development and necessary for normal development of the nervous system (Shimada et al., 2015). In a scenario related to the human syndromes MCSZ and AOA4, a series of mouse Pnkp mutant alleles were examined including the T424Gfs48X mutation and attenuated PNKP levels similar to the situation in MCSZ. Inactivation of PNKP in specific neural compartments was also examined.
In the setting of mouse development, complete loss of PNKP in the nervous system substantially impacts neural development and resulted in early postnatal lethality, underscoring the fact that PNKP mutations are hypomorphic in the human diseases. To more effectively mirror the human situation, an attenuated Pnkp allele was developed in which expression was decreased to around 10% of the normal situation. After PNKP loss, DNA damage accumulation during neural development resulted in apoptosis, predominantly in progenitors, accounting for the emergence of a microcephalic phenotype. However, there were also degenerative effects with widespread cell loss. Thus, this model encapsulated both the neurodevelopmental microcephaly in MCSZ and the cerebellar atrophy in AOA4. This mouse model also clearly demonstrated that the primary consequence of PNKP loss is a DNA repair deficiency that impacts the nervous system in a manner consistent with generation of the phenotypes in MCSZ and AOA4. Further, mice in which XRCC1 is deleted throughout the nervous system undergo seizures reflective of MCSZ, suggesting that seizures can arise from compromised SSBR (Lee et al., 2009). One caveat to modeling PNKP deficiency in the mouse is that an attempt to generate the germ-line T424Gfs48X mutation to model the human syndrome led to early embryonic lethality. This suggests that the PNKP requirements in the mouse are more stringent than that of humans. Perhaps this reflects the genetic background requirements that may contribute to the phenotypic differences between MCSZ and AOA4?
Additionally, the relative impact of PNKP loss was also assessed by restricting deletion of PNKP after birth, and in a cell- or tissue-specific manner utilizing inducible deletion of PNKP in young postnatal mice or in the glial compartment using an inducible GFAP promoter to drive cre recombinase. These models showed a particular sensitivity of the myelin-producing oligodendrocytes to PNKP loss and DNA damage accumulation (Shimada et al., 2015), consistent with myelin/white matter defects present in MCSZ and AOA4 (Bras et al., 2015; Poulton et al., 2013; Shen et al., 2010). Moreover, a requirement for PNKP to prevent widespread DNA damage accumulation in the mature brain is also apparent (Dumitrache et al, unpublished observation). Thus, the mouse provides important insight into the specific cellular impact of PNKP dysfunction and illustrates the developmental and maintenance function of this DNA repair enzyme in the nervous system.
5. The correlation between PNKP mutation and phenotype
Could the different mutations present in MCSZ and AOA4 underpin the varied clinical presentation in the respective diseases? The issue with this notion is the presence of overlapping/common mutations shared between syndromes (Figure 1 and Table 2). The T424Gfs48X mutation is present as a compound heterozygote in AOA4, rather than a homozygous mutation found in MCSZ. In AOA4 the partner mutation is a G375W, which is not found in MCSZ, although this mutation affects the kinase region, as do other causative mutations in MCSZ (Table 2). Perhaps more detailed biochemical analysis in cells from MCSZ and AOA4 individuals will address how specific mutations affect PNKP function? However, the simplest explanation is that the PNKP mutations present in AOA4 and MCSZ have similar effects on PNKP function, which is generation of an unstable protein thereby reducing needed enzyme activity.
6. Overview, conclusion and further considerations
The clinical data for individuals with MCSZ and AOA4 are clear about the absence of coincident neuropathology. Careful evaluation of AOA4 individuals revealed no microcephaly or seizure-related events, while MCSZ patients showed no signs of degenerative pathology (Bras et al., 2015; Shen et al., 2010). An exception to the exclusive neuropathology is the two affected siblings, which both contain the T424Gfs48X homozygous mutation, presenting with both microcephaly and neurodegeneration (Poulton et al., 2013). In essence, these syndromes reflect two distinct features of perturbed neural homeostasis. Microcephaly is a congenital birth defect that is the result of abnormal neural development, and as such reflects malformations that occurred prior to birth (Kaindl et al., 2010; Morris-Rosendahl and Kaindl, 2015; Woods et al., 2005). In contrast, the cerebellar atrophy resulting from neurodegeneration, and the ongoing postnatal neural cell death that eventually results in an individual being wheelchair bound, are events that manifest some months or years after birth. These neurodegenerative events may reflect the increased oxygen consumption that coincides with the commencement of respiratory metabolism and increased oxidative stress and free radical production after birth. With regard to how similar mutations in PNKP result in these two somewhat opposite scenarios is not clear from considering only the mutation(s) that disrupts normal enzyme function. Given that the disease-causing mutations in PNKP result in protein instability that reduces available enzymatic activity, it is likely this feature rather than the specific mutation underpins both microcephaly and neurodegeneration. Moreover, inspection of the mutational combinations in MCSZ and AOA4 reveal a similar general occurrence in the kinase domain (although mutations in MCSZ are also found in the phosphatase domain; Figure 1), and in regard to the common T424Gfs48X mutation, which occurs in compound heterozygous form in AOA4, the partner mutation is also in the kinase region mutation, similar to MCSZ. In other individuals, a homozygous T424Gfs48X mutation was responsible for the Dutch siblings that had both microcephaly and neurodegeneration, while the Japanese patient presenting with MCSZ had PNKP mutations in the FHA and phosphatase domain (A55S and G292R). An additional amino terminal homozygous PNKP mutation (P20S) has also been linked to one individual with encephalopathy who has seizures but not microcephaly (Carvill et al., 2013). Although unlikely, it is formally possibly that the different mutational combinations might generate distinct phenotypes. However, the presence of the identical T424Gfs48X mutation in both classes of neuropathology does support all indices of pathology linked to diminished PNKP enzymatic activity.
It is not unusual for different mutations in the same gene to lead to clinically distinct neurologic diseases. MRE11 mutations can result in the neurodegenerative syndrome ataxia telangiectasia-like disease (ATLD), while microcephaly was present in two unrelated Japanese individuals that had MRE11 mutation different to those found in ATLD (Matsumoto et al., 2011). Furthermore, a recent report describes a Korean AOA1 individual with compound heterozygous APTX mutations, who lacked the typical oculomotor apraxia (Lee et al., 2016). Mutations in the Senataxin helicase (responsible for AOA2) can result in a spectrum of other neurologic disease ranging from juvenile onset amyotrophic lateral sclerosis (ALS) to motor neuron disease and ataxia (Bennett and La Spada, 2015; Hirano et al., 2011; Rudnik-Schoneborn et al., 2012). A putative heterozygous dominant SETX mutation leading to motor neuropathy was also identified in an unaffected parent and grandparent, implying other genetic factors may contribute to the phenotype (Hirano et al., 2011).
For attenuated PNKP function to result in such different phenotypes suggests additional factors influence the relative need for enzyme activity. All AOA4 individuals are from Portugal, while those with MCSZ are from the Middle East, Turkey, Europe and the UK, indicating a broad geographical distribution for PNKP mutations. The two Dutch siblings presenting with both neurodegeneration and microcephaly had consanguineous parents, suggesting that other genes can influence the requirements for PNKP. However, AOA4 and MCSZ occurred in both consanguineous and non-consanguineous families, indicating that this feature alone doesn’t explain the variable mutant PNKP phenotype. If genetic background is an important factor, what might the genetic modifier be, and what evidence is there for this possibility? Although speculative, there are examples of multiple gene loci that might impact aspects of the MCSZ/AOA4 phenotypes. In particular, seizure related activity has been linked to defects associated with multiple genes including sodium channels (e.g. SCN1A), chromatin modifiers (e.g. CHD2) and synaptic proteins (e.g. SYNGAP1) (Carvill et al., 2013). It is possible that combinatorial mutations in other genes (DNA repair or not) can alter the specific requirements for PNKP function. For instance, polymorphisms or specific haplotypes present in certain population groups may lead to an altered impact of a PNKP mutation. Thus, the potential functional consequences of polymorphisms or missense or silent mutations across the genome may have a larger effect on protein function than predicted. In this regard, further genetic analysis (e.g. whole genome sequencing as more cases are identified) of affected individuals with PNKP mutation may provide revealing information to understand the diversity of disease phenotype.
Many DNA repair syndromes are known to be associated with transcriptional defects, and perturbations in transcription could contribute to clinical variation as happens in subgroups of xeroderma pigmentosum and related syndromes (Compe et al., 2007; Laine and Egly, 2006). PNKP has also been connected to the pathology of SCA3, due to inhibition of PNKP phosphatase activity by the repeat expansion present in SCA3 (Chatterjee et al., 2015). SCA3 is caused by CAG trinucleotide expansion and is a part of a group of SCAs characterized by abnormal CAG repeat size, is caused by Ataxin 3 (ATXN3) mutation. If this interaction is physiologically important, then PNKP function might be modified by alteration in chromatin structure or specific grouping of repetitive DNA elements.
The diverse impact of PNKP mutations towards human disease provides important insights for understanding how genome stability pathways control tissue homeostasis. The example of variability in clinical presentation of PNKP mutations is likely relevant to other genome instability syndromes. If genetic modifiers can influence the impact of repair defects then experimental manipulation of ancillary factors (e.g. perhaps those that modulate DNA repair activity such as ubiquitination or methylation) via small molecule inhibitors might be a potential approach to ameliorate the impact of DNA repair mutations. Enhanced genome data analysis will be useful to fully comprehend the impact of DNA repair-associated mutations in human disease.
Acknowledgments
PJM is supported by the NIH (NS-37956, CA-21765), the CCSG (P30 CA21765) and the American Lebanese and Syrian Associated Charities of St. Jude Children’s Research Hospital.
References
- Aguilera A, Garcia-Muse T. R loops: from transcription byproducts to threats to genome stability. Molecular cell. 2012;46:115–124. doi: 10.1016/j.molcel.2012.04.009. [DOI] [PubMed] [Google Scholar]
- Ahel I, Rass U, El-Khamisy SF, Katyal S, Clements PM, McKinnon PJ, Caldecott KW, West SC. The neurodegenerative disease protein aprataxin resolves abortive DNA ligation intermediates. Nature. 2006;443:713–716. doi: 10.1038/nature05164. [DOI] [PubMed] [Google Scholar]
- Al Tassan N, Khalil D, Shinwari J, Al Sharif L, Bavi P, Abduljaleel Z, Abu Dhaim N, Magrashi A, Bobis S, Ahmed H, Alahmed S, Bohlega S. A missense mutation in PIK3R5 gene in a family with ataxia and oculomotor apraxia. Hum Mutat. 2012;33:351–354. doi: 10.1002/humu.21650. [DOI] [PubMed] [Google Scholar]
- Ali AA, Jukes RM, Pearl LH, Oliver AW. Specific recognition of a multiply phosphorylated motif in the DNA repair scaffold XRCC1 by the FHA domain of human PNK. Nucleic acids research. 2009;37:1701–1712. doi: 10.1093/nar/gkn1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almeida KH, Sobol RW. A unified view of base excision repair: lesion-dependent protein complexes regulated by post-translational modification. DNA Repair (Amst) 2007;6:695–711. doi: 10.1016/j.dnarep.2007.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anheim M, Monga B, Fleury M, Charles P, Barbot C, Salih M, Delaunoy JP, Fritsch M, Arning L, Synofzik M, Schols L, Sequeiros J, Goizet C, Marelli C, Le Ber I, Koht J, Gazulla J, De Bleecker J, Mukhtar M, Drouot N, Ali-Pacha L, Benhassine T, Chbicheb M, M’Zahem A, Hamri A, Chabrol B, Pouget J, Murphy R, Watanabe M, Coutinho P, Tazir M, Durr A, Brice A, Tranchant C, Koenig M. Ataxia with oculomotor apraxia type 2: clinical, biological and genotype/phenotype correlation study of a cohort of 90 patients. Brain. 2009;132:2688–2698. doi: 10.1093/brain/awp211. [DOI] [PubMed] [Google Scholar]
- Anheim M, Tranchant C, Koenig M. The autosomal recessive cerebellar ataxias. N Engl J Med. 2012;366:636–646. doi: 10.1056/NEJMra1006610. [DOI] [PubMed] [Google Scholar]
- Barbot C, Coutinho P, Chorao R, Ferreira C, Barros J, Fineza I, Dias K, Monteiro J, Guimaraes A, Mendonca P, do Ceu Moreira M, Sequeiros J. Recessive ataxia with ocular apraxia: review of 22 Portuguese patients. Arch Neurol. 2001;58:201–205. doi: 10.1001/archneur.58.2.201. [DOI] [PubMed] [Google Scholar]
- Bennett CL, La Spada AR. Unwinding the role of senataxin in neurodegeneration. Discov Med. 2015;19:127–136. [PubMed] [Google Scholar]
- Bernstein NK, Williams RS, Rakovszky ML, Cui D, Green R, Karimi-Busheri F, Mani RS, Galicia S, Koch CA, Cass CE, Durocher D, Weinfeld M, Glover JN. The molecular architecture of the mammalian DNA repair enzyme, polynucleotide kinase. Molecular cell. 2005;17:657–670. doi: 10.1016/j.molcel.2005.02.012. [DOI] [PubMed] [Google Scholar]
- Bras J, Alonso I, Barbot C, Costa MM, Darwent L, Orme T, Sequeiros J, Hardy J, Coutinho P, Guerreiro R. Mutations in PNKP cause recessive ataxia with oculomotor apraxia type 4. Am J Hum Genet. 2015;96:474–479. doi: 10.1016/j.ajhg.2015.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breslin C, Caldecott KW. DNA 3’-phosphatase activity is critical for rapid global rates of single-strand break repair following oxidative stress. Mol Cell Biol. 2009;29:4653–4662. doi: 10.1128/MCB.00677-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caldecott KW. Single-strand break repair and genetic disease. Nat Rev Genet. 2008;9:619–631. doi: 10.1038/nrg2380. [DOI] [PubMed] [Google Scholar]
- Carvill GL, Heavin SB, Yendle SC, McMahon JM, O’Roak BJ, Cook J, Khan A, Dorschner MO, Weaver M, Calvert S, Malone S, Wallace G, Stanley T, Bye AM, Bleasel A, Howell KB, Kivity S, Mackay MT, Rodriguez-Casero V, Webster R, Korczyn A, Afawi Z, Zelnick N, Lerman-Sagie T, Lev D, Moller RS, Gill D, Andrade DM, Freeman JL, Sadleir LG, Shendure J, Berkovic SF, Scheffer IE, Mefford HC. Targeted resequencing in epileptic encephalopathies identifies de novo mutations in CHD2 and SYNGAP1. Nat Genet. 2013;45:825–830. doi: 10.1038/ng.2646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman JR, Taylor MR, Boulton SJ. Playing the end game: DNA double-strand break repair pathway choice. Molecular cell. 2012;47:497–510. doi: 10.1016/j.molcel.2012.07.029. [DOI] [PubMed] [Google Scholar]
- Chappell C, Hanakahi LA, Karimi-Busheri F, Weinfeld M, West SC. Involvement of human polynucleotide kinase in double-strand break repair by non-homologous end joining. EMBO J. 2002;21:2827–2832. doi: 10.1093/emboj/21.11.2827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee A, Saha S, Chakraborty A, Silva-Fernandes A, Mandal SM, Neves-Carvalho A, Liu Y, Pandita RK, Hegde ML, Hegde PM, Boldogh I, Ashizawa T, Koeppen AH, Pandita TK, Maciel P, Sarkar PS, Hazra TK. The role of the mammalian DNA end-processing enzyme polynucleotide kinase 3’-phosphatase in spinocerebellar ataxia type 3 pathogenesis. PLoS Genet. 2015;11:e1004749. doi: 10.1371/journal.pgen.1004749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Compe E, Malerba M, Soler L, Marescaux J, Borrelli E, Egly JM. Neurological defects in trichothiodystrophy reveal a coactivator function of TFIIH. Nat Neurosci. 2007;10:1414–1422. doi: 10.1038/nn1990. [DOI] [PubMed] [Google Scholar]
- D’Amours D, Desnoyers S, D’Silva I, Poirier GG. Poly(ADP-ribosyl)ation reactions in the regulation of nuclear functions. Biochem J. 1999;342(Pt 2):249–268. [PMC free article] [PubMed] [Google Scholar]
- Date H, Onodera O, Tanaka H, Iwabuchi K, Uekawa K, Igarashi S, Koike R, Hiroi T, Yuasa T, Awaya Y, Sakai T, Takahashi T, Nagatomo H, Sekijima Y, Kawachi I, Takiyama Y, Nishizawa M, Fukuhara N, Saito K, Sugano S, Tsuji S. Early-onset ataxia with ocular motor apraxia and hypoalbuminemia is caused by mutations in a new HIT superfamily gene. Nat Genet. 2001;29:184–188. doi: 10.1038/ng1001-184. [DOI] [PubMed] [Google Scholar]
- De Bont R, van Larebeke N. Endogenous DNA damage in humans: a review of quantitative data. Mutagenesis. 2004;19:169–185. doi: 10.1093/mutage/geh025. [DOI] [PubMed] [Google Scholar]
- Deans AJ, West SC. DNA interstrand crosslink repair and cancer. Nat Rev Cancer. 2011;11:467–480. doi: 10.1038/nrc3088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dizdaroglu M, Jaruga P. Mechanisms of free radical-induced damage to DNA. Free Radic Res. 2012;46:382–419. doi: 10.3109/10715762.2011.653969. [DOI] [PubMed] [Google Scholar]
- El-Khamisy SF, Saifi GM, Weinfeld M, Johansson F, Helleday T, Lupski JR, Caldecott KW. Defective DNA single-strand break repair in spinocerebellar ataxia with axonal neuropathy-1. Nature. 2005;434:108–113. doi: 10.1038/nature03314. [DOI] [PubMed] [Google Scholar]
- Frappart PO, McKinnon PJ. Ataxia-telangiectasia and related diseases. Neuromolecular Med. 2006;8:495–511. doi: 10.1385/NMM:8:4:495. [DOI] [PubMed] [Google Scholar]
- Gueven N, Chen P, Nakamura J, Becherel OJ, Kijas AW, Grattan-Smith P, Lavin MF. A subgroup of spinocerebellar ataxias defective in DNA damage responses. Neuroscience. 2007;145:1418–1425. doi: 10.1016/j.neuroscience.2006.12.010. [DOI] [PubMed] [Google Scholar]
- Hammer MB, El Euch-Fayache G, Nehdi H, Saidi D, Nasri A, Nabli F, Bouhlal Y, Maamouri-Hicheri W, Hentati F, Amouri R. Clinical and molecular findings of ataxia with oculomotor apraxia type 2 (AOA2) in 5 Tunisian families. Diagn Mol Pathol. 2012;21:241–245. doi: 10.1097/PDM.0b013e318257ad9a. [DOI] [PubMed] [Google Scholar]
- Hamperl S, Cimprich KA. The contribution of co-transcriptional RNA:DNA hybrid structures to DNA damage and genome instability. DNA Repair (Amst) 2014;19:84–94. doi: 10.1016/j.dnarep.2014.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heyer WD, Ehmsen KT, Liu J. Regulation of homologous recombination in eukaryotes. Annu Rev Genet. 2010;44:113–139. doi: 10.1146/annurev-genet-051710-150955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirano M, Quinzii CM, Mitsumoto H, Hays AP, Roberts JK, Richard P, Rowland LP. Senataxin mutations and amyotrophic lateral sclerosis. Amyotroph Lateral Scler. 2011;12:223–227. doi: 10.3109/17482968.2010.545952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Interthal H, Chen HJ, Kehl-Fie TE, Zotzmann J, Leppard JB, Champoux JJ. SCAN1 mutant Tdp1 accumulates the enzyme--DNA intermediate and causes camptothecin hypersensitivity. EMBO J. 2005;24:2224–2233. doi: 10.1038/sj.emboj.7600694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyama T, Wilson DM., 3rd DNA repair mechanisms in dividing and non-dividing cells. DNA Repair (Amst) 2013;12:620–636. doi: 10.1016/j.dnarep.2013.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature. 2009;461:1071–1078. doi: 10.1038/nature08467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jasin M, Rothstein R. Repair of strand breaks by homologous recombination. Cold Spring Harb Perspect Biol. 2013;5:a012740. doi: 10.1101/cshperspect.a012740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jilani A, Ramotar D, Slack C, Ong C, Yang XM, Scherer SW, Lasko DD. Molecular cloning of the human gene, PNKP, encoding a polynucleotide kinase 3’-phosphatase and evidence for its role in repair of DNA strand breaks caused by oxidative damage. J Biol Chem. 1999;274:24176–24186. doi: 10.1074/jbc.274.34.24176. [DOI] [PubMed] [Google Scholar]
- Kaindl AM. Autosomal recessive primary microcephalies (MCPH) Eur J Paediatr Neurol. 2014;18:547–548. doi: 10.1016/j.ejpn.2014.03.010. [DOI] [PubMed] [Google Scholar]
- Kaindl AM, Passemard S, Kumar P, Kraemer N, Issa L, Zwirner A, Gerard B, Verloes A, Mani S, Gressens P. Many roads lead to primary autosomal recessive microcephaly. Prog Neurobiol. 2010;90:363–383. doi: 10.1016/j.pneurobio.2009.11.002. [DOI] [PubMed] [Google Scholar]
- Karimi-Busheri F, Rasouli-Nia A, Allalunis-Turner J, Weinfeld M. Human polynucleotide kinase participates in repair of DNA double-strand breaks by nonhomologous end joining but not homologous recombination. Cancer research. 2007;67:6619–6625. doi: 10.1158/0008-5472.CAN-07-0480. [DOI] [PubMed] [Google Scholar]
- Katyal S, el-Khamisy SF, Russell HR, Li Y, Ju L, Caldecott KW, McKinnon PJ. TDP1 facilitates chromosomal single-strand break repair in neurons and is neuroprotective in vivo. EMBO J. 2007;26:4720–4731. doi: 10.1038/sj.emboj.7601869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, D’Andrea AD. Regulation of DNA cross-link repair by the Fanconi anemia/BRCA pathway. Genes Dev. 2012;26:1393–1408. doi: 10.1101/gad.195248.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MY, Zhang T, Kraus WL. Poly(ADP-ribosyl)ation by PARP-1: ‘PAR-laying’ NAD+ into a nuclear signal. Genes Dev. 2005;19:1951–1967. doi: 10.1101/gad.1331805. [DOI] [PubMed] [Google Scholar]
- Kobayashi J, Saito Y, Okui M, Miwa N, Komatsu K. Increased oxidative stress in AOA3 cells disturbs ATM-dependent DNA damage responses. Mutat Res Genet Toxicol Environ Mutagen. 2015;782:42–50. doi: 10.1016/j.mrgentox.2015.03.012. [DOI] [PubMed] [Google Scholar]
- Koch CA, Agyei R, Galicia S, Metalnikov P, O’Donnell P, Starostine A, Weinfeld M, Durocher D. Xrcc4 physically links DNA end processing by polynucleotide kinase to DNA ligation by DNA ligase IV. EMBO J. 2004;23:3874–3885. doi: 10.1038/sj.emboj.7600375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krokan HE, Bjoras M. Base excision repair. Cold Spring Harb Perspect Biol. 2013;5:a012583. doi: 10.1101/cshperspect.a012583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunkel TA, Erie DA. Eukaryotic Mismatch Repair in Relation to DNA Replication. Annu Rev Genet. 2015;49:291–313. doi: 10.1146/annurev-genet-112414-054722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laine JP, Egly JM. When transcription and repair meet: a complex system. Trends Genet. 2006;22:430–436. doi: 10.1016/j.tig.2006.06.006. [DOI] [PubMed] [Google Scholar]
- Le Ber I, Bouslam N, Rivaud-Pechoux S, Guimaraes J, Benomar A, Chamayou C, Goizet C, Moreira MC, Klur S, Yahyaoui M, Agid Y, Koenig M, Stevanin G, Brice A, Durr A. Frequency and phenotypic spectrum of ataxia with oculomotor apraxia 2: a clinical and genetic study in 18 patients. Brain. 2004;127:759–767. doi: 10.1093/brain/awh080. [DOI] [PubMed] [Google Scholar]
- Lee M, Kim NY, Huh JY, Kim YE, Kim YJ. Ataxia with Oculomotor Apraxia Type 1 without Oculomotor Apraxia: A Case Report. J Clin Neurol. 2016;12:126–128. doi: 10.3988/jcn.2016.12.1.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y, Katyal S, Li Y, El-Khamisy SF, Russell HR, Caldecott KW, McKinnon PJ. The genesis of cerebellar interneurons and the prevention of neural DNA damage require XRCC1. Nat Neurosci. 2009;12:973–980. doi: 10.1038/nn.2375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Lu LY, Yang CY, Wang S, Yu X. The FHA and BRCT domains recognize ADP-ribosylation during DNA damage response. Genes Dev. 2013;27:1752–1768. doi: 10.1101/gad.226357.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieber MR. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu Rev Biochem. 2010;79:181–211. doi: 10.1146/annurev.biochem.052308.093131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loizou JI, El-Khamisy SF, Zlatanou A, Moore DJ, Chan DW, Qin J, Sarno S, Meggio F, Pinna LA, Caldecott KW. The protein kinase CK2 facilitates repair of chromosomal DNA single-strand breaks. Cell. 2004;117:17–28. doi: 10.1016/s0092-8674(04)00206-5. [DOI] [PubMed] [Google Scholar]
- Lu M, Mani RS, Karimi-Busheri F, Fanta M, Wang H, Litchfeld DW, Weinfeld M. Independent mechanisms of stimulation of polynucleotide kinase/phosphatase by phosphorylated and non-phosphorylated XRCC1. Nucleic acids research. 2010;38:510–521. doi: 10.1093/nar/gkp1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madabhushi R, Pan L, Tsai LH. DNA damage and its links to neurodegeneration. Neuron. 2014;83:266–282. doi: 10.1016/j.neuron.2014.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magistretti PJ, Pellerin L. Cellular mechanisms of brain energy metabolism. Relevance to functional brain imaging and to neurodegenerative disorders. Ann N Y Acad Sci. 1996;777:380–387. doi: 10.1111/j.1749-6632.1996.tb34449.x. [DOI] [PubMed] [Google Scholar]
- Mahmood S, Ahmad W, Hassan MJ. Autosomal Recessive Primary Microcephaly (MCPH): clinical manifestations, genetic heterogeneity and mutation continuum. Orphanet J Rare Dis. 2011;6:39. doi: 10.1186/1750-1172-6-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manicklal S, Emery VC, Lazzarotto T, Boppana SB, Gupta RK. The “silent” global burden of congenital cytomegalovirus. Clin Microbiol Rev. 2013;26:86–102. doi: 10.1128/CMR.00062-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marteijn JA, Lans H, Vermeulen W, Hoeijmakers JH. Understanding nucleotide excision repair and its roles in cancer and ageing. Nat Rev Mol Cell Biol. 2014;15:465–481. doi: 10.1038/nrm3822. [DOI] [PubMed] [Google Scholar]
- Matsumoto Y, Miyamoto T, Sakamoto H, Izumi H, Nakazawa Y, Ogi T, Tahara H, Oku S, Hiramoto A, Shiiki T, Fujisawa Y, Ohashi H, Sakemi Y, Matsuura S. Two unrelated patients with MRE11A mutations and Nijmegen breakage syndrome-like severe microcephaly. DNA Repair (Amst) 2011;10:314–321. doi: 10.1016/j.dnarep.2010.12.002. [DOI] [PubMed] [Google Scholar]
- McKinnon PJ. DNA repair deficiency and neurological disease. Nat Rev Neurosci. 2009;10:100–112. doi: 10.1038/nrn2559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinnon PJ. ATM and the molecular pathogenesis of ataxia telangiectasia. Annu Rev Pathol. 2012;7:303–321. doi: 10.1146/annurev-pathol-011811-132509. [DOI] [PubMed] [Google Scholar]
- McKinnon PJ. Maintaining genome stability in the nervous system. Nat Neurosci. 2013;16:1523–1529. doi: 10.1038/nn.3537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mischo HE, Gomez-Gonzalez B, Grzechnik P, Rondon AG, Wei W, Steinmetz L, Aguilera A, Proudfoot NJ. Yeast Sen1 helicase protects the genome from transcription-associated instability. Molecular cell. 2011;41:21–32. doi: 10.1016/j.molcel.2010.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreira MC, Barbot C, Tachi N, Kozuka N, Uchida E, Gibson T, Mendonca P, Costa M, Barros J, Yanagisawa T, Watanabe M, Ikeda Y, Aoki M, Nagata T, Coutinho P, Sequeiros J, Koenig M. The gene mutated in ataxia-ocular apraxia 1 encodes the new HIT/Zn-finger protein aprataxin. Nat Genet. 2001;29:189–193. doi: 10.1038/ng1001-189. [DOI] [PubMed] [Google Scholar]
- Moreira MC, Klur S, Watanabe M, Nemeth AH, Le Ber I, Moniz JC, Tranchant C, Aubourg P, Tazir M, Schols L, Pandolfo M, Schulz JB, Pouget J, Calvas P, Shizuka-Ikeda M, Shoji M, Tanaka M, Izatt L, Shaw CE, M’Zahem A, Dunne E, Bomont P, Benhassine T, Bouslam N, Stevanin G, Brice A, Guimaraes J, Mendonca P, Barbot C, Coutinho P, Sequeiros J, Durr A, Warter JM, Koenig M. Senataxin, the ortholog of a yeast RNA helicase, is mutant in ataxia-ocular apraxia 2. Nat Genet. 2004;36:225–227. doi: 10.1038/ng1303. [DOI] [PubMed] [Google Scholar]
- Morris-Rosendahl DJ, Kaindl AM. What next-generation sequencing (NGS) technology has enabled us to learn about primary autosomal recessive microcephaly (MCPH) Mol Cell Probes. 2015;29:271–281. doi: 10.1016/j.mcp.2015.05.015. [DOI] [PubMed] [Google Scholar]
- Nakashima M, Takano K, Osaka H, Aida N, Tsurusaki Y, Miyake N, Saitsu H, Matsumoto N. Causative novel PNKP mutations and concomitant PCDH15 mutations in a patient with microcephaly with early-onset seizures and developmental delay syndrome and hearing loss. J Hum Genet. 2014;59:471–474. doi: 10.1038/jhg.2014.51. [DOI] [PubMed] [Google Scholar]
- Noyola DE, Demmler GJ, Nelson CT, Griesser C, Williamson WD, Atkins JT, Rozelle J, Turcich M, Llorente AM, Sellers-Vinson S, Reynolds A, Bale JF, Jr, Gerson P, Yow MD Houston Congenital C.M.V.L.S.G. Early predictors of neurodevelopmental outcome in symptomatic congenital cytomegalovirus infection. J Pediatr. 2001;138:325–331. doi: 10.1067/mpd.2001.112061. [DOI] [PubMed] [Google Scholar]
- O’Driscoll M, Jeggo PA. The role of double-strand break repair - insights from human genetics. Nat Rev Genet. 2006;7:45–54. doi: 10.1038/nrg1746. [DOI] [PubMed] [Google Scholar]
- O’Driscoll M, Ruiz-Perez VL, Woods CG, Jeggo PA, Goodship JA. A splicing mutation affecting expression of ataxia-telangiectasia and Rad3-related protein (ATR) results in Seckel syndrome. Nat Genet. 2003;33:497–501. doi: 10.1038/ng1129. [DOI] [PubMed] [Google Scholar]
- Ochi T, Wu Q, Blundell TL. The spatial organization of non-homologous end joining: from bridging to end joining. DNA Repair (Amst) 2014;17:98–109. doi: 10.1016/j.dnarep.2014.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pena-Diaz J, Jiricny J. Mammalian mismatch repair: error-free or error-prone? Trends Biochem Sci. 2012;37:206–214. doi: 10.1016/j.tibs.2012.03.001. [DOI] [PubMed] [Google Scholar]
- Plo I, Liao ZY, Barcelo JM, Kohlhagen G, Caldecott KW, Weinfeld M, Pommier Y. Association of XRCC1 and tyrosyl DNA phosphodiesterase (Tdp1) for the repair of topoisomerase I-mediated DNA lesions. DNA Repair (Amst) 2003;2:1087–1100. doi: 10.1016/s1568-7864(03)00116-2. [DOI] [PubMed] [Google Scholar]
- Pommier Y, Barcelo JM, Rao VA, Sordet O, Jobson AG, Thibaut L, Miao ZH, Seiler JA, Zhang H, Marchand C, Agama K, Nitiss JL, Redon C. Repair of topoisomerase I-mediated DNA damage. Prog Nucleic Acid Res Mol Biol. 2006;81:179–229. doi: 10.1016/S0079-6603(06)81005-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulton C, Oegema R, Heijsman D, Hoogeboom J, Schot R, Stroink H, Willemsen MA, Verheijen FW, van de Spek P, Kremer A, Mancini GM. Progressive cerebellar atrophy and polyneuropathy: expanding the spectrum of PNKP mutations. Neurogenetics. 2013;14:43–51. doi: 10.1007/s10048-012-0351-8. [DOI] [PubMed] [Google Scholar]
- Raichle ME, Gusnard DA. Appraising the brain’s energy budget. Proc Natl Acad Sci U S A. 2002;99:10237–10239. doi: 10.1073/pnas.172399499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds JJ, Stewart GS. A single strand that links multiple neuropathologies in human disease. Brain. 2013;136:14–27. doi: 10.1093/brain/aws310. [DOI] [PubMed] [Google Scholar]
- Reynolds JJ, Walker AK, Gilmore EC, Walsh CA, Caldecott KW. Impact of PNKP mutations associated with microcephaly, seizures and developmental delay on enzyme activity and DNA strand break repair. Nucleic acids research. 2012;40:6608–6619. doi: 10.1093/nar/gks318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard P, Feng S, Manley JL. A SUMO-dependent interaction between Senataxin and the exosome, disrupted in the neurodegenerative disease AOA2, targets the exosome to sites of transcription-induced DNA damage. Genes Dev. 2013;27:2227–2232. doi: 10.1101/gad.224923.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy R, Chun J, Powell SN. BRCA1 and BRCA2: different roles in a common pathway of genome protection. Nat Rev Cancer. 2012;12:68–78. doi: 10.1038/nrc3181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudnik-Schoneborn S, Arning L, Epplen JT, Zerres K. SETX gene mutation in a family diagnosed autosomal dominant proximal spinal muscular atrophy. Neuromuscul Disord. 2012;22:258–262. doi: 10.1016/j.nmd.2011.09.006. [DOI] [PubMed] [Google Scholar]
- Schieving JH, de Vries M, van Vugt JM, Weemaes C, van Deuren M, Nicolai J, Wevers RA, Willemsen MA. Alpha-fetoprotein, a fascinating protein and biomarker in neurology. Eur J Paediatr Neurol. 2014;18:243–248. doi: 10.1016/j.ejpn.2013.09.003. [DOI] [PubMed] [Google Scholar]
- Shen J, Gilmore EC, Marshall CA, Haddadin M, Reynolds JJ, Eyaid W, Bodell A, Barry B, Gleason D, Allen K, Ganesh VS, Chang BS, Grix A, Hill RS, Topcu M, Caldecott KW, Barkovich AJ, Walsh CA. Mutations in PNKP cause microcephaly, seizures and defects in DNA repair. Nature genetics. 2010;42:245–249. doi: 10.1038/ng.526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada M, Dumitrache LC, Russell HR, McKinnon PJ. Polynucleotide kinase-phosphatase enables neurogenesis via multiple DNA repair pathways to maintain genome stability. EMBO J. 2015;34:2465–2480. doi: 10.15252/embj.201591363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shull ER, Lee Y, Nakane H, Stracker TH, Zhao J, Russell HR, Petrini JH, McKinnon PJ. Differential DNA damage signaling accounts for distinct neural apoptotic responses in ATLD and NBS. Genes & development. 2009;23:171–180. doi: 10.1101/gad.1746609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sollier J, Cimprich KA. Breaking bad: R-loops and genome integrity. Trends Cell Biol. 2015;25:514–522. doi: 10.1016/j.tcb.2015.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stracker TH, Petrini JH. The MRE11 complex: starting from the ends. Nat Rev Mol Cell Biol. 2011;12:90–103. doi: 10.1038/nrm3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suraweera A, Becherel OJ, Chen P, Rundle N, Woods R, Nakamura J, Gatei M, Criscuolo C, Filla A, Chessa L, Fusser M, Epe B, Gueven N, Lavin MF. Senataxin, defective in ataxia oculomotor apraxia type 2, is involved in the defense against oxidative DNA damage. J Cell Biol. 2007;177:969–979. doi: 10.1083/jcb.200701042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Symington LS, Gautier J. Double-strand break end resection and repair pathway choice. Annu Rev Genet. 2011;45:247–271. doi: 10.1146/annurev-genet-110410-132435. [DOI] [PubMed] [Google Scholar]
- Takashima H, Boerkoel CF, John J, Saifi GM, Salih MA, Armstrong D, Mao Y, Quiocho FA, Roa BB, Nakagawa M, Stockton DW, Lupski JR. Mutation of TDP1, encoding a topoisomerase I-dependent DNA damage repair enzyme, in spinocerebellar ataxia with axonal neuropathy. Nat Genet. 2002;32:267–272. doi: 10.1038/ng987. [DOI] [PubMed] [Google Scholar]
- Tang H, Hammack C, Ogden SC, Wen Z, Qian X, Li Y, Yao B, Shin J, Zhang F, Lee EM, Christian KM, Didier RA, Jin P, Song H, Ming GL. Zika Virus Infects Human Cortical Neural Progenitors and Attenuates Their Growth. Cell Stem Cell. 2016 doi: 10.1016/j.stem.2016.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Loon B, Markkanen E, Hubscher U. Oxygen as a friend and enemy: How to combat the mutational potential of 8-oxo-guanine. DNA Repair (Amst) 2010;9:604–616. doi: 10.1016/j.dnarep.2010.03.004. [DOI] [PubMed] [Google Scholar]
- Vijg J, Suh Y. Genome instability and aging. Annu Rev Physiol. 2013;75:645–668. doi: 10.1146/annurev-physiol-030212-183715. [DOI] [PubMed] [Google Scholar]
- Weinfeld M, Mani RS, Abdou I, Aceytuno RD, Glover JN. Tidying up loose ends: the role of polynucleotide kinase/phosphatase in DNA strand break repair. Trends in biochemical sciences. 2011;36:262–271. doi: 10.1016/j.tibs.2011.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitehouse CJ, Taylor RM, Thistlethwaite A, Zhang H, Karimi-Busheri F, Lasko DD, Weinfeld M, Caldecott KW. XRCC1 stimulates human polynucleotide kinase activity at damaged DNA termini and accelerates DNA single-strand break repair. Cell. 2001;104:107–117. doi: 10.1016/s0092-8674(01)00195-7. [DOI] [PubMed] [Google Scholar]
- Wiederhold L, Leppard JB, Kedar P, Karimi-Busheri F, Rasouli-Nia A, Weinfeld M, Tomkinson AE, Izumi T, Prasad R, Wilson SH, Mitra S, Hazra TK. AP endonuclease-independent DNA base excision repair in human cells. Molecular cell. 2004;15:209–220. doi: 10.1016/j.molcel.2004.06.003. [DOI] [PubMed] [Google Scholar]
- Williams GJ, Hammel M, Radhakrishnan SK, Ramsden D, Lees-Miller SP, Tainer JA. Structural insights into NHEJ: building up an integrated picture of the dynamic DSB repair super complex, one component and interaction at a time. DNA Repair (Amst) 2014;17:110–120. doi: 10.1016/j.dnarep.2014.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods CG, Bond J, Enard W. Autosomal recessive primary microcephaly (MCPH): a review of clinical, molecular, and evolutionary findings. Am J Hum Genet. 2005;76:717–728. doi: 10.1086/429930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang SW, Burgin AB, Jr, Huizenga BN, Robertson CA, Yao KC, Nash HA. A eukaryotic enzyme that can disjoin dead-end covalent complexes between DNA and type I topoisomerases. Proc Natl Acad Sci U S A. 1996;93:11534–11539. doi: 10.1073/pnas.93.21.11534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoseki A, Ishihara T, Koyama A, Shiga A, Yamada M, Suzuki C, Sekijima Y, Maruta K, Tsuchiya M, Date H, Sato T, Tada M, Ikeuchi T, Tsuji S, Nishizawa M, Onodera O. Genotype-phenotype correlations in early onset ataxia with ocular motor apraxia and hypoalbuminaemia. Brain. 2011;134:1387–1399. doi: 10.1093/brain/awr069. [DOI] [PubMed] [Google Scholar]
- Yuce O, West SC. Senataxin, defective in the neurodegenerative disorder ataxia with oculomotor apraxia 2, lies at the interface of transcription and the DNA damage response. Mol Cell Biol. 2013;33:406–417. doi: 10.1128/MCB.01195-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeman MK, Cimprich KA. Causes and consequences of replication stress. Nat Cell Biol. 2014;16:2–9. doi: 10.1038/ncb2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zolner AE, Abdou I, Ye R, Mani RS, Fanta M, Yu Y, Douglas P, Tahbaz N, Fang S, Dobbs T, Wang C, Morrice N, Hendzel MJ, Weinfeld M, Lees-Miller SP. Phosphorylation of polynucleotide kinase/ phosphatase by DNA-dependent protein kinase and ataxia-telangiectasia mutated regulates its association with sites of DNA damage. Nucleic acids research. 2011;39:9224–9237. doi: 10.1093/nar/gkr647. [DOI] [PMC free article] [PubMed] [Google Scholar]