

Abstract
Despite the success of colonoscopy screening, colorectal cancer (CRC) remains one of the most common and deadly cancers, and CRC incidence is rising in some countries where screening is not routine and populations have recently switched from traditional diets to western diets. Diet and energy balance influence CRC by multiple mechanisms. They modulate the composition and function of gut microbiota, which have a prodigious metabolic capacity and can produce oncometabolites or tumor-suppressive metabolites depending, in part, on which dietary factors and digestive components are present in the GI tract. Gut microbiota also have a profound effect on immune cells in the lamina propria, which influences inflammation and subsequently CRC. Nutrient availability, which is an outcome of diet and energy balance, determines the abundance of certain energy metabolites that are essential co-factors for epigenetic enzymes and therefore impinges upon epigenetic regulation of gene expression. Aberrant epigenetic marks accumulate during CRC, and epimutations that are selected for drive tumorigenesis by causing transcriptome profiles to diverge from the cell of origin. In some instances, the above mechanisms are intertwined as exemplified by dietary fiber being metabolized by colonic bacteria into butyrate, which is both a short-chain fatty acid (SCFA) and a histone deacetylase (HDAC) inhibitor that epigenetically upregulates tumor-suppressor genes in CRC cells and anti-inflammatory genes in immune cells.
Keywords: Colorectal cancer, Gut microbiota, Oncometabolites, Tumor-suppressive metabolites, Inflammation, Warburg effect, Butyrate, Epigenetics, Histone acetylation
Influence of Diet on Colorectal Cancer
Colorectal cancer (CRC) is among the three most diagnosed and deadly cancers in both men and women in the USA as well as globally (1, 2). It is estimated that 132,700 new CRC cases were diagnosed in the USA during 2015 along with 49,700 associated deaths (2). However, the good news is that these figures have been decreasing ~3% per year over the past decade in the USA (2). This downward trend is attributed to more frequent colonoscopy screening leading to the early detection and removal of pre-cancerous adenomas (3). Although the CRC burden is stable or decreasing in the USA and other western countries, this is not the case elsewhere (4). In contrast, CRC incidence and mortality have been increasing in developing countries and newly industrialized countries such as China due to their populations adopting western diets and lifestyles. CRC rates have doubled in some Asian and Eastern European countries since the mid-1970s (4).
Notwithstanding the success of colonoscopy screening, the CRC burden could be diminished by ~50% via diet, physical activity, and weight management (5). Numerous prospective-cohort epidemiology studies have identified specific dietary and lifestyle factors that either promote or protect against CRC (Table 1) (5). For example, consumption of red meat and animal fats increase risk, whereas dietary fiber decreases risk. Energy balance is particularly important, and the obesity epidemic will undoubtedly have a significant adverse impact. Due to the Warburg effect, cancer cells become addicted to glucose: glucose transporters (GLUTs) are upregulated, glucose uptake is ≥10-fold, and aerobic glycolysis is utilized to generate ATP in a relatively inefficient manner (6). Therefore, the high blood glucose levels that occur in obese individuals with metabolic syndrome, type-2 diabetes, and even pre-diabetic insulin resistance represent a tumor-permissive environment that can metabolically drive cancer. This idea is supported by the clinical success of metformin, a diabetes drug that lowers blood glucose levels, for the prevention and treatment of CRC and other cancers (7, 8).
Table 1.
Dietary and lifestyle factors that influence CRC risk.
| Strength of Evidence | Increased CRC Risk | Decreased CRC Risk |
|---|---|---|
| Probable or Convincing | Red meat Processed meat Alcoholic drinks Obesity |
Physical activity Dietary fiber Garlic Milk Calcium |
| Suggestive | Foods containing iron Cheese Foods containining animal fats Foods containing sugars |
Nonstarchy vegetables Fruits Fish Foods containing folate Foods containing selenium Foods containing vitamin D Selenium |
| Inconclusive | Cereals (grains) and their products; potatoes; poultry; shellfish and other seafood; other dairy products; total fat; fatty acid composition; cholesterol; sugar (sucrose); coffee; tea; caffeine; total carbohydrate; starch; vitamin A; retinol; vitamin C; vitamin E; multivitamins; non-dairy sources of calcium; methionine; beta-carotene; alpha-carotene; lycopene; meal frequency; energy intake | |
Adapted from American Institute of Cancer (AICR) 2nd Annual Expert Report (5)
A positive energy balance (i.e., caloric intake > energy expenditure) that results in insulin resistance usually leads to elevated circulating levels of insulin and insulin-like growth hormone (IGF) (also known as hyperinsulinemia) as a compensatory mechanism. Increased insulin or IGF in hyperinsulinemia can drive cancer by binding to their cognate tyrosine kinase receptors and upregulating pro-oncogenic growth factor signaling pathways such as PI3K-AKT-mTOR (which can trigger MDM2 degradation of p53) and RAS-RAF-ERK (which can upregulate c-MYC expression) that stimulate cell growth and proliferation while inhibiting apoptosis (9, 10). Although CRC incidence has reached a plateau and gone down over the past decade(s) in the USA, there has been a concomitant rise in early-onset cases (e.g., diagnosis <50 years of age, which is prior to the age of routine colonoscopy screening) not associated with familial cancer syndromes such as FAP and HNPCC/Lynch syndrome (4, 11), and this trend is believed to be due to the increased prevalence of obesity. One possibility is that obesity-induced activation of RAS-RAF-ERK and PI3K-AKT-mTOR signaling in both young and old individuals accelerates carcinogenesis by substituting for somatic mutations that genetically activate these same pathways but which occur over a longer timeframe (12). This idea is compatible with KRAS, BRAF, and p53 mutations being the primary drivers of CRC progression and which are mutated more frequently than any other gene except APC (which usually initiates CRC) (13). Indeed, when CRC exome sequencing data from The Cancer Genome Atlas (TCGA) were stratified according to body mass index (BMI), obese individuals were reported to have a lower frequency of mutations for driver genes in general and for KRAS in particular (12). These findings support the idea that the selective pressure for driver mutations is lost when the corresponding genes and genetic pathways are deregulated via perturbation of their cellular milieu.
In addition to relatively direct effects of diet on CRC, as mentioned above, diet is likely to influence CRC indirectly by modulating the composition and metabolism of gut microbiota and by epigenetically regulating gene expression. These topics are covered in the next two sections.
Influence of Gut Microbiota on Colorectal Cancer
Our bodies harbor a huge number (≥1014) of microbial cells, and the lumen of the colon has the highest microbial density by far (14). Microbiome studies, which utilize metagenomic DNA sequencing approaches to catalog the diversity and abundance of bacteria in fecal samples or mucosal biopsies obtained by endoscopy, have demonstrated that the human gut microbiome is relatively stable in individuals over time except in the case of certain events such as food poisoning/infection or international travel (15, 16). This likely reflects the hegemony of long-term dietary patterns on our gut microbiome (17). Nevertheless, our gut microbiome does respond to short-term dietary interventions. For example, switching from a traditional diet that is high in plant polysaccharides including fiber and low in animal fat and processed sugar to a reciprocal western diet that is low in plant polysaccharides/fiber and high in animal fat and processed sugar (including the pervasiveness of high-fructose corn syrup) leads to a rapid shift (within a day) in the microbiome (18–20). Many observed changes are as expected and reflect natural selection or a similar process where certain bacterial clades or genera grow at the expense of others in response to an altered environment. For instance, the aforementioned dietary switch to a western diet resulted in a bloom of bile-tolerant bacteria such as Bilophilia and Bacteroides with a concomitant decline in Firmicutes such as Roseburia that metabolize plant polysaccharides (20). However, short-term dietary interventions tend to transiently affect the gut microbiome and are usually not maintained following a return to the long-term diet (17). For this reason, short-term dietary interventions are unlikely to be successful for reshaping the microbiome in a stable manner for CRC prevention as is known to be the case for weight loss. Finally, it should be noted that our knowledge is limited because diet probably has a much larger effect on the microbiome’s transcriptional output (i.e., the metatranscriptome) and metabolite production (i.e., metabolome) than the relative abundance of specific bacteria (21–24). These topics are currently being investigated and should further increase our understanding of the relationship between diet and the function of gut microbiota.
It is noteworthy that dietary fiber and red meat plus animal fats have particularly strong effects on microbiome structure because they have significant, diametrically opposed effects on CRC risk (as discussed above and listed in Table 1). This correlation supports the idea that long-term changes in our diet influence CRC via microbiome changes. Indeed, human microbiome studies have demonstrated that the bacterial community structure of the gut is often perturbed in CRC and in other disease states such as inflammatory bowel disease (IBD). Bacterial diversity is diminished in both CRC and IBD, and this is referred to as dysbiosis. Human microbiome studies have also identified significant changes in the abundance of specific bacteria in CRC cases compared to controls. For example, Escherichia coli strains containing a pks pathogenicity island, Fusobacterium nucleatum, and Providencia are overrepresented in CRC cases, whereas Lactobacillus and butyrate-producing bacteria such as Roseburia and Fecalibacterium are underrepresented (25, 26). However, metagenomic sequence data cannot establish causality because a particular microbiome change could be either a cause or a consequence of CRC. To overcome this limitation, germfree mouse models of CRC are colonized with one or more specific bacteria either enriched or depleted in human CRC cases while being maintained in gnotobiotic isolators (25). When these “humanized” mouse models have significantly different tumor burdens, Koch’s postulates are virtually fulfilled and causality is demonstrated. The bacteria can also be genetically modified in these studies as was done to demonstrate that the pks pathogenicity island, which encodes a toxin (colibactin) that induces DNA damage in the colonic epithelium, is responsible for the contribution of E. coli to CRC (27).
Gut microbiota can either promote or protect against CRC by several mechanisms. First, dietary and digestive factors are metabolized by microbiota into putative oncometabolites and tumor-suppressive metabolites (Figure 1) (28). To exemplify the promotion of carcinogenesis, red meat and bile acids produced in response to the digestion of animal fats are converted by gut bacteria into hydrogen sulfide and secondary bile acids such as DCA, respectively. To exemplify cancer prevention, plant-based polyphenols and fiber are converted by gut microbiota into equol or urolithins and short-chain fatty acids (SCFAs) such as butyrate, respectively. Second, gut microbiota affect intraepithelial lymphocytes and immune cells in the lamina propria to modulate inflammatory responses (Figure 1). Inflammation is recognized as a hallmark of cancer (29), and CRC is particularly sensitive to inflammation considering that aspirin and NSAIDs diminish CRC risk to a larger extent than any other cancer (30, 31). In further support of the microbiota-inflammation-CRC link, microbiota play a key role in IBD, and IBD patients with colonic inflammation have a 2–10 fold increased risk of CRC (32). In addition to directly affecting immune cells, the gut microbiota can influence the permeability of the colonic epithelium and barrier function (Figure 1), which, in turn, determines the extent to which luminal bacteria and bacterial antigens such as LPS and flagellin come into contact with immune cells. Furthermore, obesity is associated with increased intestinal permeability (which may involve microbiota) and inflammation (33, 34), which strongly suggests that it exacerbates CRC independent of energy balance.
Figure 1.
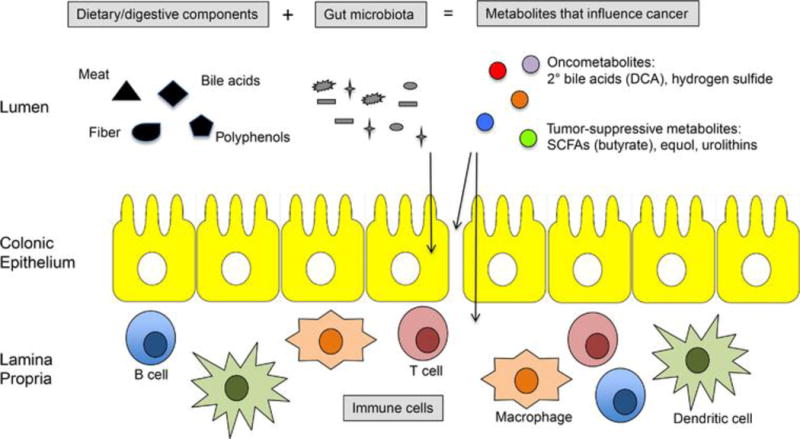
Gut microbiota influence CRC by multiple mechanisms. Schematic of colon showing 3 compartments: top, the lumen containing dietary/digestive components (black), microbiota (gray), and metabolites (colors); middle, a single layer of epithelial cells; bottom, an underlying lamina propria containing immune cells. Gut bacteria can promote CRC by metabolizing oncogenic dietary/digestive components such as meat and bile acids into oncometabolites such as secondary bile acids (e.g., DCA) and hydrogen sulfide. Conversely, gut bacteria can protect against CRC by metabolizing beneficial dietary/digestive components such as plant-based polyphenols and fiber into tumor-suppressive metabolites such as butyrate. Bacteria (and their metabolites and antigens) can have direct effects on normal or cancerous colonocytes (left arrow) or influence immune cell activation and inflammation, which can occur directly (right arrow) or by maintaining colonocyte permeability and barrier function of the epithelium (middle arrow).
Gut microbiota can also influence CRC and other cancers indirectly by affecting energy balance. The gut microbiome changes in response to diet, as discussed above, and the Bacteroides-to-Firmicutes ratio is shifted downward in obese individuals (35). The “obese microbiota” are enriched for genes involved in carbohydrate and lipid metabolism, which enables more calories to be extracted from energy-rich diets (18). To address the issue of cause versus effect, fecal microbiota from obese versus lean and normal versus malnourished donors have been transplanted into germfree mice in gnotobiotic isolators. Comparisons of FMT outcomes support the idea that gut microbiota modulate energy balance and adiposity (36–38). The gut microbiome will affect many specific components of our diets including additives and preservatives in processed foods. One example is the central role of our gut microbiota in the observation that artificial sweeteners (e.g., diet soda) induce glucose intolerance (39). Another example is emulsifiers such as carboxymethylcellulose and polsorbate-80 that can induce low-grade inflammation, obesity/metabolic syndrome, and colitis in gnotobiotic mouse models (40).
Finally, it should be emphasized that gut microbiota are not only relevant for cancer prevention but are also emerging targets for therapy. For example, irinotecan (also known as camptothecin 11) is a topoisomerase I inhibitor commonly used as a chemotherapeutic agent for the treatment of CRC. As part of normal drug metabolism, it is inactivated via glucuronidation in the liver and excreted through the GI tract, but bacterial β-glucuronidases cleave the glucuronide group as an energy source, which reactivates the active chemotherapeutic in the gut to harm or kill rapidly dividing intestinal epithelial cells and cause severe GI distress including diarrhea (41). Antibiotics does not represent a preferred treatment option because the indiscriminate killing of bacteria can be deleterious (including the onset of C. difficile infections) so pharmacologic inhibitors of bacterial β-glucuronidases were developed. Oral administration of these inhibitors is effective at alleviating the GI toxicity of irinotecan in mouse models (41), and this approach may allow the dose or duration of irinotecan to be ramped up in human CRC patients. One might expect these inhibitors to be effective in combination with additional drugs that are glucuronidated in the liver and reactivated by bacterial β-glucuronidases in the gut including other chemotherapeutics and NSAIDs (which cause intestinal bleeding when reactivated in the intestine). Another good example comes from immunotherapy, which is currently the vanguard of anti-cancer therapy. Antibodies that inhibit either CTLA-4 or PD-L1 have shown particular promise by triggering a checkpoint blockade that unleashes a robust immune response against cancer cells. Interestingly, the efficacy of anti-CTLA4 and - PD-L1 treatments is dependent on the composition of the patients’ gut microbiota and its ability to induce the maturation of dendritic cells followed by a robust response from tumor infiltrating T cells (42, 43). In addition to FMTs, specific bacteria were identified (e.g., Bifidobacterium and Bacteroides such as B. fragilis), and “therapeutic feeding” of one these bacteria improved the efficacy of immunotherapy in a mouse model (42, 43). The studies cited here focused on melanomas, but immunotherapy is being evaluated for the treatment of other cancers including CRC, and probiotics clearly represent a potential adjuvant treatment.
Influence of Epigenetics on Colorectal Cancer
Tumorigenesis is driven not only by mutations but also by dysregulated epigenomic alterations that cause the transcriptome profile of a cancer cell to diverge from the cell of origin (44). This process includes the inappropriate addition or removal of acetyl and methyl marks at specific histone residues. These histone modifications often lead to a corresponding gain or loss of DNA methylation at CpG dinucleotides, which can stably maintain the altered epigenomic state through numerous cell divisions (45, 46). Aberrant epigenetic modifications, which are sometimes referred to as “epimutations”, occur at promoters or enhancers of tumor-suppressor genes and proto-oncogenes and are often selected for during tumorigenesis. Epimutations are well documented in CRC where many genes (e.g., APC, LKB1, GATA4, p16INK4a, MLH1) and genetic pathways are common targets (47). Epigenetic inactivation of MLH1 and other mismatch repair genes results in a dramatic increase in mutations within the tumor (i.e., a mutator phenotype manifest by microsatellite instability), which demonstrates that there can be a causal relationship between epimutations and mutation burdens that contribute to genomic instability. Histone 3 lysine 9 acetylation (H3K9ac) and H3K27ac are particularly relevant because these marks are highly enriched at active gene promoters and enhancers, respectively (29, 48, 49). Furthermore, H3K27ac has been used (along with H3K4me1) to define variant enhancer loci (VEL) that are differentially acetylated/activated in normal colonic tissue versus CRC samples in humans (48). H3K9 and H3K27 residues are also noteworthy because they can be modified by either active acetyl marks or inactive methyl marks (with H3K27 methylation being catalyzed by the EZH2 component of PRC2), and this yin-yang relationship is often in a state of flux. Histone acetylation is regulated by the balance between histone acetyltransfereases (HATs) and histone deacetylases (HDACs), which add and remove acetyl groups, respectively. Non-histone proteins including p53 are also direct targets that can be relevant. An imbalance in HAT and HDAC enzymatic activities underlies a variety of cancers. For example, HDACs 1, 2, 3, and 6 are recurrently upregulated in CRC and other cancers, and genetic knockdown of these genes in tumor-derived cell lines induces cell-cycle arrest and apoptosis (50). HDAC inhibitors are being developed by biotechnology and pharmaceutical companies for anti-cancer chemotherapy, and vorinostat (also known as SAHA) and romidepsin have already been approved by the FDA (albeit for the treatment of lymphomas rather than CRC) (50).
Epigenetics also represents a mechanism that links diet and nutrient availability with regulation of gene expression to maintain homeostasis. The basis for this mechanistic link is the fact that most epigenetic enzymes utilize energy metabolites or redox factors as essential co-factors (51). DNA methyltransferases and histone methyltransferases utilize S-adenosylmethionine, a product of 1-carbon metabolism, as the methyl-group donor; histone acetyltransferases (HATs) utilize acetyl-CoA, a central component of energy metabolism, as the acetyl-group donor; class III HDACs (also known as sirtuins) are dependent on the redox factor NAD+; TET and JmjC dioxygenases require the TCA cycle intermediate α-ketoglutarate to demethylate DNA and histones, respectively. In certain cases, diet influences gene expression to an extent that influences phenotypic outcomes and disease states. For example, dietary supplementation of 1-carbon cycle micronutrients (e.g., folate, vitamin B12, choline) influences coat color, obesity, and tumorigenesis in genetically identical viable-yellow mice (51–53). And children conceived during the Dutch hunger winter of 1944–1945 and other famines have had increased incidence of schizophrenia and other ailments later in life, which in some cases have been associated with altered CpG methylation profiles of target genes such as IGF [albeit in peripheral blood cells, which may or may not be representative of the diseased tissue (e.g., CNS) or relevant to the disease state (e.g., complex behaviors)] (54). There is precedent for energy metabolites functioning in human cancer. The oncometabolite 2-hydroxyglutarate, which arises because of neomorphic IDH mutations in lymphomas and gliomas, is sufficient to promote neoplasia (55). It is generated in cancer cells at the expense α-ketoglutarate and inhibits both TETs and JmjC enzymes to alter DNA and histone methylation (55). The TCA cycle intermediates succinate and fumarate also function in this capacity in response to oncogenic mutations in genes encoding TCA cycle enzymes (56). Finally, considering the prodigious metabolic capacity of the gut microbiome, bacteria will likely convert dietary factors and digestive components into metabolites that regulate epigenetics in cancer. A recent comparison of colonic epithelial cells from germfree and conventional mice revealed surprisingly few differences in chromatin accessibility (57), but this does not rule out other possible differences such as local histone modifications. Although our knowledge in this area is limited, the next section provides a proof of principle for a bacterial metabolite in histone acetylation.
The fiber-microbiota-butyrate axis in epigenetics and CRC prevention
Dietary fiber is defined as “the edible part of plants or their extracts, or analogous carbohydrates, that are resistant to digestion and absorption in the small intestine, but are utilized after partial or complete fermentation in the large intestine by resident microbiota” (58). Fiber includes polysaccharides (e.g., resistant starch, cellulose, hemicellulose, pectins, and gums), oligosaccharides, and lignins. Although fiber has been investigated for many years, it continues to be studied and provide new insights. A particularly strong study was recently published in which native Africans and African Americans participated in a 2-week dietary intervention (59). Native Africans, who have a low rate of CRC (<5 per 100,000), switched from their traditional high-fiber diets to low-fiber western diets, while African Americans (who have a >10-fold higher rate at 65 per 100,000) switched from their western diets to high-fiber, traditional diets. The dietary changes affected the gut microbiome and led to reciprocal changes in metabolites such as SCFAs including butyrate and mucosal biomarkers of cancer risk (59).
Increased fiber consumption has the potential to decrease CRC risk by two general mechanisms that are not mutually exclusive. First, insoluble fiber speeds colonic transit and may diminish the exposure of colonic epithelial cells to ingested carcinogens such as heterocyclic amines from charred meat (Figure 2). Second, soluble fiber is fermented by certain clades of bacteria into butyrate and other potentially beneficial metabolites (Figure 2). Although dietary fiber is generally believed to diminish CRC risk, there has been a lack of consensus because of conflicting results from prospective-cohort epidemiology studies (60). These studies have been reliant on self-report questionnaires to track participants’ dietary habits for ~3 years so compliance is a concern that probably hinders reproducibility. Another limitation is that meta-analyses must be conducted carefully because different studies utilize different fiber sources, which are metabolized to different extents and yield different levels of metabolites. ≥5 microbiome studies have reported a significant decrease in butyrate-producing bacteria in human CRC cases compared to controls, but it is not clear whether this is a cause or a consequence of the disease (25, 61). Although most research using mouse models support the idea that butyrate is tumor suppressive (25, 62), a recent study suggested that butyrate promotes CRC (63). This study utilized a mouse model with impaired mismatch repair, but it is not clear whether this is sufficient to reconcile the contradictory findings (64).
Figure 2.
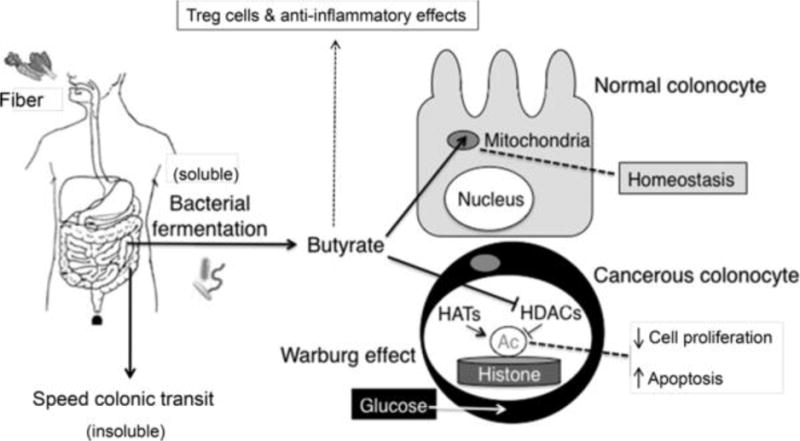
Mechanism of fiber- and butyrate-mediated tumor suppression. Insoluble fiber speeds colonic transit to minimize carciongen exposure, while soluble fiber is fermented by gut bacteria into butyrate (left). In normal colonocytes (right, top), butyrate is utilized as the primary energy source to maintain homeostasis. Because it is readily metabolized in the mitochondria via fatty acid oxidation, relatively little accumulates inside the nucleus. In cancerous colonocytes (right, bottom), glucose is the primary energy source due to the Warburg effect. Butyrate is still transported into the cell via monocarboxylate transporters but is not metabolized in the mitochondria, which allows it to accumulate in the nucleus and function as an HDAC inhibitor to epigenetically regulate genes involved in cell proliferation and apoptosis.
To investigate dietary fiber and butyrate in a highly controlled manner, a mouse model of CRC was polyassociated with several bacteria in a gnotobiotic isolator and provided control or high-fiber diets that were otherwise isocaloric and virtually identical (65, 66). The high-fiber diet was provided from weaning (prior to tumor initiation) until the time of sacrifice and had a protective effect in mice colonized with a wild-type butyrate-producing bacterium but not in mice lacking a butyrate producer (Figure 3). The same mice colonized with a mutant strain of the butyrate-producing bacterium, which harbors a small deletion in the butyryl CoA synthesis operon and produces diminished levels of butyrate, had an attenuated protective effect with an intermediate tumor burden (Figure 3). Furthermore, mice that completely lacked butyrate-producing bacteria but were provided a diet fortified with butyrate had a lower tumor burden than any of the other treatment groups (Figure 3). This is arguably the most convincing evidence that butyrate is a causal factor because it demonstrates that the fiber-microbiota tumor-suppressive effect can be recapitulated by exogenous butyrate. Future studies will need to be performed where the high-fiber, “butyrogenic” diet is provided after tumorigenesis is initiated to determine whether it is still protective.
Figure 3.
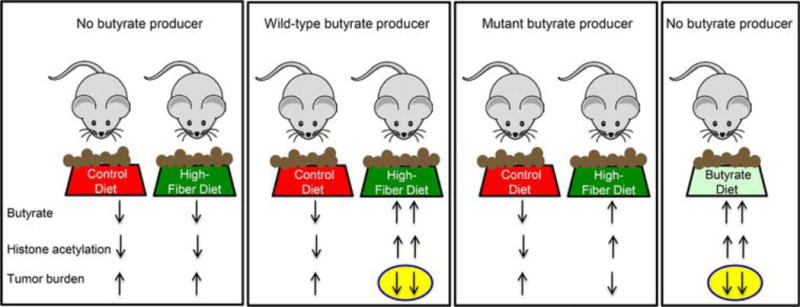
Dietary fiber protects against colorectal cancer in a microbiota- and butyrate-dependent manner in a gnotobiotic mouse model. Mice were colonized with several bacteria that either excluded or included a butyrate producer as indicated at the top. A wild-type and mutant strain of the butyrate producer was utilized in separate gnotobiotic isolators (depicted by boxes around each group of mice). In each isolator, the mice received control or high-fiber diets (6% fructo-oligosaccharides/inulin but otherwise virtually identical to the control diet) except for a butyrate-fortified diet in the isolator at the far right. Arrows at the bottom indicate relative levels of luminal butyrate along with global histone acetylation levels and tumor burden after tumorigenesis was initiated 5 months earlier. Butyrate production was attenuated, but not completely abolished, in the mutant strain when provided a high-fiber diet as denoted by one upward butyrate arrow instead of a downward arrow or two upward arrows. The yellow ovals highlight experimental conditions that yield a lower tumor burden, and this correlates with higher butyrate levels and histone acetylation levels.
The above study also explored the molecular mechanism of how butyrate functions as a dietary- and microbiota-derived tumor-suppressive metabolite (65). Unlike most cell types in the body, which utilize glucose as their primary energy source, normal colonocytes rely on butyrate for ~60–70% of their energy (67–69). As a fatty acid, butyrate undergoes β-oxidation in the mitochondria (67), and this supports energy homeostasis necessary for the rapid cell proliferation of the colonic epithelium (Figure 2). The colonic and small intestinal epithelium arguably turn over faster than any other tissue in the body (every ≤7 days) with rapid cell proliferation near the base of the crypt counterbalanced with apoptosis near the lumen. In contrast, CRC cells (and cancer cells in general) undergo the Warburg effect and switch to glucose utilization and aerobic glycolysis (6) (Figure 2). As a result of this metabolic shift, butyrate is not metabolized in the mitochondria of CRC cells to the same extent and accumulates in the nucleus where it functions as an HDAC inhibitor to epigenetically regulate gene expression (65, 70) (Figure 2). In support of this model, butyrate (detected by LC-MS) and global histone acetylation levels (detected by immunohistochemistry and western blots) were elevated in tumors from mice that were colonized with the wild-type butyrate producer and provided a high-fiber diet, and this correlated with a lower tumor burden (Figure 3). Butyrate is a well-established HDAC inhibitor (71, 72), and butyrate target genes in tumors from mice provided a high-fiber diet included Fas and p21, which promote apoptosis and inhibit cell-cycle progression, respectively. This finding is compatible with the diminished tumor burden in these mice and the idea that butyrate is a tumor-suppressive metabolite.
The tumor-suppressive mechanism(s) is probably more complicated than described above. Insoluble fibers such as cellulose are not fermented by gut microbiota and speed colonic transit as mentioned above. Soluble fibers are fermented into SCFAs other than butyrate, such as acetate and propionate, and these or other metabolites could also contribute to CRC prevention. Propionate is also an HDAC inhibitor although it is not as potent as butyrate and has broader bioavailability with less accumulation in colonocytes (73). Finally, butyrate is a pleiotropic molecule and may prevent cancer via additional mechanisms. In addition to functioning as an HDAC inhibitor, it can signal through certain G protein coupled receptors (74, 75). Butyrate may diminish tumorigenesis by attenuating inflammation. Butyrate enemas strongly ameliorate colonic inflammation associated with inflammatory bowel diseases (IBD) such as colitis and Crohn’s disease in both rodent models and human patients (72). This is noteworthy because colitis patients have up to a 10-fold increased risk of CRC as mentioned above (32, 76), which is consistent with the link between inflammation and cancer (29). A series of recent studies demonstrate that butyrate activates FoxP3 expression in naïve CD4+ T cells and dendritic cells and induces the differentiation and expansion of immunosuppressive regulatory T (Treg) cells (77–84). Another recent study demonstrates that butyrate downregulates the production of pro-inflammatory cytokines by intestinal macrophages (85). Butyrate is also known to affect colonocyte permeability and barrier function (86, 87). In other words, butyrate participates in each functional category illustrated in Figure 1. This example demonstrates that diet and gut microbiota can interact to influence multiple mechanisms relevant to cancer including epigenetics and inflammation.
Lack of consensus and unresolved issues regarding fiber and the gut microbiome
Diet may have a more modest effect on the composition and function of gut microbiota in humans than mice, and there is currently a lack of consensus regarding the effect size in humans. For example, a recently published study comparing vegans to omnivores in the USA, which included a 10-day dietary intervention trial, detected relatively subtle differences in the composition of gut microbiota (88). Furthermore, butyrate and other SCFA levels were not significantly different between the two groups although fiber intake was higher in vegans. These findings are different than what has been observed in Native Africans that still rely on a traditional, high-fiber diet (59). One possibility is that the capacity of an individual’s microbiome to ferment fiber into SCFAs might become constrained after a prolonged (i.e., years or lifetime) low-fiber diet. One might also expect transgenerational effects because an individual “inherits” their microbiota from their mother at birth and usually adopts a similar diet. Therefore, a diminished capacity to ferment fiber into SCFAs could be amplified after two or more generations of low-fiber consumption. This idea is supported by another recent study that provided a low-fiber diet to mice colonized with human microbiota and observed diminished microbiome diversity (89). Interestingly, this microbiome change was initially reversible (within one generation) but became progressively irreversible with diet-induced extinctions after several generations. In a situation like this, prebiotics (e.g., a high-fiber diet) will not be sufficient, and probiotics will be necessary to re-introduce specific gut bacteria into the gut ecosystem to produce SCFAs. In the absence of the appropriate diet (e.g., low-fiber diet), however, it might be necessary to develop something analogous to a gene-drive system (as has been proposed for eradicating malaria) to select for the re-introduced beneficial bacteria.
Other diet- and microbial-derived metabolites implicated in cancer prevention
The previously cited study identified 92 out of 361 metabolites (25%) that had significantly different concentrations in plasma from vegans versus omnivores (88). Although these changes did not correlate with microbiome changes, another study utilizing germfree mice estimated that 10% of plasma metabolites are influenced by microbiota (90). Therefore, it stands to reason that many diet- and microbe-derived metabolites will influence colorectal cancer and other cancers.
Dietary polyphenols, which include flavonoids (e.g., quercetin and kaempferol), phenolic acids, anthrocyanins, and lignins present in tea, wine, fruits, nuts, and vegetables, have received extensive attention because of their chemoprotective effects in mouse models and human epidemiology studies. Resveratrol has probably received the most attention because it is a caloric-restriction mimetic that is pleiotropic and benefits health in multiple ways. However, it is not the best example for being metabolized by gut microbiota. In contrast, ellagic acid is a polyphenol present in certain berries and nuts that is an anti-oxidant with cancer-preventive properties. Ellagic acid is metabolized by colonic microbiota into urolithins that have pro-estrogenic and anti-estrogenic activities in a context-dependent manner (91). Urolithins can also downregulate COX-2 to lower prostaglandin production and inflammation so the anti-cancer effects apparently involve multiple pathways. Another polyphenol is daidzein, which is a soy-based isoflavone metabolized by gut microbiota into equol. Some epidemiologic studies have reported correlations between equol or equol-producing bacteria and diminished breast cancer risk in women and diminished prostate cancer risk in men. However, these correlations have been observed in Asian populations but not European populations (91). It is not clear whether these ethnic disparities are due to differences in genetics, microbiota, or diet (e.g., soy consumption), and more work will be required to strengthen the link between equol and cancer prevention. Interestingly, only 30–40% of westerners are able to produce equol, whereas 60–70% of Asians are able to do so (91). Although the reason for this difference is not understood, it could be due, in part, to the relative abundance of specific bacteria. In the previously cited study, only 40% of the vegans had detectable equol in their plasma despite similar levels of isoflavone intake (88). This suggests that long-term diet and transgenerational effects may lead to irrevocable changes in the microbiome and metabolome that are relevant to both fiber/SCFA- and soy isoflavone/equol-mediated cancer susceptibility.
Cruciferous vegetables such as broccoli and cabbage contain high levels of glucosinolates. When these vegetables are uncooked and either chopped or chewed, plant-derived myrosinases convert the glucosinolates to isothiocyanates (ITC) such as sulforaphane, which is an HDAC inhibitor like butyrate, that have anti-carcinogenic properties in cell lines and mouse models and might diminish human cancer risk. The evidence is strongest for colorectal, lung, breast, prostate, and prostate although the epidemiology results are mixed. However, cruciferous vegetables are usually cooked. Although heat denatures the plant-derived myrosinases, bacteria-derived thioglucosidases convert glucosinolates into ITC in the gut to exert their beneficial effects (91).
Linoleic acid is an omega-6 polyunsaturated fatty acid (PUFA) that is a constituent of vegetable oils. Because linoleic acid is a precursor of arachadonic acid, which gives rise to prostaglandins and inflammation, high intake of vegetable oils can alter the omega-6 to omega-3 ratio and be deleterious. However, certain gut microbiota, including strains of Lactobacilli and Bifidobacteria used in probiotics, can conjugate linoleic acid (91). Not only does this bacterial conjugation diminish linoleic acid levels, but some conjugated linoleic acid isomers are reported to have anti-inflammatory and anti-carcinogenic properties.
Future of probiotics and prebiotics in cancer prevention and therapy
As our knowledge of bioactive food components and microbiota increases, it is likely that prebiotics and probiotics will play a bigger role in cancer prevention and anticancer therapy. Prebiotics are defined as indigestible food ingredients that selectively stimulate the growth and/or activity of certain gut microbiota that confer a health benefit. Probiotics correspond to the consumption of live microbiota present in foods or dietary supplements that confer a health benefit. Perhaps the best-known example of probiotics involves Lactobacillus in yogurt for promoting general GI health and preventing lactose intolerance. Streptococci and Bifidobacteria in cheeses and other foods and drinks are also commonplace. FMTs can also be considered a probiotic and are noteworthy because of their tremendous success in treating otherwise lethal Clostridium difficile infections, which usually arise because antibiotics eliminated commensal bacteria that are capable of displacing or suppressing C. difficile. Although there is not much evidence for specific microbiota in cancer prevention or treatment, this is likely to change. The most immediate measurable progress will probably involve Bifidobacterium and Bacteroides such as B. fragilis as adjuvants for immunotherapy as discussed above (42, 43). Another interesting possibility for cancer prevention or therapy involves genetically modified microbiota that provide additional value or benefit. For example, Lactobacillus strains have been engineered to overexpress the antioxidant superoxide dismutase or an anti-inflammatory protein (91). However, because even normal strains Lactobacillus have a very minor and transient impact on the gut microbiome, it might be necessary to develop a strategy to select for these microbes in the gut similar to the proposed gene-drive systems being utilized for sexually reproducing species such as mosquitoes in the case of malaria. In this regard, it is possible that CRISPR might be utilized, and this should be relatively efficient because most bacteria produce their own Cas9 so it does not need to be delivered exogenously. If successful, engineered microbiota could add to the fast-growing functional food/nutraceutical industry.
Acknowledgments
I would like to thank Dr. Aadra Bhatt for providing comments and suggestions on this manuscript. This work was supported by grants from the NIH (R01-OD-02057) and USDA (055336) to SJB.
Abbreviations
- CRC
colorectal cancer
- IBD
inflammatory bowel disease
- FMT
fecal microbiota transplant
- BMI
body mass index
- IGF
insulin-like growth factor
- NSAIDs
non-steroidal anti-inflammatory drugs
- LPS
lipopolysaccharide
- GLUTs
glucose transporters
- ATP
adenosine triphosphate
- TCA
tricarboxylic acid
- FDA
food and drug administration
- SCFA
short-chain fatty acid
- EZH2
enhancer of zeste 2
- PRC2
Polycomb repressive complex 2
- HDACs
histone deacetylases
- HATs
histone acetyltransferases
- DCA
dichloroacetate
- TCGA
the cancer genome atlas
- FAP
familial adenomatous polyposis
- HNPCC
hereditary nonpolyposis colorectal cancer
- SCFAs
short-chain fatty acids
- PUFA
polyunsaturated fatty acid
- ITC
isothiocynates
Footnotes
The author has declared no conflict of interest.
References
- 1.American Cancer Society. Cancer Facts and Figures 2015. Atlanta, Ga: American Cancer Society; 2015. [Google Scholar]
- 2.http://seer.cancer.gov/statfacts/html/colorect.html.
- 3.Levin B, Lieberman DA, McFarland B, Andrews KS, Brooks D, Bond J, Dash C, Giardiello FM, Glick S, Johnson D, et al. Screening and surveillance for the early detection of colorectal cancer and adenomatous polyps, 2008: a joint guideline from the American Cancer Society, the US Multi-Society Task Force on Colorectal Cancer, and the American College of Radiology. Gastroenterology. 2008;134(5):1570–95. doi: 10.1053/j.gastro.2008.02.002. [DOI] [PubMed] [Google Scholar]
- 4.Haggar FA, Boushey RP. Colorectal cancer epidemiology: incidence, mortality, survival, and risk factors. Clin Colon Rectal Surg. 2009;22(4):191–7. doi: 10.1055/s-0029-1242458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.http://www.aicr.org/research/research_science_policy_report.html.
- 6.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324(5930):1029–33. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang ZJ, Zheng ZJ, Kan H, Song Y, Cui W, Zhao G, Kip KE. Reduced risk of colorectal cancer with metformin therapy in patients with type 2 diabetes: a meta-analysis. Diabetes Care. 2011;34(10):2323–8. doi: 10.2337/dc11-0512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Morales DR, Morris AD. Metformin in cancer treatment and prevention. Annu Rev Med. 2015;66:17–29. doi: 10.1146/annurev-med-062613-093128. [DOI] [PubMed] [Google Scholar]
- 9.Hursting SD, Berger NA. Energy balance, host-related factors, and cancer progression. J Clin Oncol. 2010;28(26):4058–65. doi: 10.1200/JCO.2010.27.9935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Khandekar MJ, Cohen P, Spiegelman BM. Molecular mechanisms of cancer development in obesity. Nature Reviews Cancer. 2011;11(12):886–95. doi: 10.1038/nrc3174. [DOI] [PubMed] [Google Scholar]
- 11.Bailey CE, Hu CY, You YN, Bednarski BK, Rodriguez-Bigas MA, Skibber JM, Cantor SB, Chang GJ. Increasing disparities in the age-related incidences of colon and rectal cancers in the United States, 1975–2010. JAMA Surg. 2015;150(1):17–22. doi: 10.1001/jamasurg.2014.1756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bordonaro M, Lazarova D. Hypothesis: Obesity Is Associated with a Lower Mutation Threshold in Colon Cancer. J Cancer. 2015;6(9):825–31. doi: 10.7150/jca.12352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wood LD, Parsons DW, Jones S, Lin J, Sjoblom T, Leary RJ, Shen D, Boca SM, Barber T, Ptak J, et al. The genomic landscapes of human breast and colorectal cancers. Science. 2007;318(5853):1108–13. doi: 10.1126/science.1145720. [DOI] [PubMed] [Google Scholar]
- 14.Sekirov I, Russell SL, Antunes LC, Finlay BB. Gut microbiota in health and disease. Physiol Rev. 2010;90(3):859–904. doi: 10.1152/physrev.00045.2009. [DOI] [PubMed] [Google Scholar]
- 15.David LA, Materna AC, Friedman J, Campos-Baptista MI, Blackburn MC, Perrotta A, Erdman SE, Alm EJ. Host lifestyle affects human microbiota on daily timescales. Genome Biology. 2014;15(7):R89. doi: 10.1186/gb-2014-15-7-r89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Faith JJ, Guruge JL, Charbonneau M, Subramanian S, Seedorf H, Goodman AL, Clemente JC, Knight R, Heath AC, Leibel RL, et al. The long-term stability of the human gut microbiota. Science. 2013;341(6141):1237439. doi: 10.1126/science.1237439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xu Z, Knight R. Dietary effects on human gut microbiome diversity. The British Journal of Nutrition. 2015;113(Suppl):S1–5. doi: 10.1017/S0007114514004127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Turnbaugh PJ, Backhed F, Fulton L, Gordon JI. Diet-induced obesity is linked to marked but reversible alterations in the mouse distal gut microbiome. Cell Host & Microbe. 2008;3(4):213–23. doi: 10.1016/j.chom.2008.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Turnbaugh PJ, Ridaura VK, Faith JJ, Rey FE, Knight R, Gordon JI. The effect of diet on the human gut microbiome: a metagenomic analysis in humanized gnotobiotic mice. Science Translational Medicine. 2009;1(6):6ra14. doi: 10.1126/scitranslmed.3000322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.David LA, Maurice CF, Carmody RN, Gootenberg DB, Button JE, Wolfe BE, Ling AV, Devlin AS, Varma Y, Fischbach MA, et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature. 2014;505(7484):559–63. doi: 10.1038/nature12820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Louis P, Hold GL, Flint HJ. The gut microbiota, bacterial metabolites and colorectal cancer. Nature Reviews Microbiology. 2014;12(10):661–72. doi: 10.1038/nrmicro3344. [DOI] [PubMed] [Google Scholar]
- 22.Sharon G, Garg N, Debelius J, Knight R, Dorrestein PC, Mazmanian SK. Specialized metabolites from the microbiome in health and disease. Cell Metabolism. 2014;20(5):719–30. doi: 10.1016/j.cmet.2014.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Franzosa EA, Morgan XC, Segata N, Waldron L, Reyes J, Earl AM, Giannoukos G, Boylan MR, Ciulla D, Gevers D, et al. Relating the metatranscriptome and metagenome of the human gut. Proceedings of the National Academy of Sciences of the United States of America. 2014;111(22):E2329–38. doi: 10.1073/pnas.1319284111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Integrative HMPRNC. The Integrative Human Microbiome Project: dynamic analysis of microbiome-host omics profiles during periods of human health and disease. Cell Host & Microbe. 2014;16(3):276–89. doi: 10.1016/j.chom.2014.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bultman SJ. Emerging roles of the microbiome in cancer. Carcinogenesis. 2014;35(2):249–55. doi: 10.1093/carcin/bgt392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Burns MB, Lynch J, Starr TK, Knights D, Blekhman R. Virulence genes are a signature of the microbiome in the colorectal tumor microenvironment. Genome Medicine. 2015;7(1):55. doi: 10.1186/s13073-015-0177-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Arthur JC, Perez-Chanona E, Muhlbauer M, Tomkovich S, Uronis JM, Fan TJ, Campbell BJ, Abujamel T, Dogan B, Rogers AB, et al. Intestinal inflammation targets cancer-inducing activity of the microbiota. Science. 2012;338(6103):120–3. doi: 10.1126/science.1224820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hullar MA, Burnett-Hartman AN, Lampe JW. Gut microbes, diet, and cancer. Cancer Treatment and Research. 2014;159:377–99. doi: 10.1007/978-3-642-38007-5_22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 30.Bosetti C, Rosato V, Gallus S, Cuzick J, La Vecchia C. Aspirin and cancer risk: a quantitative review to 2011. Ann Oncol. 2012;23(6):1403–15. doi: 10.1093/annonc/mds113. [DOI] [PubMed] [Google Scholar]
- 31.Cuzick J, Otto F, Baron JA, Brown PH, Burn J, Greenwald P, Jankowski J, La Vecchia C, Meyskens F, Senn HJ, et al. Aspirin and non-steroidal anti-inflammatory drugs for cancer prevention: an international consensus statement. The Lancet Oncology. 2009;10(5):501–7. doi: 10.1016/S1470-2045(09)70035-X. [DOI] [PubMed] [Google Scholar]
- 32.Itzkowitz SH, Harpaz N. Diagnosis and management of dysplasia in patients with inflammatory bowel diseases. Gastroenterology. 2004;126(6):1634–48. doi: 10.1053/j.gastro.2004.03.025. [DOI] [PubMed] [Google Scholar]
- 33.Teixeira TF, Collado MC, Ferreira CL, Bressan J, Peluzio Mdo C. Potential mechanisms for the emerging link between obesity and increased intestinal permeability. Nutr Res. 2012;32(9):637–47. doi: 10.1016/j.nutres.2012.07.003. [DOI] [PubMed] [Google Scholar]
- 34.Bastard JP, Maachi M, Lagathu C, Kim MJ, Caron M, Vidal H, Capeau J, Feve B. Recent advances in the relationship between obesity, inflammation, and insulin resistance. Eur Cytokine Netw. 2006;17(1):4–12. [PubMed] [Google Scholar]
- 35.Ley RE, Turnbaugh PJ, Klein S, Gordon JI. Microbial ecology: human gut microbes associated with obesity. Nature. 2006;444(7122):1022–3. doi: 10.1038/4441022a. [DOI] [PubMed] [Google Scholar]
- 36.Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. 2006;444(7122):1027–31. doi: 10.1038/nature05414. [DOI] [PubMed] [Google Scholar]
- 37.Smith MI, Yatsunenko T, Manary MJ, Trehan I, Mkakosya R, Cheng J, Kau AL, Rich SS, Concannon P, Mychaleckyj JC, et al. Gut microbiomes of Malawian twin pairs discordant for kwashiorkor. Science. 2013;339(6119):548–54. doi: 10.1126/science.1229000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ridaura VK, Faith JJ, Rey FE, Cheng J, Duncan AE, Kau AL, Griffin NW, Lombard V, Henrissat B, Bain JR, et al. Gut microbiota from twins discordant for obesity modulate metabolism in mice. Science. 2013;341(6150):1241214. doi: 10.1126/science.1241214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Suez J, Korem T, Zeevi D, Zilberman-Schapira G, Thaiss CA, Maza O, Israeli D, Zmora N, Gilad S, Weinberger A, et al. Artificial sweeteners induce glucose intolerance by altering the gut microbiota. Nature. 2014;514(7521):181–6. doi: 10.1038/nature13793. [DOI] [PubMed] [Google Scholar]
- 40.Chassaing B, Koren O, Goodrich JK, Poole AC, Srinivasan S, Ley RE, Gewirtz AT. Dietary emulsifiers impact the mouse gut microbiota promoting colitis and metabolic syndrome. Nature. 2015;519(7541):92–6. doi: 10.1038/nature14232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wallace BD, Wang H, Lane KT, Scott JE, Orans J, Koo JS, Venkatesh M, Jobin C, Yeh LA, Mani S, et al. Alleviating cancer drug toxicity by inhibiting a bacterial enzyme. Science. 2010;330(6005):831–5. doi: 10.1126/science.1191175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vetizou M, Pitt JM, Daillere R, Lepage P, Waldschmitt N, Flament C, Rusakiewicz S, Routy B, Roberti MP, Duong CP, et al. Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science. 2015;350(6264):1079–84. doi: 10.1126/science.aad1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sivan A, Corrales L, Hubert N, Williams JB, Aquino-Michaels K, Earley ZM, Benyamin FW, Lei YM, Jabri B, Alegre ML, et al. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science. 2015;350(6264):1084–9. doi: 10.1126/science.aac4255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Baylin SB, Jones PA. A decade of exploring the cancer epigenome - biological and translational implications. Nature reviews Cancer. 2011;11(10):726–34. doi: 10.1038/nrc3130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Archer SY, Hodin RA. Histone acetylation and cancer. Curr Opin Genet Dev. 1999;9(2):171–4. doi: 10.1016/s0959-437x(99)80026-4. [DOI] [PubMed] [Google Scholar]
- 46.Sandoval J, Esteller M. Cancer epigenomics: beyond genomics. Curr Opin Genet Dev. 2012;22(1):50–5. doi: 10.1016/j.gde.2012.02.008. [DOI] [PubMed] [Google Scholar]
- 47.Carmona FJ, Esteller M. Epigenomics of human colon cancer. Mutat Res. 2010;693(1–2):53–60. doi: 10.1016/j.mrfmmm.2010.07.007. [DOI] [PubMed] [Google Scholar]
- 48.Akhtar-Zaidi B, Cowper-Sal-lari R, Corradin O, Saiakhova A, Bartels CF, Balasubramanian D, Myeroff L, Lutterbaugh J, Jarrar A, Kalady MF, et al. Epigenomic enhancer profiling defines a signature of colon cancer. Science. 2012;336(6082):736–9. doi: 10.1126/science.1217277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Scanlan PD, Shanahan F, Clune Y, Collins JK, O’Sullivan GC, O’Riordan M, Holmes E, Wang Y, Marchesi JR. Culture-independent analysis of the gut microbiota in colorectal cancer and polyposis. Environmental Microbiology. 2008;10(3):789–98. doi: 10.1111/j.1462-2920.2007.01503.x. [DOI] [PubMed] [Google Scholar]
- 50.West AC, Johnstone RW. New and emerging HDAC inhibitors for cancer treatment. The Journal of Clinical Investigation. 2014;124(1):30–9. doi: 10.1172/JCI69738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Donohoe DR, Bultman SJ. Metaboloepigenetics: interrelationships between energy metabolism and epigenetic control of gene expression. Journal of Cellular Physiology. 2012;227(9):3169–77. doi: 10.1002/jcp.24054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cooney CA, Dave AA, Wolff GL. Maternal methyl supplements in mice affect epigenetic variation and DNA methylation of offspring. The Journal of Nutrition. 2002;132(8 Suppl):2393S–400S. doi: 10.1093/jn/132.8.2393S. [DOI] [PubMed] [Google Scholar]
- 53.Waterland RA, Jirtle RL. Transposable elements: targets for early nutritional effects on epigenetic gene regulation. Mol Cell Biol. 2003;23(15):5293–300. doi: 10.1128/MCB.23.15.5293-5300.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Painter RC, Roseboom TJ, Bleker OP. Prenatal exposure to the Dutch famine and disease in later life: an overview. Reprod Toxicol. 2005;20(3):345–52. doi: 10.1016/j.reprotox.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 55.Losman JA, Kaelin WG., Jr What a difference a hydroxyl makes: mutant IDH, (R)-2-hydroxyglutarate, and cancer. Genes & Development. 2013;27(8):836–52. doi: 10.1101/gad.217406.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang T, Cai G, Qiu Y, Fei N, Zhang M, Pang X, Jia W, Cai S, Zhao L. Structural segregation of gut microbiota between colorectal cancer patients and healthy volunteers. The ISME Journal. 2012;6(2):320–9. doi: 10.1038/ismej.2011.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Camp JG, Frank CL, Lickwar CR, Guturu H, Rube T, Wenger AM, Chen J, Bejerano G, Crawford GE, Rawls JF. Microbiota modulate transcription in the intestinal epithelium without remodeling the accessible chromatin landscape. Genome Research. 2014;24(9):1504–16. doi: 10.1101/gr.165845.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lim CC, Ferguson LR, Tannock GW. Dietary fibres as “prebiotics”: implications for colorectal cancer. Molecular nutrition & food research. 2005;49(6):609–19. doi: 10.1002/mnfr.200500015. [DOI] [PubMed] [Google Scholar]
- 59.O’Keefe SJ, Li JV, Lahti L, Ou J, Carbonero F, Mohammed K, Posma JM, Kinross J, Wahl E, Ruder E, et al. Fat, fibre and cancer risk in African Americans and rural Africans. Nat Commun. 2015;6:6342. doi: 10.1038/ncomms7342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ferguson LR, Harris PJ. The dietary fibre debate: more food for thought. Lancet. 2003;361(9368):1487–8. doi: 10.1016/S0140-6736(03)13219-9. [DOI] [PubMed] [Google Scholar]
- 61.Bultman SJ. Molecular pathways: gene-environment interactions regulating dietary fiber induction of proliferation and apoptosis via butyrate for cancer prevention. Clinical Cancer Research. 2014;20(4):799–803. doi: 10.1158/1078-0432.CCR-13-2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Perrin P, Pierre F, Patry Y, Champ M, Berreur M, Pradal G, Bornet F, Meflah K, Menanteau J. Only fibres promoting a stable butyrate producing colonic ecosystem decrease the rate of aberrant crypt foci in rats. Gut. 2001;48(1):53–61. doi: 10.1136/gut.48.1.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Belcheva A, Irrazabal T, Robertson SJ, Streutker C, Maughan H, Rubino S, Moriyama EH, Copeland JK, Kumar S, Green B, et al. Gut microbial metabolism drives transformation of MSH2-deficient colon epithelial cells. Cell. 2014;158(2):288–99. doi: 10.1016/j.cell.2014.04.051. [DOI] [PubMed] [Google Scholar]
- 64.Bultman SJ, Jobin C. Microbial-derived butyrate: an oncometabolite or tumor-suppressive metabolite? Cell Host & Microbe. 2014;16(2):143–5. doi: 10.1016/j.chom.2014.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Donohoe DR, Holley D, Collins LB, Montgomery SA, Whitmore AC, Hillhouse A, Curry KP, Renner SW, Greenwalt A, Ryan EP, et al. A Gnotobiotic Mouse Model Demonstrates That Dietary Fiber Protects against Colorectal Tumorigenesis in a Microbiota- and Butyrate-Dependent Manner. Cancer Discovery. 2014;4(12):1387–97. doi: 10.1158/2159-8290.CD-14-0501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sebastian C, Mostoslavsky R. Untangling the fiber yarn: butyrate feeds warburg to suppress colorectal cancer. Cancer Discovery. 2014;4(12):1368–70. doi: 10.1158/2159-8290.CD-14-1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Donohoe DR, Garge N, Zhang X, Sun W, O’Connell TM, Bunger MK, Bultman SJ. The microbiome and butyrate regulate energy metabolism and autophagy in the mammalian colon. Cell Metabolism. 2011;13(5):517–26. doi: 10.1016/j.cmet.2011.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Roediger WE. Role of anaerobic bacteria in the metabolic welfare of the colonic mucosa in man. Gut. 1980;21(9):793–8. doi: 10.1136/gut.21.9.793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Roediger WE. Utilization of nutrients by isolated epithelial cells of the rat colon. Gastroenterology. 1982;83(2):424–9. [PubMed] [Google Scholar]
- 70.Donohoe DR, Collins LB, Wali A, Bigler R, Sun W, Bultman SJ. The Warburg effect dictates the mechanism of butyrate-mediated histone acetylation and cell proliferation. Molecular Cell. 2012;48(4):612–26. doi: 10.1016/j.molcel.2012.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Davie JR. Inhibition of histone deacetylase activity by butyrate. The Journal of Nutrition. 2003;133(7 Suppl):2485S–93S. doi: 10.1093/jn/133.7.2485S. [DOI] [PubMed] [Google Scholar]
- 72.Hamer HM, Jonkers D, Venema K, Vanhoutvin S, Troost FJ, Brummer RJ. Review article: the role of butyrate on colonic function. Alimentary Pharmacology & Therapeutics. 2008;27(2):104–19. doi: 10.1111/j.1365-2036.2007.03562.x. [DOI] [PubMed] [Google Scholar]
- 73.Waldecker M, Kautenburger T, Daumann H, Busch C, Schrenk D. Inhibition of histone-deacetylase activity by short-chain fatty acids and some polyphenol metabolites formed in the colon. The Journal of Nutritional Biochemistry. 2008;19(9):587–93. doi: 10.1016/j.jnutbio.2007.08.002. [DOI] [PubMed] [Google Scholar]
- 74.Goncalves P, Martel F. Butyrate and colorectal cancer: the role of butyrate transport. Current Drug Metabolism. 2013;14(9):994–1008. doi: 10.2174/1389200211314090006. [DOI] [PubMed] [Google Scholar]
- 75.Thangaraju M, Cresci GA, Liu K, Ananth S, Gnanaprakasam JP, Browning DD, Mellinger JD, Smith SB, Digby GJ, Lambert NA, et al. GPR109A is a G-protein-coupled receptor for the bacterial fermentation product butyrate and functions as a tumor suppressor in colon. Cancer Research. 2009;69(7):2826–32. doi: 10.1158/0008-5472.CAN-08-4466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Terzic J, Grivennikov S, Karin E, Karin M. Inflammation and colon cancer. Gastroenterology. 2010;138(6):2101–14 e5. doi: 10.1053/j.gastro.2010.01.058. [DOI] [PubMed] [Google Scholar]
- 77.Arpaia N, Campbell C, Fan X, Dikiy S, van der Veeken J, deRoos P, Liu H, Cross JR, Pfeffer K, Coffer PJ, et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature. 2013;504(7480):451–5. doi: 10.1038/nature12726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Furusawa Y, Obata Y, Fukuda S, Endo TA, Nakato G, Takahashi D, Nakanishi Y, Uetake C, Kato K, Kato T, et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature. 2013;504(7480):446–50. doi: 10.1038/nature12721. [DOI] [PubMed] [Google Scholar]
- 79.Singh N, Gurav A, Sivaprakasam S, Brady E, Padia R, Shi H, Thangaraju M, Prasad PD, Manicassamy S, Munn DH, et al. Activation of Gpr109a, receptor for niacin and the commensal metabolite butyrate, suppresses colonic inflammation and carcinogenesis. Immunity. 2014;40(1):128–39. doi: 10.1016/j.immuni.2013.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Smith PM, Howitt MR, Panikov N, Michaud M, Gallini CA, Bohlooly YM, Glickman JN, Garrett WS. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science. 2013;341(6145):569–73. doi: 10.1126/science.1241165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hegazy AN, Powrie F. MICROBIOME. Microbiota RORgulates intestinal suppressor T cells. Science. 2015;349(6251):929–30. doi: 10.1126/science.aad0865. [DOI] [PubMed] [Google Scholar]
- 82.Ohnmacht C, Park JH, Cording S, Wing JB, Atarashi K, Obata Y, Gaboriau-Routhiau V, Marques R, Dulauroy S, Fedoseeva M, et al. MUCOSAL IMMUNOLOGY. The microbiota regulates type 2 immunity through RORgammat(+) T cells. Science. 2015;349(6251):989–93. doi: 10.1126/science.aac4263. [DOI] [PubMed] [Google Scholar]
- 83.Sefik E, Geva-Zatorsky N, Oh S, Konnikova L, Zemmour D, McGuire AM, Burzyn D, Ortiz-Lopez A, Lobera M, Yang J, et al. MUCOSAL IMMUNOLOGY. Individual intestinal symbionts induce a distinct population of RORgamma(+) regulatory T cells. Science. 2015;349(6251):993–7. doi: 10.1126/science.aaa9420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Park J, Kim M, Kang SG, Jannasch AH, Cooper B, Patterson J, Kim CH. Short-chain fatty acids induce both effector and regulatory T cells by suppression of histone deacetylases and regulation of the mTOR-S6K pathway. Mucosal Immunol. 2015;8(1):80–93. doi: 10.1038/mi.2014.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Chang PV, Hao L, Offermanns S, Medzhitov R. The microbial metabolite butyrate regulates intestinal macrophage function via histone deacetylase inhibition. Proceedings of the National Academy of Sciences of the United States of America. 2014;111(6):2247–52. doi: 10.1073/pnas.1322269111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kelly CJ, Zheng L, Campbell EL, Saeedi B, Scholz CC, Bayless AJ, Wilson KE, Glover LE, Kominsky DJ, Magnuson A, et al. Crosstalk between Microbiota-Derived Short-Chain Fatty Acids and Intestinal Epithelial HIF Augments Tissue Barrier Function. Cell Host & Microbe. 2015;17(5):662–71. doi: 10.1016/j.chom.2015.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Peng L, Li ZR, Green RS, Holzman IR, Lin J. Butyrate enhances the intestinal barrier by facilitating tight junction assembly via activation of AMP-activated protein kinase in Caco-2 cell monolayers. The Journal of Nutrition. 2009;139(9):1619–25. doi: 10.3945/jn.109.104638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wu GD, Compher C, Chen EZ, Smith SA, Shah RD, Bittinger K, Chehoud C, Albenberg LG, Nessel L, Gilroy E, et al. Comparative metabolomics in vegans and omnivores reveal constraints on diet-dependent gut microbiota metabolite production. Gut. 2016;65(1):63–72. doi: 10.1136/gutjnl-2014-308209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Sonnenburg ED, Smits SA, Tikhonov M, Higginbottom SK, Wingreen NS, Sonnenburg JL. Diet-induced extinctions in the gut microbiota compound over generations. Nature. 2016;529(7585):212–5. doi: 10.1038/nature16504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wikoff WR, Anfora AT, Liu J, Schultz PG, Lesley SA, Peters EC, Siuzdak G. Metabolomics analysis reveals large effects of gut microflora on mammalian blood metabolites. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(10):3698–703. doi: 10.1073/pnas.0812874106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Bultman SJ. The microbiome and its potential as a cancer preventive intervention. Seminars in Oncology. 2016;43(1):97–106. doi: 10.1053/j.seminoncol.2015.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]