

Abstract
Herein an intramolecular Ni-catalyzed (4+2) coupling between cyclobutanones and allenes via C–C cleavage is reported, which provides a distinct approach to access [3.2.2] bicyclic scaffolds that are challenging to prepare through conventional approaches. The reaction is efficient, chemoselective and pH/redox neutral. Room temperature conditions and low catalyst loadings can be adopted. Excellent enantioselectivity is also achieved.
Keywords: allene, cycloaddition, nickel catalysis, cyclobutanone, C–C activation
Graphical Abstract
An intramolecular Ni-catalyzed (4+2) coupling between cyclobutanones and allenes via C–C cleavage is reported, which provides a unique approach to access [3.2.2] bicyclic scaffolds. The reaction is efficient, chemoselective and pH/redox neutral. Excellent enantioselectivity is also achieved.
Transition metal (TM)-catalyzed C–C cleavage reactions have recently emerged as attractive synthetic methods that offer unusual strategic disconnections for preparation of complex scaffolds.[1] In particular, the couplings between a strained ketone and an unsaturate unit via C–C activation provide efficient access to various ring systems. Two complementary approaches have been developed to assist the coupling (Scheme 1): one involves oxidative addition of a TM, e.g. RhI, Ni0, Ru0, into a strained C–C bond, followed by 2π insertion to complete the cycloaddition (path a);[2] the other approach, pioneered by Murakami and co-workers,[3a] utilizes a cyclometalation pathway to first form a five-membered metallacycle between the carbonyl group and the unsaturated unite, followed by β-carbon elimination and reductive elimination to furnish the ring (path b).[3]
Scheme 1.

Two C-C activation modes with cyclobutanones
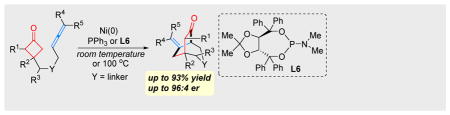
To date, a number of unsaturated units, including alkenes,[2b,c,e,f.i] alkynes,[2a,d,h] 1,3-dienes,[3d,g] ketones/ aldehydes[2k] and imines,[2l] have been employed to couple with strained ketones through one or both pathways. In contrast, the use of allenes as a 2π unit would deliver an additional olefin moiety into the product for further derivatization,[4] which unfortunately, has received much less attention. The challenge is likely due to that 1) allenes are generally less stable and tend to undergo dimerization catalyzed by TMs,[5] and 2) control of the chemoselectivity in the allene-mediated reactions is nontrivial.[6] Recently, we reported the first Rh-catalyzed cyclobutanone-allene coupling via path a involving oxidative addition into C–C bonds (Scheme 2a); however, instead of giving the desired (4+2) product, an unexpected (4+1) addition was found as the major reaction pathway, in which allenes serve as a one-carbon unit.[7] Stimulated by this result, an intriguing question arose: how would allenes behave in a cyclometalation/β-carbon elimination pathway (path b)? In other words, can (4+2) products be exclusively formed with allenes as a 2π unit in a C–C cleavage/coupling reaction? Driven by these questions, herein, we describe our systematic efforts on the development of a Ni-catalyzed chemo- and enantioselective intramolecular (4+2) coupling between cyclobutanones and allenes via C–C cleavage (Scheme 2b). This method provides a unique and efficient entry to functionalized [3.2.2] hetero- and carbocycles that are commonly found in natural products and other bioactive compounds (Figure 1).
Scheme 2.

Coupling of cyclobutanones with allenes
Figure 1.

Representative natural products or pharmaceutical compounds containing [3.2.2] bicyclic skeleton.
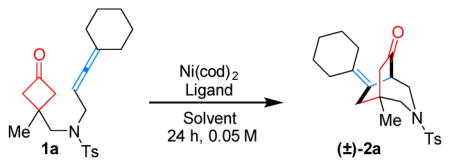
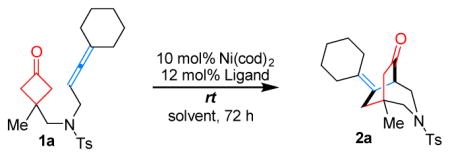
Our study began using allene 1a as the model substrate with Ni(cod)2 as the precatalyst (Table 1). While bidentate ligands gave full conversions of the starting material, a complex mixture of unidentifiable products was obtained (entries 1–4). Given that the Ni(II) intermediate after cyclometalation would require an open coordination site for subsequent β-carbon elimination, the chelation of bidentate ligands would inhibit ligand dissociation; thus, it was envisioned that use of monodentate ligands should avoid this issue. Indeed, when PPh3 was employed, which is known to undergo fast ligand dissociation,[8] the desired (4+2) product (2a) bearing a 3-aza[3.3.2] scaffold was formed in 92% yield (entry 5). Other monodentate ligands, such as PCy3, also afforded the desired product but in a slightly lower yield (entry 6). Halving the ligand loading did not affect the reaction efficiency, suggesting that only one phosphine is needed per metal (entry 7). A survey of solvent effects suggested toluene to be optimal (entries 8 and 9). Note that the catalyst loading can be further reduced to 5 and 2.5 mol% without significantly diminishing the yields (entries 10 and 11). In addition, the reaction run at room temperature also gave an excellent yield with a longer reaction time (entry 12).
Table 1.
Selected optimization conditions with allene 1a
| ||||||
|---|---|---|---|---|---|---|
| Entry | Ni (mol%) | Ligand (mol%) | Temp (°C) | Solvent | Conv.(%) | Yield (%)[a] |
| 1 | 10% | dppp (12) | 100 | Tol | 100 | 0 |
| 2 | 10% | dppb (12) | 100 | Tol | 100 | 0 |
| 3 | 10% | dppe (12) | 100 | Tol | 100 | 0 |
| 4 | 10% | BINAP (12) | 100 | Tol | 100 | 0 |
| 5 | 10% | PPh3 (24) | 100 | Tol | 100 | 92 |
| 6 | 10% | PCy3 (24) | 100 | Tol | 100 | 80 |
| 7 | 10% | PPh3 (12) | 100 | Tol | 100 | 92 |
| 8 | 10% | PPh3 (12) | 100 | THF | 100 | 85 |
| 9 | 10% | PPh3 (12) | 100 | 1,4-dioxane | 100 | 86 |
| 10 | 5% | PPh3 (6) | 100 | Tol | 100 | 91 |
| 11 | 2.5% | PPh3 (3) | 100 | Tol | 75 | 71 |
| 12[b] | 10% | PPh3 (12) | 25 | Tol | 100 | 91 |
Determined by 1H NMR using mesitylene as the internal standard. PCy3 = tricyclohexylphosphine. cod = 1,5-cyclooctadiene.
72 h.
With the optimized conditions (Table 1, entries 7 and 12) in hand, the scope of the Ni-catalyzed (4+2) coupling was explored. First, 1,1,3-trisubstituted allenes were found to be excellent coupling partners (Table 2, substrates 1a–1h). Both cycloalkyl and gem-dialkyl substitution were well tolerated. When unsymmetrical allenes were employed, mixtures of E/Z isomers were obtained (1g and 1h). For substrate 1h, with a pendant benzyl ether substituent, lower reactivity (40% conversion) was observed under the standard conditions. However, when the reaction concentration was increased to 0.15 M, the desired [3.2.2] bicycle 2h was isolated in 81% yield. While attempts to use 1,3,3-trisubstituted and tetrasubstituted allenes (1l and 1m) were unfruitful likely due to the steric hindrance in the cyclometalation step, 1,3-disubstituted allenes (1i and 1j) proved to be suitable substrates. The diminished yields were attributed to the instability of the more exposed allene moiety under the reaction conditions. Not surprisingly, when mono-substituted allene 1k was tested, only allene dimers were detected.
Table 2.
Substrate Scope on allene[a]

|
Run on a 0.1 mmol scale with 10 mol% Ni(cod)2 and 12 mol% PPh3 in 2.0 mL toluene at 100 °C for 24 h.
Room temperature for 72 h.
Number in parentheses is the yield based on recovered starting material (brsm).
0.7 mL toluene.
Next, variations on the cyclobutanone part were examined (Table 3). Besides methyl group (2a), other alkyl and aryl groups, such as ethyl (2n), propyl (2o), phenyl (2p), benzyl (2q) and benzyl ether (2r), were found compatible for this transformation. Due to the mild reaction conditions, an unprotected primary alcohol remained intact (2s). Cyclobutanone 1t with hydrogen at the C3 position is also a competent substrate. In addition, cyclobutanone 2u with a C2 substituent underwent selective C–C cleavage at the less hindered position offering the desired [3.2.2] bicycle with a 6:1 d.r.
Table 3.
Substrate Scope on cyclobutanone[a]

|
Run on a 0.1 mmol with 10 mol% Ni(cod)2 and 12 mol% PPh3 in 2.0 mL toluene at 100 °C for 24 h.
Room temperature for 72 h.
Number in parentheses is brsm yield.
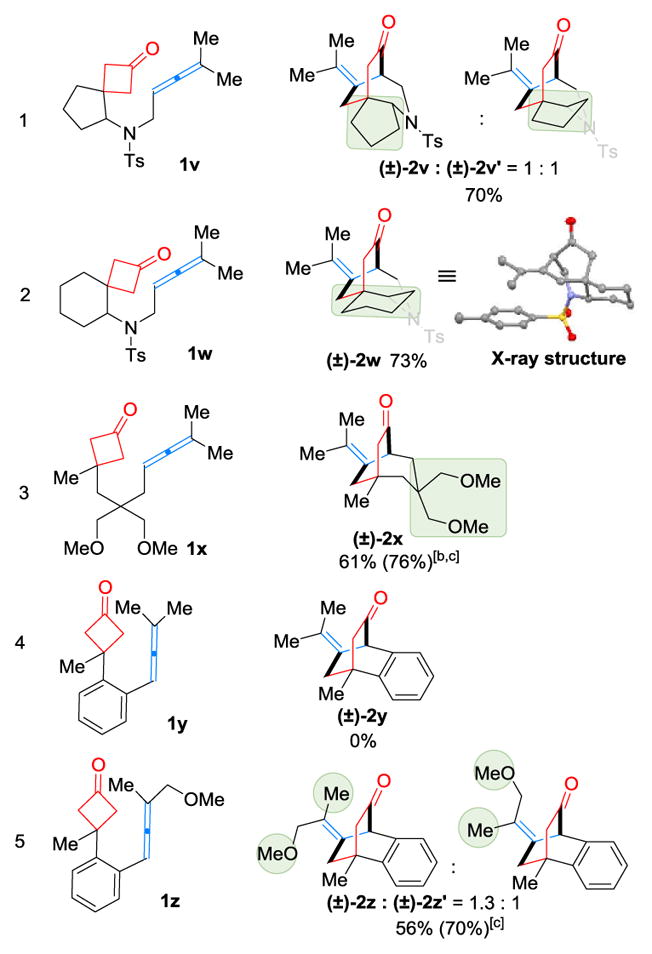
In addition, substrates 1v and 1w, containing a spirocyclic center, were successfully employed to construct tricyclic scaffolds in good yields (entries 1 and 2, Table 4). While the cyclopentyl-based substrate (1v) showed no diastereoselectivity, the cyclohexane one (1w) nevertheless afforded a single diastereomer, whose structure was confirmed by X-ray crystallography.[9] The nearly complete diastereoselectivity for 1w suggested a highly ordered β-carbon elimination process possibly controlled by the chair conformation of the cyclohexane ring. Besides nitrogen tethers, carbon linkages were also investigated (entries 3–5, Table 4). While the sp3 carbon linker (1x) gave the desired (4+2) product in a good yield,[10] it is surprising that substrate 1y, bearing an arene backbone, was found inactive. We reasoned that, although the arene linkage brings the cyclobutanone and allene closer to each other, the rigid backbone also causes the coordination of nickel with the two moieties more difficult due to the steric repulsion between the allene substituent (Me) and the metal. Thus, we further hypothesized that introduction of a weak coordinating group to the allene substituent would enhance its binding with nickel, and consequently provide better reactivity. Indeed, when the MeO-substituted analogue (1z) was employed, the desired benzo [2.2.2] bicycle was afforded in a reasonably good yield.
Table 4.
Substrate Scope on Backbone and Linkage[a]
|
Run on a 0.1 mmol scale in a sealed 4 mL vial, using 10 mol% Ni(cod)2 and 12 mol% PPh3 in toluene (2.0 mL) at 100 °C for 24 h.
Number in parentheses is brsm yield.
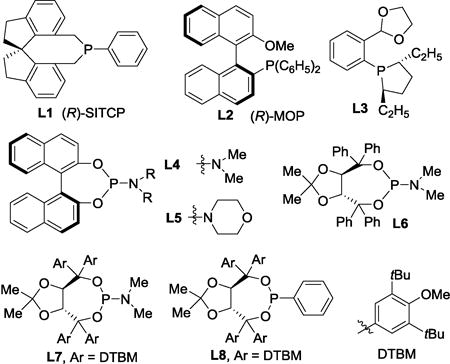
To control the absolute stereochemistry of the [3.2.2] products, the enantioselective variant of the reaction was explored (Table 5). A range of chiral monodentate phosphine ligands were examined (entries 1–6), and the highest e.r. was obtained with TADDOL-derived phosphoramidite L6 (entry 6).[11] Interestingly, the enantioselectivity is highly sensitive to the reaction concentration (entries 7–10);[12] ultimately, a 79% yield and 96:4 e.r. were achieved when increasing the concentration from 0.03 to 1.0 M (entry 10). Further, use of mesitylene as solvent gave higher yield (entry 13). Other TADDOL-derived phosphoramidite ligands such as L7 and L8 were also tested and proved less efficient than L6 (entries 14 and 15).
Table 5.
Selected Optimization for the Enantioselective Reaction[a]
| |||||
|---|---|---|---|---|---|
| Entry | Ligand | Solvent | Concentration | Yield[b] | e.r.[c] |
| 1 | L1 | toluene | 0.03 M | <5% | --[d] |
| 2 | L2 | toluene | 0.03 M | 62% | 54.5:45.5 |
| 3 | L3 | toluene | 0.03 M | 32% | 66.5:33.5 |
| 4 | L4 | toluene | 0.03 M | 35% | 69:31 |
| 5 | L5 | toluene | 0.03 M | 89% | 53:47 |
| 6 | L6 | toluene | 0.03 M | 51% | 76:24 |
| 7 | L6 | toluene | 0.015 M | 59% | 62.5:37.5 |
| 8 | L6 | toluene | 0.1 M | 60% | 88:12 |
| 9 | L6 | toluene | 0.4 M | 61% | 95:6 |
| 10 | L6 | toluene | 1.0 M | 79% | 96:4 |
| 11 | L6 | THF | 1.0 M | 52% | 88:12 |
| 12 | L6 | 1,4-dioxane | 1.0 M | 75% | 95:5 |
| 13 | L6 | mesitylene | 1.0 M | 82% | 96:4 |
| 14 | L7 | toluene | 1.0 M | 60% | 71.5:28.5 |
| 15 | L8 | toluene | 1.0 M | 90% | 66.5:33.5 |
Run on a 0.03 mmol scale.
Determined by crude 1H NMR using 1,1,2,2-tetrachloroethane as the internal standard.
Determined by HPLC analysis using a chiral stationary phase.
Not determined. (R)-SITCP = (11aR)-(+)-5,6,10,11,12,13-Hexahydro-5-phenyl-4H-diindeno[7,1-cd:1,7-ef] phosphocin, (R)-MOP = (R)-(+)-2-(diphenylphosphino)-2′-methoxy-1,1′-binaphthyl, DTBM = 3,5-di-tert-butyl-4-methoxyphenyl.
The scope of the enantioselective (4+2) cyclization was next studied (Table 6). Two important observations were made: 1) substrates with cycloalkyl and di-alkyl substituted allenes all provided good yields and high e.r. (2a–2g); 2) changing the substituent at the C3 position of cyclobutanone did not significantly disturb the enantioselectivity. The absolute stereochemistry was assigned based on the X-ray crystallographic structure of product 2aa.[9]
Table 6.
Substrate Scope of the Enantioselective (4+2) Cyclization[a]

|
Run on a 0.1 mmol scale. Number in parentheses is brsm yield.
The Ni-catalyzed (4+2) cyclization is also scalable, and the product (2a) can undergo many facile transformations to access other structures or functional groups (Scheme 3). Application of this method in complex molecule synthesis is ongoing in our laboratory.
Scheme 3.

Synthetic Applications
Supplementary Material
Acknowledgments
This project was supported by NIGMS (R01GM109054) and the Welch Foundation (F 1781). G.D. is a Searle Scholar and Sloan fellow. Johnson Matthey is acknowledged for a generous donation of Rh salts. Prof. Dr. M. C. Young is thanked for proofreading the manuscript. We also thank Dr. V. Lynch and Dr. M. C. Young for X-ray structures.
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- 1.For recent reviews on transition-metal-mediated C–C activation, see: Jones WD. Nature. 1993;364:676.Murakami M, Ito Y. Top Organomet Chem. 1999;3:97.Rybtchinski B, Milstein D. Angew Chem. 1999;111:918. doi: 10.1002/(SICI)1521-3773(19990401)38:7<870::AID-ANIE870>3.0.CO;2-3.Angew Chem Int Ed. 1999;38:870.van der Boom ME, Milstein D. Chem Rev. 2003;103:1759. doi: 10.1021/cr960118r.Jun CH. Chem Soc Rev. 2004;33:610. doi: 10.1039/b308864m.Satoh T, Miura M. Top Organomet Chem. 2005;14:1.Jun CH, Park JW. Top Organomet Chem. 2007;24:117.Necas D, Kotora M. Curr Org Chem. 2007;11:1566.Kondo T, Mitsudo TA. Chem Lett. 2005;34:1462.Ruhland K. Eur J Org Chem. 2012:2683.Korotvicka A, Necas D, Kotora M. Curr Org Chem. 2012;16:1170.Seiser T, Saget T, Tran DN, Cramer N. Angew Chem. 2011;123:7884. doi: 10.1002/anie.201101053.Angew Chem Int Ed. 2011;50:7740.Murakami M, Matsuda T. Chem Commun. 2011;47:1100. doi: 10.1039/c0cc02566f.Dermenci A, Coe PW, Dong G. Org Chem Front. 2014;1:567. doi: 10.1039/c4qo00053f.Dong G, editor. Topics in Current Chemistry. Springer-Verlag; Berlin: 2014. C–C bond activation; p. 346.Chen F, Wang T, Jiao N. Chem Rev. 2014;114:8613. doi: 10.1021/cr400628s.Souillart L, Cramer N. Chem Rev. 2015;115:9410. doi: 10.1021/acs.chemrev.5b00138.Shaw MH, Bower JF. Chem Commun. 2016;52:10817. doi: 10.1039/c6cc04359c.
- 2.For selected examples, see: South MS, Liebeskind LS. J Am Chem Soc. 1984;106:4181.Murakami M, Itahashi T, Ito Y. J Am Chem Soc. 2002;124:13976. doi: 10.1021/ja021062n.Kondo T, Taguchi Y, Kaneko Y, Niimi M, Mitsudo T. Angew Chem Int Ed. 2004;43:5369. doi: 10.1002/anie.200461002.Auvinet AL, Harrity JPA. Angew Chem Int Ed. 2011;50:2769. doi: 10.1002/anie.201007598.Xu T, Dong G. Angew Chem Int Ed. 2012;51:7567. doi: 10.1002/anie.201202771.Angew Chem. 2012;124:7685.Xu T, Ko HM, Savage NA, Dong G. J Am Chem Soc. 2012;134:20005. doi: 10.1021/ja309978c.Ko HM, Dong G. Nat Chem. 2014;6:739. doi: 10.1038/nchem.1989.Chen PH, Xu T, Dong G. Angew Chem. 2014;126:1700. doi: 10.1002/anie.201310100.Angew Chem Int Ed. 2014;53:1674.Souillart L, Cramer N, Parker E. Angew Chem. 2014;126:3045. doi: 10.1002/anie.201311009.Angew Chem, Int Ed. 2014;53:3001.Parker E, Cramer N. Organometallics. 2014;33:780.Souillart L, Cramer N. Angew Chem. 2014;126:9794. doi: 10.1002/anie.201405834.Angew Chem Int Ed. 2014;53:9640.Deng L, Xu T, Li H, Dong G. J Am Chem Soc. 2016;138:369. doi: 10.1021/jacs.5b11120.Kondo T, Niimi M, Nomura M, Wada K, Mitsudo T. Tetrahedron Lett. 2007;48:2837.
- 3.For the seminal work, see: Murakami M, Ashida S, Matsuda T. J Am Chem Soc. 2005;127:6932. doi: 10.1021/ja050674f.. For selected examples, see: Murakami M, Ashida S. Chem Commun. 2006:643. doi: 10.1039/b611522e.Liu L, Ishida N, Murakami M. Angew Chem Int Ed. 2012;51:2485. doi: 10.1002/anie.201108446.Julia-Hernandez F, Ziadi A, Nishimura A, Martin R. Angew Chem Int Ed. 2015;54:9537. doi: 10.1002/anie.201503461.Kumar P, Louie J. Org Lett. 2012;14:2026. doi: 10.1021/ol300534j.Kumar P, Zhang K, Louie J. Angew Chem Int Ed. 2012;51:8602. doi: 10.1002/anie.201203521.Angew Chem. 2012;124:8730.Thakur A, Facer ME, Louie J. Angew Chem Int Ed. 2013;52:12161. doi: 10.1002/anie.201306869.Angew Chem. 2013;125:12383.
- 4.For selected reviews on allene-mediated cycloadditions, see: López F, Mascareñas JL. Chem Soc Rev. 2014;43:2904. doi: 10.1039/c4cs00024b.Croatt MP, Wender PA. Eur J Org Chem. 2010:19.
- 5.a) Hong X, Stevens MC, Liu P, Wender PA, Houk KN. J Am Chem Soc. 2014;136:17273. doi: 10.1021/ja5098308. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Miura T, Biyajima T, Toyoshima T, Murakami M. Beilstein J Org Chem. 2011;7:578. doi: 10.3762/bjoc.7.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.a) Tran YS, Kwon O. J Am Chem Soc. 2007;129:12632. doi: 10.1021/ja0752181. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Alcarazo M, Stork T, Anoop A, Thiel W, Fürstner A. Angew Chem Int Ed. 2010;49:2542. doi: 10.1002/anie.200907194. [DOI] [PubMed] [Google Scholar]; c) Toyoshima T, Miura T, Murakami M. Angew Chem Int Ed. 2011;50:10436. doi: 10.1002/anie.201105077. [DOI] [PubMed] [Google Scholar]; d) Jung E, Nishimura N. Org Lett. 2001;3:2113. doi: 10.1021/ol016073k. [DOI] [PubMed] [Google Scholar]
- 7.Zhou X, Dong G. J Am Chem Soc. 2015;137:13715. doi: 10.1021/jacs.5b09799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hartwig JF. Organotransition Metal Chemistry: From Bonding to Catalysis. University Science Books; Sausalito: 2010. [Google Scholar]
- 9.CCDC. 1504411–1504414 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from the Cambridge Crystallographic Data Centre via. www.ccdc.ac.uk/data_request/cif.
- 10.The corresponding malonate-tethered substrate gave no desired product, and the reason is unclear.
- 11.For a recent reviews on TADDOL-type phosphoramidite ligands, see: Teichert JF, Feringa BL. Angew Chem, Int Ed. 2010;49:2486. doi: 10.1002/anie.200904948.Angew Chem. 2010;122:2538.
- 12.At high concentration, substrate 1a only dissolved slowly during the course of the reaction. The resulting transient higher catalyst/substrate ratio may account for the higher enantioselectivity.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.