

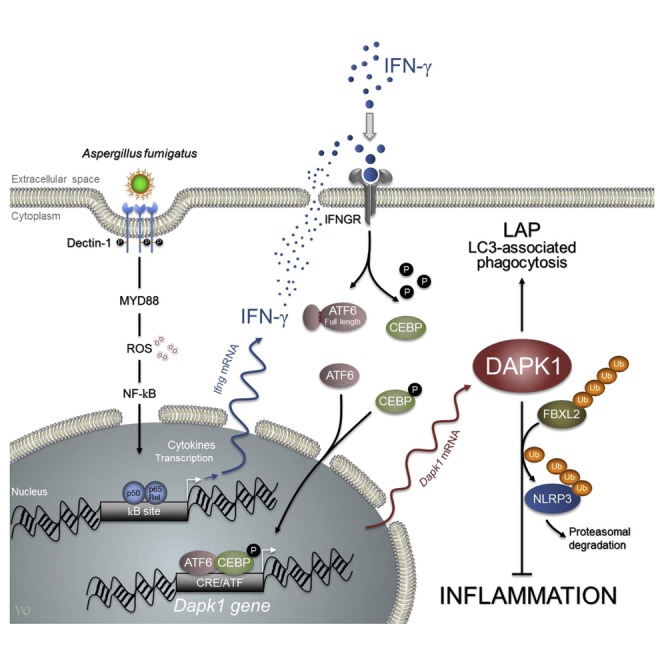
Summary
Defects in a form of noncanonical autophagy, known as LC3-associated phagocytosis (LAP), lead to increased inflammatory pathology during fungal infection. Although LAP contributes to fungal degradation, the molecular mechanisms underlying LAP-mediated modulation of inflammation are unknown. We describe a mechanism by which inflammation is regulated during LAP through the death-associated protein kinase 1 (DAPK1). The ATF6/C/EBP-β/DAPK1 axis activated by IFN-γ not only mediates LAP to Aspergillus fumigatus but also concomitantly inhibits Nod-like receptor protein 3 (NLRP3) activation and restrains pathogenic inflammation. In mouse models and patient samples of chronic granulomatous disease, which exhibit defective autophagy and increased inflammasome activity, IFN-γ restores reduced DAPK1 activity and dampens fungal growth. Additionally, in a cohort of hematopoietic stem cell-transplanted patients, a genetic DAPK1 deficiency is associated with increased inflammation and heightened aspergillosis susceptibility. Thus, DAPK1 is a potential drugable player in regulating the inflammatory response during fungal clearance initiated by IFN-γ.
Graphical Abstract
Highlights
-
•
IFN-γ restrains inflammation during fungal clearance via DAPK1
-
•
DAPK1 promotes noncanonical autophagy and NLRP3 proteasomal degradation
-
•
DAPK1 deficiency predicts infection and inflammation in CGD or transplanted patients
-
•
IFN-γ therapy restores defective DAPK1
Defects in noncanonical autophagy increase inflammatory pathology during fungal infection. Oikonomou et al. find that the kinase DAPK1 induced by IFN-γ promotes both noncanonical autophagy and NLRP3 inflammasome proteasomal degradation in response to Aspergillus fumigatus. By restoring DAPK1, IFN-γ may assist fungal clearance while restraining inflammation in mice and humans.
Introduction
Phagocytes fight pathogens using canonical and noncanonical autophagy pathways (Codogno et al., 2011). Also known as LC3-associated phagocytosis (LAP), this form of noncanonical autophagy is a unique pathway that links signaling during phagocytosis with optimal degradation of the phagocytosed cargo via recruitment of the LC3-PE conjugation system required for lysosomal fusion and maturation of the LAPosome (Mehta et al., 2014). LAP, but not canonical autophagy, plays a critical role in the degradation of engulfed Aspergillus conidia (Akoumianaki et al., 2016, de Luca et al., 2014, Kanayama et al., 2015b, Kyrmizi et al., 2013, Ma et al., 2012, Martinez et al., 2015). Accordingly, mice defective for LAP exhibit increased fungal burden. These mice, however, also exhibited increased pathological inflammation and pro-inflammatory cytokine levels. Thus, as observed in murine and human chronic granulomatous disease (CGD), in which LAP is defective (de Luca et al., 2014), the inflammation and infectious susceptibility are all regulated by LAP. By allowing efficient pathogen clearance and/or degradation of inflammatory mediators (Lapaquette et al., 2015), cassettes of autophagy proteins may play a broad role in cellular and immune homeostasis (Subramani and Malhotra, 2013). Indeed, defects in autophagic machinery have been linked with aberrant host defense and inflammatory diseases (Lapaquette et al., 2015, Levine et al., 2011, Netea-Maier et al., 2016). For LAP, in particular, emerging evidence suggests that this pathway also regulates, among others (Mehta et al., 2014), dead cell clearance and inflammation (Martinez et al., 2016). Thus, understanding the molecular mechanisms underlying LAP ability to modulate the inflammatory response during autophagy may have therapeutic implications.
IFN-γ is an essential cytokine in the protective host response against fungi (Romani, 2011) and has been implicated as a treatment option in invasive fungal infections (Delsing et al., 2014, Dignani et al., 2005). In addition to its immunometabolic activity (Cheng et al., 2016, Romani et al., 2008), IFN-γ also induces degradative autophagy (Al-Zeer et al., 2009, Gutierrez et al., 2004) via recruitment of immunity-related GTPase (Al-Zeer et al., 2009, Gutierrez et al., 2004, Kim et al., 2011, Singh et al., 2006) as well as the death-associated protein kinase 1 (DAPK1) (Gade et al., 2012). DAPK1 is a cell death-inducing Ca2+/calmodulin-regulated serine/threonine kinase, originally identified as an activator of IFN-γ-induced cell death (Gozuacik et al., 2008). DAPK1-induced expression by IFN-γ occurs through the transcription factor C/EBP-β in association with the key endoplasmic reticulum (ER) stress-activated transcription factor, ATF6 (Gade et al., 2012, Kalvakolanu and Gade, 2012). Recent findings have highlighted additional roles of this kinase beyond cell death (Lin et al., 2010) that include a negative regulation of inflammation (Lai and Chen, 2014) and attenuation of a variety of inflammatory responses in lung and airway injury (Nakav et al., 2012). Thus, DAPK1 could be an attractive player in the regulation of the inflammatory response during the process of fungal clearance initiated by IFN-γ.
In the present study, we have evaluated whether DAPK1 may contribute to dampening the inflammatory response during fungal LAP. To this purpose, we have resorted to a combination of basic and translational approaches involving selected deficient mice as well as human immunodeficiencies to establish that the ATF6/C/EBP-β/DAPK1 axis activated by IFN-γ not only mediates LAP to A. fumigatus but also regulates Nod-like receptor protein 3 (NLRP3) activation during infection. Genetic or functional DAPK1 deficiency increased inflammation and susceptibility to aspergillosis in high-risk patients. However, DAPK1 activity could be restored by IFN-γ. Thus, recognizing specific autophagy hallmarks triggered during infection may provide important insights into how autophagy and inflammation are integrated and reciprocally regulated to provide the optimal fungal clearance in the relative absence of immunopathology.
Results
Aspergillus fumigatus Conidia Induce IFN-γ-Dependent DAPK1 Expression In Vitro and In Vivo
In order to evaluate whether DAPK1 is induced in response to A. fumigatus, we monitored DAPK1 expression by RT-PCR, immunoblotting, and immunofluorescence staining either in vitro in RAW264.7 cells exposed to live conidia or in vivo in mice intranasally infected with the fungus. DAPK1 gene (Figure 1A) and protein (Figures 1B and 1C) expression was greatly upregulated at 4–6 hr of exposure to conidia, remained elevated till 10–12 hr, and declined thereafter. Germination of the fungus appeared to be required for DAPK1 expression to occur, being maximally induced by swollen conidia (Figure 1D). In vivo, DAPK1 gene (Figure 1E) and protein (Figure 1F) expression was increased during the first week of the infection in C57BL/6 mice. Consistent with the observation that DAPK1 is induced by IFN-γ (Gade et al., 2012), IFN-γ exposure significantly enhanced DAPK1 expression in RAW264.7 cells (Figures 1A–1C), and DAPK1 expression was defective in Ifng−/− mice in vivo (Figures 1E and 1F) and Ifng−/− macrophages in vitro (Figure 1G). Of interest, exogenous IFN-γ restored DAPK1 expression in IFN-γ deficiency in vivo (Figures 1E and 1F) and in vitro (Figure 1G), a finding suggesting that signaling through the IFN receptor is required for DAPK1 activation.
Figure 1.

A. fumigatus Conidia Induce IFN-γ-Dependent DAPK1 Expression In Vitro and In Vivo
(A–C) DAPK1 gene expression (A) and production by immunoblotting (B) and immunofluorescence (C) in RAW264.7 cells pulsed with live A. fumigatus conidia in the presence of rIFN-γ.
(D) DAPK1 expression in C57BL/6 lung macrophages after exposure to A. fumigatus heat-inactivated resting conidia (RC), swollen conidia (SC), or hyphae.
(E and F) DAPK1 gene (E) and protein (F) expression in lung of mice either uninfected (0 dpi, days post-infection) or infected with A. fumigatus conidia and treated with rIFN-γ.
(G) Immunoblotting of DAPK1 in purified lung macrophages exposed to live A. fumigatus conidia in vitro in the presence of rIFN-γ. Dapk1 gene expression was assessed by real-time qPCR and normalized on Gapdh.
For immunoblotting, normalization was performed on mouse β-actin (the corresponding pixel densities are indicated). For immunofluorescence, nuclei were counterstained with DAPI. Photographs were taken with a high-resolution microscope (Olympus BX51). Scale bars, 25 μm. DAPK1 fluorescence intensity was measured with the ImageJ software; BF, brightfield. Data (mean values ± SD) represent pooled results or representative images (immunofluorescence and immunoblotting) from three experiments. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001, rIFN-γ treated versus untreated RAW264.7 cells and rIFN-γ treated versus untreated Ifng−/− mice. None, unpulsed and/or untreated cells. See also Figure S1.
Because innate and non-innate cells are capable of producing IFN-γ in response to fungi (Romani, 2011), a paracrine action on myeloid cells for DAPK1 induction is likely. However, upon looking for IFN-γ expression and production by macrophages in response to the fungus, we found that IFN-γ gene expression increased in more than 85% of RAW264.7 cells after fungal exposure (Figure 2A) and was paralleled by increased protein production, as revealed by immunofluorescence (Figure 2B) and immunoblotting (Figure 2C). Similar results were obtained with purified lung macrophages (Figures 2D and 2E). The production of IFN-γ was functional, as evidenced by the upregulated expression of IP10 gene (Figure 2F), and occurred in response to Dectin-1 and MyD88 signaling (Figures 2G and 2H) via NF-kB (Figure 2I). Because DAPK1 expression paralleled that of IFN-γ (Figures 2J and 2K), these results suggest that IFN-γ production upon Dectin-1/MyD88 recognition of the fungus may autocrinally activate DAPK1 in phagocytic cells.
Figure 2.

A. fumigatus Conidia Induce IFN-γ Production In Vitro and In Vivo
(A–C) IFN-γ gene expression (A) and production by immunofluorescence (B) and immunoblotting (C) in RAW264.7 cells pulsed with live A. fumigatus conidia.
(D and E) IFN-γ production by immunoblotting (D) and ELISA (E) in C57BL/6 lung macrophages exposed to live A. fumigatus conidia.
(F) IP10 gene expression in RAW264.7 cells pulsed with live conidia in the presence of rIFN-γ.
(G and H) IFN-γ production by immunoblotting (G) and ELISA (H) in lung macrophages from C57BL/6, Dectin1−/−, or Myd88−/− mice exposed to conidia.
(I) Immunoblotting of IFN-γ in RAW264.7 cells exposed to conidia and treated with the NF-kB inhibitor, SN50.
(J and K) Dapk1 gene (J) and protein (K) expression in lung macrophages stimulated as above. Gene expression was assessed by real-time qPCR (normalization was performed on Gapdh).
For immunoblotting, normalization was performed on mouse β-actin or Gapdh (the corresponding pixel density is indicated). For immunofluorescence, nuclei were counterstained with DAPI. Photographs were taken with a high-resolution microscope (Olympus BX51). Scale bars, 100 μm. IFN-γ fluorescence intensity was measured with the ImageJ software. Data (mean values ± SD) represent pooled results or representative images (immunofluorescence and immunoblotting) from three experiments. ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001, conidia-pulsed versus unpulsed (none) or rIFN-γ treated versus untreated cells and knockouts versus wild-type mice.
DAPK1 Is Activated through the ATF6-C/EBP-β Pathway
The transcription factor C/EBP-β regulates transcription of DAPK1 in response to IFN-γ, in collaboration with ATF6, a key ER stress-activated transcription factor (Gade et al., 2012, Gade et al., 2014, Kalvakolanu and Gade, 2012). ER stress causes dissociation of ATF6 from its inhibitor, allowing its translocation from the ER membrane to the Golgi apparatus, where site-specific endoproteolysis generates transcriptionally active ATF6, permitting its nuclear entry and target gene regulation (Shen and Prywes, 2005). To evaluate the involvement of ATF6 and C/EBP-β in IFN-γ-dependent DAPK1 activation, we assessed ATF6 cleavage and C/EBP-β phosphorylation in RAW264.7 stimulated with IFN-γ and exposed to swollen conidia. Similar to tunicamycin, used as positive control (Kozutsumi et al., 1988), the cleaved form of ATF6 was observed upon stimulation and was enhanced by IFN-γ, as revealed by immunoblotting (Figure S1A, available online), immunofluorescence staining (Figure S1B), and nuclear translocation (Figure S1C). Conidia also activated C/EBP-β phosphorylation (Figure S1D) and its nuclear translocation (Figure S1E), an activity potentiated by IFN-γ. All together, these results demonstrated that A. fumigatus conidia activated the IFN-γ-ATF6-C/EBP-β/DAPK1 pathway.
DAPK1 Contributes to LC3-Dependent Aspergillus Autophagy
Increased accumulation of the autophagy marker GFP-LC3 in punctae representing the autophagosome membrane has been reported upon DAPK1 expression (Levin-Salomon et al., 2014). To evaluate the contribution of DAPK1 to A. fumigatus-induced LAP, we measured the LC3 punctae and the LC3B-II/LC3B-I ratio in RAW264.7 cells transiently transfected with the EGFP-LC3 plasmid, pulsed with swollen conidia, and depleted of DAPK1 by means of small interfering RNA (siRNA) or a chemical inhibitor. In line with previous observations (De Luca et al., 2012, Kyrmizi et al., 2013), the number of cells with punctate dots containing EGFP-LC3 increased (Figures 3A and 3B) upon phagocytosis of the fungus and was dependent on DAPK1, as did the ratio of LC3-II to LC3-I (Figure 3C). Not only did DAPK1 co-localize with LC3 in phagosomes containing conidia in more than 80% of cells, but not inert beads, which do not activate LAP (Martinez et al., 2015, Sanjuan et al., 2007) (Figure 3D), but its functional inhibition, while not affecting fungal phagocytosis (data not shown), also decreased the number of LC3 punctae and the LC3B-II/LC3B-I ratio (Figures 3A–3C). The involvement of DAPK1 appeared to be selective for LAP, as DAPK1 co-localized with the LAP components Rubicon, Beclin-1, and Atg7 (Martinez et al., 2015, Sanjuan et al., 2007) upon phagocytosis of conidia, but not beads, as revealed by cell immunofluorescence staining (Figures 3D and S2) and immunoblotting of purified phagosomes (Figures 3E and 3F). Consistent with the requirement of NOX2 for LAP (Martinez et al., 2015), silencing of Rubicon and NOX2, in addition to Beclin-1 and Atg7, with specific siRNA abrogated DAPK1's recruitment on phagosomes (Figures 3G and S6), suggesting that retention of DAPK1 on Aspergillus-containing phagosomes depends on the essential LAP protein Rubicon and additional autophagy proteins. Confirming the involvement of DAPK1 in LAP to the fungus, DAPK1 inhibition did not affect rapamycin-induced autophagy (Figures S3A and S3B), a finding confirming that DAPK1 is not involved in starvation-induced autophagy (Gozuacik and Kimchi, 2006). This finding is important, given that IFN-γ could activate autophagy through tryptophan starvation via the indoleamine 2,3-dioxygenase (IDO)1 enzyme (Metz et al., 2012). In this regard, we found that IFN-γ-induced autophagy was defective in Indo−/− macrophages and not modified by the DAPK1 inhibitor (Figure S3C). In contrast, LC3-dependendent phagocytosis of the fungus was observed in Indo−/− macrophages in vitro (Figure S3D), and DAPK1 expression increased in Indo−/− mice upon infection in vivo (Figure S3E). Blocking DAPK1 greatly increased the fungal burden and the dissemination in infected Indo−/− mice (Figure S3F), as well as the inflammatory cell recruitment in the lung and the bronchoalveolar lavage fluid (Figures S3G and S3H), a finding indicating that the starvation-induced autophagy and LAP are distinct, yet complementary, pathways in fungal clearance. Altogether, these results point to DAPK1 as an important effector molecule mediating IFN-γ-dependent LAP and the ensuing fungal clearance.
Figure 3.

DAPK1 Contributes to LC3-Dependent Aspergillus Autophagy
(A and B) GFP-LC3 punctae accumulation (A) (immunofluorescence) and percentage of GFP-LC3-positive cells (B) in RAW-GFP-LC3 cells after 1 hr stimulation with live A. fumigatus conidia + rIFN-γ in the presence of the DAPK inhibitor, Dapk1-specific siRNA (siDapk1), or negative control (Mock).
(C) LC3B-II/LC3B-I expression in RAW264.7 cells stimulated as above.
(D) Immunofluorescence imaging of RAW-GFP-LC3 or RAW264.7 cells pulsed with A. fumigatus conidia or inert beads in the presence of rIFN-γ.
(E–G) Purified phagosomes from RAW264.7 cells were pulsed with A. fumigatus conidia or inert beads for 1 hr in the presence of rIFN-γ or the indicated siRNAs to evaluate DAPK1 (E and G), Rubicon, Atg7, Beclin-1, and LC3B-II (F) expression by immunoblotting.
For immunofluorescence, nuclei were counterstained with DAPI. For immunoblotting, normalization was performed on mouse Gapdh (the corresponding ratio is indicated). Photographs were taken with a high-resolution microscope (Olympus BX51). Scale bar, 25 μm. BF, brightfield. Numbers refer to co-localization coefficients to quantify the overlap degree of DAPK1 and Beclin-1, Rubicon and Atg7. Data (mean values ± SD) represent pooled results or representative images (immunofluorescence and immunoblotting) from three experiments. ∗∗∗p < 0.001, DAPK inhibitor or siDapk1 treated versus untreated RAW-GFP-LC3 cells. None, unpulsed and untreated cells. See also Figure S2.
DAPK1 Activates the Proteasomal Degradation Pathway during LAP to Inhibit NLPR3
These results were corroborated by strong acidification (Figure 4A) and LAMP-1 (lysosomal-associated membrane protein 1) positivity (Figure 4B) in conidia-containing vacuoles, indicating fusion with lysosomes. Interestingly, and quite surprisingly, both acidification and LAMP-1 positivity were unaffected, if not increased, upon DAPK1 inhibition (Figures 4A and 4B), a finding suggesting that the lysosomal degradative pathway of fungal clearance was relatively independent from DAPK1.
Figure 4.

DAPK1 Activates the Proteosomal Degradation Pathway during LAP
(A and B) Acridine orange (A) and Lamp-1 (B) immunofluorescence staining in RAW264.7 cells pulsed with A. fumigatus conidia + rIFN-γ in the presence of the DAPK inhibitor. Acridine orange emits red or green fluorescence in acidic or neutral environments, respectively.
(C–E) Ubiquitin (C), SQSTM1/p62 immunoblotting (D), and SQSTM1/p62 (E) immunofluorescence staining in cells pulsed as above.
(F) Fluorometric quantification of proteosomal activity in RAW264.7 cells, lung macrophages, and neutrophils stimulated as above. Succ-LLVY-AMC, proteasome substrate that releases AMC-tagged peptide substrate in the presence of proteolytic activity.
For immunoblotting, normalization was performed on mouse β-actin (the corresponding pixel density is indicated). For immunofluorescence, nuclei were counterstained with DAPI. Photographs were taken with a high-resolution microscope (Olympus BX51). Scale bars, 100 and 25 μm. Arrows indicate conidia. Lamp-1 fluorescence intensity was measured with the ImageJ software. Data (mean values ± SD) represent pooled results or representative images (immunofluorescence and immunoblotting) from three experiments. ∗∗∗∗p < 0.0001, DAPK inhibitor treated versus untreated cells. None, unpulsed and untreated cells.
The ubiquitin- and adaptor p62-dependent autophagic targeting of damaged membranes that occurs during pathogen phagocytosis has been shown to help to control detrimental downstream signaling during bacterial invasion into host cells (Choi et al., 2014, Dupont et al., 2009, Zhao et al., 2008). Given the pleiotropic cellular effects of DAPK1 beyond kinase activity (Lin et al., 2010), which include degradation through ankyrin repeats (Shiloh et al., 2014), we tested whether DAPK1 could control the activation of the proteasomal degradation pathway during fungal clearance. We have recently found that this pathway crucially regulates NLRP3 inflammasome activity in response to the fungus that serves to curb inflammation (Iannitti et al., 2016). We found decreased cellular ubiquitination (Figure 4C), increased p62 expression (Figures 4D and 4E), and proteasomal degradation (Figure 4F) upon inhibition of DAPK1 in RAW264.7 cells or ex vivo phagocytes exposed to conidia in the presence of IFN-γ, a finding suggesting that the ubiquitin/proteasomal pathway is activated during fungal LAP and that DAPK1 is an active player. We looked therefore for NLRP3 expression and activity in RAW264.7 cells exposed to the fungus in vitro as well as in infection in vivo upon DAPK1 inhibition. We found that NLRP3 expression and caspase-1 cleavage were increased in vitro (Figures 5A and 5B). Ubiquitin-mediated proteasomal processing of NLRP3 involves the FBXO3 E3 ligase component that targets FBXL2, which robustly catalyzes NLRP3 ubiquitination (Han et al., 2015). The increased NLRP3 activity upon DAPK1 inhibition correlated with decreased NLRP3 ubiquitination (Figures 5C and S4) and decreased binding of FBXL2 to NLRP3 (Figure 5D). In vivo, DAPK1 inhibition with the chemical inhibitor or siRNA (Figures S5 and S6) was associated with increased NLRP3 expression (Figure 5E), caspase-1 cleavage (Figure 5F), IL-1β/IL-18 production and Mpo expression (Figure 5G), increased fungal load (Figure 5H), and tissue inflammation (Figure 5I), likely due to defective autophagy (Figure 5J). These effects were not observed in Nlrp3−/− mice treated with the DAPK1 inhibitor (Figures 5K and 5L), confirming that NLRP3 mediates the downstream effects on inflammation of DAPK1. Despite the ability of DAPK1 to regulate cell apoptosis via Bcl-2 expression (Akar et al., 2008), lung apoptosis was apparently unaffected upon DAPK1 inhibition (Figure 5H). These findings indicate that DAPK1 activation during LAP may restrain pathogenic NLRP3 activity in response to the fungus, likely impacting local inflammation.
Figure 5.

DAPK1 Activates FBXL2-Dependent NLRP3 Degradation
(A and B) NLRP3 (A) and caspase-1 (B) expression in RAW264.7 cells stimulated with A. fumigatus conidia + rIFN-γ in the presence of the DAPK inhibitor.
(C and D) Ubiquitin (C) and FBXL2 (D) immunoprecipitation followed by NLRP3, FBXL2, and ubiquitin immunoblotting.
(E–J) C57BL/6 mice were infected with A. fumigatus live conidia, treated with the DAPK1 inhibitor, and assessed (at 3 dpi) for NLRP3 expression (E), caspase-1 cleavage (F), cytokine production (ELISA on lung homogenates), Mpo expression on total lung cells (G), fungal burden (H), lung pathology (by PAS staining and Tunel assay) (I), and LC3B-II/LC3B-I ratio on total lung cells (J).
(K and L) Fungal burden (K) and lung pathology (L) (PAS staining) in Nlrp3−/− mice infected and treated as above.
For immunoblotting, normalization was performed on mouse β-actin or Gapdh and corresponding pixel density or ratio is depicted. For immunofluorescence, nuclei were counterstained with DAPI. Photographs were taken with a high-resolution microscope (Olympus BX51). Scale bars, 100 μm. Data (mean values ± SD) represent pooled results or representative images (immunofluorescence and immunoblotting) from three experiments. ∗∗p < 0.01, DAPK inhibitor treated versus untreated RAW264.7 cells or mice. Naive, uninfected mice. See also Figures S4 and S5.
IFN-γ Restores Defective DAPK1 in CGD
The above results prompted us to evaluate whether the ability of IFN-γ to activate DAPK1 could be therapeutically exploited. To this purpose, we assessed DAPK1 expression and its modulation by IFN-γ in murine and human CGD, in which both defective autophagy and increased inflammasome activity have been described (de Luca et al., 2014). DAPK1 gene and protein expression was defective in p47phox−/− infected mice (Figures 6A and 6B) and in vitro in both p47phox−/− lung macrophages (Figures 6C–6E) and monocytes from CGD patients (Figure 6F). IFN-γ protein expression (Figure 6G) and production (Figure 6H) were also defective in CGD cells, a finding indicating that reactive oxygen species (ROS) are upstream signals for IFN-γ induction. Of great interest, treatment with IFN-γ restored DAPK1 expression (Figures 6A and 6B), reduced the fungal growth (Figure 6I), and decreased IL-1β production (Figure 6J) in infection. Similarly, in vitro, IFN-γ restored DAPK1 expression (from <5% to up 90% double positive cells after treatment) (Figures 6C–6E) and decreased inflammation in murine (Figure 6K) and human CGD mononuclear cells (Figures 6F, 6L, and 6M), a finding suggesting the drugability of DAPK1 in CGD by IFN-γ therapy. Incidentally, our unpublished observations suggest that anakinra, known to decrease NLRP3 activity and restore LAP in both murine and human CGD cells (de Luca et al., 2014), also activates DAPK1.
Figure 6.

IFN-γ Restores Defective DAPK1 in CGD
(A and B) C57BL/6 and p47phox−/− mice were infected with A. fumigatus conidia, treated with rIFN-γ, and assessed at the indicated days post-infection (dpi) for Dapk1 gene (A) and protein (B) expression.
(C and D) Dapk1 gene (C) and protein (D) expression in lung macrophages from C57BL/6 and p47phox−/− mice after pulsing with A. fumigatus conidia in the presence of rIFN-γ.
(E and F) LC3B and DAPK1 expression in macrophages (E) from p47phox−/− mice and monocytes (F) from CGD patients stimulated as in (C).
(G and H) IFN-γ expression (G) and production (H) on purified lung macrophages exposed to A. fumigatus conidia in vitro.
(I and J) Fungal burden (I) and IL-1β production (J) (ELISA on lung homogenates) evaluated at 3 dpi in C57BL/6 and p47phox−/− mice infected and treated as above.
(K) IL-1β production (ELISA on culture supernatant) in lung macrophages from C57BL/6 and p47phox−/− treated as in (C).
(L and M) NLRP3 (L) and caspase-1 cleavage (M) in monocytes from healthy control (Ct) or CGD patients pulsed with A. fumigatus conidia in the presence of rIFN-γ.
For immunoblotting, normalization was performed on mouse β-actin or Gapdh and corresponding pixel density or ratio is depicted. For immunofluorescence, nuclei were counterstained with DAPI. Photographs were taken with a high-resolution microscope (Olympus BX51). Scale bars, 25 μm. Data (mean values ± SD) represent pooled results or representative images (immunofluorescence and immunoblotting) from three experiments. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001, rIFN-γ treated versus untreated cells or mice. None, unpulsed and untreated cells.
DAPK1 Polymorphisms Affect Susceptibility to Aspergillosis in Hematopoietic Transplanted Patients
Although aspergillosis remains a major cause of morbidity and mortality in CGD patients (Marciano et al., 2015), it is a much more common problem in hematopoietic stem cell transplantation (HSCT) patients (Harrison et al., 2015). We therefore assessed the impact of DAPK1 in this setting by analyzing five DAPK1 SNPs in a cohort of HSCT patients. In the univariate analysis, applying a general genetic model, none of the DAPK1 SNPs significantly associated with an increased incidence of infection, either in recipients or in donors (Table S1). However, the DAPK1 rs1964911 SNP, located on the promoter region, showed a trend for association in recipients (p = 0.066). Indeed, by testing the association of rs1964911 using a recessive genetic model, the C/C genotype turned out to be significantly associated with an increased incidence of aspergillosis in recipients (30.2% for C/C and 16.2% for A carriers, p = 0.029) (Figure 7A), but not in donors (24.2% for C/C and 16.7% for A carriers, p = 0.219). The analysis of DAPK1 rs1964911 recessive recipient/donor combination further confirmed the effect of C/C genotype in increasing the risk of infection, especially when present simultaneously in both recipients and donors (42.1% for C/C pairs versus 17.4% for A carriers pairs, p = 0.029) (Figure 7B). The association of C/C genotype with increased risk of infection remained statistically significant after adjusting for HLA-matching status, underlying disease, and antifungal prophylaxis (donors, RR = 1.873, p = 0.036; donors/recipients combined, RR = 2.184, p = 0.027). Functional studies in vitro revealed that both DAPK1 gene (Figure 7C) and protein (Figures 7D and 7E) were defective in peripheral mononuclear cells from patients with the C/C genotype, while NLRP3 expression (Figure 7F) and IL-1β production (Figure 7G) were increased. Thus, genetic deficiency of DAPK1 may contribute to the risk of invasive aspergillosis in HSCT patients.
Figure 7.

DAPK1 rs1964911 Is Associated with Increased Risk of Invasive Aspergillosis in HSCT Recipients
(A and B) Cumulative incidence of invasive aspergillosis at 24 months in recipients (C/C versus A carriers, p = 0.029) (A) and cumulative incidence of invasive aspergillosis at 24 months in recipient/donor combination (B) (full model, p = 0.029).
(C–G) DAPK1 gene expression (C) and production (D and E), NLRP3 expression (F), and IL-1β production (G) in macrophages isolated from peripheral mononuclear blood cells of subjects carrying diverse genotypes at rs1964911 stimulated with A. fumigatus conidia.
Normalization was performed on Gapdh for real-time PCR and on human β-actin for immunoblotting. For immunofluorescence, nuclei were counterstained with DAPI. Photographs were taken with a high-resolution microscope (Olympus BX51). Scale bars, 25 μm. Data (mean values ± SD) represent pooled results or representative images (immunofluorescence and immunoblotting) from three experiments. ∗∗p < 0.01, ∗∗∗p < 0.001, CC versus A+ genotype. None, unstimulated cells. See also Table S1.
Discussion
The results of the present study identify a host defense pathway controlled by IFN-γ signaling. We found that IFN-γ not only mediated LAP to A. fumigatus but also regulated NLRP3 activation during infection. The importance of LAP for fungal clearance (Kyrmizi et al., 2013, Martinez et al., 2015, Sprenkeler et al., 2016) and its subversion by fungal melanin (Akoumianaki et al., 2016) have been recently described. However, inflammation has to be controlled under microbial autophagy in the lung to prevent pulmonary overreaction (Kanayama et al., 2015a). We found the DAPK1 may serve this function. The DAPK1 interactome has shown that DAPK1, by participating in several signaling cascades by means of its catalytic activity and protein-protein interactions (Bialik and Kimchi, 2014), mediates various cellular effects. This may explain the complex role of DAPK1 in the regulation of inflammation, which is likely dependent on cell types and environment (Lai and Chen, 2014). Indeed, while DAPK1 deficiency attenuated inflammasome-induced peritonitis (Chuang et al., 2011), it increased LPS-induced inflammation in the lung, an activity dependent upon its inflammation-suppressive functions rather than the prevention of the inflammatory cell recruitment (Nakav et al., 2012). Consistently, DAPK1 was shown to activate the translational silencing of various inflammatory genes in response to IFN-γ (Mukhopadhyay et al., 2008). Our study expands upon the molecular mechanisms by which DAPK1 regulates the inflammatory response in the lung by showing that it inhibits NLRP3 activity by promoting its proteasomal degradation via FBXL2, as reported (Han et al., 2015). This finding is only apparently in contrast with what was observed in 293T cells, where DAPK1 served a structural role in the assembly of the NLRP3 inflammasome (Chuang et al., 2011). Indeed, the DAPK1 requirement for NLRP3 activation was stimulus dependent; NLRP3 activity was ablated upon DAPK deficiency in response to TLR2 and TLR7, but not TLR4 signaling (Chuang et al., 2011). As a matter of fact, LPS-induced intestinal inflammation was increased in DAPK1 deficiency in mice (Nakav et al., 2012) and humans where enhanced severity of gut inflammation and ulcerative colitis was observed (Kuester et al., 2010). Promotion of NLRP3 activity was meant to contribute to DAPK1-induced cell death (Chuang et al., 2011), an activity that we have not seen in the lung. Therefore, DAPK1 activation initiated by IFN-γ may serve a unique role in the lung by tightly regulating inflammasome activity during fungal LAP through a mechanism that is shared with autophagy (Shi et al., 2012).
The ubiquitin-proteasomal pathway appeared to be involved in this process, for both cellular ubiquitination and adaptor p62 expression were decreased or increased, respectively, after DAPK1 inhibition. That the ubiquitin-like conjugation system of the autophagy pathway, but not the canonical degradative autophagy process, is required for IFN-γ-induced LAP in intracellular infection has been reported (Choi et al., 2014). Interestingly, we have found that conidia may undergo ubiquitination during phagocytosis (Figure 4A), a finding suggesting the occurrence of xenophagy, a process that targets intravacuolar microbes for ubiquitination followed by recruitment of, among others, p62 adaptor protein and LC3, and leading to autophagosome fusion with lysosomes for degradation (Choi et al., 2014, Deretic et al., 2013). In this regard, we have already shown that inhibitors of both the lysosomal and proteasomal pathways negatively affected the ability of phagocytes to control intracellular fungal growth (Iannitti et al., 2016). Precisely how the ubiquitin-proteasomal degradative pathway is activated by DAPK1 to inhibit NLRP3, and whether the activation of the immunoproteasome by IFN-γ (Aki et al., 1994) is also involved, remains to be determined. It is nevertheless of interest that the ankyrin repeats of DAPK1 may bind E3 ubiquitin ligases (Jin et al., 2002) and that the immunoproteasome is implicated in fungal pathogenicity (Mundt et al., 2016) and lung responses to infections (Keller et al., 2015).
IFN-γ, produced by innate and adaptive lymphocytes, plays a critical role in mediating protection against fungi (Romani, 2011). The results of the present study show that macrophages are alternative sources of IFN-γ produced upon Dectin-1/MyD88 stimulation and ROS signaling, a finding suggesting that early production of IFN-γ by macrophages may occur in the relative absence of innate or adaptive immune cells and, as such, be exploitable in selected immunodeficiencies. Combined with antifungal drug therapy, immunotherapy with IFN-γ represents a treatment option in patients at risk for fungal infections (The International Chronic Granulomatous Disease Cooperative Study Group, 1991, Armstrong-James et al., 2010, Bodasing et al., 2002, Delsing et al., 2014, Dignani et al., 2005, Stevens et al., 2006). The results of the present study increase our understanding of the immunopharmacology of IFN-γ in infection treatment that now includes an activity on DAPK1. This may have therapeutic implications given that DAPK expression and activity are deregulated in a variety of diseases including cancer (Li et al., 2015). We have found that DAPK1 activity was defective in CGD but restored by IFN-γ. We have also found that genetic DAPK1 deficiency increased inflammation and susceptibility of HSCT patients to aspergillosis. These findings, while reinforcing the therapeutic utility of IFN-γ in human CGD, also suggest a possible IFN-γ recommendation in transplanted patients.
In conclusion, it appears that IFN-γ, through its multitasking activity encompassing immune tolerance via the IDO1 pathway (Romani et al., 2008), immunometabolic activity (Cheng et al., 2016, Leentjens et al., 2012), and, now, optimal microbial clearance through the DAPK1-LAP pathway, fulfills the requirement of an ideal candidate player at the host-fungi interface.
Experimental Procedures
Detailed experimental procedures can be found in the Supplemental Experimental Procedures.
Mice
Mice, 8–10 weeks old, were purchased from Charles River or bred at the Animal Facility of Perugia.
Fungal Strains, Infections, and Treatments
For infection, mice received intranasal instillation of A. fumigatus conidia. A total of 20,000 U/mouse IFN-γ was given daily starting the day of the infection until the sacrifice. DAPK inhibitor was administered intranasally at the dose of 500 μg/kg for 5 days starting 3 days before the infection. For Dapk1 silencing, mice received intranasal administration of 10 mg/kg Dapk1 siRNA.
Cell Preparation, Culture, and Transfection
RAW264.7 and RAW-GFP-LC3 cells were grown in supplemented RPMI medium as described (De Luca et al., 2012). Alveolar macrophages from mice lung were isolated after plastic adherence at 37°C. Cells were pretreated overnight with 200 U/mL rIFN-γ before pulsing with A. fumigatus resting or swollen conidia. For DAPK1 inhibition, cells were pretreated with 3 μM DAPK inhibitor as described (Usui et al., 2012). Rapamycin was used at 50 μM. For NF-kB inhibition, SN50 was used at 18 μM for 1 hr before A. fumigatus pulsing. For in vitro silencing, cells were transfected with 40 nM specific siRNAs before A. fumigatus or inert bead stimulation.
Immunoblotting and Immunoprecipitation
For immunoprecipitation, the lysates were incubated with anti-ubiquitin, anti-FBXL2 antibody, and Protein G Sepharose beads. For immunoblotting, the lysates were incubated with the following antibodies: DAP kinase 1, ATF6; C/EBPβ and p-C/EBPβ, LC3B, SQSTM1/p62, FBXL2, HA, ubiquitin, caspase-1 p10, IFN-γ, Lamin B1, NLRP3, and caspase-1. Phagosomes from RAW264.7 cells were obtained as described (Martinez et al., 2015) and the proteins were probed with DAP kinase 1, LC3B, Beclin-1, Rubicon, and Atg7.
ELISA and Real-Time PCR
Cytokines in lung homogenates or cell culture supernatants was determined by specific ELISAs (R&D Systems). Real-time RT-PCR was performed using SYBR Green chemistry (Biorad).
Immunofluorescence and Acridine Orange Staining
Fixed cells were incubated with DAPK1, ATF6, C/EBPβ, p-C/EBPβ, IFN-γ, LC3B, SQSTM1/p62, LAMP1, NLRP3, and ubiquitin antibodies. Acridine orange was used to detect the cellular acidic compartment by measuring the red fluorescence emission.
Human Studies
Isolation and Culture of Monocytes from Human PBMCs
Monocytes were isolated from peripheral blood mononuclear cells (PBMCs) of healthy donors or two CGD patients, harboring the mutations c.736C>T, p.Q246X and whole CYBB gene deletion (69.84 kb). Cells were cultured for 4 hr with live A. fumigatus conidia in the presence of 200 U/mL rIFN-γ before assays.
Patients
The genetic study included 277 patients undergoing allogeneic HSCT and their respective donors (who were related in 98% of cases). Patient characteristics are summarized in Table S1.
SNP Selection and Genotyping
Five DAPK1 SNPs complied with the selection criteria: rs1964911, rs4878104, rs4877365, rs7025760, and rs1056719. Genotyping was performed using KASPar assays.
Statistical Analysis
Data are expressed as mean ± SD. p values were calculated by a one-way ANOVA (Bonferroni’s post hoc test). For single comparison, p values were calculated by a two-tailed Student’s t test. The probability of IA according to DAPK1 variants was determined with the use of the cumulative incidence method, and DAPK1 status among patients with and without infection was compared with the use of Gray’s test. Cumulative incidences were computed with R software.
Author Contributions
V.O. and G.R. performed most of the in vitro experiments; M.B. performed murine in vivo experiments; M. Pariano performed some immunofluorescence experiments; M. Puccetti and C.G. performed SNP analysis and V.N. analyzed and compiled the SNP data; L.A., A.C., L.P., A.S., and A.F. followed patients and provided clinical samples; and V.O., S.M., C.A.P., P.R., A.V., F.A., and L.R. designed the experiments, analyzed the data, and wrote the paper.
Acknowledgments
This study was supported by the Specific Targeted Research Project FunMeta (ERC-2011-AdG-293714 to L.R.).
Published: November 23, 2016
Footnotes
Supplemental Information includes Supplemental Experimental Procedures, six figures, and one table and can be found with this article online at http://dx.doi.org/10.1016/j.chom.2016.10.012.
Supplemental Information
References
- Akar U., Chaves-Reyez A., Barria M., Tari A., Sanguino A., Kondo Y., Kondo S., Arun B., Lopez-Berestein G., Ozpolat B. Silencing of Bcl-2 expression by small interfering RNA induces autophagic cell death in MCF-7 breast cancer cells. Autophagy. 2008;4:669–679. doi: 10.4161/auto.6083. [DOI] [PubMed] [Google Scholar]
- Aki M., Shimbara N., Takashina M., Akiyama K., Kagawa S., Tamura T., Tanahashi N., Yoshimura T., Tanaka K., Ichihara A. Interferon-gamma induces different subunit organizations and functional diversity of proteasomes. J. Biochem. 1994;115:257–269. doi: 10.1093/oxfordjournals.jbchem.a124327. [DOI] [PubMed] [Google Scholar]
- Akoumianaki T., Kyrmizi I., Valsecchi I., Gresnigt M.S., Samonis G., Drakos E., Boumpas D., Muszkieta L., Prevost M.C., Kontoyiannis D.P. Aspergillus cell wall melanin blocks LC3-associated phagocytosis to promote pathogenicity. Cell Host Microbe. 2016;19:79–90. doi: 10.1016/j.chom.2015.12.002. [DOI] [PubMed] [Google Scholar]
- Al-Zeer M.A., Al-Younes H.M., Braun P.R., Zerrahn J., Meyer T.F. IFN-gamma-inducible Irga6 mediates host resistance against Chlamydia trachomatis via autophagy. PLoS ONE. 2009;4:e4588. doi: 10.1371/journal.pone.0004588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong-James D., Teo I.A., Shrivastava S., Petrou M.A., Taube D., Dorling A., Shaunak S. Exogenous interferon-gamma immunotherapy for invasive fungal infections in kidney transplant patients. Am. J. Transplant. 2010;10:1796–1803. doi: 10.1111/j.1600-6143.2010.03094.x. [DOI] [PubMed] [Google Scholar]
- Bialik S., Kimchi A. The DAP-kinase interactome. Apoptosis. 2014;19:316–328. doi: 10.1007/s10495-013-0926-3. [DOI] [PubMed] [Google Scholar]
- Bodasing N., Seaton R.A., Shankland G.S., Pithie A. Gamma-interferon treatment for resistant oropharyngeal candidiasis in an HIV-positive patient. J. Antimicrob. Chemother. 2002;50:765–766. doi: 10.1093/jac/dkf206. [DOI] [PubMed] [Google Scholar]
- Cheng S.C., Scicluna B.P., Arts R.J., Gresnigt M.S., Lachmandas E., Giamarellos-Bourboulis E.J., Kox M., Manjeri G.R., Wagenaars J.A., Cremer O.L. Broad defects in the energy metabolism of leukocytes underlie immunoparalysis in sepsis. Nat. Immunol. 2016;17:406–413. doi: 10.1038/ni.3398. [DOI] [PubMed] [Google Scholar]
- Choi J., Park S., Biering S.B., Selleck E., Liu C.Y., Zhang X., Fujita N., Saitoh T., Akira S., Yoshimori T. The parasitophorous vacuole membrane of Toxoplasma gondii is targeted for disruption by ubiquitin-like conjugation systems of autophagy. Immunity. 2014;40:924–935. doi: 10.1016/j.immuni.2014.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang Y.T., Lin Y.C., Lin K.H., Chou T.F., Kuo W.C., Yang K.T., Wu P.R., Chen R.H., Kimchi A., Lai M.Z. Tumor suppressor death-associated protein kinase is required for full IL-1β production. Blood. 2011;117:960–970. doi: 10.1182/blood-2010-08-303115. [DOI] [PubMed] [Google Scholar]
- Codogno P., Mehrpour M., Proikas-Cezanne T. Canonical and non-canonical autophagy: variations on a common theme of self-eating? Nat. Rev. Mol. Cell Biol. 2011;13:7–12. doi: 10.1038/nrm3249. [DOI] [PubMed] [Google Scholar]
- De Luca A., Iannitti R.G., Bozza S., Beau R., Casagrande A., D’Angelo C., Moretti S., Cunha C., Giovannini G., Massi-Benedetti C. CD4(+) T cell vaccination overcomes defective cross-presentation of fungal antigens in a mouse model of chronic granulomatous disease. J. Clin. Invest. 2012;122:1816–1831. doi: 10.1172/JCI60862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Luca A., Smeekens S.P., Casagrande A., Iannitti R., Conway K.L., Gresnigt M.S., Begun J., Plantinga T.S., Joosten L.A., van der Meer J.W. IL-1 receptor blockade restores autophagy and reduces inflammation in chronic granulomatous disease in mice and in humans. Proc. Natl. Acad. Sci. USA. 2014;111:3526–3531. doi: 10.1073/pnas.1322831111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delsing C.E., Gresnigt M.S., Leentjens J., Preijers F., Frager F.A., Kox M., Monneret G., Venet F., Bleeker-Rovers C.P., van de Veerdonk F.L. Interferon-gamma as adjunctive immunotherapy for invasive fungal infections: a case series. BMC Infect. Dis. 2014;14:166. doi: 10.1186/1471-2334-14-166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deretic V., Saitoh T., Akira S. Autophagy in infection, inflammation and immunity. Nat. Rev. Immunol. 2013;13:722–737. doi: 10.1038/nri3532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dignani M.C., Rex J.H., Chan K.W., Dow G., deMagalhaes-Silverman M., Maddox A., Walsh T., Anaissie E. Immunomodulation with interferon-gamma and colony-stimulating factors for refractory fungal infections in patients with leukemia. Cancer. 2005;104:199–204. doi: 10.1002/cncr.21142. [DOI] [PubMed] [Google Scholar]
- Dupont N., Lacas-Gervais S., Bertout J., Paz I., Freche B., Van Nhieu G.T., van der Goot F.G., Sansonetti P.J., Lafont F. Shigella phagocytic vacuolar membrane remnants participate in the cellular response to pathogen invasion and are regulated by autophagy. Cell Host Microbe. 2009;6:137–149. doi: 10.1016/j.chom.2009.07.005. [DOI] [PubMed] [Google Scholar]
- Gade P., Ramachandran G., Maachani U.B., Rizzo M.A., Okada T., Prywes R., Cross A.S., Mori K., Kalvakolanu D.V. An IFN-γ-stimulated ATF6-C/EBP-β-signaling pathway critical for the expression of death associated protein kinase 1 and induction of autophagy. Proc. Natl. Acad. Sci. USA. 2012;109:10316–10321. doi: 10.1073/pnas.1119273109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gade P., Manjegowda S.B., Nallar S.C., Maachani U.B., Cross A.S., Kalvakolanu D.V. Regulation of the death-associated protein kinase 1 expression and autophagy via ATF6 requires apoptosis signal-regulating kinase 1. Mol. Cell. Biol. 2014;34:4033–4048. doi: 10.1128/MCB.00397-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gozuacik D., Kimchi A. DAPk protein family and cancer. Autophagy. 2006;2:74–79. doi: 10.4161/auto.2.2.2459. [DOI] [PubMed] [Google Scholar]
- Gozuacik D., Bialik S., Raveh T., Mitou G., Shohat G., Sabanay H., Mizushima N., Yoshimori T., Kimchi A. DAP-kinase is a mediator of endoplasmic reticulum stress-induced caspase activation and autophagic cell death. Cell Death Differ. 2008;15:1875–1886. doi: 10.1038/cdd.2008.121. [DOI] [PubMed] [Google Scholar]
- Gutierrez M.G., Master S.S., Singh S.B., Taylor G.A., Colombo M.I., Deretic V. Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell. 2004;119:753–766. doi: 10.1016/j.cell.2004.11.038. [DOI] [PubMed] [Google Scholar]
- Han S., Lear T.B., Jerome J.A., Rajbhandari S., Snavely C.A., Gulick D.L., Gibson K.F., Zou C., Chen B.B., Mallampalli R.K. Lipopolysaccharide primes the NALP3 inflammasome by inhibiting its ubiquitination and degradation mediated by the SCFFBXL2 E3 ligase. J. Biol. Chem. 2015;290:18124–18133. doi: 10.1074/jbc.M115.645549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison N., Mitterbauer M., Tobudic S., Kalhs P., Rabitsch W., Greinix H., Burgmann H., Willinger B., Presterl E., Forstner C. Incidence and characteristics of invasive fungal diseases in allogeneic hematopoietic stem cell transplant recipients: a retrospective cohort study. BMC Infect. Dis. 2015;15:584. doi: 10.1186/s12879-015-1329-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iannitti R.G., Napolioni V., Oikonomou V., De Luca A., Galosi C., Pariano M., Massi-Benedetti C., Borghi M., Puccetti M., Lucidi V. IL-1 receptor antagonist ameliorates inflammasome-dependent inflammation in murine and human cystic fibrosis. Nat. Commun. 2016;7:10791. doi: 10.1038/ncomms10791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Y., Blue E.K., Dixon S., Shao Z., Gallagher P.J. A death-associated protein kinase (DAPK)-interacting protein, DIP-1, is an E3 ubiquitin ligase that promotes tumor necrosis factor-induced apoptosis and regulates the cellular levels of DAPK. J. Biol. Chem. 2002;277:46980–46986. doi: 10.1074/jbc.M208585200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalvakolanu D.V., Gade P. IFNG and autophagy: a critical role for the ER-stress mediator ATF6 in controlling bacterial infections. Autophagy. 2012;8:1673–1674. doi: 10.4161/auto.21403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanayama M., He Y.W., Shinohara M.L. The lung is protected from spontaneous inflammation by autophagy in myeloid cells. J. Immunol. 2015;194:5465–5471. doi: 10.4049/jimmunol.1403249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanayama M., Inoue M., Danzaki K., Hammer G., He Y.W., Shinohara M.L. Autophagy enhances NFκB activity in specific tissue macrophages by sequestering A20 to boost antifungal immunity. Nat. Commun. 2015;6:5779. doi: 10.1038/ncomms6779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller I.E., Vosyka O., Takenaka S., Kloß A., Dahlmann B., Willems L.I., Verdoes M., Overkleeft H.S., Marcos E., Adnot S. Regulation of immunoproteasome function in the lung. Sci. Rep. 2015;5:10230. doi: 10.1038/srep10230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim B.H., Shenoy A.R., Kumar P., Das R., Tiwari S., MacMicking J.D. A family of IFN-γ-inducible 65-kD GTPases protects against bacterial infection. Science. 2011;332:717–721. doi: 10.1126/science.1201711. [DOI] [PubMed] [Google Scholar]
- Kozutsumi Y., Segal M., Normington K., Gething M.J., Sambrook J. The presence of malfolded proteins in the endoplasmic reticulum signals the induction of glucose-regulated proteins. Nature. 1988;332:462–464. doi: 10.1038/332462a0. [DOI] [PubMed] [Google Scholar]
- Kuester D., Guenther T., Biesold S., Hartmann A., Bataille F., Ruemmele P., Peters B., Meyer F., Schubert D., Bohr U.R. Aberrant methylation of DAPK in long-standing ulcerative colitis and ulcerative colitis-associated carcinoma. Pathol. Res. Pract. 2010;206:616–624. doi: 10.1016/j.prp.2010.05.004. [DOI] [PubMed] [Google Scholar]
- Kyrmizi I., Gresnigt M.S., Akoumianaki T., Samonis G., Sidiropoulos P., Boumpas D., Netea M.G., van de Veerdonk F.L., Kontoyiannis D.P., Chamilos G. Corticosteroids block autophagy protein recruitment in Aspergillus fumigatus phagosomes via targeting dectin-1/Syk kinase signaling. J. Immunol. 2013;191:1287–1299. doi: 10.4049/jimmunol.1300132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai M.Z., Chen R.H. Regulation of inflammation by DAPK. Apoptosis. 2014;19:357–363. doi: 10.1007/s10495-013-0933-4. [DOI] [PubMed] [Google Scholar]
- Lapaquette P., Guzzo J., Bretillon L., Bringer M.A. Cellular and molecular connections between autophagy and inflammation. Mediators Inflamm. 2015;2015:398483. doi: 10.1155/2015/398483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leentjens J., Kox M., Koch R.M., Preijers F., Joosten L.A., van der Hoeven J.G., Netea M.G., Pickkers P. Reversal of immunoparalysis in humans in vivo: a double-blind, placebo-controlled, randomized pilot study. Am. J. Respir. Crit. Care Med. 2012;186:838–845. doi: 10.1164/rccm.201204-0645OC. [DOI] [PubMed] [Google Scholar]
- Levin-Salomon V., Bialik S., Kimchi A. DAP-kinase and autophagy. Apoptosis. 2014;19:346–356. doi: 10.1007/s10495-013-0918-3. [DOI] [PubMed] [Google Scholar]
- Levine B., Mizushima N., Virgin H.W. Autophagy in immunity and inflammation. Nature. 2011;469:323–335. doi: 10.1038/nature09782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y., Zhu M., Zhang X., Cheng D., Ma X. Clinical significance of DAPK promoter hypermethylation in lung cancer: a meta-analysis. Drug Des. Devel. Ther. 2015;9:1785–1796. doi: 10.2147/DDDT.S78012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y., Hupp T.R., Stevens C. Death-associated protein kinase (DAPK) and signal transduction: additional roles beyond cell death. FEBS J. 2010;277:48–57. doi: 10.1111/j.1742-4658.2009.07411.x. [DOI] [PubMed] [Google Scholar]
- Ma J., Becker C., Lowell C.A., Underhill D.M. Dectin-1-triggered recruitment of light chain 3 protein to phagosomes facilitates major histocompatibility complex class II presentation of fungal-derived antigens. J. Biol. Chem. 2012;287:34149–34156. doi: 10.1074/jbc.M112.382812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marciano B.E., Spalding C., Fitzgerald A., Mann D., Brown T., Osgood S., Yockey L., Darnell D.N., Barnhart L., Daub J. Common severe infections in chronic granulomatous disease. Clin. Infect. Dis. 2015;60:1176–1183. doi: 10.1093/cid/ciu1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez J., Malireddi R.K., Lu Q., Cunha L.D., Pelletier S., Gingras S., Orchard R., Guan J.L., Tan H., Peng J. Molecular characterization of LC3-associated phagocytosis reveals distinct roles for Rubicon, NOX2 and autophagy proteins. Nat. Cell Biol. 2015;17:893–906. doi: 10.1038/ncb3192. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Martinez J., Cunha L.D., Park S., Yang M., Lu Q., Orchard R., Li Q.Z., Yan M., Janke L., Guy C. Noncanonical autophagy inhibits the autoinflammatory, lupus-like response to dying cells. Nature. 2016;533:115–119. doi: 10.1038/nature17950. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Mehta P., Henault J., Kolbeck R., Sanjuan M.A. Noncanonical autophagy: one small step for LC3, one giant leap for immunity. Curr. Opin. Immunol. 2014;26:69–75. doi: 10.1016/j.coi.2013.10.012. [DOI] [PubMed] [Google Scholar]
- Metz R., Rust S., Duhadaway J.B., Mautino M.R., Munn D.H., Vahanian N.N., Link C.J., Prendergast G.C. IDO inhibits a tryptophan sufficiency signal that stimulates mTOR: a novel IDO effector pathway targeted by D-1-methyl-tryptophan. OncoImmunology. 2012;1:1460–1468. doi: 10.4161/onci.21716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay R., Ray P.S., Arif A., Brady A.K., Kinter M., Fox P.L. DAPK-ZIPK-L13a axis constitutes a negative-feedback module regulating inflammatory gene expression. Mol. Cell. 2008;32:371–382. doi: 10.1016/j.molcel.2008.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mundt S., Basler M., Buerger S., Engler H., Groettrup M. Inhibiting the immunoproteasome exacerbates the pathogenesis of systemic Candida albicans infection in mice. Sci. Rep. 2016;6:19434. doi: 10.1038/srep19434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakav S., Cohen S., Feigelson S.W., Bialik S., Shoseyov D., Kimchi A., Alon R. Tumor suppressor death-associated protein kinase attenuates inflammatory responses in the lung. Am. J. Respir. Cell Mol. Biol. 2012;46:313–322. doi: 10.1165/rcmb.2011-0181OC. [DOI] [PubMed] [Google Scholar]
- Netea-Maier R.T., Plantinga T.S., van de Veerdonk F.L., Smit J.W., Netea M.G. Modulation of inflammation by autophagy: consequences for human disease. Autophagy. 2016;12:245–260. doi: 10.1080/15548627.2015.1071759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romani L. Immunity to fungal infections. Nat. Rev. Immunol. 2011;11:275–288. doi: 10.1038/nri2939. [DOI] [PubMed] [Google Scholar]
- Romani L., Fallarino F., De Luca A., Montagnoli C., D’Angelo C., Zelante T., Vacca C., Bistoni F., Fioretti M.C., Grohmann U. Defective tryptophan catabolism underlies inflammation in mouse chronic granulomatous disease. Nature. 2008;451:211–215. doi: 10.1038/nature06471. [DOI] [PubMed] [Google Scholar]
- Sanjuan M.A., Dillon C.P., Tait S.W., Moshiach S., Dorsey F., Connell S., Komatsu M., Tanaka K., Cleveland J.L., Withoff S., Green D.R. Toll-like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature. 2007;450:1253–1257. doi: 10.1038/nature06421. [DOI] [PubMed] [Google Scholar]
- Shen J., Prywes R. ER stress signaling by regulated proteolysis of ATF6. Methods. 2005;35:382–389. doi: 10.1016/j.ymeth.2004.10.011. [DOI] [PubMed] [Google Scholar]
- Shi C.S., Shenderov K., Huang N.N., Kabat J., Abu-Asab M., Fitzgerald K.A., Sher A., Kehrl J.H. Activation of autophagy by inflammatory signals limits IL-1β production by targeting ubiquitinated inflammasomes for destruction. Nat. Immunol. 2012;13:255–263. doi: 10.1038/ni.2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiloh R., Bialik S., Kimchi A. The DAPK family: a structure-function analysis. Apoptosis. 2014;19:286–297. doi: 10.1007/s10495-013-0924-5. [DOI] [PubMed] [Google Scholar]
- Singh S.B., Davis A.S., Taylor G.A., Deretic V. Human IRGM induces autophagy to eliminate intracellular mycobacteria. Science. 2006;313:1438–1441. doi: 10.1126/science.1129577. [DOI] [PubMed] [Google Scholar]
- Sprenkeler E.G., Gresnigt M.S., van de Veerdonk F.L. LC3-associated phagocytosis: a crucial mechanism for antifungal host defence against Aspergillus fumigatus. Cell. Microbiol. 2016;18:1208–1216. doi: 10.1111/cmi.12616. [DOI] [PubMed] [Google Scholar]
- Stevens D.A., Brummer E., Clemons K.V. Interferon- gamma as an antifungal. J. Infect. Dis. 2006;194(Suppl 1):S33–S37. doi: 10.1086/505357. [DOI] [PubMed] [Google Scholar]
- Subramani S., Malhotra V. Non-autophagic roles of autophagy-related proteins. EMBO Rep. 2013;14:143–151. doi: 10.1038/embor.2012.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The International Chronic Granulomatous Disease Cooperative Study Group A controlled trial of interferon gamma to prevent infection in chronic granulomatous disease. N. Engl. J. Med. 1991;324:509–516. doi: 10.1056/NEJM199102213240801. [DOI] [PubMed] [Google Scholar]
- Usui T., Okada M., Hara Y., Yamawaki H. Death-associated protein kinase 3 mediates vascular inflammation and development of hypertension in spontaneously hypertensive rats. Hypertension. 2012;60:1031–1039. doi: 10.1161/HYPERTENSIONAHA.112.200337. [DOI] [PubMed] [Google Scholar]
- Zhao Z., Fux B., Goodwin M., Dunay I.R., Strong D., Miller B.C., Cadwell K., Delgado M.A., Ponpuak M., Green K.G. Autophagosome-independent essential function for the autophagy protein Atg5 in cellular immunity to intracellular pathogens. Cell Host Microbe. 2008;4:458–469. doi: 10.1016/j.chom.2008.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.