

Abstract
Background
Although recent studies have greatly advanced understanding of deep molluscan phylogeny, placement of some taxa remains uncertain as different datasets support competing class-relationships. Traditionally, morphologists have placed Monoplacophora, a group of morphologically simple, limpet-like molluscs as sister group to all other conchiferans (shelled molluscs other than Polyplacophora), a grouping that is supported by the latest large-scale phylogenomic study that includes Laevipilina. However, molecular datasets dominated by nuclear ribosomal genes support Monoplacophora + Polyplacophora (Serialia). Here, we evaluate the potential of mitochondrial genome data for resolving placement of Monoplacophora.
Results
Two complete (Laevipilina antarctica and Vema ewingi) and one partial (Laevipilina hyalina) mitochondrial genomes were sequenced, assembled, and compared. All three genomes show a highly similar architecture including an unusually high number of non-coding regions. Comparison of monoplacophoran gene order shows a gene arrangement pattern not previously reported; there is an inversion of one large gene cluster. Our reanalyses of recently published polyplacophoran mitogenomes show, however, that this feature is also present in some chiton species. Maximum Likelihood and Bayesian Inference analyses of 13 mitochondrial protein-coding genes failed to robustly place Monoplacophora and hypothesis testing could not reject any of the evaluated placements of Monoplacophora.
Conclusions
Under both serialian or aculiferan-conchiferan scenarios, the observed gene cluster inversion appears to be a convergent evolution of gene arrangements in molluscs. Our phylogenetic results are inconclusive and sensitive to taxon sampling. Aculifera (Polyplacophora + Aplacophora) and Conchifera were never recovered. However, some analyses recovered Serialia (Monoplacophora + Polyplacophora), Diasoma (Bivalvia + Scaphopoda) or Pleistomollusca (Bivalvia + Gastropoda). Although we could not shed light on deep evolutionary traits of Mollusca we found unique patterns of gene arrangements that are common to monoplacophoran and chitonine polyplacophoran species but not to acanthochitonine Polyplacophora.
Graphical abstract
Complete mitochondrial genome of Laevipilina antarctica
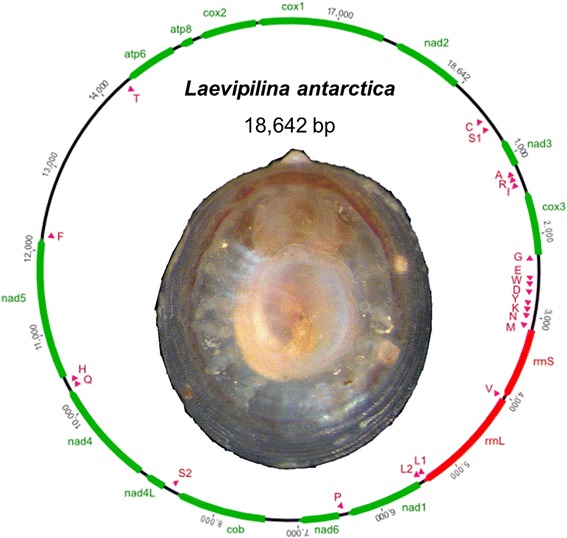
Electronic supplementary material
The online version of this article (doi:10.1186/s12862-016-0829-3) contains supplementary material, which is available to authorized users.
Keywords: Mollusca, Mitogenome, Monoplacophora, Serialia, Aculifera, Conchifera, Gene arrangement, Phylogeny, Evolution
Background
Mollusca, comprising eight extant classes, has high diversity and an origin that dates back more than 540 million years [1, 2]. One of the most enigmatic classes, Monoplacophora, was thought to be extinct since the Palaeozoic until a living exemplar of Neopilina galatheae was found during the Galathea expedition in 1952 [3]. The significance of “living fossil” monoplacophorans for deep molluscan systematics was soon recognized [3], and Monoplacophora (with about 30 recent members called Tryblidia [4]) were central in several palaeontological, morphological and cladistic analyses (e.g., [5–8]) that tried to resolve the phylogeny of Mollusca. These analyses resulted in a number of different phylogenetic placements being hypothesized for Monoplacophora. Under the Conchifera/Aculifera hypothesis, Monoplacophora were traditionally viewed as the sister group to all other conchiferans with and as the sister group of Aplacophora (Caudofoveata + Solenogastres; [9]).
Early molecular analyses based on nuclear ribosomal DNA did not include monoplacophorans [10, 11]. Later analysis of a data set dominated by nuclear ribosomal genes and including all eight extant molluscan classes placed Monoplacophora within Polyplacophora, Serialia [12]. The single 28S sequence from Laevipilina antarctica used in that study was a chimera between monoplacophoran and chiton 28S [13], subsequent studies based on the same markers but free of contamination recovered Monoplacophora as sister to Polyplacophora but retained the term Serialia [2, 13, 14]. However, relationships among molluscan classes in these studies were unconventional, recovering Serialia as sister group to bivalves and gastropods, and clustering scaphopods together with aplacophorans and cephalopods. The Serialia hypothesis, which is based on ribosomal DNA dominated data, is provocative, since it challenges traditional taxonomic text-book hypotheses.
Both Aculifera and Conchifera are strongly supported by phylogenomic studies [15–17] and became a new paradigm in molluscan systematics [18–22]; but see [23–25]. Schrödl and Stöger [26] recently emphasized that there is some conflict between the consensus topology (Fig. 1 in [26]), and any of the several phylogenomic [15–17, 27–30] and other nuclear sequence sets [31, 32]. All these molecular datasets cover substantial sequence data, but represent a limited taxon sampling. Smith et al. [16] present the first phylogenomic study including representatives of all eight molluscan classes, and thus it directly addressed placement of Monoplacophora [16, 17]. Although the authors detected many sites in their dataset with weak signal for Serialia and some sites with strong signal for Serialia, the sister group relationship of the one sampled monoplacophoran species and Cephalopoda is clearly favored [16, 17]. A more recent phylogenomic analysis [33] placed the sole monoplacophoran representative employed as the most basal lineage of conchiferans, albeit with low nodal support, but in line with some traditional morphological hypotheses.
Fig. 1.
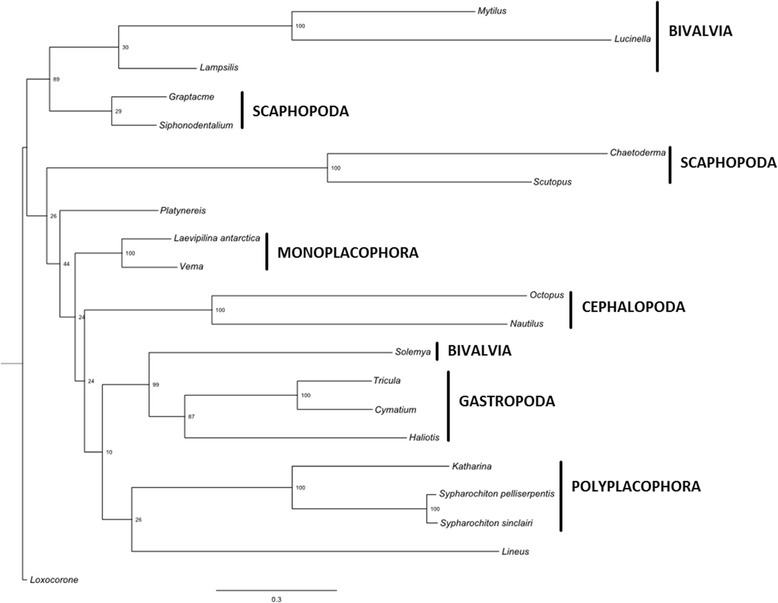
Preferred Maximum Likelihood tree based on the large amino acid dataset and inferred with RAxML-HPC executing 500 bootstrap replicates under the CAT approximation for rate heterogeneity. Loxocorone was used to root the tree
An alternative to studying multiple genes is exploring the information content of mitochondrial (mt) genomes [34]. In Metazoa, mitochondrial genomes usually consist of a highly conserved set of 13 protein-coding genes (PCGs), two ribosomal RNAs (rRNAs), and 22 transfer RNAs (tRNAs) [35, 36]. Furthermore, metazoan mtDNA includes at least one (sometimes more in molluscs) non-coding region of which the largest typically contains the control region, the site of initiation for transcription and/or replication [10]. All known mt genomes in molluscs are circular, with orthologs readily identifiable, making them easy to compare. Analyses of mitochondrial protein-coding genes have been successfully used to resolve phylogenetic relationships as for example the affiliation of Sipuncula and Annelida [37–39]. Although the analysis of mitochondrial sequence data provides good resolution in some molluscan subgroups, e.g. Bivalvia [40] or Cephalopoda [41, 42], the resolution for deep molluscan class-relationships is generally poor [35, 43]. Even the analysis of all protein-coding genes of 96 available mt genomes covering six molluscan classes (lacking Monoplacophora and Solenogastres) lacked sufficient phylogenetic signal to robustly resolve relationships among the major lineages of Mollusca [44]. The known problem of increased rates of sequence evolution [45] in some subclades such as bivalves and scaphopods [46] in addition to the Precambrian split of Mollusca from the closest outgroups [2, 30] not surprisingly leads to long-branch attraction ([44]. Taxa showing massive gene rearrangements also show faster nucleotide evolution [26, 34, 44], creating analytical challenges. Stöger and Schrödl [44] recommended analyses of a more representative molluscan taxon set, with fast-evolving taxa at both sequence and gene rearrangement level excluded from analyses. Osca et al. [47] followed this strategy, excluding bivalves and including a second caudofoveate taxon, Scutopus ventrolineatus, resulting in an aculiferan/conchiferan topology, although with low support in Maximum Likelihood (ML) analyses. Support for Aculifera is strong for their Bayesian topology, but the clade is nested within outgroup taxa. Plazzi et al. [48] published the first mitogenome of Protobranchia, which are putatively basal lineage of bivalves. This genome appears more conservative relative to the inferred ancestral molluscan and lophotrochozoan arrangements compared to other bivalves, which show greater rearrangement [34, 44]. More recently, mitogenomes of five further chiton species were published in 2014 [49, 50]. According to the authors [49], gene orders are highly congruent with the earlier published mt genome of Katharina [35], showing a plesiomorphic arrangement for lophotrochozoans, but this interpretation is not correct.
Here we contribute to the class-level taxon sampling of molluscan mitochondrial genomes by sequencing two Recent monoplacophorans (Laevipilina antarctica and Vema ewingi) and an almost complete mitogenome of Laevipilina hyalina. By generating the first mitogenomes for Monoplacophora our aims were 1) to explore the origin of the enigmatic Monoplacophora, 2) to evaluate whether or not a more balanced taxon excluding rapidly-evolving taxa improved resolution of deep molluscan phylogeny and 3) to compare monoplacophoran gene arrangements with a lophotrochozoan ground pattern [34].
Results
General structure/architecture of the monoplacophoran mitogenomes
Mitogenomes of Vema ewingi and L. antarctica are 17,910 bp and 18,642 bp in length, respectively. Both genomes include the complete set of 37 bilaterian mitochondrial genes: 13 protein-coding genes (PCGs), two ribosomal RNAs (rRNAs), and 22 transfer RNAs (tRNAs). Distribution of PCGs between the two strands is almost equal: ATP synthase subunits (atp6, atp8) and cytochrome c oxidase subunits (cox1, cox2, cox3), as well as nad2 and nad3 are located on the plus strand, whereas NADH dehydrogenase subunits (nad1, nad4, nad4L, nad5, nad6) and cytochrome b (cob) are on the minus strand. Ribosomal genes, rrnS and rrnL, as well as most of the tRNAs (15 in L. antarctica, 16 in Vema ewingi) are located on the plus strand. Only seven tRNAs in L. antarctica and six in Vema ewingi can be found on the opposite (minus) strand. Long-PCR fragments of L. hyalina were assembled into 1 contig totaling 15,102 bp and comprising 12 PCGs (atp8 is missing), both rRNAs and 16 of 22 tRNAs (trnT, trnC, trnW, trnG, trnH, trnE are missing). We detected two copies of trnK in L. hyalina. One copy with a lower e-value (5.223e-05) is located within the tRNA complex DYKNM and the second trnK with an e-value of 0.6443 is adjacent to cox2. In comparison, that trnK with a lower e-value is more probable. Both copies of trnK show typical cloverleaf secondary structures, similar to that of the two other monoplacophoran trnK structures, and the typical anticodon for lysine (UUU), so both copies are potentially functional. All PCGs that could be detected by MITOS are evenly distributed between both strands whereas rrnS and rrnL are exclusively located on the positive strand. Twelve tRNAs can be found on the plus strand, five are on the minus strand.
For L. antarctica, the GC content of the complete mitochondrial genome is 35.5%. GC content of individual PCGs ranges between 33.9% in atp8 and 39.8% in cox2 and values for ribosomal RNAs are slightly below the average of the complete genome with 34.4% for rrnS and 31.7% for rrnL. Transfer RNAs show considerable variation in their GC content with values ranging from 16.1% (trnH) to 46.8% (trnY). The GC content of the complete mitochondrial genome of Vema ewingi is 36.7% with a GC content of PCGs between 33.9% (nad3) and 40.4% (nad6). Both ribosomal RNAs have a value of 33.8% and tRNAs range between 17.5% (trnH) and 55.6% (trnY). GC content of the mitogenome of L. hyalina is 38.8%. GC content of PCGs is minimum 36.0% in nad3 and maximum 45.8% in cox2. For ribosomal RNAs the GC content is 37.5% for rrnS and 34.3% for rrnL, within tRNAs range from 22.7% in trnS2 to 50.0% in trnY.
Based on the MITOS results, we identified 28 non-coding regions (NCR) within the mitogenome of L. antarctica. Six are less than 10 bp long, 16 are between 10 and 100 bp in length and only six are larger than 100 bp. The largest NCR between trnF and trnT is 2012 bp long and contains a pattern with the regular expression TATA[TC]ATATATA[GT]A[CT][AT][TA][AT][TCG][GC], we refer to that pattern hereinafter as motif 1. Motif 1 includes an (AT)6 repetition (see Table 1). Moreover, some repetitive motifs occur in that NCR (not shown). Motif 1 is additionally detected within the NCR between trnG and trnE (181 bp) with (AT)7. A second motif with the regular expression CCTCGAAATCGTTGCATC (motif 2, Table 1), is visible in the NCR between nad2 and trnC (478 bp). Moreover the NCR between trnF and trnT includes remains of atp6. In the NCR between nad6 and cob MITOS detects residual sequence parts of nad6.
Table 1.
Table shows motifs 1 and 2, their location in the mitogenome and the specific motif sequence
| Motif no. | Occurrence | NCR border | Starting position within NCR | Motif sequence |
|---|---|---|---|---|
| 1 | L. antarctica | trnG/trnE | 55 | TATATATATATAGATATATG |
| 1 | Vema ewingi | trnG/trnE | 78 | TATATATATATATACATATG |
| 1 | L. antarctica | trnF/trnT | 893 | TATATATATATAGACTATCG |
| 1 | Vema ewingi | trnF/trnT | 898 | TATACATATATATACTTAGC |
| 2 | L. antarctica | nad2/trnC | 23 | CCTCGAAATCGTTGCATC |
| 2 | Vema ewingi | nad2/trnS1 | 22 | CCTCGAAATCGTTGCATC |
In Vema ewingi we found 27 non-coding regions; five regions are less than 10 bp long, 18 are 10-100 bp long and four are larger than 100 bp. The largest NCR between trnF and trnT (2287 bp) as well as the NCR located between trnG and trnE (151 bp) contain motif 1, which is already described for L. antarctica. Between trnF and trnT the motif contains (AT)6 with a discontinuity of one (CA), and between trnG and trnE we count (AT)9 (Table 1). Motif 2 was detected in the NCR between nad2 and trnS1 (108 bp) (Table 1). Moreover, repetitive motifs are visible in this largest NCR between trnF and trnT of Vema ewingi (not shown).
Within the partial mitogenome of L. hyalina we found 21 NCRs, one of which is less than 10 bp long. Sixteen regions are 10 to 100 bp and four are more than 100 bp in length. Within the NCR between cox1 and trnK (299 bp) motif 1 with (AT)10 is visible.
The largest NCRs of L. antarctica and Vema ewingi are located between trnF and trnT in both mtDNAs and in both NCRs the congruent motif 1 which includes AT-repetitions occurs at almost the same relative positions (Table 1). This motif 1 is recovered in a second NCR in each mitogenome again at congruent relative positions. Motif 2 can be found in NCR between nad2 and trnC in L. antarctica and in NCR between nad2 and trnS1 of Vema ewingi. This motif 2 is located at almost identical relative positions (Table 1). Neither comparisons of these two NCRs to the BLAST nucleotide database results in any similarities to gene regions of other taxa nor are the 2D-foldings informative, which were computed in Geneious with default parameters.
Comparing the relative gene borders of the non-coding regions of the three monoplacophoran species, we discovered 13 NCRs that are embedded between the same genes in all three monoplacophoran genomes (Fig. 3). This number might be even higher since we do not know all NCR borders of L. hyalina. Identical positions of NCRs relative to gene order between L. antarctica and Vema ewingi are 11 whereas only one NCR has the same position between both Laevipilina species (Fig. 3). All three monoplacophoran species appear to share two NCRs with the cephalopod Nautilus [51]. This is NCR between cox1 and nad2, and NCR between nad1 and trnP (Fig. 3).
Fig. 3.

Gene arrangements of selected molluscan taxa; arrangements are annotated with MITOS and linearized and rotated to cox1 for display reasons. Gene lengths of coding and non-coding regions correspond to relative lengths of the genomes. The directions of the genes are given by arrows. Green arrows indicate protein coding genes (PCGs); red arrows indicate ribosomal genes (rRNAs); pink arrows indicate transfer RNAs (tRNAs), which are named corresponding to the one-letter code. Sypharochiton sinclairi and S. pelliserpentis showed identical gene order
We detected six overlapping regions that occur in all three monoplacophoran mt genomes. These overlaps are located between gene pairs trnY/trnK, trnM/rrnS, rrnS/trnV, rrnL/trnL1, and trnP/nad6 (Fig. 3). Two pairs, trnV/rrnL and rrnL/trnL1, are overlapping with more than 25nts according to the MITOS annotation output.
Gene order within Monoplacophora
Gene arrangements of L. antarctica and Vema ewingi are shown in Fig. 3. They appear in two clusters (cluster means a group of genes in the following), this is trnT-atp6-atp8-cox2-cox1-nad2-trnC-trnS1-nad3-trnA-trnR-trnI-cox3-trnG on one strand and trnE-trnW-trnD-trnY-trnK-trnN-trnM-rrnS-trnV-rrnL-trnL1-trnL2-nad1-trnP-nad6-cob-trnS2-nad4L-nad4-trnQ-trnH-nad5-trnF on the opposite strand for L. antarctica (Fig. 3). The difference in Vema ewingi is the position of trnC, which is not located between nad2 and trnS1 as in L. antarctica, but is found within the tRNA complex GEWDCYKNM. The two gene clusters, nad4/nad4L and atp6/atp8 are known to appear adjacent to each other in many animals [40, 52], which is detected here, too.
Within the partial mt genome of L. hyalina we observed a very similar gene order and orientation as in L. antarctica and Vema ewingi, although there are some differences (aside from missing genes). TrnC as well as tRNAs GEW are missing in the cluster GEWD[C]YKNM in the gene order of L. hyalina (Fig. 3). Though trnK is present within that complex, a second trnK with a much more reliable e-value appears adjacent to cox2. TrnH, adjacent to trnQ in Vema and L. antarctica, is missing in L. hyalina, as well as trnF and atp8.
The gene order in monoplacophoran PCGs and rRNA genes investigated herein is highly similar, therefore we summarize these arrangements and refer to it as the monoplacophoran plesiomorphic state.
Gene order within Polyplacophora
In addition to the mitogenome of the black chiton Katharina [35] five more chiton mitogenomes are available now [49, 50]. The three acanthochitonine mt gene arrangements (Cryptochiton, Cyanoplax, Nuttalina) are in line with the Katharina arrangement except the two tRNA complexes KARNI and MCYWQGE, which are present in Katharina and Cryptochiton. Both complexes appear in inversed orders in Nuttalina and Cyanoplax. Although mitogenomes of the chitonine taxa Sypharochiton pelliserpentis and S. sinclairi have already been published their gene order is not thoroughly examined [49]. The authors claim that the gene arrangements of their chitonine species resemble that of other chitons, but did not show the actual arrangement, so we have reexamined these mitogenomes (Fig. 3). Both Sypharochiton mitogenomes are congruent to each other in their gene arrangement but contra [49] the gene order is not “almost identical to that found in Katharina tunicata” ([49], Fig. 3 herein). The genes of Sypharochiton are arranged in the two clusters of genes that are already described for Monoplacophora (Fig. 3). Moreover, these two clusters have identical orientation as in the monoplacophoran arrangement (Table 4). Differences to the monoplacophoran gene order are restricted to the two tRNA complexes: one is INRAK in Sypharochiton, the second is EGQWYCM, which are exactly inverse to the Katharina order (Fig. 3, Table 4), but congruent to the order of Nuttalina and Cyanoplax.
Table 4.
Directions of PCGs and rRNAs in the two clusters; tRNAs are not considered. Based on the lophotrochozoan ground pattern [34] we find two evolutionary lines. One is evident in Katharina, as well as in Octopus, with an inversion of PCGs in cluster 2. From this derived arrangement we can infer the Nautilus gene order with a “simple” translocation of rRNAs. The second line is an inversion of cluster 1 of the lophotrochozoan ground pattern, which leads to the monoplacophoran (and the Sypharochiton) pattern of gene arrangement. We could not detect this arrangement of PCGs in another lophotrochozoan group so far (see e.g., [80])
| Cluster 1: cox3-nad3-nad2-cox1-cox2-atp8-atp6 |
Cluster 2: rrnS-rrnL-nad1-nad6-cob-nad4L-nad4-nad5 |
Remarks | |
|---|---|---|---|
| Lophotrochozoan ground pattern (Bernt et al. [34]) | → | → | |
| Monoplacophoran plesiomorphic state | ← | → | Cluster 1 missing atp8 in L. hyalina as it was not sampled |
| L. antarctica | ← | → | |
| Vema ewingi | ← | → | |
| L. hyalina | ← | → | Cluster 1 misses atp8 |
| Sypharochiton spp. | ← | → | |
| K. tunicata | → | ← |
Phylogenetic analyses
Our initial taxon set based on the amino acid alignment of all protein-coding genes includes 18 molluscs and three lophotrochozoan outgroup taxa (Table 5, Fig. 1, Additional file 1: Figure S1). The entoproct Loxocorone was used to root the tree as it represents the most distant related of the non-mollusc taxa employed [33]. Maximum Likelihood (ML) analysis of this taxon set recovers Mollusca as non-monophyletic with Platynereis (Annelida) and Lineus (Nemertinea) nested within Mollusca. Monoplacophora, Polyplacophora, Caudofoveata, and Cephalopoda were recovered monophyletic with maximal bootstrap support (bs) whereas support for gastropod monophyly was moderate (bs = 87%) and support for scaphopod monophyly was weak (bs = 29%). Relationships among higher level taxa were generally poorly supported. Also, Scaphopoda together with three non-protobranch bivalves form a moderately well-supported clade (bs = 89%; Fig. 1, Table 2).
Table 5.
Table shows all taxa that were used in this study with their corresponding GenBank accession numbers
| Class | Taxon | GenBank acc. no. | |
|---|---|---|---|
| Outgroup taxa | Annelida | Platynereis dumerilii | NC 000931 |
| Entprocta | Loxocorone allax | NC 010431 | |
| Nemertea | Lineus viridis | NC 012889 | |
| Mollusca | Bivalvia | Lampsilis ornata | NC 005335 |
| Lucinella divaricata | NC 013275 | ||
| Mytilus edulis | NC 006161 | ||
| Solemya velum | NC 017612 | ||
| Caudofoveata | Chaetoderma nitidulum | NC 013846 | |
| Scutopus ventrolineatus | KC 757645 | ||
| Cephalopoda | Nautilus macromphalus | NC 007980 | |
| Octopus vulgaris | NC 006353 | ||
| Gastropoda | Cymatium parthenopeum | NC 013247 | |
| Haliotis rubra | NC 005940 | ||
| Tricula hortensis | NC 013833 | ||
| Monoplacophora | Laevipilina antarctica | KY 244020 | |
| Laevipilina hyalina | KY 284344 | ||
| Vema ewingi | KY 244019 | ||
| Polyplacophora | Katharina tunicata | NC 001636 | |
| Sypharochiton pelliserpentis | NC 024174 | ||
| Sypharochiton sinclairi | NC 024173 | ||
| Scaphopoda | Graptacme eborea | NC 006162 | |
| Siphonodentalium lobatum | NC 005840 |
Table 2.
Table gives an overview on all Maximum Likelihood (pre-) analyses and the resulting molluscan hypotheses; taxon sets aa-1 – aa-11 are based on amino acis data, taxon sets nuc-1 – nuc-11 are based on nucleotide datasets; main analyses based on aa-1 and aa-2 were additionally analyzed with Phylobayes which is indicated in the first column; Numbers are bootstrap support values of the corresponding hypothesis that appeared in that analysis, numbers followed by “pp” are posterior probabilities of the Phylobayes analysis; “-“means that the hypothesis did not appear in that analysis
| Taxon set | Inclusion/exclusion of taxa | Monophyletic Mollusca | Aculifera | Conchifera | “Diasoma” (non-protobranch Bivalvia + Scaphopoda) | Monoplacophora + Cephalopoda | “Pleistomollusca” (Solemya + Gastropoda) | Serialia |
|---|---|---|---|---|---|---|---|---|
| aa-1 | initial and largest taxon set, comprising 18 molluscan taxa and 3 lophotrochozoan outgroup taxa | - | - | - | 81 | - | 100 | - |
| aa-1 Phylobayes | initial and largest taxon set, comprising 18 molluscan taxa and 3 lophotrochozoan outgroup taxa, analyzed with Phylobayes | - | - | - | - | - | Bivalvia + Gastropoda: 0.99 pp | - |
| aa-2 | initial taxon set except non-protobranch Bivalvia and outgroups reduced to Platynereis (Annelida) only | 0 | - | - | - | - | 100 | - |
| aa-2 Phylobayes | initial taxon set except non-protobranch Bivalvia and outgroups reduced to Platynereis (Annelida)only, analyzed with Phylobayes | - | - | - | - | - | 0.96 pp | - |
| aa-3 | initial taxon set except non-protobranch Bivalvia, Scaphopoda and outgroups reduced toPlatynereis (Annelida) only | 0 | - | - | - | - | 100 | - |
| aa-4 | aa-1 without any outgroups | - | - | - | 8 | - | - | - |
| aa-5 | aa-2 without any outgroups | - | - | - | - | - | - | - |
| aa-6 | aa-3 without any outgroups | - | - | - | - | - | - | - |
| aa-7 | aa-1 plus Scutopus | - | - | - | - | 11 | 99 | - |
| aa-8 | aa-2 plus Scutopus | 0 | - | - | - | - | - | - |
| aa-9 | aa-1 plus Scutopus and two Sypharochiton species | - | - | - | - | - | 99 | - |
| aa-10 | aa-2 plus Scutopus and two Sypharochiton species | 0 | - | - | - | - | 99 | - |
| aa-11 | aa-10 Solemya excluded | 0 | - | - | - | - | - | - |
| nuc-1 | initial and largest taxon set, comprising 18 molluscan taxa and 3 lophotrochozoan outgroup taxa | - | - | - | - | - | - | - |
| nuc-2 | initial taxon set except non-protobranch Bivalvia and outgroups reduced to Platynereis (Annelida) only | 0 | - | - | - | - | - | 100 |
| nuc-3 | initial taxon set except non-protobranch Bivalvia, Scaphopoda and outgroups reduced to Platynereis(Annelida) only | 0 | - | - | - | - | - | 56 |
| nuc-4 | nuc-1 without any outgroups | - | - | - | 92 | - | - | 100 |
| nuc-5 | nuc-2 without any outgroups | - | - | - | - | - | - | - |
| nuc-6 | nuc-3 without any outgroups | - | - | - | - | - | - | 100 |
| nuc-7 | nuc-1 plus Scutopus | - | - | - | - | - | - | 100 |
| nuc-8 | nuc-2 plus Scutopus | 0 | - | - | - | - | - | 100 |
| nuc-9 | nuc-1 plus Scutopus and two Sypharochiton species | - | - | - | - | - | - | - |
| nuc-10 | nuc-2 plus Scutopus and two Sypharochiton species | 0 | - | - | - | - | - | - |
| nuc-11 | nuc-10 Solemya excluded | 0 | - | - | - | - | - | - |
Phylobayes analysis of this dataset recovered a topology that is unresolved at its base. All classes of Mollusca except Scaphopoda were recovered monophyletic with strong support (posterior probabilities, pp = 0.99-1.00). Pleistomollusca was also strongly supported (pp = 0.99) and Monoplacophora was recovered sister to Caudofoveata (albeit with weak support by Bayesian standards, pp = 0.84; Additional file 1: Figure S1, Table 2).
Exclusion of the outgroup taxa Lineus and the more distant outgroup Loxocorone and the reduction of bivalve taxa to the protobranch taxon Solemya, which is the most basal bivalve group, lead to a ML topology with a strongly supported Pleistomollusca (bs = 99) and a moderately supported sister group relationship of Caudofoveata and Scaphopoda (bs = 73; Fig. 2). Phylobayes analysis of this trimmed down dataset (Additional file 1: Figure S1, Table 2) yielded similar results with Scaphopoda (pp = 0.72) being the most weakly supported class and Pleistomollusca recovered (pp = 0.96).
Fig. 2.
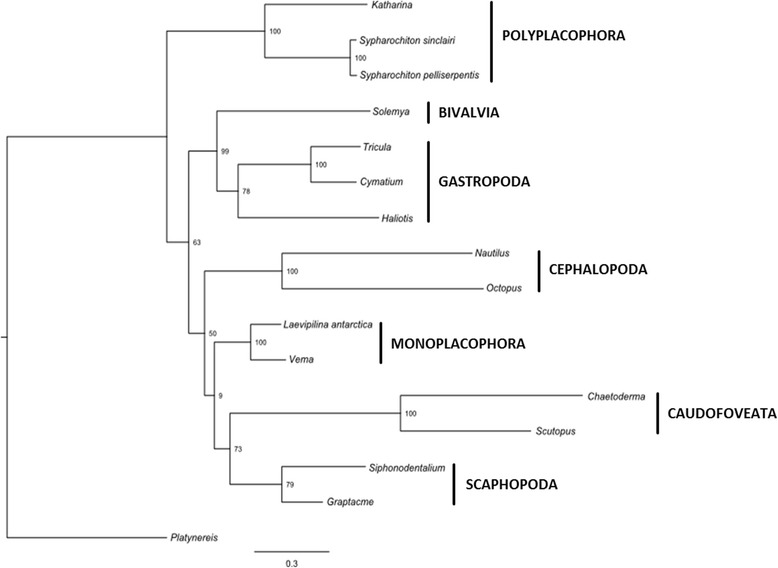
Maximum Likelihood tree based on the amino acid dataset without the two outgroup taxa Lineus and Loxocorone and the reduction of bivalve taxa to the protobranch taxon Solemya. Tree was inferred with RAxML-HPC executing 500 bootstrap replicates under the CAT approximation for rate heterogeneity. Platynereis was used to root the tree
In our test on saturation of the alignments TreSpEx calculated cox1 as the least saturated and nad6 as the most saturated. There is a gradual decline in the slope value from the best to the worst so cutting out particular genes does probably not improve the tree topology. BaCoCa measures rate heterogeneity and again detects cox1 as the “best” gene but there is a gradual decline. Platynereis and Nautilus are the most compositionally heterogeneous taxa in the datasets but not extremely so. Overall, we were not able to identify certain genes or taxa that are particularly problematic.
Hypothesis testing using the Shimodaira-Hasegawa (SH) test and the Approximately Unbiased (AU) test failed to reject Aculifera, Conchifera, Monoplacophora as the sister taxon to the rest of Conchifera, Monoplacophora sister to Cephalopoda, Serialia, or Testaria as being significantly less likely than the most likely tree recovered in either of the two ML analyses (Table 3). Hypothesis testing was performed on both main datasets (aa-1 and aa-2 in Table 2).
Table 3.
Results of SH and AU hypothesis testing
| Analysis | Constraint | Log-likelihood | AU-test p-value | SH-test p-value |
|---|---|---|---|---|
| aa-1 | Unconstrained | −99817.64 | 0.852 | 0.935 |
| aa-1 | Aculifera monophyletic | −99837.74 | 0.113 | 0.392 |
| aa-1 | Conchifera monophyletic | −99854.34 | 0.113 | 0.113 |
| aa-1 | Monoplacophora sister to rest of Conchifera | −99859.16 | 0.069 | 0.089 |
| aa-1 | Monoplacophora sister to Cephalopoda | −99825.77 | 0.467 | 0.704 |
| aa-1 | Serialia monophyletic | −99839.30 | 0.062 | 0.354 |
| aa-1 | Testaria monophyletic | −99860.90 | 0.053 | 0.083 |
| aa-2 | Unconstrained | −99817.64 | 0.854 | 0.940 |
| aa-2 | Aculifera monophyletic | −99837.74 | 0.130 | 0.390 |
| aa-2 | Conchifera monophyletic | −99854.34 | 0.118 | 0.120 |
| aa-2 | Monoplacophora sister to rest of Conchifera | −99859.16 | 0.066 | 0.092 |
| aa-2 | Monoplacophora sister to Cephalopoda | −99825.77 | 0.449 | 0.703 |
| aa-2 | Serialia monophyletic | −99839.30 | 0.063 | 0.353 |
| aa-2 | Testaria monophyletic | −99860.90 | 0.053 | 0.083 |
Discussion
Gene order
The gene arrangement of Monoplacophora revealed herein is either highly conserved or the taxa here recently diverged from each other. L. antarctica and Vema ewingi differ only in the position of trnC which is adjacent to trnS1 in L. antarctica but is embedded in the tRNA complex GEWDCYKNM in Vema ewingi (Fig. 3).
Presence of two conserved gene blocks was confirmed in these monoplacophoran species (Table 4). One conserved block, rrnS-rrnL-nad1-nad6-cob, was defined previously for Lophotrochozoa [34], and the second block that is putatively conserved in Lophotrochozoa, nad4L-nad4-trnH-nad5, could be detected adjacent to rrnS-rrnL-nad1-nad6-cob (shown as combined cluster 2 in Table 4), although in a somehow aberrant appearance concerning tRNAs, since in L. antarctica and Vema ewingi there is trnQ nested between nad4 and trnH and in L. hyalina trnH is missing (Fig. 3). The part of yet another lophotrochozoan conserved gene block (cox3-nad3-nad2-cox1-cox2-atp8-atp6) usually appears in the forward direction. In our monoplacophoran species the part nad2-cox1-cox2-atp8-atp6 is inverted (Table 4). In L. hyalina atp8 is missing but trnK is included. Presence of these conserved lophotrochozoan gene blocks and a relatively high percentage of divergence between the PCGs of the monoplacophoran species (22.4%) leads to the assumption that gene order in Monoplacophora is conserved.
A potential synapomorphy for Mollusca [44], aggregation of trnG-trnE with the tRNA complex MCYWQ, is present in Monoplacophora, although the complex is reversed in its order (Fig. 3). A second tRNA complex that appears frequently in Lophotrochozoa is KARNI [44]. Within our monoplacophoran taxa we instead find ARI which is also present in the caudofoveate Chaetoderma. A clade of caudofoveates and monoplacophorans is recovered by some of our sequence analyses, but not by any other analyses including nuclear data (for review see [26]); we thus assume that congruency in the tRNA order ARI is convergent.
Focusing on the gene order of protein-coding genes (PCGs) and ribosomal RNAs, the ancestral state for both PCG clusters is forward in the lophotrochozoan ground pattern (cluster 1 and 2, see Table 4). Within Mollusca, the order of PCGs that is observed in Katharina and other Acanthochitonina [35, 50] is hypothesized to represent the ancestral arrangement for at least molluscs, since this arrangement is recurring with no or almost no modifications in other molluscan classes [44]. In reference to the lophotrochozoan pattern, we show that the orientation of cluster 1 of the Acanthochitonina gene order is ancestral, whereas cluster 2 is derived (Table 4). This order is opposite in Monoplacophora: Their gene orders reflect a derived orientation for cluster 1, but the plesiomorphic state for cluster 2, which appears to be a unique condition among lophotrochozoans. We confirm a plesiomorphic gene arrangement in Acanthochitonina but a monoplacophoran-like derived gene order in Chitonina (Table 4). Rearrangements of PCG clusters are considered to be rare events, and thus are given high phylogenetic significance [51, 52]. Accordingly, the uniquely derived arrangement of cluster 2 could be interpreted as a synapomorphy, supporting Serialia; because of the undisputed monophyly of Polyplacophora, the heterogeneous arrangement within chitons implies homoplasy. Unfortunately, no information is available on mitogenomes of the Lepidopleurida, the morphologically most plesiomorphic chiton clade [53, 54]. Under the Aculifera-Conchifera concept we find this derived condition of gene order in some but not all members of both major clades, also implying convergence within Mollusca. Such convergent rearrangements of large PCG complexes have rarely been detected in invertebrates [52] but not in vertebrates [55]. One such example is known from Caenogastropoda, which shares a congruent gene order of PCGs with the nemertean Lineus [44]. We could not find any similar examples within molluscs in the literature and we anticipate that denser sampling may reveal more cases.
Gene architecture
Mitogenome lengths of L. antarctica and Vema ewingi are consistent with other molluscan mitochondrial genomes, which range between 13.6 kb in Biomphalaria (Gastropoda) to 31.5 kb in Placopecten (Bivalvia) [44]. Nevertheless, both range at the upper bound of animal mtDNA length, which is typically less than 20 kb [56]. Both mitogenomes contain the complete gene complement of a typical bilaterian mitogenome [52]. L. hyalina lacks atp8 and six tRNAs. Atp8 is conserved in just a short fragment at the 5′ region [36, 57], which makes it rather difficult to identify. That might explain the absence of atp8 in L. hyalina since that gene is not located at the boundaries of the contig sequence that was used as input for MITOS, where we would expect missing data in an incomplete mitochondrial genome.
We detected two copies of trnK in L. hyalina, both highly similar to the trnK of L. antarctica and Vema ewingi in their structure. Duplication of tRNAs is not uncommon and has been reported before (e.g., [37]). A partial inversion of at least cox1-cox2-trnK of a conserved lophotrochozoan gene complex could explain the duplicated trnK detected in L. hyalina, since MITOS additionally detected relics of cox1 in a row with trnK and cox2 in that individual arrangement. This could also indicate a tandem duplication random loss event.
The three monoplacophoran mitogenomes analyzed herein exhibit almost the same number of non-coding regions; 21 in the incomplete mtDNA of L. hyalina and 27 and 28 in Vema ewingi and L. antarctica, respectively. Several non-coding regions are larger than 100 bp, distributed throughout the genomes and differing substantially in lengths within the same genome. This occurs frequently in molluscan mitogenomes. For example, in the class Gastropoda, some families possess many small NCRs [58, 59], as well as in Cephalopoda, which show intergenic regions that may be longer than 900 bp [51]. Katharina (Polyplacophora) also has several NCRs [35], and the bivalve taxon Placopecten contains NCRs up to 10,000 bp [57]. Almost half of the NCRs in Monoplacophora are located between the same genes in all three mtDNAs. L. antarctica shares more relative gene boundaries of NCRs with Vema ewingi than with L. hyalina; this is unexpected since it suggests a closer relationship of L. antarctica to Vema ewingi than to L. hyalina, but this might also be due to information missing in L. hyalina. The congruent relative location of two NCRs found in Nautilus and Monoplacophora with two identical or even highly similar sequence motifs might be synapomorphic and thus indicate common ancestry for monoplacophorans and cephalopods as it is proposed by Smith and colleagues [16, 17]; however, the motifs are very short and could also be either plesiomorphic or convergent.
Each of the two complete mitogenomes of L. antarctica and Vema ewingi has its largest NCR between trnF and trnT (see Fig. 3). These NCRs are 2012 bp and 2287 bp long respectively and both contain the AT-rich motif 1 that is almost identical in both mitogenomes concerning nucleotide composition, length, and position within the NCR (Table 1). A very similar motif is visible in L. hyalina in the NCR between trnK and cox1 (Table 1). The long and unassigned regions could be the potential origins of transcription of our monoplacophoran mtDNAs since AT-rich motifs are usually evidence for the control region of mitogenomes [59, 60]. Several other repetitive motifs are visible in the largest NCRs of L. antarctica and Vema ewingi, which provide even more evidence that this region is the control region. Motif 1 is repeated between trnG and trnE in L. antarctica and Vema ewingi, again with almost congruent starting points and very similar positions within the NCRs (Table 1, Fig. 3). We hypothesize that the initiation region was partially duplicated to have two starting points for the replication process which would lead to an increased transcription rate as was suggested for cephalopods before [61]. Although we found evidence for the potential control region in L. hyalina, too, we were not able to detect its duplication in this incomplete mt genome.
MITOS annotated fragmentary cox1 in L. hyalina and parts of atp6 in L. antarctica in the potential initiation regions. These protein-coding gene fragments are located near their functional copies. A possible scenario could be that part of the mitogenome, consisting minimally of the respective PCGs, was duplicated, and this is still visible in both Laevipilina individuals through residual PCG fragments. These duplicated copies might be in the process of being lost. Whether in Vema ewingi the loss is already finished, or the duplication event never took place is not clear. Nevertheless, we identified a region of accelerated rearrangement rate and this is third indication for locating the origin of replication in these NCRs in Laevipilina. Such a control region is usually described as the longest non-coding region within the mitogenome that is rich in AT, often including repetitive motifs, and seems to be a hotspot for rearrangements [59, 62]. The existence of duplicated control regions or parts thereof could be seen as a similarity for Monoplacophora and Cephalopoda (see [63]), since this feature is not known from other molluscs so far but is observed in other metazoan mitogenomes [64–66].
The second repetitive sequence motif (motif 2), is found in L. antarctica in the unassigned region between nad2 and trnC as well as in Vema ewingi in the non-coding part between nad2 and trnS1 (Table 1). This motif starts in both NCRs at almost the same position. Unassigned regions are known to be extremely variable because they do not underlie any selective pressure. Independent evolution of two identical 18 bp long nucleotide motifs in the same position is unlikely, so this motif is probably an apomorphy inherited from the common ancestor of these two taxa.
Phylogeny
Several phylogenetic approaches resulted in ambiguous topologies, which were sensitive to taxon sampling. Neither nucleotide nor amino acid taxon sets supported Aculifera (Polyplacohora + Aplacophora) or Conchifera (comprising all other shell-bearing classes), in contrast to Osca and colleagues [47] (see Table 2). A trend in amino acid analyses is the repeated recovery of a highly supported Pleistomollusca, whereas nucleotide based analyses supported Serialia (Table 2). In the data set with 3 non-molluscan outgroups, neither the amino acid nor nucleotide datasets supported the monophyly of Mollusca, which is, however, well-established [2, 15, 16, 27, 28, 32]. Molluscan non-monophyly is a common result of phylogenetic analyses based on mt protein coding genes [34, 44, 46] which was unaffected by the addition of more taxa here (Table 2).
Analyses recovered a monophyletic Monoplacophora and tended to support monophyly of other molluscan classes, except for bivalves. Non-protobranch bivalves have longer branches and rearranged gene orders compared to the protobranch Solemya. Such high levels of gene rearrangements were suggested to be linked with high rates of nucleotide substitution [26, 34, 44].
In amino acid datasets, the lamellibranch bivalves cluster as the sister group to scaphopods, but Solemya clusters with gastropods (Fig. 1, Table 2). The latter relationship was also recovered by Plazzi et al. 2013 [48] but was interpreted as an artifact due to limited phylogenetic signal in the bivalve lineage of Opponobranchia (including Nuculida and Solemyida). Solemya is the only bivalve in our dataset that has its genes arranged on both strands, a fact that leads to different substitution skew between plus and minus strands of the mt genome. Such differences in nucleotide composition might influence phylogenetic analyses and could be an explanation for our diphyletic clustering of bivalve taxa [67, 68].
Pruning non-protobranch bivalves recovers Solemya as the sister group to gastropods, i.e. a taxon Pleistomollusca ([15], Fig. 2; Additional files 1 and 2: Figures S1 and S2) in most amino acid analyses. Excluding the remaining protobranch bivalve, Solemya, from our analyses did not result in an aculiferan topology (Table 2). That is in contrast to Osca et al. [47] who excluded Bivalvia and recovered Aculifera (although Solenogastres was not sampled) either with poor support (ML) or with strong support but not as part of a monophyletic Mollusca (BI). In the taxon set in Osca et al. [47], Conchifera were lacking Bivalvia, which were pruned, and Monoplacophora.
As Osca et al. [47] recovered Aculifera and Conchifera we expected that adding further, taxa such as protobranchs and monoplacophorans might be beneficial to resolve further aspects of deep molluscan evolution. Within this study we employed different taxon sets to explore the robustness of the data. However, the diversity of topologies recovered herein is striking and suggests there is limited phylogenetic signal in this data. By modifying datasets we recovered several formerly proposed and currently disregarded hypotheses of higher taxa, but never the preferred Aculifera or Conchifera [47].
Conclusion
This mitogenomic study includes three members of two monoplacophoran genera. Our phylogenetic results of analyzing the protein coding supermatrix of 13 genes of 18 selected molluscan taxa across 7 of 8 classes stay ambiguous. Common and highly accepted molluscan hypotheses as the Aculifera or Conchifera concepts never appear in any of our phylogenetic permutations.
Our finding of unique protein gene arrangements in Monoplacophora and chitonine but not acanthochitonine Polyplacophora is remarkable because it may support the Serialia hypothesis, which is in conflict with the Aculifera/Conchifera hypothesis, but more likely it represents a plesiomorphic genome structure for molluscs. Any topology would imply convergent evolution of identical PCG clusters within Mollusca. On one hand, this clearly weakens the significance of supposedly rare gene rearrangement events and single genome level characters. On the other hand, this demonstrates the existence of further genome level characters that may become useful if mitogenomes are explored densely over molluscan (and other) taxa. Unfortunately, phylogenetic analyses of the mtDNA provided little information for resolving mollusc phylogeny. Furthermore, we need to expand our yet limited knowledge on mitochondrial evolution and data from the molluscan class Solenogastres (=Neomeniomorpha) is still lacking. High throughput sequencing as used here is a powerful and accurate way to add further mitogenomes of taxa that are small or with limited material available.
Methods
Preparation of Vema ewingi
Vema ewingi was collected on R/V “Dimitry Mendellev” at 8°S 81°W in 5800 m depth. DNA was extracted and purified using the Qiagen DNeasy kit (Qiagen, Hilden, Germany) following the manufacturer’s protocol. DNA concentration was measured using a Qubit with the double-stranded DNA broad range kit. DNA quality was evaluated using a 1% SB agarose gel. Gel electrophoresis revealed that the DNA was degraded with an average fragment size of around 500 bp. However, some large fragments of DNA up to around 10,000 bp were present.
An Illumina Nextera (Illumina, San Diego, CA, USA) library was prepared following the manufacturer’s protocol. However, the resulting library had a low size distribution because the template DNA was degraded. Additional attempts were made to prepare Nextera libraries using more template DNA than recommended by the Illumina protocol. This produced better quality libraries based on size distribution with the optimal library using four times the recommend amount or 200 ng total.
Sequencing was conducted using a 2 × 250 bp paired-end (PE) v2 kit on the Illumina MiSeq at Auburn University. The Vema libraries were sequenced in parallel with libraries for other projects with around eight dual-indexed libraries sequenced at a time. Several attempts at sequencing various Vema Nextera libraries were made using different amounts of template DNA, combining all of the Vema genomic data collected to that point, and assembling the paired-end reads using Ray 2.2.0 with a k-mer of 31 on the Auburn University SkyNet server never yielded a complete mitochondrial genome.
Therefore, we abandoned the Nextera approach and prepared libraries using the NEB Next Ultra kit (New England Biolabs, Ipswich, MA, USA) for Illumina sequencing. As the DNA was already degraded to an average size of around 500 bp, no shearing was necessary. End-repair, adapter ligation, and barcode incorporation via PCR were conducted following the manufacturer’s protocol. As above, sequencing was conducted using a 2 × 250 bp paired-end (PE) v2 kit on the Illumina MiSeq at Auburn University. Again, around eight indexed libraries were sequenced at a time and after two runs, a complete mitochondrial genome could be assembled for Vema.
In order to identify the complete mitochondrial genome, the assembly was searched against a nucleotide BLAST database constructed from the complete mitochondrial genome of Katharina tunicata (Polyplacophora) using BLASTN and TBLASTX using an e-value cutoff of 0.01.
Preparation of Laevipilina antarctica
Total genomic DNA was extracted from a piece of tissue of one specimen of Laevipilina antarctica (ZSM-Mol-20090330, DNABANK-Mol-MS-016), which was collected during the expedition with R/V Polarstern in Antarctica, using the NucleoSpin Tissue Kit (Macherey-Nagel, Düren, Germany) following the instructions in [69].
Ten nanogram of DNA was used for multiple strand replacement based DNA amplification using the illustra GenomiPhi V2 DNA Amplification Kit (GE Healthcare Life Sciences, Freiburg, Germany) using the manufacturers instruction followed by standard ethanol precipitation. Subsequently the DNA was purified using the Qiagen MinElute system (Qiagen, Hilden, Germany), DNA concentration was determined using the Qubit® 2.0 Fluorometer. 1 μg of DNA was used to create a standard fragment DNA sequencing library with the TruSeq DNA Sample Preparation Kit v2 (Illumina, San Diego, CA, USA); the experimental average insert size was 250 bp. Two lanes of 101 bp paired-end-reads were sequenced on the Ilumina HiSeq2000 system. About 90 Gigabasepairs (Gbp) were obtained. These were filtered for quality, PCR duplicates, and adaptor sequences and corrected using SOAPfilter_v2.0 (https://github.com/tanghaibao/jcvi-bin/blob/master/SOAP/SOAPfilter_v2.0) using default settings. We subsetted 5–200 million paired reads in K-mer iterations of 23–99 and using various parameters for mitogenome assembly using SOAPdenovo2 [70]. The best assembly of the complete mitogenome was discovered using 50 million paired reads and settings other than default –R –u.
Preparation of Laevipilina hyalina
Total genomic DNA was extracted from a single specimen collected off California [13] using the Qiagen DNeasy kit (Qiagen, Hilden, Germany), following manufacturer’s protocols. Standard PCR protocols were used to generate sequences from Cytochrome c oxidase I (COI), 16S rDNA (16S) (see [13]) plus Cytochrome oxidase B (cob) using universals 424f + 876R [71] and Cytochrome c oxidase III (COIII) [72]. All amplifications were done using illustra PuReTaq Ready-To-Go PCR Beads (GE Healthcare Life Sciences, Freiburg, Germany) following the manufacturer’s protocols. PCR products were cleaned using USB ExoSAP-IT, and sequenced by Retrogen Inc. (San Diego, CA, USA). Sequencher v4 was used to inspect and trim sequences. Sequences from these mitochondrial genes were used to design Laevipilina-specific primers for long PCR amplification. The Primer3 algorithm was used to design these primers [73].
Various primer combinations were tested, and a final set of MCOIf + MCytbr (5′-ATTGGCTGGGGCAGTTACTA-3′ + 5′-TGTGGAGAGGGGTAACAAGG-3′) and MCOX3f + MCOIR (5′-GATGTTTCGGTTGGGATACG-3′ + 5′-AAAGGAACCCGCTCAAGAGT-3′) resulted in two overlapping fragments (approximately 7 kb and 3 kb respectively). All long PCR products were amplified using Platinum Taq DNA Polymerase High Fidelity (Invitrogen, Waltham, MA, USA) following the manufacturer’s specifications. The PCR products were visualized on 1% agarose gels run at 80 V for 90 min. PCR products were cleaned using GelElute Extraction kit (5 Prime, South San Francisco, CA, USA) and outsourced to Engencore (Selah Genomics, Greenville, SC, USA) for sequencing and assembly with the Roche 454 platform and Newbler v2.3.
Annotation of mitogenomic consensus sequences
Mitogenomic sequences were filtered from the whole genome assemblies via BLAST searches and by alignment to known sequences of mitochondrial genes. The MITOS web server [74] was used to annotate mitogenomic data of L. antarctica and Vema ewingi as well as the partial consensus sequence of L. hyalina. Mitogenomic consensus sequences of the bivalve Solemya velum (NC_017612 [48]), the caudofoveate Scutopus ventrolineatus (KC_757645 [47]) as well as Sypharochiton pelliserpentis (KJ_534307 [49]) and S. sinclairi (KJ_534306 [49]) were downloaded from GenBank and newly annotated via the MITOS web server as well. Recommended default parameters [74] and the invertebrate mitochondrial genetic code (translation Table 5) were used for all annotations of protein coding genes, transfer and ribosomal RNAs. Annotated single sequences were imported in Geneious version 6.1.7 to work on GC content, extract and examine non-coding regions as well as overlaps, to visualize secondary structures of tRNAs of special interest (default parameters in Geneious were used), and to compile the different datasets for phylogenetic analyses (please see section “Phylogenetic analyses” for details). As Tomita et al. [75] proposed for non-coding regions in Loligo, we conducted BLAST searches of all non-coding regions larger than 100 bp of our three monoplacophoran genomes to find possible similarities to other mt genomes but we did not find any noticeable hits. Moreover, we checked the largest NCRs (>2 kb) of L. antarctica and Vema ewingi for group II transposons. This phenomenon was found in the annelid Nephtys [76] but also in insects [77] and might give an explanation for the unusually long NCRs in our species. We conducted DNA foldings of the non-coding sequences via the Mfold web server under default options, but could not find any similarities to the described secondary structure of Nephtys which is described as a central core with six radiating helical domains [76]. Both NCRs were compared to the Dfam database [78], but no hits were detected.
MITOS detected genes atp6, cob, cox3, nad3, and nad4 in the L. hyalina consensus sequence divided in two parts, nad2 in three parts. The parts of atp6, cob, and cox3 are overlapping (atp6, cob) or are at least adjacent (cox3) and therefore were combined manually; nad2, nad3, and nad4 do actually have non-annotated nucleotides in reverse order between the annotated gene parts. These non-annotated parts turned out to be reverse complement parts and were corrected by hand in Geneious version 6.1.7.
Annotated gene arrangements of all three monoplacophoran species were compared to each other and to other molluscan taxa (Katharina tunicata (NC_001636 [35]), Sypharochiton spp. (KJ_534306, KJ_534307 [49]), Nautilus macromphalus (NC_007980 [51]), Octopus vulgaris (NC_006353 [61]) by eye. Furthermore we searched for sequence motifs that occur in more than one monoplacophoran species with MEME Suite version 4.9.1 via the MEME web server [79].
Phylogenetic analyses
Newly generated data for Vema ewingi and Laevipilina antarctica and reannotated mt data of Solemya velum [48], Scutopus ventrolineatus [47], Sypharochiton pelliserpentis and S. sinclairi [49] were added to a taxon-subset of the 13 mitogenomic protein coding genes (PCGs) from Stöger and Schrödl [44], comprising 18 molluscan and three lophotrochozoan outgroup taxa (Table 5). Due to visibly exceptionally long branches and unusual attraction of outgroup taxa to ingroups in previous studies [34, 44, 46, 80] and in own pre-analyses, we excluded all outgroup taxa except Platynereis (Annelida) that showed a short branch in pre-analyses with more outgroup taxa, and excluded all bivalve taxa but the basal protobranch Solemya [81]. To reduce potential long-branch attraction artifacts that are already known from previous studies (e.g., [47]), we removed the two scaphopod taxa Graptacme and Siphonodentalium. Moreover, we also ran analyses based on nucleotide (nuc) and amino acid (aa) datasets of all taxon sets without any outgroups. All single nucleotide PCG sets were translated into amino acids using the invertebrate mitochondrial genetic code. Single nucleotide and amino acid datasets of PCGs were aligned using MAFFT version 7.017 [82] implemented in Geneious under the E-INS-i algorithm with a gap open penalty of 3. In pre-analyses we masked all single gene-alignments (nuc and aa) with Aliscore version of 5th February 2008 [83, 84] by running 10.000.000.000 replicates. Hypervariable positions were trimmed with Alicut version 2.0 [83, 84]. Moreover, we ran pre-analyses where we eliminated poorly aligned and hypervariable regions of all aa single alignments via Gblocks [85] since this program is more restrictive than Aliscore. In Gblocks we applied default options except for the atp8 alignment because this dataset would have been subsequently eliminated completely and we wanted to include the complete set of protein-coding genes; for atp8 alignments we chose all options using a less stringent selection. The Gblocks masked single alignments were tested for best fitting evolutionary models with ProtTest version 2.4 [86] by choosing from those models that are available in RAxML (DAYHOFF, DCMUT, JTT, MTREV, WAG, RTREV, CPREV, VT, BLOSUM62, and MTMAM). We further tried to improve the aa single alignments by refining the MAFFT-alignment via Muscle version 3.8 [87]. The resulting nucleotide and amino acid individual PCG-gene alignments under the different treatments were concatenated in Geneious with the following order: atp6, atp8, cob, cox1, cox2, cox3, nad1, nad2, nad3, nad4, nad4L, nad5, nad6. All atp8 alignments produced herein are missing the sequence for Mytilus, since this taxon lacks the atp8 gene [43]. All Maximum Likelihood (ML) analyses were performed with the program RAxML-HPC [88], executing 500 bootstrap replicates under the CAT approximation for rate heterogeneity and the GTR model. Masking with Aliscore or Gblocks or no masking procedure as well as partitioning the concatenated dataset or not did not make any difference in the resulting tree topology and will not be discussed below. Further analyses of selected concatenated alignments were carried out with the program SplitsTree version 4 [89] to test for potential conflicts of the data.
For the two preferred datasets (aa-1, aa-2) we carried out additional analyses with Phylobayes MPI on the CIPRES Science Gateway (https://cushion3.sdsc.edu/portal2/) using the CAT-GTR model and running 4 chains for each of the datasets. Analysis of datset aa-1 was executed for 79.839, respectively aa-2 for 105.593 generations until stationarity was reached. Burn-in was set to 2000 for each of the chains. Maxdiff for aa-1 was 0,07, for aa-2 it was 0,1.
Competing phylogenetic hypotheses run on the two main datasets aa-1 and aa-2 were evaluated using the Shimodaira Hasegawa test [90] and the Approximately Unbiased test [91] in RAxML 8.2.4 [92] and Consel [90]. The PROTGAMMAGTR model was used for these analyses.
Since the phylum Mollusca diverged in the Cambrian or earlier, non-phylogenetic signal in the molecular datasets could lead to anomalous topologies due to compositional biases, substitution saturation or increased substitution rates [93, 94]. Therefore we tested our preferred single gene alignments (amino acid only) for saturation and rate heterogeneity with the programs TreSpEx [95] and BaCoCa [96].
Acknowledgements
Specimens were collected during the ANDEEP-SYSTCO cruise on RV Polarstern, as well as during a collection trip on RV Dimitry Mendellev and on RV Robert Gordon Sproul. Thanks go to Enrico Schwabe, Greg Rouse and Nikita Kutcheruk who helped to collect the material. We thank the Ludwig-Maximilians-University Graduate Center (stipend to IS), the German Research Foundation Priority Programs 1174 (SCHR667/9-1) and 1158 (SCHR667/15-1) for financial support, the US National Science Foundation for funding to Kevin Kocot (DEB-1210518), Scripps Institution of Oceanography for support, Liz Borda for advice in the lab, and Yuanning Li, Nina Mikkelsen, and Pam Brannock for help in the lab.
Availability of data
The main datasets and tree topologies of this article will be available at TreeBase (http://purl.org/phylo/treebase/phylows/study/TB2:S20256). New mitogenomic sequence data of the three monoplacophoran species generated herein will be deposited at Genbank (see Table 5 for accession numbers).
Authors’ contributions
IS performed analyses and drafted the manuscript; AJP, KMK and NGW carried out the molecular lab work and genome assemblies; DI, KMH, NGW and MS did the field work and provided monoplacophorans; KMH, NGW and MS conceived the study and contributed to writing the paper. All authors contributed to read and approved the final manuscript version.
Competing interests
The authors declare that they have no competing interests.
Ethics approval and consent to participate
The authors confirm that all experiments conducted in this study comply with institutional, national, or international guidelines.
Abbreviations
- aa
Amino acid
- atp6-8
ATP synthase subunits 6–8
- bp
Base pair
- cob
Cytochrome oxidase b
- cox1-3
Cytochrome c oxidase subunits 1–3
- Gbp
Gigabase pair
- ML
Maximum likelihood
- mt
mitochondrial
- nad1-6
4L, NADH dehydrogenase subunits 1–6, 4L
- NCR
Non-coding region
- nuc
Nucleotide
- PCG
Protein-coding gene
- PCR
Polymerase chain reaction
- PE
Paired-end
- rRNA
Ribosomal RNA
- rrnL
Large ribosomal RNA
- rrnS
Small ribosomal RNA
- tRNA
Transfer RNA
- trnX
Transfer RNA for amino acid X (denoted by the one-letter IUPAC code)
Additional files
Bayesian Inference tree based on the large amino acid dataset. The tree was inferred with Phylobayes running four chains and 79.839 generations until stationarity was reached. Loxocorone was used to root the tree. (PDF 2 kb)
Bayesian Inference tree based on the amino acid dataset without the two outgroup taxa Lineus and Loxocorone and the reduction of bivalve taxa to the protobranch taxon Solemya. The tree was inferred with Phylobayes running four chains and 105.593 generations until stationarity was reached. Platynereis was used to root the tree. (PDF 2 kb)
Contributor Information
I. Stöger, Email: Isabella.Stoeger@zsm.mwn.de
K. M. Kocot, Email: kmkocot@ua.edu
A. J. Poustka, Email: poustka@molgen.mpg.de, Email: poustka@dcgms.de
N. G. Wilson, Email: Nerida.Wilson@museum.wa.gov.au
D. Ivanov, Email: ivanovdl@zmmu.msu.ru
K. M. Halanych, Email: ken@auburn.edu
M. Schrödl, Email: Michael.Schroedl@zsm.mwn.de
References
- 1.Parkhaev PY. The early molluscan radiation. In: Ponder WF, Lindberg DR, editors. Phylogeny and evolution of the Mollusca. Berkeley: University of California Press; 2008. pp. 33–69. [Google Scholar]
- 2.Stöger I, Sigwart JD, Kano Y, Knebelsberger T, Marshall BA, Schwabe E, et al. The continuing debate on deep molluscan phylogeny: Evidence for Serialia (Mollusca, Monoplacophora + Polyplacophora). Biomed Res Int. 2013;http://dx.doi.org/10.1155/2013/407072 [DOI] [PMC free article] [PubMed]
- 3.Lemche H. A new living deep-sea mollusc of the Cambro-Devonian class Monoplacophora. Nature. 1957;179:413–416. doi: 10.1038/179413a0. [DOI] [Google Scholar]
- 4.Ruthensteiner B, Schröpel V, Haszprunar G. Anatomy and affinities of Micropilina minuta Warén, 1989 (Monoplacophora: Micropilinidae). J Moll Stud. 2010;doi:10.1093/mollus/eyq013
- 5.Runnegar B, Pojeta J., Jr Molluscan phylogeny: the paleontological viewpoint. Science. 1974;186:311–317. doi: 10.1126/science.186.4161.311. [DOI] [PubMed] [Google Scholar]
- 6.Wingstrand KG. On the anatomy and relationships of recent Monoplacophora. Galathea Report. 1985;16:1–94. [Google Scholar]
- 7.Haszprunar G. Is the Aplacophora monophyletic? A cladistic point of view. Am Malacol Bull. 2000;15:115–130. [Google Scholar]
- 8.Salvini-Plawen L. The significance of the Placophora for molluscan phylogeny. Venus. 2006;65:1–17. [Google Scholar]
- 9.Haszprunar G, Schander C, Halanych KM. Relationships of the higher molluscan taxa. In: Ponder WF, Lindberg DR, editors. Towards a phylogeny of Mollusca. Berkeley: University of California Press; 2008. pp. 19–32. [Google Scholar]
- 10.Winnepenninckx B, Backeljau T, De Wachter R. Investigation of molluscan phylogeny on the basis of 18S rRNA sequences. Mol Biol Evol. 1996;13:1306–1317. doi: 10.1093/oxfordjournals.molbev.a025577. [DOI] [PubMed] [Google Scholar]
- 11.Passamaneck YJ, Schander C, Halanych KM. Investigation of molluscan phylogeny using large-subunit and small-subunit nuclear rRNA sequences. Mol Phylogenet Evol. 2004;32:25–38. doi: 10.1016/j.ympev.2003.12.016. [DOI] [PubMed] [Google Scholar]
- 12.Giribet G, Okusu A, Lindgren AR, Huff SW, Schrödl M, Nishiguchi ML. Evidence for a clade composed of mollusks with serially repeated structures: monoplacophorans are related to chitons. P Natl Acad Sci USA. 2006;doi:10.1073/pnas.0602578103 [DOI] [PMC free article] [PubMed]
- 13.Wilson NG, Rouse GW, Giribet G. Assessing the molluscan hypothesis Serialia (Monoplacophora + Polyplacophora) using novel molecular data. Mol Phylogenet Evol. 2010;doi:10.1016/j.ympev.2009.07.028 [DOI] [PubMed]
- 14.Kano Y, Kimura S, Kimura T, Warén A. Living Monoplacophora: morphological conservatism or recent diversification? Zool Scr. 2012;doi:10.1111/j.1463-6409.2012.00550.x
- 15.Kocot KM, Cannon JT, Todt C, Citarella MR, Kohn AB, Meyer A, et al. Phylogenomics reveals deep molluscan relationships. Nature. 2011;doi:10.1038/nature10382 [DOI] [PMC free article] [PubMed]
- 16.Smith SA, Wilson NG, Goetz FE, Feehery C, Andrade SCS, Rouse GW, et al. Resolving the evolutionary relationships of mollusks with phylogenomic tools. Nature. 2011;doi:10.1038/nature10526 [DOI] [PubMed]
- 17.Smith SA, Wilson NG, Goetz FE, Feehery C, Andrade SCS, Rouse GW, et al. Corrigendum: Resolving the evolutionary relationships of mollusks with phylogenomic tools. Nature. 2013;doi:10.1038/nature11736 [DOI] [PubMed]
- 18.Telford MJ, Budd GE. Invertebrate evolution: bringing order to the molluscan chaos. Curr Biol. 2011;doi:10.1016/j.cub.2011.10.029 [DOI] [PubMed]
- 19.Kocot KM. Recent advances and unanswered questions in deep molluscan phylogenetics. Am Malacol Bull. 2013;http://dx.doi.org/10.4003/006.031.0112
- 20.Giribet G. On Aculifera: A review of hypotheses in tribute to Christopher Schander. J Nat Hist. 2014;48:2739–2749. doi: 10.1080/00222933.2014.958112. [DOI] [Google Scholar]
- 21.Sigwart JD, Todt C, Scheltema AH. Who are the Aculifera? J Nat Hist. 2014;http://dx.doi.org/10.1080/00222933.2014.964788
- 22.Vinther J. The origins of molluscs. Front Palaeont. 2014;doi:10.1111/pala.12140
- 23.Haszprunar G, Wanninger A. Molluscs. Curr Biol. 2012;doi:10.1016/j.cub.2012.05.039 [DOI] [PubMed]
- 24.Haszprunar G, Ruthensteiner BR. Monoplacophora (Tryblidia) - some unanswered questions. Am Malacol Bull. 2013;http://dx.doi.org/10.4003/006.031.0111
- 25.Lv S-P, Steiner G. The Testaria concept (Polyplacophora + Conchifera) updated. J Nat Hist. 2014;48:2751–2772. doi: 10.1080/00222933.2014.964787. [DOI] [Google Scholar]
- 26.Schrödl M, Stöger I. A review on deep molluscan phylogeny: old markers, integrative approaches, persistent problems. J Nat Hist. 2014;doi:10.1080/00222933.2014.963184
- 27.Dunn CW, Hejnol A, Matus DQ, Pang K, Browne WE, Smith SA, et al. Broad phylogenomic sampling improves resolution of the animal tree of life. Nature. 2008;doi:10.1038/nature06614 [DOI] [PubMed]
- 28.Meyer A, Witek A, Lieb B. Selecting ribosomal protein genes for invertebrate phylogenetic inferences: how many genes to resolve the Mollusca? Methods Ecol Evol. 2011;doi:10.1111/j.2041-210X.2010.00052.x
- 29.Struck TH, Wey-Fabrizius AR, Golombek A, Hering L, Weigert A, Bleidorn C, et al. Platyzoan paraphyly based on phylogenomic data supports a non-coelomate ancestry of Spiralia. Mol Phylogenet Evol. 2014;doi:10.1093/molbev/msu143 [DOI] [PubMed]
- 30.Zapata F, Wilson NG, Howison M, Andrade SCS, Jörger KM, Schrödl M, et al. Phylogenomic analyses of deep gastropod relationships reject. Orthogastropoda. 2014;http://dx.doi.org/10.1101/007039 [DOI] [PMC free article] [PubMed]
- 31.Lieb B, Todt C. Hemocyanin in mollusks - a molecular survey and new data on caudofoveate hemocyanin gene sequences. Mol Phylogenet Evol. 2008;doi:10.1016/j.ympev.2008.06.005 [DOI] [PubMed]
- 32.Vinther J, Sperling EA, Briggs DEG, Peterson KJ. A molecular palaeobiological hypothesis for the origin of aplacophoran mollusks and their derivation from chiton-like ancestors. Proc R Soc Lond B. 2012;doi:10.1098/rspb.2011.1773 [DOI] [PMC free article] [PubMed]
- 33.Kocot KM, Struck TH, Merkel J, Waits DS, Todt C, Brannock PM, et al. Phylogenomics of Lophotrochozoa with consideration of systematic error. Syst Biol. 2016;doi:10.1093/sysbio/syw079 [DOI] [PubMed]
- 34.Bernt M, Bleidorn C, Braband A, Dambach J, Donath A, Fritzsch G, et al. A comprehensive analysis of bilaterian mitochondrial genomes and phylogeny. Mol Phylogenet Evol. 2013d;doi:10.1016/j.ympev.2013.05.002 [DOI] [PubMed]
- 35.Boore JL, Brown WM. Complete DNA sequence of the mitochondrial genome of the black chiton. Katharina tunicata Genetics. 1994;138:423–443. doi: 10.1093/genetics/138.2.423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gissi C, Iannelli F, Pesole G. Evolution of the mitochondrial genome of Metazoa as exemplified by comparison of congeneric species. Heredity. 2008;doi:10.1038/hdy.2008.62 [DOI] [PubMed]
- 37.Zhong M, Struck TH, Halanych KM. Phylogenetic information from three mitochondrial genomes of Terebelliformia (Annelida) worms and duplication of the methionine tRNA. Gene. 2008;doi:10.1016/j.gene.2008.02.020 [DOI] [PubMed]
- 38.Shen X, Ma X, Ren J, Zhao F. A close phylogenetic relationship between Sipuncula and Annelida evidenced from the complete mitochondrial genome sequence of Phascolosoma esculenta. BMC Genomics. 2009;doi:10.1186/1471-2164-10-136 [DOI] [PMC free article] [PubMed]
- 39.Aguado MT, Glasby CJ, Schroeder PC, Weigert A, Bleidorn C. The making of a branching annelid: an analysis of complete mitochondrial genome and ribosomal data of Ramisyllis multicaudata. Scientific Reports. 2015;doi:10.1038/srep12072. [DOI] [PMC free article] [PubMed]
- 40.Doucet-Beaupré H, Breton S, Chapman EG, Blier PU, Bogan AE, Stewart DT, et al. Mitochondrial phylogenomics of the Bivalvia (Mollusca): searching for the origin and mitogenomic correlates of doubly uniparental inheritance of mtDNA. BMC Evol Biol. 2010;doi:10.1186/1471-2148-10-50 [DOI] [PMC free article] [PubMed]
- 41.Akasaki T, Nikaido M, Tsuchiya K, Segawa S, Hasegawa, M, Okada N. Extensive mitochondrial gene arrangements in coleoid Cephalopoda and their phylogenetic implications. Mol Phylogenet Evol. 2006;doi:10.1016/j.ympev.2005.10.018 [DOI] [PubMed]
- 42.Allcock AL, Cooke IR, Strugnell JM. What can the mitochondrial genome reveal about higher-level phylogeny of the molluscan class Cephalopoda? Zool J Linn Soc. 2011;doi:10.1111/j.1096-3642.2010.00656.x
- 43.Boore JL, Medina M, Rosenberg LA. Complete sequences of two highly rearranged molluscan mitochondrial genomes, those of the scaphopod Graptacme eborea and of the bivalve Mytilus edulis. Mol Biol Evol. 2004;doi:10.1093/molbev/msh090 [DOI] [PubMed]
- 44.Stöger I, Schrödl M. Mitogenomics does not resolve deep molluscan relationships (yet?). Mol Phylogenet Evol. 2013;doi:10.1016/j.ympev.2012.11.017 [DOI] [PubMed]
- 45.Van de Peer Y. Phylogenetic inference based on distance methods. In: Lemey P, Salemi M, Vandamme A-M, editors. The phylogenetic handbook. Cambridge: Cambridge University Press; 2009. pp. 157–158. [Google Scholar]
- 46.Yokobori S-i, Iseto T, Asakawa S, Sasaki T, Shimizu N, et al. Complete nucleotide sequences of mitochondrial genomes of two solitary entprocts, Loxocorone allax and Loxosomella aloxiata: Implications for lophotrochozoan phylogeny. Mol Phylogenet Evol. 2008;doi:10.1016/j.ympev.2008.02.013 [DOI] [PubMed]
- 47.Osca D, Irisarri I, Todt C, Grande C, Zardoya R. The complete mitochondrial genome of Scutopus ventrolineatus (Mollusca: Chaetodermomorpha) supports the Aculifera hypothesis. BMC Evol Biol. 2014;doi:10.1186/s12862-014-0197-9 [DOI] [PMC free article] [PubMed]
- 48.Plazzi F, Ribani A, Passamonti M. The complete mitochondrial genome of Solemya velum (Mollusca: Bivalvia) and its relationships with Conchifera. BMC Genomics. 2013;doi:10.1186/1471-2164-14-409 [DOI] [PMC free article] [PubMed]
- 49.Veale AJ, Williams L, Tsai P, Thakur V, Lavery S. The complete mitochondrial genomes of two chiton species (Sypharochiton pelliserpentis and Sypharochiton sinclairi) obtained using Illumina next generation sequencing. MDN. 2014;doi:10.3109/19401736.2014.905846 [DOI] [PubMed]
- 50.Irisarri I, Eernisse DJ, Zardoya R. Molecular phylogeny of Acanthochitona (Mollusca: Polyplacophora: Chitonida): three new mitochondrial genomes, rearranged gene orders and systematics. J Nat Hist. 2014;doi:10.1080/00222933.2014.963721
- 51.Boore JL. The complete sequence of the mitochondrial genome of Nautilus macromphalus (Mollusca: Cephalopoda). BMC Genomics. 2006;doi:10.1186/1471-2164-7-182 [DOI] [PMC free article] [PubMed]
- 52.Boore JL. Animal mitochondrial genomes. Nucleic Acids Res. 1999;27:1767–1780. doi: 10.1093/nar/27.8.1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sigwart JD, Schwabe E, Saito H, Samadi S, Giribet G. Evolution in the deep sea: combined analysis of the earliest-derived living chitons using molecules and morphology (Mollusca, Polyplacophora, Lepidopleurida). Invertebr Syst. 2011;doi:http://dx.doi.org/10.1071/IS10028.
- 54.Sigwart JD, Stöger I, Knebelsberger T, Schwabe E. Chiton phylogeny (Mollusca : Polyplacophora) and the placement of the enigmatic species Choriplax grayi (H. Adams & Angas). Invertebr Syst. 2013;doi:http://dx.doi.org/10.1071/IS13013.
- 55.Edwards SV, Jennings WB, Shedlock AM. Phylogenetics of modern birds in the era of genomics. Proc R Soc B. 2005;doi:10.1098/rspb.2004.3035 [DOI] [PMC free article] [PubMed]
- 56.Vallès Y, Boore JL. Lophotrochozoan mitochondrial genomes. Integr Comp Biol. 2006;doi:10.1093/icb/icj056 [DOI] [PubMed]
- 57.Smith DR, Snyder M. Complete mitochondrial DNA sequence of the scallop Placopecten magellanicus: evidence of transposition leading to an uncharacteristically large mitochondrial genome. J Mol Evol. 2007;doi:10.1007/s00239-007-9016-x [DOI] [PubMed]
- 58.Grande C, Templado J, Zardoya R. Evolution of gastropod mitochondrial genome arrangements. BMC Evol Biol. 2008;doi:10.1186/1471-2148-8-61 [DOI] [PMC free article] [PubMed]
- 59.Rawlings TA, MacInnis MJ, Bieler R, Boore JL, Collins TM. Sessile snails, dynamic genomes: gene rearrangements within the mitochondrial genome of a family of caenogastropod molluscs. BMC Genomics. 2010;doi:10.1186/1471-2164-11-440 [DOI] [PMC free article] [PubMed]
- 60.Bernt M, Braband A, Schierwater B, Stadler PF. Genetic aspects of mitochondrial genome evolution. Mol Phylogenet Evol. 2013c;doi:10.1016/j.ympev.2012.10.020 [DOI] [PubMed]
- 61.Yokobori S-i, Fukuda N, Nakamura M, Aoyama T, Oshima T. Long-term conservation of six duplicated structural genes in cephalopod mitochondrial genomes. Mol Biol Evol. 2004;doi:10.1093/molbev/msh227 [DOI] [PubMed]
- 62.Boore JL, Brown WM. Big trees from little genomes: mitochondrial gene order as a phylogenetic tool. Curr Opin Genet Dev. 1998;8:668–674. doi: 10.1016/S0959-437X(98)80035-X. [DOI] [PubMed] [Google Scholar]
- 63.Yokobori S-i, Lindsay DJ, Yoshida M, Tsuchiya K, Yamagishi A, Maruyama T, Oshima T. Mitochondrial genome structure and evolution in the living fossil vampire squid, Vampyroteuthis infernalis, and extant cephalopods. Mol Phylogenet Evol. 2007;doi:10.1016/j.ympev.2007.05.009. [DOI] [PubMed]
- 64.Kumazawa Y, Ota H, Nishida M, Ozawa T. The complete nucleotide sequence of a snake (Dinodon semicarinatus) mitochondrial genome with two identical control regions. Genetics. 1998;150:313–329. doi: 10.1093/genetics/150.1.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Eberhard JR, Wright TF, Bermingham E. Duplication and concerted evolution of the mitochondrial control region in the parrot genus Amazona. Mol Biol Evol. 2001;18:1330–1342. doi: 10.1093/oxfordjournals.molbev.a003917. [DOI] [PubMed] [Google Scholar]
- 66.Lavrov DV, Boore JL, Brown WM. Complete mtDNA sequences of two millipedes suggest a new model for mitochondrial gene rearrangements: duplication and nonrandom loss. Mol Biol Evol. 2002;19:163–169. doi: 10.1093/oxfordjournals.molbev.a004068. [DOI] [PubMed] [Google Scholar]
- 67.Hassanin A, Léger N, Deutsch J. Evidence for multiple reversals of asymmetric mutational constraints during the evolution of the mitochondrial genome of Metazoa, and consequences for phylogenetic inferences. Syst Biol. 2005;doi:10.1080/10635150590947843 [DOI] [PubMed]
- 68.Dreyer H, Steiner G. The complete sequences and gene organisation of the mitochondrial genomes of the heterodont bivalves Acanthocardia tuberculata and Hiatella arctica—and the first record for a putative Atpase subunit 8 gene in marine bivalves. Front Zool. 2006;doi:10.1186/1742-9994-3-13 [DOI] [PMC free article] [PubMed]
- 69.Knebelsberger T, Stöger I. DNA Extraction, Preservation and Amplification. In: Kress WJ, Erickson DL, editors. Methods in Molecular Biology, DNA BARCODES: METHODS AND PROTOCOLS. Berlin: Humana Press Springer; 2012. pp. 311–338. [DOI] [PubMed] [Google Scholar]
- 70.Luo R, Liu B, Xie Y, Li Z, Huang W, Yuan J, et al. SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. GigaScience. 2012;doi:10.1186/2047-217X-1-18 [DOI] [PMC free article] [PubMed]
- 71.Von Nickisch-Rosenegk M, Brown WM, Boore JL. Complete sequence of the mitochondrial genome of the tapeworm Hymenolepis diminuta: gene arrangements indicate that platyhelminths are eutrochozoans. Mol Biol Evol. 2001;18:721–730. doi: 10.1093/oxfordjournals.molbev.a003854. [DOI] [PubMed] [Google Scholar]
- 72.Boore JL, Brown WM. Mitochondrial genomes of Galathealinum, Helobdella, and Platynereis: sequence and gene arrangement comparisons indicate that Pogonophora is not a phylum and Annelida and Arthropoda are not sister taxa. Mol Biol Evol. 2000;17:87–106. doi: 10.1093/oxfordjournals.molbev.a026241. [DOI] [PubMed] [Google Scholar]
- 73.Koressaar T, Remm M. Enhancements and modifications of primer design program Primer3. Bioinformatics. 2007;23:1289–1291. doi: 10.1093/bioinformatics/btm091. [DOI] [PubMed] [Google Scholar]
- 74.Bernt M, Donath A, Jühling F, Externbrink F, Florentz C, Fritzsch G, et al. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol Phylogenet Evol. 2013a;doi:10.1016/j.ympev.2012.08.023 [DOI] [PubMed]
- 75.Tomita K, Yokobori S-i, Oshima T, Ueda T, Watanabe K. The cephalopod Loligo bleekeri mitochondrial genome: Multiplied noncoding regions and transposition of tRNA genes. J Mol Evol. 2002;doi:10.1007/s00239-001-0039-4. [DOI] [PubMed]
- 76.Vallès Y, Halanych KM, Boore JL. Group II Introns break new boundaries: Presence in a bilaterian’s genome. PLOS One. 2008;doi:10.1371/journal.pone.0001488.t001 [DOI] [PMC free article] [PubMed]
- 77.White TB, Lambowitz AM. The retrohoming of linear group II intron RNAs in Drosophila melanogaster occurs by both DNA Ligase 4–dependent and –independent mechanisms. PLOS Genet. 2012;doi:10.1371/journal.pgen.1002534 [DOI] [PMC free article] [PubMed]
- 78.Wheeler TJ, Clements J, Eddy SR, Hubley R, Jones TA, Jurka J, et al. Dfam: a database of repetitive DNA based on profile hidden Markov models. Nucleic Acids Res. 2013;doi:10.1093/nar/gks1265 [DOI] [PMC free article] [PubMed]
- 79.Bailey TL, Bodén M, Buske FA, Frith M, Grant CE, Clementi L, et al. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 2009;doi:10.1093/nar/gkp335 [DOI] [PMC free article] [PubMed]
- 80.Podsiadlowski L, Braband A, Struck TH, von Döhren J, Bartolomaeus T. Phylogeny and mitochondrial gene order variation in Lophotrochozoa in the light of new mitogenomic data from Nemertea. BMC Genomics. 2009;doi:10.1186/1471-2164-10-364 [DOI] [PMC free article] [PubMed]
- 81.González VL, Andrade SCS, Bieler R, Collins TM, Dunn CW, Mikkelsen PM, et al. A phylogenetic backbone for Bivalvia: an RNA-seq approach. Proc R Soc B. 2015;doi:10.1098/rspb.2014.2332 [DOI] [PMC free article] [PubMed]
- 82.Katoh K, Kazuharu M, Kuma K–i, Miyata T. MAFFT: a novel method for rapid multiple sequence alignment based on fast fourier transform. Nucleic Acids Res. 2002;30:3059–3066. doi: 10.1093/nar/gkf436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Misof B, Misof K. A Monte Carlo approach successfully identifies randomness in multiple sequence alignments: a more objective means of data exclusion. Syst Biol. 2009;doi:10.1093/sysbio/syp006 [DOI] [PubMed]
- 84.Kück P, Meusemann K, Dambach J, Thormann B, von Reumont B, Wägele JW. Parametric and non-parametric masking of randomness in sequence alignments can be improved and leads to better resolved trees. Front Zool. 2010;7:10. doi: 10.1186/1742-9994-7-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Castresana J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol. 2000;17:540–552. doi: 10.1093/oxfordjournals.molbev.a026334. [DOI] [PubMed] [Google Scholar]
- 86.Abascal F, Zardoya R, Posada D. ProtTest: Selection of best-fit models of protein evolution. Bioinformatics. 2005;doi:10.1093/bioinformatics/bti263 [DOI] [PubMed]
- 87.Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;doi:10.1093/nar/gkh340 [DOI] [PMC free article] [PubMed]
- 88.Stamatakis A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics. 2006;doi:10.1093/bioinformatics/btl446 [DOI] [PubMed]
- 89.Huson DH, Bryant D. Application of phylogenetic networks in evolutionary studies. Mol Biol Evol. 2006;doi:10.1093/molbev/msj030 [DOI] [PubMed]
- 90.Shimodaira H, Hasegawa M. CONSEL: for assessing the confidence of phylogenetic tree selection. Bioinformatics. 2001;17:1246–1247. doi: 10.1093/bioinformatics/17.12.1246. [DOI] [PubMed] [Google Scholar]
- 91.Shimodaira H. An approximately unbiased test of phylogenetic tree selection. Syst Biol. 2002;51:492–508. doi: 10.1080/10635150290069913. [DOI] [PubMed] [Google Scholar]
- 92.Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;doi:10.1093/bioinformatics/btu033 [DOI] [PMC free article] [PubMed]
- 93.Philippe H, Brinkmann H, Lavrov DV, Littlewood DTJ, Manuel M, Wörheide G, et al. Resolving difficult phylogenetic questions: why more sequences are not enough. PLoS Biol. 2011;doi:10.1371/journal.pbio.1000602 [DOI] [PMC free article] [PubMed]
- 94.Nosenko T, Schreiber F, Adamska M, Adamski M, Eitel M, Hammel J, et al. Deep metazoan phylogeny: when different genes tell different stories. Mol Phylogenet Evol. 2013;doi:10.1016/j.ympev.2013.01.010 [DOI] [PubMed]
- 95.Struck TH. TreSpEx – Detection of misleading signal in phylogenetic reconstructions based on tree information. Evol Bioinformatics. 2014;10:51–67. doi: 10.4137/EBO.S14239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kück P, Struck TH. BaCoCa – a heuristic software tool for the parallel assessment of sequence biases in hundreds of gene and taxon partitions. Mol Phylogenet Evol. 2014;70:94–98. doi: 10.1016/j.ympev.2013.09.011. [DOI] [PubMed] [Google Scholar]