

Abstract
Background
C3 glomerulopathy (C3G) includes both C3 glomerulonephritis (C3GN) and dense deposit disease (DDD) and is defined by C3-dominant deposits on immunofluorescence. Dysfunction of the alternative pathway (AP) of complement is central to the pathophysiology of C3G and young patients often harbor genetic alterations of AP mediators. Recently, a link between C3G and paraproteinemia has been established. We performed this study to better characterize older patients with C3G where this association is more frequently seen.
Methods
Fourteen biopsies from 12 patients meeting diagnostic criteria for C3G were identified in patients > 49 years of age from 2005 to 2015 after exclusion of cases containing masked monotypic immunoglobulin deposits. Pathologic and clinical features were reviewed.
Results
The median age was 63.5 years and 75% of patients were male. All had renal insufficiency at presentation. Kidney biopsy showed DDD in three patients and C3GN in the remainder. Serum protein electrophoresis revealed a paraprotein in 10 patients, 8 of which had a plasma cell dyscrasia on bone marrow biopsy. A membranoproliferative pattern of glomerular injury was seen in 64% of biopsies, while mesangial proliferative and endocapillary proliferative patterns were seen less frequently. Among patients with at least 1 year of follow-up (n = 9), five were on renal replacement therapy, three showed stable (but impaired) kidney function and one demonstrated improvement.
Conclusions
C3G is an uncommon but important cause of kidney injury in older adults and associates with a high prevalence of paraproteinemia. In adult patients with C3G, prognosis is guarded as most patients showed either progression to end-stage kidney disease or stable but impaired kidney function.
Keywords: alternative pathway, C3 glomerulopathy, monoclonal gammopathy
Introduction
The emergence of C3 glomerulopathy (C3G) as a diagnostic category of glomerular disease has resulted in a shift in the classification of membranoproliferative glomerulonephritis (MPGN) [1, 2]. Prior to the introduction of C3G, classification was based on light microscopic and ultrastructural findings, whereas current practice utilizes immunofluorescence staining patterns to determine whether glomerular injury is immune complex mediated or complement mediated. Since the first reports linking C3G with dysregulation of the alternative pathway (AP) of complement, the search for a cause has primarily focused on genetic alterations of mediators of AP and autoantibodies to these mediators [3].
Recently a link between paraproteinemia and C3G has been observed. Sethi et al. [4] reported that 10 of the 14 patients with dense deposit disease (DDD) >49 years of age had paraproteinemia. A link between paraproteinemia and C3 glomerulonephritis (C3GN) has also been suggested. Bridoux et al. [5] reported a series of six C3GN patients with monoclonal gammopathy. Similarly, Zand et al. [6] reported a prevalence of monoclonal gammopathy of 31% in 32 patients with C3GN. Many of the patients in these studies with paraprotein-related C3G were older adults, as would be expected given the higher prevalence of monoclonal gammopathy in this population. However, detailed clinicopathologic characterization of C3G in older adult patients has not been performed. We therefore conducted the following study to better understand C3G in older adult patients, including the association with underlying paraproteinemia and/or other forms of hematopoietic neoplasia.
Materials and methods
We reviewed the University of Utah Department of Pathology archives from 2005 to 2015. Cases included patients >49 years of age with biopsy characteristics that fulfilled the diagnostic criteria for glomerulonephritis (GN) with dominant C3 set forth by the C3G consensus report [7]. C3 staining was considered dominant if the observed staining intensity was at least two orders of magnitude greater than other immunoreactants (scale: 0–4+). Cases favored to be infection related were excluded if there was a recent or a concomitant infection with clinical and serologic recovery without relapse.
Light, immunofluorescence and electron microscopy findings were reviewed, as were clinical data from the time of biopsy and during follow-up. Pathologic features that were noted include the number of glomeruli present on the biopsy, light microscopic pattern of glomerular injury, degree of glomerular and tubulointerstitial scarring, immunofluorescence staining for immunoglobulins (Igs) and complement factors and ultrastructural localization and appearance of deposits. Where tissue was available, additional immunofluorescence staining was performed for C4d on the frozen tissue and for IgG, κ light chain and λ light chain on the paraffin-embedded tissue following predigestion with proteinase K to evaluate for masked deposits [8]. Cases found to have masked monotypic immunoglobulin deposits were also excluded from the cohort.
Clinical data were obtained from the electronic health record and included kidney function at the time of the biopsy and follow-up including serum creatinine, degree of proteinuria, presence of hematuria, bone marrow biopsy reports and clinical notes. Nephrotic-range proteinuria was defined as proteinuria >3.5 g/day, chronic kidney disease (CKD) stage 2 was defined as 60–89 mL/min/1.73 m2, stage 3 as 30–59 mL/min/1.73 m2, stage 4 as 15–29 mL/min/1.73 m2 and end-stage renal disease (ESRD) as <15 mL/min/1.73 m2. Estimated glomerular filtration rate (eGFR) was determined using the Modification of Diet in Renal Disease (MDRD) study equation. Serologic studies were reviewed, including serum complement levels and serum protein electrophoresis (SPEP)/immunofixation electrophoresis (IFE).
The University of Utah Institutional Review Board approved this study.
Results
Study cohort
From 2005 to 2015, 740 kidney biopsies were identified in patients ≥50 years of age. Twenty-four biopsies from 22 patients showed a GN with dominant C3 staining by routine immunofluorescence microscopy on frozen tissue. Of these, seven were chosen to represent infection-related GN due to recent or concomitant infection with clinical and serologic recovery. Two patients were excluded due to complete absence of follow-up data. One additional patient was found to have monotypic IgG κ deposits unmasked following immunofluorescence staining on the paraffin-embedded tissue block and was excluded from the final cohort. In all, 14 biopsies from 12 patients met the criteria for C3G and were included in this study, representing ∼2% of biopsies in patients >49 years of age during the study period. Three biopsies were diagnostic of DDD, while the remaining biopsies showed C3GN.
Clinical and laboratory data
Presenting and follow-up clinical data are shown in Table 1. The median age at the time of the diagnostic kidney biopsy was 63.5 years (range 52–90 years) and nine (75%) patients were male. All patients presented with hematuria and kidney insufficiency. The degree of proteinuria was highly variable [median 1.8 g/g of creatinine (range 0.1–10)]. Nephrotic-range proteinuria was present in 4 of the 11 patients with available data. The median presenting creatinine was 2 mg/dL (range 1.2–6.9). Complement C3 levels were low in 3 of the 10 patients (30%) with available data and C4 levels were normal in all 8 patients with available data. Evaluation of the AP of complement was performed in two patients. One showed no disease-causing mutations, risk alleles or autoantibodies, while the other carried two copies each of complement factor H (CFH) risk alleles p.Val62 and p.His402 without other disease-causing mutations or autoantibodies.
Table 1.
Clinical data
| Patient | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Age/sex | 63/M | 67/F | 63/M | 52/M | 54/M | 68/M | 64/M | 60/M | 90/F | 55/F | 75/M | 77/M |
| Presenting SCr/eGFR | 2.8/23 | 2/25 | 2.3/29 | 2.4/29 | 1.9/37 | 1.9/35 | 1.5/47 | 1.8/39 | 2.5/18 | 1.2/47 | n/a | 6.9/8 |
| Proteinuria (g/day) | 7 | 3 | 10 | 4.5 | <1 | 1.1 | >3.5 | <1 | 1.7 | 0.8 | n/a | 1.8 |
| Hematuria | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y |
| Complement C3 | 55 | 140 | 106 | 111 | n/a | Low | n/a | 99 | 109 | 105 | Normal | low |
| SPEP/IFE | IgG κ | IgG κ | IgG κ | IgG κ | IgG λ | – | IgG κ | – | IgG κ | IgG κ | IgA κ | IgG κ |
| BM biopsy | MGRS | Polyclonal plasmacytosis | MM | MM | MM | n/a | MGRS | n/a | n/a | MM | MGRS | MGRS |
| Follow-up time (months) | 120 | 2 | 24 | 36 | 66 | 17 | 2 | 10 | 36 | 36 | 40 | 24 |
| Follow-up SCr/eGFR | ESRD | 1.83/27 | ESRD | ESRD | ESRD | 1.9/35 | eGFR: 47 | 1.8/37 | 2.4/19 | 0.9/64 | 1.8/37 | ESRD |
SCr, serum creatinine (mg/dL); eGFR, estimated glomerular filtration rate (mL/min/1.73 m2); SPEP, serum protein electrophoresis; IFE, immunofixation electrophoresis; BM, bone marrow; MM, multiple myeloma; MGRS, monoclonal gammopathy of renal significance.
SPEP results were available in all 12 patients. Ten (83%) had a paraprotein, including all three patients with DDD. Of the patients with paraproteinemia, eight had IgG κ, one had IgG λ and one had IgA κ. Nine patients underwent further hematologic workup. Four were subsequently diagnosed with monoclonal gammopathy of renal significance and four with multiple myeloma (MM), two of which were considered smoldering. One patient with a faint IgG κ band on SPEP/IFE had a bone marrow biopsy showing a polyclonal plasmacytosis. In contrast, four of the seven patients with presumed infection-related GN had SPEP/IFE and only one showed a faint λ-restricted band without the associated heavy chain.
Pathologic characteristics
Detailed pathologic features are listed in Table 2. The median number of glomeruli present was 18.5 (range 9–32). An MPGN pattern of glomerular injury was prominent in nine (64%) biopsies (Figure 1), while a mesangial proliferative GN was seen in three (21%) cases and an endocapillary proliferative/exudative pattern was present in the remaining two (14%) cases. Two patients had follow-up biopsies. One (patient 1) had a recurrence of C3GN in his kidney allograft with a focal exudative GN (original biopsy showed MPGN). The other repeat biopsy (patient 10) showed a persistent MPGN with progression to DDD. The degree of global glomerular sclerosis was variable with a median of 36% (range 0–59%). Segmental sclerosis was observed in five cases and a crescentic form of glomerular injury was identified in two cases and was focal in both, involving 5 and 20% of glomeruli, respectively. The degree of interstitial fibrosis and tubular atrophy was mild (<25%) in four biopsies, moderate (25–50%) in six and severe (>50%) in four. None of the patients showed evidence of other paraprotein-related disease such as amyloidosis or light chain cast nephropathy.
Table 2.
Pathologic features
| Patient | 1 |
2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 |
11 | 12 | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Glomeruli, n | 17 | 20 | 32 | 18 | 10 | 21 | 14 | 19 | 10 | 17 | 24 | 9 | 22 | 31 |
| Pattern of injury | MPGN | FPGN | MPGN | MPGN | MPGN | MPGN | MPGN | MesPGN | MesPGN | MPGN | MPGN | MPGN | MesPGN | DPGN |
| Global sclerosis, % | 20 | 0 | 56 | 39 | 20 | 57 | 29 | 42 | 40 | 59 | 4 | 33 | 5 | 48 |
| IFTA, % | 50 | 0 | 30 | 70 | 30 | 30 | 30 | 30 | 10 | 60 | 20 | 30 | 20 | 70 |
| IF microscopy (frozen) | C3 (3+) IgG (tr) |
C3 (3+) IgM (tr) |
C3 (3+) IgM (1+) |
C3 (3+) IgM (1+) |
C3 (3+) | C3 (3+) | C3 (2+) | C3 (3+) | C3 (3+) | C3 (3+) IgM (tr) |
C3 (3+) | C3 (3+) | C3 (3+) | C3 (3+) IgM (<1+) |
| IF microscopy (paraffin, IgG/κ/λ) | n/a | Neg | Neg | Neg | Neg | Neg | Neg | Neg | Neg | Neg | Neg | Neg | Neg | Neg |
| C4d | n/a | MES (< 1+) |
Neg | MES (1+) | MES (1+) | Neg | n/a | MES (<1+) |
MES (1+) |
MES (< 1+) |
Neg | Neg | Neg | Neg |
| Ultrastructural localization of deposits | MES SEN SEP IN |
MES SEN IN |
MES SEN SEP |
MES SEN IN |
MES SEN SEP IN |
DDD | MES SEN |
MES SEP IN |
MES SEN SEP IN |
MES SEN SEP IN |
MES SEN SEP |
DDD | DDD | MES SEN SEP |
MPGN, membranoproliferative glomerulonephritis; FPGN, focal proliferative glomerulonephritis; DPGN, diffuse proliferative glomerulonephritis; MesPGN, mesangial proliferative glomerulonephritis; IF, immunofluorescence; IFTA, interstitial fibrosis and tubular atrophy; MES, mesangial; SEN, subendothelial; SEP, subepithelial; IN, intramembranous; DDD, dense deposit disease.
Fig. 1.
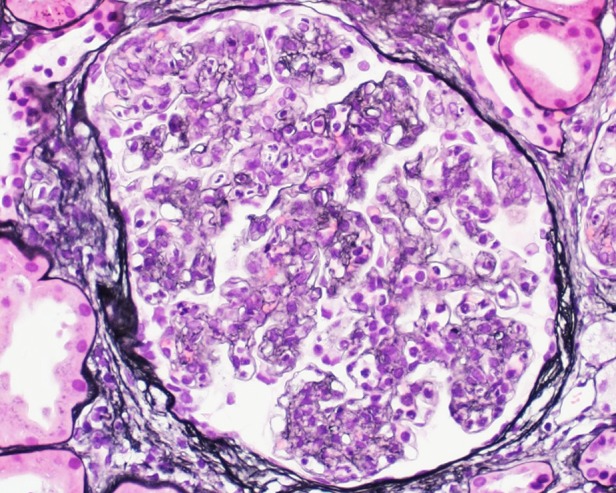
A membranoproliferative pattern of glomerular injury was the predominant pattern of injury in 64% of biopsies as seen in this 52-year-old male (patient 4), with lobular accentuation of the glomerular tuft, mesangial and endocapillary hypercellularity and segmental glomerular basement membrane duplication. Serum protein electrophoresis showed IgG κ, with bone marrow biopsy revealing smoldering myeloma. Three years after presentation, he progressed to ESRD requiring HD.
By immunofluorescence microscopy, all biopsies showed dominant C3 staining as defined by the C3 consensus report [7] (Figure 2A), with 13 biopsies showing 3+ staining for C3 and 1 demonstrating an intensity of 2+. Staining for Igs was seen in six biopsies with trace IgG in one case, 1+ IgM in three cases and trace IgM in two cases. Where tissue was available, additional immunofluorescence microscopy was performed on the frozen sample for C4d and on the paraffin-embedded sample for IgG, κ light chain and λ light chain following predigestion with proteinase K. C4d staining was performed by indirect immunofluorescence on 12 biopsies and showed the absence of staining in 6 biopsies and segmental mesangial staining (trace to 1+) in 6 biopsies (Figure 2B). Immunofluorescence on paraffin-embedded tissue was performed on at least one biopsy from every patient. All 12 included patients showed either no staining or nonspecific staining of serum without glomerular deposits. One patient (who was subsequently excluded) showed no staining for Ig or light chains on the frozen tissue but paraffin immunofluorescence revealed mesangial staining for IgG and κ light chain without staining for λ light chain, consistent with masked monotypic immunoglobulin deposits. This patient was subsequently diagnosed with stage IV mantle cell lymphoma, which was κ-restricted.
Fig. 2.

All biopsies showed dominant C3 staining by routine immunofluorescence microscopy as seen in this 64-year-old male (patient 7) with an IgG κ paraprotein and bone marrow biopsy showing monoclonal gammopathy of undetermined significance. Additional staining for C4d showed only trace mesangial staining in this patient (B).
Ultrastructural examination was performed on all biopsies and identified elongated ‘sausage-like’ hyperosmiophilic intramembranous deposits in three cases, defining DDD (Figure 3A). Other intramembranous deposits not meeting the threshold of DDD were identified in seven of the remaining cases of C3GN (Figure 3B). Mesangial deposits were identified in all 14 biopsies and subendothelial deposits were present in 11 biopsies. Subepithelial hump-shaped deposits were seen in nine biopsies (Figure 3C), with none of the DDD cases showing this finding. One biopsy showed vague substructural organization with a variegated appearance within the deposits (Patient 2, Figure 3D).
Fig. 3.

(A) Ultrastructural examination identified three patients with elongated hyperosmiophilic intramembranous deposits replacing the lamina densa, defining dense deposit disease. (B) Seven patients with C3 GN also demonstrated segmental intramembranous deposits. (C) Subepithelial hump-like deposits were identified in 64% of patients and were most commonly segmental in distribution. (D) One patient (patient 2) showed vague substructural organization with a variegated appearance within deposits.
Treatment
Information regarding postbiopsy therapy is available for 8 of the 12 patients. Of the four patients with MM (patients 3, 4, 5, 10), all received therapy including bortezomib and dexamethasone. Patient 5 also received cyclophosphamide and patient 10 ultimately showed a hematologic response to therapy and underwent stem cell transplantation (SCT). None of the patients with monoclonal gammopathy without myeloma were treated with chemotherapy. Two patients (2 and 8) were treated symptomatically with angiotensin receptor blockers. Patient 1 initiated therapy with eculizumab shortly after his diagnosis of recurrent C3G posttransplantation, with subsequent improvement of proteinuria from 1.5 to 0.3 g/g 3 weeks after initiation of therapy. Patient 9 declined therapy.
Renal outcome
The median length of follow-up was 30 months (range 2–120). Of those with at least 1 year of follow-up (n = 9), five (56%) patients developed ESRD, four of whom were on chronic hemodialysis (HD) at last follow-up. One patient received a kidney transplant after 4 years on HD and developed biopsy-proven recurrence 18 months after transplant. Of the remaining four with at least 1-year of follow-up, two had stable eGFR, one had improved eGFR and one had a follow-up eGFR of 37 mL/min/1.73 m2 (this patient did not have a baseline eGFR available). Of the patient's with DDD, one was HD dependent at last follow-up (5.5 years after initial presentation) and one had eGFR of 37 mL/min/1.73 m2 at the time of death, 3 years after the initial diagnosis. One patient initially presented with acute kidney injury and biopsy showed a proliferative GN with dominant C3 deposits with a subsequent biopsy 1 year later showing DDD. She was found to have MM, for which she received chemotherapy and SCT. At the last follow-up she had improvement of kidney function with a serum creatinine of 0.9 mg/dL 1-year post-SCT and 3 years after the initial diagnosis of GN. Of the patients with C3GN, one received a kidney transplant with subsequent recurrent disease, three are dependent on HD, and the remaining five patients had stable CKD at the last follow-up. Of the four patients with myeloma, three progressed to ESRD while one had improvement of kidney function following aggressive therapy with hematologic response and stem cell transplant. Five patients had monoclonal gammopathy, without meeting criteria for a diagnosis of MM. Two of these patients progressed to ESRD at the last follow-up, while the other three had stable CKD.
Discussion
The observation that GN with isolated C3 deposits is often associated with inherited and/or acquired abnormalities in the AP of complement has altered how nephropathologists and nephrologists diagnose and manage this unique family of diseases [9]. As we approach the end of the first decade of the C3G era, the heterogeneity of these lesions has become more apparent. Whereas acquired disease has historically been associated with autoantibody formation, other mechanisms for AP dysregulation in adults have been identified. Infectious stimuli can stimulate the AP of complement and lead to deposition of complement factors within glomeruli, as seen in infection-related GN. Indeed, some reports have shown a high prevalence of elevated anti-streptolysin O titers in patients with C3G and a link between genetic alterations of AP and atypical postinfectious GN (PIGN) has been demonstrated [10–12]. This relationship suggests that PIGN may represent a self-limiting form of C3G. In addition to infection-related and genetic forms of C3G, this and other reports highlight a potential connection between C3G and paraproteinemia.
Monotypic Igs are associated with a number of deposition-related glomerular diseases including amyloidosis, fibrillary GN, immunotactoid glomerulopathy, cryoglobulinemic GN, monoclonal IG deposition disease, Waldenstrom macroglobulinemic GN and proliferative GN with monoclonal IgG deposits (PGNMID) [13, 14]. C3G is a recent addition to the list of glomerulopathies tied to hematopoietic neoplasia. Mechanistically, Meri et al. [15] and Jokiranta et al. [16] showed an interaction between monoclonal λ light chain and CFH resulting in activation of the AP of complement. This suggests that in some cases the presence of a monoclonal protein may promote AP activation. The relationship between paraproteinemia and C3G is one of great clinical importance, as the diagnostic workup in older patients should focus on determining the presence of a plasma cell dyscrasia or other lymphoproliferative disorder, which if present should be the primary target of therapy.
In this study we show that C3G is an uncommon but important cause of kidney injury in patients ≥50 years of age. These patients represent a distinct subgroup within C3G with a strong association with monoclonal gammopathy. Eighty-three percent of patients in our study demonstrated monoclonal gammopathy (of renal significance) or MM, compared with only 18% of patients <50 years reported by Zand et al. [6]. In addition, the high association with monoclonal gammopathy greatly exceeds that of the general population in this age, which is estimated at 3.2% [17]. Interestingly, none of the patients in this study showed other lesions typically associated with dysproteinemia, suggesting unique pathogenic properties of these paraproteins. Further studies are required to better understand the specific interactions with mediators of AP of complement.
Along with the association with monoclonal gammopathy, older patients with C3G showed a guarded prognosis with 42% of patients requiring renal replacement therapy and an additional 50% with stable but impaired kidney function at the last follow-up. Only one patient in our cohort demonstrated improvement of renal function and this followed therapy of her underlying MM with hematologic response followed by SCT. The observed poor renal outcomes in older patients with C3G, particularly those associated with paraproteinemia, are consistent with previous reports [4–6].
Recent literature has promoted the use of immunofluorescence staining for C4d on native biopsies and utilization of immunofluorescence on paraffin-embedded tissue as potentially useful ancillary studies in the evaluation of proliferative GN [8, 18]. Staining for C4d showed absent to only weak mesangial staining in all of the biopsies, providing further evidence that the GN in our patients is primarily driven by AP of complement dysfunction [18]. Immunofluorescence studies on paraffin-embedded tissue failed to reveal masked monotypic deposits. Our study is the first to include only cases of C3G after excluding cases with masked monotypic Ig deposits, thus ensuring that only true cases of C3G are studied.
In summary, we report a series of 12 patients ≥50 years of age with C3G. Eighty-three percent of these patients had paraproteinemia. The renal prognosis in these cases was poor in >50% with at least 1-year of follow-up and the one patient who received a kidney allograft experienced a recurrence of disease. A limitation of this study is that complete AP of complement testing was not available for all patients. Nevertheless, our data suggest that the diagnosis of C3G in older adults should prompt thorough investigation for an underlying plasma cell dyscrasia or other lymphoproliferative disorder and is associated with variable long-term renal outcome.
Conflict of interest statement
None declared.
Acknowledgements
This study was previously presented in abstract form at the 2016 annual meeting of the US and Canadian Academy of Pathology, which was held in Seattle, WA, USA and has not otherwise been published.
K.L.R. receives support from Career Development Award IK2 CX000537 from the US Department of Veterans Affairs Clinical Sciences Research and Development Service and Robert Wood Johnson Foundation (Harold Amos Medical Faculty Development Award).
References
- 1.Hou J, Markowitz GS, Bomback AS et al. . Toward a working definition of C3 glomerulopathy by immunofluorescence. Kidney Int 2014; 85: 450–456 [DOI] [PubMed] [Google Scholar]
- 2.Sethi S, Nester CM, Smith RJ. Membranoproliferative glomerulonephritis and C3 glomerulopathy: resolving the confusion. Kidney Int 2012; 81: 434–441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Servais A, Noel LH, Roumenina LT et al. . Acquired and genetic complement abnormalities play a critical role in dense deposit disease and other C3 glomerulopathies. Kidney Int 2012; 82: 454–464 [DOI] [PubMed] [Google Scholar]
- 4.Sethi S, Sukov WR, Zhang Y et al. . Dense deposit disease associated with monoclonal gammopathy of undetermined significance. Am J Kidney Dis 2010; 56: 977–982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bridoux F, Desport E, Fremeaux-Bacchi V et al. . Glomerulonephritis with isolated C3 deposits and monoclonal gammopathy: a fortuitous association? Clin J Am Soc Nephrol 2011; 6: 2165–2174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zand L, Kattah A, Fervenza FC et al. . C3 glomerulonephritis associated with monoclonal gammopathy: a case series. Am J Kidney Dis 2013; 62: 506–514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pickering MC, D'Agati VD, Nester CM et al. . C3 glomerulopathy: consensus report. Kidney Int 2013; 84: 1079–1089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Larsen CP, Messias NC, Walker PD et al. . Membranoproliferative glomerulonephritis with masked monotypic immunoglobulin deposits. Kidney Int 2015; 88: 867–873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Servais A, Fremeaux-Bacchi V, Lequintrec M et al. . Primary glomerulonephritis with isolated C3 deposits: a new entity which shares common genetic risk factors with haemolytic uraemic syndrome. J Med Genet 2007; 44: 193–199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Medjeral-Thomas NR, O'Shaughnessy MM, O'Regan JA et al. . C3 glomerulopathy: clinicopathologic features and predictors of outcome. Clin J Am Soc Nephrol 2014; 9: 46–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sethi S, Fervenza FC, Zhang Y et al. . Atypical postinfectious glomerulonephritis is associated with abnormalities in the alternative pathway of complement. Kidney int 2013; 83: 293–299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.De Vriese AS, Sethi S, Van Praet J et al. . Kidney disease caused by dysregulation of the complement alternative pathway: an etiologic approach. J Am Soc Nephrol 2015; 26: 2917–2929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bridoux F, Leung N, Hutchison CA et al. . Diagnosis of monoclonal gammopathy of renal significance. Kidney Int 2015; 87: 698–711 [DOI] [PubMed] [Google Scholar]
- 14.Markowitz GS. Dysproteinemia and the kidney. Adv Anat Pathol 2004; 11: 49–63 [DOI] [PubMed] [Google Scholar]
- 15.Meri S, Koistinen V, Miettinen A et al. . Activation of the alternative pathway of complement by monoclonal lambda light chains in membranoproliferative glomerulonephritis. J Exp Med 1992; 175: 939–950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jokiranta TS, Solomon A, Pangburn MK et al. . Nephritogenic lambda light chain dimer: a unique human miniautoantibody against complement factor H. J Immunol 1999; 163: 4590–4596 [PubMed] [Google Scholar]
- 17.Kyle RA, Therneau TM, Rajkumar SV et al. . Prevalence of monoclonal gammopathy of undetermined significance. N Engl J Med 2006; 354: 1362–1369 [DOI] [PubMed] [Google Scholar]
- 18.Sethi S, Nasr SH, De Vriese AS et al. . C4d as a diagnostic tool in proliferative GN. J Am Soc Nephrol 2015; 26: 2852–2859 [DOI] [PMC free article] [PubMed] [Google Scholar]