

Abstract
The Prolyl Hydroxylase Domain protein (PHD):Hypoxia Inducible Factor (HIF) pathway is the main pathway by which changes in oxygen concentration are transduced to changes in gene expression. In mammals, there are three PHD paralogues, and PHD2 has emerged as a particularly critical one for regulating HIF target genes such as ERYTHROPOIETIN (EPO), which controls red cell mass and hematocrit. PHD2 is distinctive among the three PHDs in that it contains an N-terminal MYND type zinc finger. We have proposed that this zinc finger binds a Pro-Xaa-Leu-Glu (PXLE) motif found in proteins of the HSP90 pathway to facilitate HIF-α hydroxylation. Targeting it may provide a means of specifically inhibiting this PHD isoform. Here, we screened a library of chemical compounds for their capacity to inhibit the zinc finger of PHD2. We identified compounds that, in vitro, can inhibit PHD2 binding to a PXLE-containing peptide and induce activation of HIF. Injection of one of these compounds into mice induces an increase in hematocrit. This study offers proof of principle that inhibition of the zinc finger of PHD2 can provide a means of selectively targeting PHD2 to activate the HIF pathway.
Keywords: PHD2, Drug Discovery, High-throughput screening, EGLN1, Hypoxia Inducible Factor
Graphical abstract
The zinc finger of PHD2 has been shown to have a critical role in the oxygen dependent control of hypoxia inducible factors. Using a novel high throughput screen in conjunction with cell and animal models, we identify small molecule inhibitors of this domain. Furthermore we show that one of these small molecules leads to a hallmark of HIF activation, increased red cell mass.
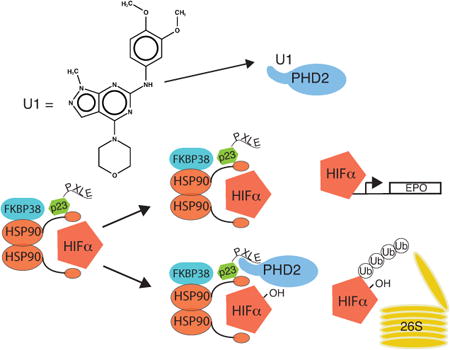
The HIF family of transcription factors mediate a wide variety of cellular responses to low oxygen through activation of numerous genes [1–3]. They were first identified as key components in the hypoxia-associated transcriptional stimulation of the EPO gene, which produces the key hormone regulating red cell development. Subsequently, the HIFs have been shown to underlie hypoxia-associated changes in glucose metabolism, iron and glucose transport, angiogenesis, and vascular tone through a broad range of downstream targets [4–6]. The HIF family is composed of multiple α and a single β subunit. HIF-α protein stability is inversely related to oxygen concentration through oxygen-dependent, posttranslational prolyl hydroxylation [7–9]. This hydroxylation is catalyzed by a family of Prolyl Hydroxylase Domain (PHD) proteins which recognize short sequence motifs for hydroxylation[1]. This modification leads to HIF-α degradation via hydroxylproline-dependent recognition by the von Hippel Lindau protein-containing E3 ubiquitin ligase complex and subsequent degradation by the 26S proteasome [7,9]. Conversely, HIF-β protein is expressed ubiquitously and is not degraded in this manner but serves as a common binding partner for HIF-α during transcriptional activation.
There are three PHD isoforms which hydroxylate HIF-α. Among these, PHD2 (also known as EGLN1) appears to be particularly important for the control of red cell mass, as mutations in the catalytic domain of PHD2 have been associated with development of erythrocytosis[10,11]. PHD1 and PHD3 are also important in other biologic contexts, and in certain tissues they appear to have redundancy with PHD2 [12–16]. There are a number of pathological conditions in which HIF activation may be a desirable outcome and PHD inhibition may be of use. Among these are anemia associated with chronic kidney disease and chemotherapy, decreased vascularity associated with peripheral artery disease, and other ischemic diseases[17]. In this regard, significant effort has focused on developing inhibitors that target the catalytic domain of PHD2, such as by mimicking the cofactor 2-oxoglutarate [18]. The latter strategy was originally utilized to inhibit collagen prolyl hydroxylases. In fact, there are more than sixty 2-oxoglutarate dependent dioxygenases [19]. Given this, selective inhibition of a particular 2-oxoglutarate dependent dioxygenase is a considerable challenge.
PHD2, in addition to its catalytic domain, has a highly conserved MYND type zinc finger domain which associates with components of the HSP90 pathway by binding to a PXLE motif present in the latter proteins, which include p23, FKBP38, and HSP90 itself [20]. The HIF-α's are client proteins of HSP90 and this association gives rise to a model in which PHD2 is recruited to HSP90 to facilitate early interaction with HIF subunits, thereby contributing to the efficient hydroxylation of HIF-α under oxygen replete conditions [20,21]. In support of this model, we have recently found that mutations that ablate the zinc finger of PHD2 lead to increased red cell mass and serum Epo levels, hallmarks of HIF stabilization [22]. Pharmacologic targeting of this non-catalytic domain may circumvent potential off-target effects that might be associated with targeting its active site. Of note, our approach would be predicted to have the opposite effect of HSP90 inhibitors, which are being investigated for their capacity to inhibit HSP90 mediated folding (as opposed to hydroxylation) of HIF-α[23].
To this end, we designed a screen to identify specific inhibitors of the zinc finger domain of PHD2, which should block its association with the HSP90 pathway. We predict that compounds acting in this way will stabilize HIF-α subunits which normally rely on PHD2 for its degradation. Furthermore, in vivo, we predict that we may potentially attain a greater degree of specificity in comparison to current strategies.
We employed an AlphaScreen Assay examining the interaction between a biotinylated peptide containing four copies of the PXLE motif (4×PXLE) and full length recombinant Flag-PHD2 present in baculovirus-infected Sf9 cell extracts (Figure 1A). The interaction was monitored with the use of streptavidin-coated donor beads and anti-Flag antibody-coated acceptor beads. In pilot studies, we observe a readily detectable signal in the presence of peptide and Flag-PHD2 that is absent when the latter is omitted (Figure 1B). The signal is essentially abolished by the inclusion of the metal chelator EDTA, consistent with this signal depending on the integrity of the zinc finger of PHD2. Furthermore, Flag-PHD1, which has a catalytic domain homologous to that of PHD2 but lacks a MYND-type zinc finger, fails to interact with the 4×PXLE peptide. Addition of a competitor non-biotinylated 4×PXLE peptide inhibits the interaction in a concentration-dependent manner (Figure 1C). Using this peptide as a positive control inhibitor yields an assay Z' factor of 0.72 (Figure 1D), indicative of a reliable signal to noise ratio.
Figure 1.
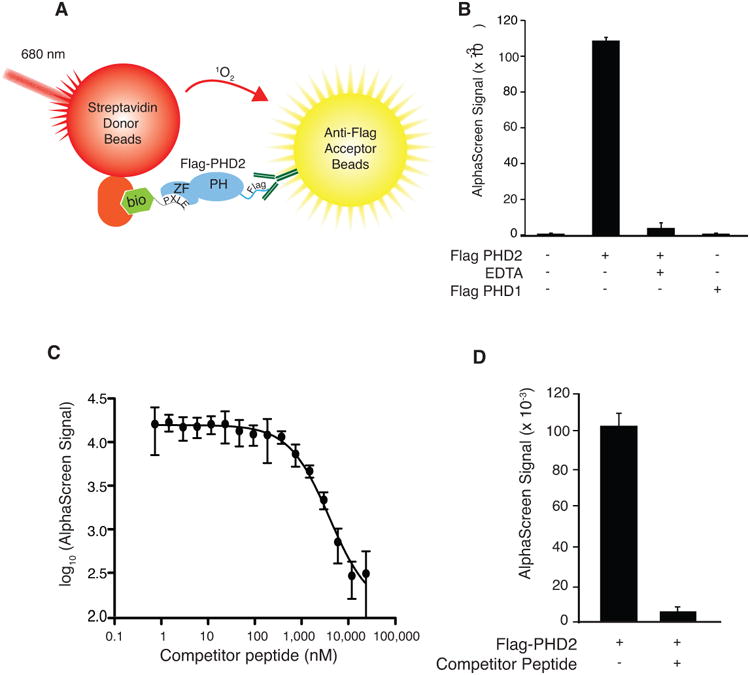
A) Schematic representation of AlphaScreen assay format showing excitation of streptavidin-conjugated donor bead at 680 nm, production of singlet oxygen (1O2), and emission by anti-Flag-conjugated acceptor beads at 615 nm when a complex of biotinylated (bio) 4×PLXLE containing peptide and Flag-PHD2 is formed. ZF = zinc finger, PH = prolyl hydroxylase domain. B) AlphaScreen employing a biotinylated 4×PXLE peptide. Flag-PHD2 was omitted, EDTA (1 mM) was included, or Flag-PHD2 was substituted by Flag-PHD1 as indicated. n=3 per group. Error bars represent standard deviation. C) Inhibition of AlphaScreen signal from interaction of Flag-PHD2 (present in Sf9 extracts) and the biotinylated 4×PXLE peptide by non-biotinylated 4×PXLE (competitor) peptide with the identical amino acid sequence. n=3 per group. Error bars represent standard deviation. D) Pilot experiment for determining Z' parameter using competitor peptide. n=384. Error bars represent standard deviation.
To identify potential inhibitors using this assay, we screened a library of ∼50,000 drug-like molecules. Compounds were arrayed in 384 well micro-titer plates at a final concentration of 5 μM in DMSO in a multiplex format. Five candidate compounds were distributed per well and each compound was represented in two independent wells. Each plate contained wells with positive and negative controls, namely, non-biotinylated 4×PXLE peptide or DMSO vehicle, respectively. Compound responses were normalized on a plate by plate basis to these controls for calculation of relative inhibition.
Initial hits were identified by comparing mean Z-scores for each compound after plate level normalization. We next removed lead compounds that are known to be pan-assay interference compounds (PAINS) [24]. Similarly, we removed a number of compounds that, while not specifically identified as PAINS, were identified in public databases (pubchem) as having broad spectrum inhibition in unrelated fluorescent or HTS assays. We also did not pursue compounds which caused precipitation of protein or assay buffer components. Next, we obtained dose-response curves for 11 putative hits using identical assay parameters as in the initial screen. Positive dose response relationships were seen for 6 compounds (Figure S1). To our knowledge, there have been no prior reports of biological activity for any of these compounds.
Two of these compounds (denoted U and V) had similar sub-structures (Tanimoto score = 0.52), pointing to a likely structure-activity relationship with their shared scaffold and arguing against these being non-specific inhibitors (Figure 2A). Initial library compounds were screened in silico for structural similarity to the common scaffold for U and V, but no others were identified with significant similarity. The other remaining lead compounds were more diverse in their structure and did not show significant scaffold similarity (Figure S1).
Figure 2.

A) Structures of lead compounds U and V. B) Compound U (10 μM) inhibits interaction of PHD2 and the PXLE-containing protein FKBP38 in a mammalian two-hybrid assay. C) Compound U (10 μM) shows no effect on a control Gal4-VP16 fusion protein. B and C) n=4 and Error bars represent standard deviation. ** = P < 0.01, n.s. = not significant
In an orthogonal screen, we employed a mammalian two-hybrid assay. One partner consisted of the Gal4 DNA binding domain fused to PHD2. The other partner consisted of the VP16 activation domain fused to FKBP38, a PXLE-containing HSP90 cochaperone previously identified as a PHD2 interacting protein in immunoprecipitation experiments [20]. The complex of Gal4-PHD2 and VP16-FKBP38 was used to drive expression of a luciferase reporter construct. Compound U inhibited (∼52%) the interaction of full length PHD2 with FKBP38, but had no effect in a control experiment using the Gal4 DNA binding domain directly fused to a VP16 activation domain (Figures 2B and C), thus supporting the results observed in the AlphaScreen based assay.
We subsequently examined a number of structural derivatives of compounds U (12 total). These derivatives maintained the central pyrazolo[3,4-d] pyrimidine ring structure common to compounds U and V and consisted of mainly functional group substitutions to the amino benzene ring. Many of these behaved similarly to the parent compound U (data not shown). Two compounds in which the morpholine ring was substituted by other functional groups showed slightly reduced activity relative to the parent compounds by mammalian two-hybrid assay (data not shown).
One compound, U1 (Figure 3A), showed significantly more potent inhibition in the mammalian two-hybrid assay examining the interaction of FKBP38 with either PHD2 (1-63) (Figure 3B) or full length PHD2 (data not shown). PHD2 (1-63) is an N-terminal fragment that contains the zinc finger, consistent with the zinc finger being the PXLE-binding domain. U1 (10 μM) also inhibited the interaction of PHD2 with the 4×PXLE peptide in the AlphaScreen assay to a degree comparable to a 10-fold excess of non-biotinylated competitor peptide (Figure 3C). As a specificity control for this experiment, we investigated the interaction of the 4×PXLE peptide with ANKMY2. ANKMY2 is a protein that contains a MYND-type zinc finger similar to that found in PHD2 and, like PHD2, it binds to the PXLE-containing protein FKBP38 [25].
Figure 3.

A) Structure of compound U1. B) U1 is more effective at inhibiting N-terminal PHD2 [Gal4-PHD2 (1-63)] interaction with FKBP38 (VP16-FKBP38) than the parent compound, U. C-E) AlphaScreen assays of compound U1 for effects on binding of C) human PHD2, D) human ANKMY2, and E) murine Phd2 (1-125) present in Sf9 extracts to a biotinylated 4×PXLE peptide (100 nM). The final DMSO concentration was 1%. A 10-fold excess of unlabeled competitor peptide (1 μM) was included as a positive control in each set of assays. U1 is present in each experiment at 10 μM. F) AlphaScreen assay for effect of U1 on binding of purified GST-PHD2 (1-63) to purified (His)6-3×Flag-p23. Unlabeled competitor peptide (10 μM) was included as a positive control. n=4 per group for all experiments, and error bars represent SD. * = P < 0.05, ** = P < 0.01, n.s. = not significant
We find that ANKMY2 can bind to the 4×PXLE peptide and that non-biotinylated 4×PXLE peptide is effective at disrupting this interaction (Figure 3D). In contrast to the PHD2:4×PXLE interaction, the ANKMY2:4×PXLE interaction is not inhibited by compound U1, supporting our hypothesis that U1 has specificity for PHD2 and not simply all MYND zinc finger-containing proteins. Unexpectedly, U1 did not inhibit binding of the zinc finger of murine Phd2 to the 4×PXLE peptide by AlphaScreen (Figure 3E). While the zinc finger region that binds to the PXLE motif is highly conserved through evolution, there are amino acid differences within and flanking the murine and human PHD2 zinc fingers (Figure S2). The zinc finger (amino acids 21-58 in both human and mouse PHD2) is 97% identical between the human and mouse proteins, whereas exon 1 (amino acids 1-297 in human PHD2, 1-274 in mouse Phd2) is 71% identical between the two. Whether these differences account for the differential effects of U1 on the mouse and human PHD2 zinc fingers remains to be determined.
The initial AlphaScreens were performed using PHD2 present in Sf9 cellular lysate. Using AlphaScreen, we were also able to detect the direct interaction of purified GST-PHD2 (1-63), which contains the zinc finger, and purified 3×Flag-p23 (Figure 3F). p23, like FKBP38, contains a PXLE motif that binds PHD2[20]. Similar to the previous results, U1 was effective at disrupting this signal, with an estimated IC50 of 1.34 uM. Indeed, high levels of U1 (≥ 8 μM) yielded signals indistinguishable from background GST-PHD2 (1-63) alone.
Evaluation of HIF stabilization by U1 in cells is potentially complicated by the observation that many cells also express PHD1 and PHD3, raising the possibility of redundancy among the PHD paralogues. To circumvent this issue, we employed CRISPR technology [26] to knock out the PHD1 and PHD3 genes from the CML-derived cell line, HAP1. In brief, cells were cotransfected with plasmids encoding for Cas9 and two guide RNAs targeting exons encoding the catalytic domains of PHD1 and PHD3 (third and second exons respectively, Figure S3A). Cells were subjected to puromycin selection, and then individual clones were isolated. Clones were screened by Surveyor assay to identify deletions present in mutated alleles. We identified a positive clone with cleavage patterns consistent with mutations at both loci (Figure S3B). One clone (PHD1/3 DKO)-identified in this way was confirmed by Sanger sequencing to have 3 and 14 bp deletions on the two alleles of PHD1 and 17 and 10 bp deletions on the two alleles of PHD3 (Figure S3C); the first of these deletions disrupts a splice site, while the other three lead to frame shift mutations. Real-time RT-PCR was performed both on PHD1/3 DKO and parental HAP1 cells (Figure S3D) show a near-complete loss of PHD1 mRNA in PHD1/3 DKO cells (over 100-fold reduction) and low to nearly undetectable levels of PHD3 mRNA in both PHD1/3 DKO and WT HAP1 cells (10,000-fold less than that of PHD1). Loss of expression at the protein level was confirmed by western blotting, which showed loss of PHD1 and PHD3 expression compared to WT (Figure S3E). We transfected the PHD1/3 DKO cell with a luciferase reporter gene driven by two copies of a Hypoxia Response Element (HRE), treated with dimethyloxaloylglycine (DMOG), a non-metabolizable 2-oxoglutarate analogue and inhibitor of PHDs, compound U1, or both, and then exposed cells to either normoxia or hypoxia (2% O2). Hypoxia, as expected, activates the HRE reporter gene, and importantly both DMOG and U1 activate it further (Figure 4A). Interestingly, the combination of both results in an additive effect. Treatment of the PHD1/3 DKO cells, even for short times (2 hours) was sufficient to induce stabilization of HIF-1α in a manner similar to desferrioxamine (DFO), an active site iron chelator, under mild hypoxic stress (4% oxygen) (Figure 4B).
Figure 4.
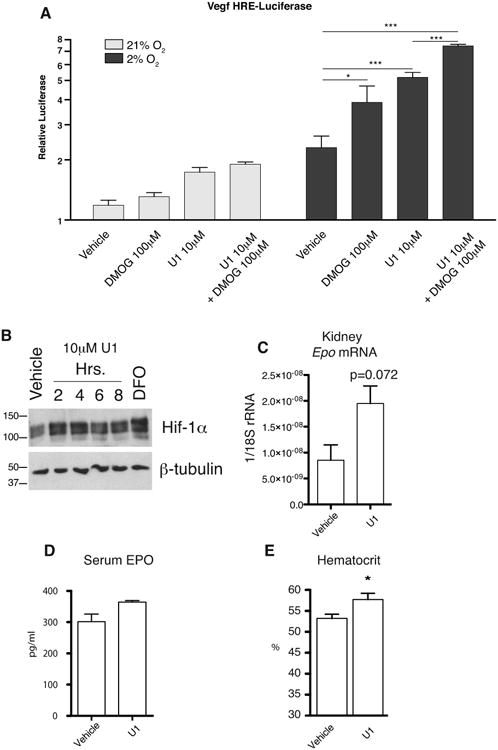
A) Compound U1 is effective at inducing HRE-dependent luciferase activity in PHD1/3 DKO cells under hypoxia (2% oxygen). B) U1 increases levels of HIF1α in PHD1/3 DKO cells equivalent to DFO (10 μM) treatment under mild hypoxia (4% oxygen). Phd2 Hum/Hum mice show a trend towards increased C) renal Epo mRNA and D) serum Epo 6 hours after a single IP dose (20 mg/kg) of compound U1. E) Phd2 Hum/Hum mice show increased hematocrit following 4 days of twice-daily injection with compound U1 at a dose of 20 mg/kg/day as compared to vehicle control. Error bars represent SD. n=5 per group A) or 4 per group C to E). * = P < 0.05, *** = P < 0.001
Our recent work using a mouse model with a knockin PHD2 zinc finger mutation [22] has suggested that abrogation of the PHD2 zinc finger function may be an effective means of inducing increases in hematocrit through HIF stabilization. Numerous groups have attempted to identify active site prolyl hydroxylase inhibitors as a means of treating anemia associated with CKD and chemotherapy regimens. To test the efficacy of compound U1 in a murine model and to accommodate the specificity of compound U1 for human over murine PHD2, we generated a new knockin mouse line bearing a humanized Phd2 zinc finger (Phd2 Hum/Hum), achieved by replacing the coding sequence of exon 1 of the murine Phd2 gene with that of the human PHD2 gene (Figure S4A-C). In vitro, U1 inhibited the PHD2 chimera that is produced from human PHD2 exon 1 and mouse Phd2 exons 2 to 5 using the mammalian two hybrid assay (Figure S5A-B). In pilot experiments, we injected Phd2 Hum/Hum mice with compound U1 intraperitoneally at a dose of 20 mg/kg. One hour later, we detected it in serum at high nanomolar levels (410 ± 157 nM, n=3) by LC-MS (data not shown), and at six hours we observed trends towards increased renal Epo mRNA and serum EPO protein, though neither reached statistical significance (Figure 4C-D). We then injected Phd2 Hum/Hum for four consecutive days with U1 at 20 mg/kg/day and found a statistically significant increase in hematocrit compared to vehicle-injected controls (Figure 4E). As a control, injection of wild type C57BL/6 mice with U1 did not produce an increase in hematocrit (Figure S5C-E). This provides further evidence that the PHD2 zinc finger serves a positive regulatory function and that inhibition of its interaction with the HSP90 pathway is a viable means to induce increases in hematocrit.
In summary, we have identified compounds that can disrupt the interaction of the zinc finger of PHD2 with a PXLE containing peptide in vitro, provided evidence that these compounds can disrupt this interaction in cells and activate the HIF pathway, and shown that one of these compounds can induce an increase in hematocrit in a mouse bearing a humanized Phd2 gene in vivo. Collectively, the results support a critical role for the zinc finger of PHD2 and show that targeting it has the capacity to induce HIF activity.
Experimental Section
Plasmids
pcDNA3-Gal4-PHD2 was constructed by subcloning the 1.3 kb BamH I/Xba I fragment of pcDNA3-Flag-PHD2 [27] into the BamH I/Xba I site of pcDNA3-Gal4. pcDNA3-Gal4-PHD2 (1-63) was constructed by subcloning the 0.2 kb BamH I/Not I fragment of pGEX-PHD2 (1-63) [20] into the BamH I/Not I site of pcDNA3-Gal4. pcDNA3-Gal4-hmPHD2 (humanized mouse PHD2, consisting of amino acids 1-297 of human PHD2 fused to amino acids 275-400 of mouse Phd2) was constructed in several steps. First, pcDNA5/FRT/TO-HA-PHD2 was constructed by subcloning the 0.6 kb Hind III/Xho I fragment of pcDNA3-HA-PHD2[20] into to Hind III/Xho I site of pcDNA5/FRT/TO-Flag-PHD2[20]. Then, we substituted the coding sequence of the 12 C-terminal amino acids of human PHD2 (NKPSDSVGKDVF) with the coding sequence of the 9 C-terminal amino acids of mouse Phd2 (KPNSVSKDV) in pcDNA5/FRT/TO-HA-PHD2 by overlapping PCR to create pcDNA5/FRT/TO-HA-hmPHD2. Finally, the 1.3 kb BamH I/Xba I band from pcDNA5/FRT/TO-HA-hmPHD2 was subcloned into the BamH I/Xba I site of pcDNA3-Gal4.
pcDNA3-Gal4-ANKMY2 was constructed as follows. First, the 1.3 kb coding sequence of ANKMY2 was PCR amplified from IMAGE clone 5185501 using the following two primers: 5′-GAT CGG ATC CAG ATG GTT CAC ATA AAG AAA GGC-3′ and 5′-GTA CTC TAG ATT ACT CCT CAG ACA CCT GTG G-3′. The product was digested with BamH I/Xba I and subcloned into the BamH I/Xba I site of pcDNA3-HA to generate pcDNA3-HA-ANKMY2. Then, the 1.3 kb BamH I/Xba I fragment of pcDNA3-HA-ANKMY2 was subcloned into the BamH I/Xba I site of pcDNA3-Gal4. pVP16-FKBP38 was constructed by subcloning the 1.2 kb BamH I/Xba I fragment of pcDNA3-HA-FKBP38 [20] into the BamH I/Xba I site of pVP16. pcDNA3-Gal4-VP16 was constructed by subcloning an oligonucleotide duplex consisting of the following sequences: 5′-GAT CCC GGC CGA CGC CCT GGA CGA CTT CGA CCT GGA CAT GCT GCC TGC TGA TGC TCT CGA TGA TTT CGA TCT AGA TAT GCT CCC GGG TAA CTA AT-3′ and 5′-CTA GAT TAG TTA CCC GGG AGC ATA TCT AGA TCG AAA TCA TCG AGA GCA TCA GCA GGC AGC ATG TCC AGG TCG AAG TCG TCC AGG GCG TCG GCC GG-3′ into the BamH I/Xba I site of pcDNA3- Gal4. The sources of G5E1b-Luc and pRL-TK have been described [27,28].
The gRNA expression construct MLM3636 was a gift from Dr. Keith Joung (Addgene plasmid # 43860). pMLM3636-PHD1gRNA2 was constructed by subcloning into the BsmBI site of MLM3636 an oligonucleotide duplex consisting of the following sequences: 5′-ACA CCG AAC TGG GAC GTT AAG GTA GG-3′ and 5′- AAA ACC TAC CTT AAC GTC CCA GTT CG-3′. pMLM3636-PHD3gRNA2 was similarly constructed using an oligonucleotide duplex consisting of the following sequences: 5′-ACA CCC ACC GTT GGG GTT GTC CAC GG-3′ and 5′-AAA ACC GTG GAC AAC CCC AAC GGT GG-3′. The Cas9 expression construct pHL-EF1a-SphcCas9-iP-A was a gift from Dr. Akitsu Hotta (Addgene plasmid # 60599).
Baculovirus
pFastBac-HT-FlagANKMY2 was prepared by excising the coding sequence of pFastBac-HT-FlagPHD3 [27] by digestion with BamH I/Xba I and replacing it with that of pcDNA3-HA-ANKMY2. pFastBac-HT-Flag-mPhd2 (1- 125) was constructed by subcloning the 0.4 kb BamH I fragment of pcDNA3-Flag-mPhd2 [29] into the BamH I site of pFastBac-HT-FlagPHD1[27]. pFastBac-HT-3×Flag-p23 was constructed by subcloning the 0.6 kb Nco I/Xho I fragment of pcDNA5/FRT/TO-3×Flag-p23 [20] into the Nco I/Xho I site of pFastBac-HTc. Baculoviruses for (His)6-FlagANKMY2, (His)6-Flag-mPhd2 (1-125), and (His)6-3×Flag-p23 were then prepared using the Bac-to-Bac system (Invitrogen). Baculoviruses for (His)6-FlagPHD2 and (His)6-FlagPHD1 have been described[27].
Proteins
GST-PHD2 (1-63) was purified from E. coli transformed with pGEX-PHD2 (1-63) [20] by affinity chromatography on GST-agarose (GE Healthcare Life Sciences). (His)6-3×Flag-p23 was purified from baculovirus-infected Sf9 cells using Ni-NTA-agarose (Qiagen).
AlphaScreen
An AlphaScreen to identify compounds that can disrupt the interaction between the zinc finger of PHD2 and the PXLE motif was conducted at The Wistar Institute Molecular Screening Facility. We obtained, from GenScript, an N-terminally biotinylated peptide consisting of the following sequence (PXLE motif underlined): YEDELPDLEEELPDLEDELPDLEEELPDLE. The concentration of peptide stock solutions was determined based on A280 measurements using a molar absorption of 1280 M-1cm-1, owing to the single tyrosine residue[30]. We incubated extracts of Sf9 cells previously infected with baculovirus expressing (His)6-Flag-PHD2 [27] with this peptide for 30 min at 20 °C in 20 mMHepes, pH 8, 100 mMKCl, 0.1 % Triton X-100, 1 μM zinc. Next, this mixture was added to wells previously spotted with compound, vehicle, or non-biotinylated peptide controls. We then added a mixture of Streptavidin Donor beads and Anti-Flag Acceptor beads, and incubated an additional 60 min at 20 °C in the dark. We employed the peptide, donor beads, and acceptor beads at concentrations of 100 nM, 8 μg/ml, and 8 μg/ml, respectively, in a final assay volume of 20 μl. Assays were conducted in white ProxiPlate-384 Plus shallow well microplates, and read on a PerkinElmer EnVision Xcite Multilabel plate reader.
In some experiments, we employed baculovirus expressing (His)6-Flag-PHD1 [27], (His)6-Flag-ANKMY2, or (His)6-Flag-mPhd2 (1-63). In additional experiments, we employed Anti-Flag Donor beads and Anti-GST Acceptor beads, and purified (His)6-3×Flag-p23 and GST-PHD2 (1-63). For Z′ calculations, we performed the assay on a 384 well plate with 100 nM biotinylated peptide in the absence (one half of the plate) or presence (the other half of the plate) of 120 μM of non-biotinylated competitor peptide, on two consecutive days which yielded a Z′ of 0.72. For screening, we tested 50,000 compounds from a combination of Discovery Chemistry and Target Specific Chemistry libraries from ChemDiv. Compounds were screened at a concentration of 5 μM in DMSO.
Mammalian Two-Hybrid Assay
HEK293FT cells (Invitrogen) were maintained in DMEM supplemented with 10% FBS, 100 units/ml penicillin, and 100 μg/ml streptomycin. Gal4 and VP16 fusion plasmids were cotransfected into HEK293FT cells using Lipofectamine 2000 (Invitrogen) based on manufacturer's protocols. G5E1b-Luc reporter and pRL-TK normalization plasmids were included in all transfections. Four hours after transfection, compounds or vehicle controls (DMSO) were added to wells and cells were incubated for an additional 16 hours. Following incubation, cells were lysed and assayed for firefly luciferase/renilla luciferase using the Dual-Luciferase assay reporter system (Promega) on a Wallac LB9507 luminometer.
CRISPR Gene Knock-out
Human, CML-derived HAP1 cells (Horizon Discovery) were used for generation of a PHD1/PHD3 double knockout cell line. Cells were grown in Iscove's DMEM supplemented with 10% FBS, 100 units/ml penicillin, and 100 μg/ml streptomycin. Cells were cotransfected with a combination of pHL-EF1a-SphcCas9-iP-A, pMLM3636-PHD1gRNA2, and pMLM3636-PHD3gRNA2. Twelve hours after transfection, cells were selected using 10 μg/ml puromycin. Following 48 hours of selection, cells were serial diluted to isolate clonal populations which were then screened for mutation of PHD1 and PHD3.
Initial clone screening was done by Surveyor Mutation Detection Kit (Integrated DNA Technologies) following the manufacturer's protocol. PCR primers used for heteroduplex production from the PHD1 gene were PHD1 ex3 screen 5′: 5′-GGT TTG GGG TAT CTT GAA ACA A-3′, and PHD1 ex3 screen 3′: 5′-GGG AAA GTG TTC ATT ACA AGG C-3′. For the PHD3 gene, the PCR primers were PHD3 ex2 screen 5′: 5′-GGC ATA CAA ACA GGT TAT GCA A-3′, and PHD3 ex2 screen 3′: 5′-TTA CCT TCC CTT CAT CAA CCA C-3′. Following mutation detection, Sanger sequencing was performed on these PCR products to confirm mutation. Real-Time PCR, described later, was used to confirm loss of transcript abundance relative to HAP1 control cells.
HRE Luciferase Assays
(vHRE)2-Luc, which contains two copies of the HRE from the VEGFA gene, was constructed by subcloning into the Kpn I/Xho I site of pGL3-Promoter (Promega) an oligonucleotide duplex consisting of the following two sequences: 5′- CCA CAG TGC ATA CGT GGG CTC CAA CAG GTC CTC TTC CAC AGT GCA TAC GTG GGC TCC AAC AGG TCC TCT TC-3 and 5′- TCG AGA AGA GGA CCT GTT GGA GCC CAC GTA TGC ACT GTG GAA GAG GAC CTG TTG GAG CCC ACG TAT GCA CTG TGG GTA C-3′. (vHRE)2-Luc and pRL-TK were transfected into PHD1/3 DKO Hap1 cells using Lipofectamine 2000. For 12 hours immediately following transfection, cells were maintained under either 21% or 2% oxygen and treated with vehicle (DMSO 0.25%), dimethyloxalylglycine (DMOG 100 μM), compound U1 (10μM), or the combination of DMOG and U1. Hypoxic exposure was performed in an Invivo2 Hypoxia workstation (Ruskinn). Following treatment, cells were lysed and analyzed using the Dual-Luciferase assay reporter system (Promega) as described above.
Western Blotting
Protein extracts were prepared by lysing cells in 20 mM HEPES, 150 mM NaCl, 1% Triton X-100, 10% glycerol supplemented with mammalian protease inhibitor cocktail (Sigma P8340). Monoclonal antibodies to Hif-1α(Clone 54) were obtained from BD Biosciences. Anti-β-tubulin mouse MAb (E7) was generated by Michael Klymkowsky and was obtained from the Developmental Studies Hybridoma Bank developed under the auspices of the NICHD and maintained by the Department of Biology, University of Iowa, Iowa City, IA. Goat anti-mouse IgG secondary antibodies conjugated to alkaline phosphatase (sc-2008) were obtained from Santa Cruz Biotechnology. CDP-Star was employed as substrate.
Phd2Hum Knock-In Mouse Gene Targeting
The construct for generating a Phd2 allele bearing a humanized PHD2 exon 1 coding sequence (Phd2Hum) was prepared by recombineering[31]. In brief, a minitargeting vector was constructed in the vector pL452 [32]. The minitargeting vector contained genomic DNA encompassing exon 1 of the mouse Phd2 gene but with the coding sequence of murine exon 1 (amino acids 1-274) replaced by the corresponding sequence of human PHD2 exon 1 (amino acids 1-297). This vector also contained a neomycin selection cassette flanked by loxP sites, and additional sequences downstream of exon 1. A retrieval plasmid was constructed in the vector pMC1-DTA [33]. This retrieval plasmid contained sequences that flank 12 kb of genomic DNA sequence at the mouse Phd2 locus, as well as a diphtheria toxin A negative selection cassette. This retrieval plasmid was used to capture, by recombineering, 12 kb of mouse Phd2 genomic DNA containing exon 1 from C57BL/6 bacterial artificial chromosome (BAC) clone RP23-356I16 (Invitrogen). The resulting product was then used, in the second recombineering step with the minitargeting vector, to generate the final targeting vector. The targeting vector contains a 8.3 kb 5′ arm containing the 5′ untranslated region of murine Phd2 exon 1, the human PHD2 exon 1 coding sequence, a neomycin selection cassette flanked by loxP sites, and a 3.7 kb 3′ arm (Figures S3A). The presence of the human PHD2 exon 1 coding sequence and the integrity of exon 1 were confirmed by DNA sequencing.
EAP6 C57BL/6 ES cells were electroporated with the Phd2Hum targeting vector, and selected using G418 by the Perelman School of Medicine Gene Targeting Core Facility. Screening was performed by Southern blotting. Digoxigenin-labeled probes were generated by PCR using a PCR DIG Probe Synthesis Kit (Roche). For the 5′ probe (0.40 kb), the primers were 5′- TTA TCA TTA TCA ATT GGT TCT G -3′ and 5′-TGT TCT CTG AAC CCC TCG TGT GCC -3′. For the 3′ probe (0.40 kb), the primers were 5′- TCT CTG TGC TGC GGT TAA TTA GTC-3′ and 5′- ACA TTA TGA CTC CTA ACA ATA GCG -3′. For both probes, BAC clone BAC RP23-356I16 was employed as the template. Southern blotting was performed using DIG Easy Hyb, DIG wash and block buffer set, anti-digoxigenin-AP conjugates, and CDP-Star substrate (all from Roche).
A total of 192 clones were screened by Southern blotting. Southern blotting with a 5′ probe revealed a clone with the expected 16.4 kb band for the Phd2Hum allele upon hybridization with Hind III digested DNA in addition to the wild type 8.9 kb band (Figures S3B, lane 2). Southern blotting with a 3′ probe revealed the expected 16.4 kb band for Phd2Hum, upon hybridization with Hind III digested DNA in addition to the wild type 5.4 kb band (Figures S3B, lane 7). Sequencing confirmed the presence of the desired sequence for the Phd2Hum (data not shown) allele. This clone was injected into Balb/c blastocysts by the Perelman School of Medicine Transgenic Core Facility to produce chimeras. Chimeric male mice were then mated with C57BL/6J-Tyrc-2J female mice, and germline transmission (as assessed by coat color and then PCR) was obtained. Southern blotting confirmed germline transmission (Figure S3B, lanes 4 and 9). Mice with germline transmission of the knock-in allele were then mated with C57BL/6-Gt(ROSA)26Sortm16(Cre)Arte mice (Taconic) to delete the neomycin cassette (Figure S3B, lanes 5 and 10), followed by further crossing with C57BL/6 mice to segregate the Cre allele, thereby creating Phd2 Hum/+mice. Phd2 Hum/+ mice were maintained on a C57BL/6 background. Phd2 Hum/Hum mice were obtained by Phd2 Hum/+ intercrosses.
PCR Genotyping
DNA was isolated from mouse tails [34]. The following primers were employed for genotyping Phd2 with the humanized exon 1 coding sequence: CSint1-1 5′ = 5′- CCA GAC ACT GTG TTT AGC TAG GAC -3′ and CSint1-1 3′ = 5′- AAA CCA GGA AGC CAC AGA AGC AGC-3′. The wild type allele produces a PCR product of 0.30 kb, whereas the knock-in mutant alleles produces a PCR product of 0.40 kb (Figure S3C).
Mouse Injections
U1 stocks were stored in DMSO in amber vials. Formulations for injection were diluted to 1 mg/ml in a solution of 5% DMSO, 40% PEG 400, and 2% Tween-80 [35] and warmed to 25 °C prior to injection. Phd2 Hum mice were injected intraperitoneally with formulations of compound U1 at either 20 mg/kg or, alternatively, twice daily at 10 mg/kg (20 mg/kg/day) for extended experiments. Control mice were injected with equivalent volumes of vehicle.
LC-MS
Serum samples from mice injected with U1 or vehicle control were spiked with internal standards, and proteins were precipitated with cold methanol (80%). Methanolic supernatants were dried by SpeedVac (Heto) followed by dissolution in 50% acetonitrile, 0.1% formic acid. Separation and detection was performed by The Wistar Institute Proteomics Facility using a reverse phase nanoflow LC-MS/MS containing a 15 cm column packed with Magic C18 5 micron beads eluting onto an LTQ Orbitrap XL Mass Spectrometer (Thermo Scientific). Compound levels were determined by comparing peaks areas of extracted ion chromatograms relative to a spiked internal standards (Compound U) and to those from standard curves generated previously using spiked control serum and identical extraction and detection methods.
Real-Time PCR
RNA was isolated from either cells or approximately 125 mm3 sections kidney and used as source material. Details of RNA isolation, cDNA synthesis and Real-Time PCR cycling conditions have been described previously [29]. Primer sequences for Epo cDNA detection have also been described previously [36].
Serum Epo Analysis
Serum samples were derived from terminal blood draws from the inferior vena cava of vehicle or U1-injected mice. Blood was allowed to clot for 1 hour at room temperature prior to centrifugation at 4000 × g for 15 minutes at 4 °C. Serum supernatants were analyzed by ELISA using the rodent Quantikine Epo Immunoassay kit (R&D Systems MEP00B). Absorbance was read on a Sunrise microplate reader (Tecan).
Hematology Analysis
Retro-orbital blood samples for hematology analyses were collected in Microvette 100 LH heparin-coated collection tubes (Sarstedt). Hematocrit was measured manually using a CritSpin microhematocrit centrifuge (StatSpin).
Supplementary Material
Acknowledgments
This work was supported by a Transdisciplinary Awards Program in Translational Medicine and Therapeutics (TAPITMAT) grant from the University of Pennsylvania Institute for Translational Medicine and Therapeutics to F.S.L. and P.M.L., and grants R01-DK567611 and R33-HL120751 from the NIH to F.S.L. P.R.A. was supported by grant T32-DK007780 from the NIH. We thank Dr. Tobias Raabe of the University of Pennsylvania Perelman School of Medicine Gene Targeting facility for performing the ES cell electroporation, Dr. Jean Richa of the University of Pennsylvania Perelman School of Medicine Transgenic core facility for conducting injections of targeted ES cells into blastocysts, Dr. David Schultz of The Wistar Institute Molecular Screening Facility for guidance on performing the AlphaScreen, and Dr. Hsin-Yao Tang of The Wistar Institute Proteomics Facility for performing the LC-MS analysis of compound U1.
References
- 1.Kaelin WG, Jr, Ratcliffe PJ. Mol Cell. 2008;30:393–402. doi: 10.1016/j.molcel.2008.04.009. [DOI] [PubMed] [Google Scholar]
- 2.Majmundar AJ, Wong WJ, Simon MC. Mol Cell. 2010;40:294–309. doi: 10.1016/j.molcel.2010.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Semenza GL. Annual Review of Pathology: Mechanisms of Disease. 2014;9:47–71. doi: 10.1146/annurev-pathol-012513-104720. [DOI] [PubMed] [Google Scholar]
- 4.Haase VH. Am J Physiol Renal Physiol. 2010;299:F1–F13. doi: 10.1152/ajprenal.00174.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang GL, Semenza GL. J Biol Chem. 1995;270:1230–1237. doi: 10.1074/jbc.270.3.1230. [DOI] [PubMed] [Google Scholar]
- 6.Lendahl U, Lee KL, Yang H, Poellinger L. Nat Rev Genet. 2009;10:821–832. doi: 10.1038/nrg2665. [DOI] [PubMed] [Google Scholar]
- 7.Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M, Salic A, Asara JM, Lane WS, Kaelin WG. Science. 2001;292:464–468. doi: 10.1126/science.1059817. [DOI] [PubMed] [Google Scholar]
- 8.Jaakkola P, Mole DR, Tian YM, Wilson MI, Gielbert J, Gaskell SJ, von Kriegsheim A, Hebestreit HF, Mukherji M, Schofield CJ, et al. Science. 2001;292:468–472. doi: 10.1126/science.1059796. [DOI] [PubMed] [Google Scholar]
- 9.Yu F, White SB, Zhao Q, Lee FS. PNAS. 2001;98:9630–9635. doi: 10.1073/pnas.181341498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gardie B, Percy MJ, Hoogewijs D, Chowdhury R, Bento C, McMullin MF, Schofield CJ, Lee FS, Richard S, Arsenault P, et al. Hypoxia. 2014;2014:71–90. doi: 10.2147/HP.S54455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee FS, Percy MJ. Annu Rev Pathol. 2011;6:165–192. doi: 10.1146/annurev-pathol-011110-130321. [DOI] [PubMed] [Google Scholar]
- 12.Minamishima YA, Kaelin WG., Jr Science. 2010;329:407. doi: 10.1126/science.1192811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Minamishima YA, Moslehi J, Padera RF, Bronson RT, Liao R, Kaelin WG. Mol Cell Biol. 2009;29:5729–5741. doi: 10.1128/MCB.00331-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rytkönen KT, Williams TA, Renshaw GM, Primmer CR, Nikinmaa M. Mol Biol Evol. 2011;28:1913–1926. doi: 10.1093/molbev/msr012. [DOI] [PubMed] [Google Scholar]
- 15.Lee S, Nakamura E, Yang H, Wei W, Linggi MS, Sajan MP, Farese RV, Freeman RS, Carter BD, Kaelin WG, et al. Cancer Cell. 2005;8:155–167. doi: 10.1016/j.ccr.2005.06.015. [DOI] [PubMed] [Google Scholar]
- 16.Zhang Q, Gu J, Li L, Liu J, Luo B, Cheung HW, Boehm JS, Ni M, Geisen C, Root DE, et al. Cancer Cell. 2009;16:413–424. doi: 10.1016/j.ccr.2009.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chan MC, Holt-Martyn JP, Schofield CJ, Ratcliffe PJ. Mol Aspects Med. 2016;47-48:54–75. doi: 10.1016/j.mam.2016.01.001. [DOI] [PubMed] [Google Scholar]
- 18.Rabinowitz MH. J Med Chem. 2013;56:9369–9402. doi: 10.1021/jm400386j. [DOI] [PubMed] [Google Scholar]
- 19.Guengerich FP. J Biol Chem. 2015;290:20700–20701. doi: 10.1074/jbc.R115.675652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Song D, Li LS, Heaton-Johnson KJ, Arsenault PR, Master SR, Lee FS. J Biol Chem. 2013;288:9662–9674. doi: 10.1074/jbc.M112.440552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Minet E, Mottet D, Michel G, Roland I, Raes M, Remacle J, Michiels C. FEBS Lett. 1999;460:251–256. doi: 10.1016/s0014-5793(99)01359-9. [DOI] [PubMed] [Google Scholar]
- 22.Arsenault PR, Song D, Chung YJ, Khurana TS, Lee FS. Mol Cell Biol. 2016;36:2328–2343. doi: 10.1128/MCB.00090-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Proia DA, Bates RC. Cancer Res. 2014;74:1294–1300. doi: 10.1158/0008-5472.CAN-13-3263. [DOI] [PubMed] [Google Scholar]
- 24.Baell JB, Holloway GA. J Med Chem. 2010;53:2719–2740. doi: 10.1021/jm901137j. [DOI] [PubMed] [Google Scholar]
- 25.Saita S, Shirane M, Ishitani T, Shimizu N, Nakayama KI. J Biol Chem. 2014;289:25639–25654. doi: 10.1074/jbc.M114.558635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM. Science. 2013;339:823–826. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huang J, Zhao Q, Mooney SM, Lee FS. J Biol Chem. 2002;277:39792–39800. doi: 10.1074/jbc.M206955200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhao Q, Lee FS. Biochemistry. 2003;42:3627–3634. doi: 10.1021/bi027117f. [DOI] [PubMed] [Google Scholar]
- 29.Arsenault PR, Pei F, Lee R, Kerestes H, Percy MJ, Keith B, Simon MC, Lappin TRJ, Khurana TS, Lee FS. J Biol Chem. 2013;288:33571–33584. doi: 10.1074/jbc.M113.482364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gill SC, von Hippel PH. Analytical Biochemistry. 1989;182:319–326. doi: 10.1016/0003-2697(89)90602-7. [DOI] [PubMed] [Google Scholar]
- 31.Copeland NG, Jenkins NA, Court DL. Nature Reviews Genetics. 2001;2:769–779. doi: 10.1038/35093556. [DOI] [PubMed] [Google Scholar]
- 32.Liu P, Jenkins NA, Copeland NG. Genome Res. 2003;13:476–484. doi: 10.1101/gr.749203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yagi T, Ikawa Y, Yoshida K, Shigetani Y, Takeda N, Mabuchi I, Yamamoto T, Aizawa S. PNAS. 1990;87:9918–9922. doi: 10.1073/pnas.87.24.9918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Land T, Rouault TA. Mol Cell. 1998;2:807–815. doi: 10.1016/s1097-2765(00)80295-6. [DOI] [PubMed] [Google Scholar]
- 35.Tejwani RW, Joshi HN, Varia SA, Serajuddin ATM. J Pharm Sci. 2000;89:946–950. doi: 10.1002/1520-6017(200007)89:7<946::AID-JPS12>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 36.Li X, Sutherland S, Takeda K, Fong GH, Lee FS. Blood Cells Mol Dis. 2010;45:9–19. doi: 10.1016/j.bcmd.2010.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.