

Abstract
The entomopoxvirus from Amsacta moorei serves as the prototype of the group B entomopoxviruses. One of the interesting genes found in Amsacta moorei entomopoxvirus (AmEPV) is a superoxide dismutase (sod) (open reading frame AMV255). Superoxide dismutases (SODs) catalyze the conversion of superoxide radicals to hydrogen peroxide and oxygen. Many vertebrate poxviruses contain a sod gene, but to date, none have been demonstrated to be active. There are three families of SODs, characterized by their metal ion-binding partners, Fe, Mn, or Cu and Zn. Poxvirus enzymes belong to the Cu-Zn SOD family. Unlike inactive vertebrate poxvirus SODs, AMVSOD contains all the amino acids necessary for function. We expressed and purified a 6X-His-tagged version of the AMVSOD in Escherichia coli. The recombinant AMVSOD demonstrates superoxide dismutase activity both in an in situ gel assay and by stopped flow spectrophotometry. The kcat/Km for AMVSOD is 4 × 107 M−1s−1. In infected cells, the AMVSOD protein behaves as a dimer and is catalytically active; however, disruption of the gene in AMEPV has little or no effect on growth of the virus in cell culture. An analysis of mRNA expression indicates that AMVsod is expressed late during infection of Lymantria dispar (Ld652) cells and produces a discrete nonpolydisperse transcript. Characterization of protein expression with a monoclonal antibody generated against AMVSOD confirms that the AMVSOD protein can be classified as a late, postreplicative gene. Therefore, AMVSOD is the first example of an active poxvirus SOD.
Free oxygen radicals have been implicated in a number of diseases, including neurodegenerative disorders and lung disease, as well as in the process of aging (32, 36, 38). Superoxide dismutases (SODs) catalyze the dismutation of O2− to H2O2 and O2, rendering the potentially harmful superoxide anion less hazardous. SODs require a metal cofactor for function and can be grouped by the bound metal ion (5, 19, 60). Iron-containing SODs have been found in prokaryotes and some plants. Manganese SODs are found in both prokaryotes and eukaryotes and are localized primarily to the mitochondria. The third class represents SODs which require both copper and zinc as cofactors (Cu-Zn SODs). Cu-Zn SODs are homodimers in eukaryotes and are located predominantly in the cytoplasm. In most prokaryotes, Cu-Zn SODs exist as homodimers found in the periplasm. However, Escherichia coli is a notable exception in which the Cu-Zn SOD is a monomer (8). Human Cu-Zn SOD, encoded by the SOD1 gene, has been implicated as the source of some cases of familial amyotrophic lateral sclerosis (16, 47). The eukaryotic extracellular SOD is a subset of the Cu-Zn variety but functions as a tetramer rather than the usual dimer found in eukaryotes.
The most extensively studied of the Cu-Zn SODs is the bovine SOD, which is readily isolated from blood (39). Cu-Zn SODs are among the fastest enzymes known, with kcat/Km values of 2.3 × 109 M−1 s−1 for bovine SOD (33) and 8.5 ± 0.8 × 109 M−1 s−1 for the bacterial enzyme from Photobacterium legionathi (54). Functional bovine SOD is a homodimer with an intrasubunit disulfide bond within the monomer (1). Each monomer binds one Cu and one Zn ion, and copper has been demonstrated to be essential for full function of the enzyme (39).
The Poxviridae family of viruses includes members that infect a wide range of hosts from mammals to arthropods (17). These viruses are known to encode a number of immunomodulatory proteins, such as serpins and cytokine receptors, which function to disable host defense mechanisms (49). Many of the poxviruses, including variola virus, the causative agent of smallpox, and vaccinia virus, which is used for immunization against smallpox, contain genes exhibiting sequence homology to those for Cu-Zn SODs. Two recent studies examined the role of SOD in vaccinia virus and the leporipoxviruses Shope fibroma virus and myxoma virus (4, 13). In both cases, the protein produced from the sod gene was found to be nonfunctional as a superoxide dismutase. Vaccinia virus SOD is missing most of the metal-binding residues as well as the catalytic arginine. Although enzymatically inactive, the vaccinia virus SOD is produced and packaged into the core of the mature virion, but deletion of the gene does not affect the growth or virulence of the virus (4). Myxoma virus and Shope fibroma virus are closely related, and their SODs are more similar to bovine SOD than is vaccinia virus SOD; however, the leporipoxvirus SODs do not contain the catalytic arginine and lack one of the cysteines necessary for intrasubunit disulfide bond formation. As with the vaccinia virus SOD, the protein is packaged into the mature virion, and deletion of the myxoma virus sod gene has only a slight effect on the spread of the virus in rabbits, with no difference in final disease state (13). More recently, Teoh et al. (55) demonstrated that the Shope fibroma virus SOD complexes with the host cellular copper chaperone and thus may inhibit cellular enzyme function by preventing the addition of the required copper to the cellular SOD enzyme.
The entomopoxvirus isolated from the red hairy caterpillar Amsacta moorei (AmEPV) has been sequenced and can be grown in tissue culture. Open reading frame AMV255 of AmEPV is homologous by BLAST searches to genes encoding Cu-Zn SODs (6, 9). From sequence analysis we determined that unlike the vertebrate poxvirus SODs, all critical residues for function were present in the AMVSOD and we therefore predicted that the AMVSOD would be active as a superoxide dismutase. We demonstrate here that AmEPV does indeed produce a functional Cu-Zn SOD. Furthermore, we show that although the SOD is active, the sod gene is not required for growth in cell culture. This is the first demonstration of an active SOD from a poxvirus and may represent another means used by poxviruses to disable host defense mechanisms.
MATERIALS AND METHODS
Cell and virus culture.
AmEPV was propagated in the Lymantria dispar cell line Ld652 (21, 26). Ld652 cells were maintained in Sf900II serum-free medium (Gibco, Carlsbad, Calif.) supplemented with 10% heat-treated fetal bovine serum (Gibco), 50 IU of penicillin, and 50 μg of streptomycin per ml (Cellgro, Herndon, Va.). For virus production, spinner flasks of Ld652 cells at a density of 5 × 105 cells/ml were infected with AmEPV at a multiplicity of infection of 0.1 to 0.5. Cells were maintained at 5 × 105 cells/ml by the addition of medium until the virus load dropped the cell density below 5 × 105/ml. The virus was harvested when the cell density reached 2 to 3 × 105 and the viability of the cells was 50 to 60%. The cells and virus were centrifuged at 1,000 × g for 10 min to remove intact cells and cellular debris. The resulting supernatant contained the extracellular virus and was stored at 4°C. Virus titers were determined via plaque assays modified from that described previously (26). Briefly, the modifications included the use of 1.3× TC100 (Gibco) containing 13.3% fetal bovine serum and 1.3% SeaPlaque agarose (Cambrex, East Rutherford, N.J.) as the overlay medium and increasing the absorption time to 2 h of rocking at room temperature and an additional 2 h stationary at 28°C.
Protein expression.
The AMV255 open reading frame was amplified from wild-type AmEPV genomic DNA with primers FS369, containing a HindIII site, and FS370, containing an XhoI restriction site (Table 1) (Sigma-Genosys, The Woodlands, Tex.). The 0.481-kb DNA fragment containing the sod gene was digested with XhoI and HindIII, as was the vector, pET28b (Novagen, Madison, Wis.). pET28b DNA was then treated with shrimp alkaline phosphatase (U.S. Biochemicals, Cleveland, Ohio). All DNA fragments were gel purified with Qiagen's gel extraction kit (Valencia, Calif.). Purified sod and pET28b DNAs were ligated to produce plasmid p28-AMVsod. The resulting clone encodes two N-terminal epitope tags, a 6XHis tag and a T7 tag. Plasmid p28-AMVsod was transformed into E. coli strains BL21(DE3), BL21(DE3) pLysS, Rosetta pLysS (all from Novagen), BL21 Star (Invitrogen, Carlsbad, Calif.), and BL21(DE3) RIL (Stratagene, La Jolla, Calif.) for protein expression.
TABLE 1.
Primers for DNA amplification
| Primer | Sequence 5′ → 3′ | Restriction site |
|---|---|---|
| FS369 | CAGCCAAGCTTATGAAAGCTATATGTGTTAT | HindIII |
| FS370 | GACCGCTCGAGTTAATCTTTTGCAATACCAAT | XhoI |
| IDT 37 | ATCGATCTGCAGGAGCAAGGGCGAGGAACTGTT | PstI |
| IDT 38 | CACAGTGAATTCTCACTTGTACAGCTCGTCCA | EcoRI |
| IDT 84 | GCATCAGGATCCTATAAATAATATAAATTCTT | BamHI |
| IDT 85 | GCATCGAAGCTTAGTGAATATATGTATAAATC | HindIII |
| IDT 86 | GCATCGAGGTACCAATTCTCACTTGTACAGCTC | Acc65I |
| IDT 87 | GCATCGAGGTACCTACTGTCGAAACATCTAACG | Acc65I |
Overnight cultures of p28-AMVsod in expression strains were grown in Luria broth (LB) containing 30 μg of kanamycin per ml and 1% glucose. For protein production, LB medium was inoculated with 1/20th volume of overnight culture and grown at 37°C until the optical density at 600 nm was 0.6 to 0.8, at which time the medium was supplemented with 0.5 mM CuCl2 and 0.1 mM ZnCl2 and protein expression was induced with 1 mM isopropylthiogalactopyranoside (IPTG). Induction was allowed to proceed for 2 h.
Expression of the recombinant protein, His-SOD, and its solubility were determined by sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE) followed by immunoblot and detection with the His tag antibody (Novagen). For large-scale preparation of His-SOD protein, 3 liters of p28-AMVsod in E. coli Rosetta pLysS was grown. The cells were pelleted by centrifugation for 15 min at 4,400 × g. The resulting cell pellet was resuspended in CelLytic II B (Sigma, St. Louis, Mo.) following the manufacturer's instructions and then frozen in a dry ice-ethanol bath. Subsequently, the lysate was thawed on ice and treated with 5 μg of DNase I per ml (Sigma) for 15 min with intermittent mixing. The lysate was clarified by centrifugation at 22,000 × g for 15 min, and the supernatant was stored at −80°C. Before affinity chromatography, the supernatant was filtered through a 0.45-μm filter.
Protein purification.
The His-SOD protein was purified from E. coli extracts by passing the extract over a 5-ml chelating column (Amersham Biosciences, Piscataway, N.J.) charged with Ni2+ per the manufacturer's instructions. The purification was performed with a Quad Tech Duo Flow (Bio-Rad, Hercules, Calif.) chromatography system. All chromatography buffers contained 0.02 M sodium phosphate buffer, pH 7.4, and 0.5 M NaCl with different amounts of imidazole. Prior to loading, the column was washed with buffer containing 10 mM imidazole (binding buffer). After the protein was applied to the column, the column was washed with 5 column volumes of 50 mM imidazole buffer, and the protein was eluted from the column with a 10 column volume gradient of imidazole from 10 to 300 mM. Alternate fractions were analyzed on SDS-10% PAGE gels. Fractions identified by Coomassie staining and immunoblotting as containing the His-SOD protein were combined and concentrated with Amicon CentriPlus 3,000-kDa-cutoff concentrators (Millipore, Billerica, Mass.) per the manufacturer's instructions.
SOD gel in situ activity assay.
Protein extracts were mixed with an equal volume of 2× loading buffer (34) without reducing agents. The proteins were not further denatured before being subjected to electrophoresis on either native or SDS-polyacrylamide gels. Gels containing SDS were washed three times in water for 10 min each before assaying (14). The gels were then incubated in a solution of 2-mg/ml nitroblue tetrazolium (NBT) in the dark for 20 min before changing the solution to one containing 10 mg of riboflavin and 4.5 g of putrescine per liter in 36 mM potassium phosphate buffer, pH 7.8, and incubation with illumination from a 75-W bulb with shaking until sufficient contrast between clear areas and the blue background was obtained (10). Gels were subsequently dried.
Monoclonal antibody production.
The AMVSOD monoclonal antibody was produced via immunization of BALB/c mice with purified His-SOD protein at the University of Florida Hybridoma Core Facility with standard techniques. Antibodies were screened by enzyme-linked immunosorbent assay with the recombinant protein as well as by Western blots with uninfected and AmEPV-infected Ld652 cell protein extracts. Monoclonal antibody 2B8-1C9 was isolated as an AMVSOD-specific antibody.
Immunoblots.
Proteins were mixed with an equal volume of 2× loading buffer with no reducing agent and denatured at 95°C for 5 min. After separation by 10% PAGE, the proteins were blotted to nitrocellulose (Micron Separations, Westboro, Mass.) at 0.2 A for 1 h with a semidry blotter. The blots were blocked overnight at 4°C in 5% milk dissolved in 0.05 M Tris, pH 7.5, 0.15 M NaCl, 0.1% Tween 20 (TBST). All remaining steps were carried out at room temperature. For blots with the His tag antibody (Novagen), the blots were washed twice in TBST for 5 min each and reblocked in a solution of 3% bovine serum albumin in TBS (TBST without Tween 20). Subsequently the blots were incubated with the His tag monoclonal antibody at a dilution of 1:1,000 in 3% bovine serum albumin-TBS for 1 h. The blot was then washed three times for 10 min each in 0.02 M Tris (pH 7.5)-0.5 M NaCl-0.05% Tween 20-0.2% Triton X-100 (TBST+T) before incubation with a goat anti-mouse immunoglobulin-horseradish peroxidase conjugate (Southern Biotechnology Associates, Birmingham, Ala.), diluted 1:2,000 in 5% milk-TBST, and incubated for 1 h.
Following incubation with the secondary antibody, the blots were washed three times for 10 min each in TBST+T. Two further washes of 10 min each were performed in TBST, and detection was via the ECL+ chemiluminescent substrate (Amersham Biosciences) and exposure to X-ray film. For blots with the AMVSOD monoclonal antibody 2B8-1C9, the blot was blocked overnight in 5% milk-TBST at 4°C and then washed briefly in TBST before incubation with the antibody. The antibody was used at a dilution of 1:1,000 in 5% milk-TBST incubated for 90 min, and then the blot was washed with TBST. The remaining steps were the same as detailed above, with all washes in TBST.
Determination of Cu-Zn content.
The amount of Cu and Zn in the purified protein was determined with inductively coupled plasma mass spectroscopy at ABC Research Corporation, Gainesville, Fla. The assay used 1 mg of protein.
Stopped-flow spectrophotometry.
Initial velocities for catalysis by His-SOD were measured with the stopped-flow method of McClune and Fee (37). Potassium superoxide (Sigma Aldrich) was dissolved in dry dimethyl sulfoxide. An approximately equimolar amount of 18-crown-6 ether was added to increase superoxide solubility. The decrease in absorption of superoxide at 250 nm (ɛ250 = 2,000 M−1 cm−1) (44) was observed with a dual mixing stopped-flow spectrophotometer (SX18.MV; Applied Photophysics, Leatherhead, Surrey, United Kingdom).
Instrumental dead time was minimized to approximately 4 ms by using two dilutions in sequence. In the first step, superoxide in dimethyl sulfoxide from a 250-μl syringe was mixed with an aqueous solution at pH 11 containing 2 mM 3-cyclohexylamino-1-propanesulfonic acid (CAPS) and 1 mM EDTA from a 2.5-ml syringe. After a 500-ms delay for premixing, this solution was rapidly mixed in a 1:1 ratio with a solution containing 0.5 μM enzyme, 200 mM buffer, and 1 mM EDTA. Total Cu occupancy, determined by inductively coupled plasma-mass spectrometry, was used to adjust the total active enzyme concentration. The dimethyl sulfoxide concentration was 4.5% by volume. After averaging six to eight catalytic traces, the initial 10% of the progress curve was fit to the sum of a zero-order (catalyzed) and second-order (uncatalyzed) equation. The error was typically less than 10%.
Isolation of total RNA from infected cells.
Ld652 cells were seeded at a density of 1.5 × 106/well in six-well dishes and infected at a multiplicity of infection of 10 with wild-type AmEPV. After 3 h, the medium was removed and replaced with fresh medium. Cells were harvested at 3, 6, 9, 12, 15, 18, 24, and 48 h and pelleted at 700 × g. The pellet was washed with phosphate-buffered saline. The cells were then resuspended in 350 μl of RLT buffer (Qiagen) and processed with a Qiashredder. Samples were stored at −80°C. Total RNA was isolated with the RNeasy kit (Qiagen) following the manufacturer's instructions and stored at −80°C.
Northern blots.
Three micrograms of total RNA per sample was subjected to electrophoresis on a 1.5% agarose-formaldehyde gel. The RNA was blotted via capillary action overnight to a Hybond N+ membrane (Amersham Biosciences). After UV cross-linking per the manufacturer's instructions, the blot was prehybridized in QuickHyb (Stratagene) for 30 min at 42°C. Three different oligonucleotides located near the 5′ end, middle, and 3′ end of the sod gene were end labeled with [γ-32P]ATP for use as probes (Table 2). Upon the addition of 2.5 × 106 cpm of labeled probe per ml, the blots were hybridized for 45 min at 42°C. Blots were washed with 2× SSC (1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate)-0.1% SDS at room temperature and then at 50°C and exposed to film or a Phosphorimager screen (Storm; Molecular Dynamics). All other blots were performed by the same procedure with the oligonucleotides listed in Table 2.
TABLE 2.
Oligonucleotide probes
| Gene | Sequence, 5′ → 3′ | Position in the gene, antisense strand (bp) |
|---|---|---|
| AMV255 sod 5′ | TCCATTAACTTTTCCGGTCA | 38-20 |
| AMV255 sod middle | TTTCGTCTGCATACACATTACC | 268-247 |
| AMV255 sod 3′ | TCCGCCAGAATTACCACTTG | 420-401 |
| AMV016 thymidine kinase | GCCAGAAAACATAGGACCAATTAT | 39-16 |
| AMV199 DNA ligase | GAGTTGTGTCATTCTTGGTTCTAAT | 651-627 |
| AMV147 p4B | TCCGCCTATCGAATCCAAAA | 1343-1325 |
| AMV139 p4A | TGCGACGTAATCCCACTAAAA | 997-977 |
| AMV054 RAP94 | TTTGGATTTGCGTCTGGATA | 203-184 |
Protein extraction from infected cells.
For activity assays, Ld652 cells were infected with AmEPV at a multiplicity of infection of 0.15, and the infected cells were harvested at 48 h postinfection. After centrifugation for 5 min at 700 × g, the cell pellet was resuspended in cold phosphate-buffered saline and repelleted. The proteins were isolated by lysing the cells with NP-40 lysis buffer (1% NP-40, 50 mM Tris, pH 7.5, 150 mM NaCl, 2 mM EDTA) and one freeze-thaw cycle. Insoluble proteins were precipitated by centrifugation for 5 min at 16,000 × g at 4°C, and the supernatant containing soluble proteins was retained. For some experiments, proteins were further purified by extraction with a mixture of 37.5% chloroform-62.5% ethanol added to a final concentration of 40% (vol/vol) of protein. After vortexing, the mixture was incubated at room temperature for 10 min and then separated via a 10-min centrifugation at 16,000 × g at room temperature. The upper fraction was transferred to a new tube, and the remaining ethanol was removed from the samples by evaporation in a Speed-Vac concentrator (Thermo Savant, Milford, Mass.) for 10 min without the addition of heat.
The following modifications were used to isolate protein for gel filtration chromatography. A spinner culture of Ld652 cells containing approximately 108 cells was infected with wild-type AmEPV at a multiplicity of infection of 0.04. The cells were harvested after 72 h by centrifugation for 15 min at 1000 × g. The cell pellet was resuspended in a modified NP-40 lysis buffer (0.5% NP-40, 100 mM Tris, pH 8, 100 mM NaCl) and frozen in a dry ice-ethanol bath. After thawing, the protein solution was extracted as described above with chloroform-ethanol. Following evaporation of the ethanol, the proteins were further concentrated with Micron 10K filters per the manufacturer's instructions (Millipore).
Gel filtration chromatography.
A Superdex HR 75 10/30 column (Amersham) was first equilibrated with Tris-buffered saline and then calibrated with molecular size standards from Amersham as directed by the manufacturer. Purchased bovine Cu-Zn SOD (Calbiochem, EMD Biosciences, La Jolla, Calif.) was also used. Approximately 400 μg of wild-type AmEPV protein in a volume of 250 μl was separated on the column. Half-milliliter fractions were collected, and 25 μl of each fraction was used for Western analysis. Fractions containing the AMVSOD were identified via immunoblotting with monoclonal antibody 2B8-1C9 at a dilution of 1:500. Kav values for each standard and for AMVSOD were calculated with the formula (Ve − VO)/(Vt − V0), where Ve is the elution volume, V0 is the void volume determined by blue dextran exclusion (7.84 ml), and Vt is the the total column volume (24 ml). The molecular weight of the size standards was graphed on a logarithmic scale against the Kav values to generate a standard curve.
Construction of plasmids for sod gene inactivation.
Briefly, the strategy to inactivate the AMVsod gene involved inserting the green fluorescent protein (GFP) gene as a marker under the control of the AmEPV spheroidin (sph) promoter into the sod gene, thus disrupting the gene and simultaneously generating a small deletion. All primer sequences are listed in Table 1. The initial step in the process was to construct a plasmid with GFP expressed from the AMVsph promoter. GFP was amplified from pTRUF5 (61) with primers IDT 37 and IDT 38, containing PstI and EcoRI restriction sites, respectively. After digestion with EcoRI and PstI, the PCR product was ligated into EcoRI- and PstI-digested plasmid pDU20 (25), which contains promoter sequences from sph, placing GFP under the control of the sph promoter and producing plasmid pDU20-GFP. The construct was verified by sequencing.
A fragment of 2.46 kb was amplified from genomic wild-type AmEPV DNA with primers IDT 84 and IDT 85, and the fragment contained the sod gene and approximately 1 kb of 5′ and 3′ flanking sequence. The amplified product was digested with BamHI and HindIII and ligated to BamHI- and HindIII-digested pBlueScript II KS+ (Stratagene), producing plasmid pBS SOD+1kb. The 2.46-kb insert was sequenced after cloning and verified to match the published AmEPV sequence (9).
In order to construct the disruption plasmids, the sph-GFP cassette was amplified from pDU20-GFP with primers IDT 86 and IDT 87, which both contain Acc65I sites. After digestion with Acc65I, the fragment was ligated to BsrGI-digested pBS SOD+1kb. This resulted in deletion of 85 bp of the sod gene. Plasmids with the sph-GFP cassette inserted in either orientation were obtained. Plasmid pSOD KO GFP 1 contains sph-GFP in the same transcriptional orientation as the sod gene, and pSOD KO GFP 2 has sph-GFP in the opposite orientation based on restriction enzyme analysis. These plasmids were not sequenced but do express GFP.
sod deletions in AmEPV.
For the production of recombinant viruses containing an inactive sod gene, each plasmid (pSOD KO GFP 1 and pSOD KO GFP 2) was independently transfected into wild-type AmEPV-infected Ld652 cells with the following conditions. Ld652 cells (106) were infected at a multiplicity of infection of 0.5 and transfected 4 h later with 4 μg of DNA and 12 μl of Cellfectin transfection reagent (Invitrogen). Transfections were allowed to proceed for 19.5 h, at which time the medium was replaced. Virus was harvested from the infection-transfection at 48 h posttransfection and plaqued on Ld652 cells at dilutions of 10−2 and 10−3. Fluorescent plaques were picked, and pure recombinant viruses were obtained after five to eight rounds of plaque purification. Viruses were examined for correct construction with PCR primers within the sod gene. Viral DNA for PCR was isolated with the Qiagen DNeasy kit.
Comparative modeling of AMVSOD domains.
Theoretical models of AMVSOD domains were generated with the programs implemented by the Automated Protein Modeling Server (Swiss-Model) (48). Residues 1 to 152 of AMVSOD were queried with BLAST2P (6) to identify homologous proteins of known structure deposited in the Protein Data Bank. ProModII was used to build atomic models (24, 42). Energy minimization was conducted with Gromos96 (11, 53). Root mean squared deviations and distances were calculated with LSQKAB with CCP4 (43). Figures were generated with SETOR (18).
RESULTS
Sequence alignments.
Examination of the completely sequenced AmEPV genome (9) revealed the presence of an open reading frame with sequence similarity to genes encoding Cu-Zn SODs. This open reading frame, designated AMV255, encodes a protein of 152 amino acids with a predicted size of 16.2 kDa. BLAST (6) searches indicate that AMV255 has high levels of similarity to mammalian and viral SODs. Sequence alignment to the bovine SOD (accession no. P00442) (52) revealed that all of the key residues necessary for function of the enzyme are present, specifically, the copper and zinc binding amino acids, the catalytic arginine, and the location of the cysteines used for intrasubunit disulfide bond formation in bovine SOD are all present (Fig. 1). As noted previously (12, 13, 50), the vaccinia virus and myxoma virus SODs are both clearly missing key residues required for full function. We hypothesized from the sequence alignments that the SOD from AmEPV should be an enzymatically active protein, and we chose to express AMVSOD in E. coli and analyze the purified protein for SOD activity.
FIG. 1.

Alignment of sod genes from viral and cellular sources. The sequences were aligned with AlignX software (Invitrogen-Informax). The sequences used were AmEPV AMV255 (AMV255, GenBank accession number NP_065037), bovine SOD (P00442), human SOD (P00441), vaccinia virus SOD (strain Wyeth, AAK76404), myxoma virus SOD (MYX, NP_051845), Shope fibroma virus SOD (SFV, strain Kasza, NP_052020). The key metal binding residues are indicated by ∧, intrastrand disulfide linkages by * and the catalytic arginine by #. The ∼ represents residues at the dimer interface for bovine SOD. AMV255 contains all of the key elements necessary for function compared with bovine SOD; however, the vaccinia virus, myxoma virus, and Shope fibroma virus sod genes have alterations to the gene which would render them inactive (4, 12, 13).
Purification of recombinant SOD.
After examining the protein expression from p28-AMVsod in several E. coli strains, we determined that the highest levels of soluble protein expression were obtained with the Rosetta pLysS strain, which contains an additional plasmid that encodes the tRNA genes for several of the codons that are rarely used by E. coli. In the case of the AMVsod gene, 17% of the codons in the gene are considered rare codons in E. coli. In our initial experiments, we expressed the protein without supplementation of the medium with Cu or Zn, and while the yields were very high, the resulting protein exhibited low levels of activity (data not shown). When His-SOD protein expression was induced in the presence of supplemental Cu and Zn, less protein was produced and the density of the E. coli cells failed to increase after 2 h of induction, indicating that the protein, when fully active, might be toxic to the cells.
The purified protein produced in the presence of additional metals was subjected to inductively coupled plasma-mass spectrometry in order to determine the percentage of copper and zinc present in the preparation. The results indicated that Cu was present at 42% and Zn at 102%. When the purified protein was subjected to electrophoresis on denaturing polyacrylamide gels in the absence of reducing agents, several different protein bands were seen, including one at 20 kDa, the predicted size of the monomeric His-SOD protein (Fig. 2A). The major bands that can be seen in the Coomassie-stained gel were also immunoreactive with the His tag antibody, indicating that they represent multiple forms of the protein (Fig. 2B).
FIG. 2.

Purification and activity assay for His-SOD. His-SOD protein isolated from E. coli was electrophoresed on 10% polyacrylamide-SDS gels (A, B, and C) and a native polyacrylamide gel (D). The size standards refer to panels A, B, and C. (A) A Coomassie-stained gel of denatured purified protein. (B) Immunoblot with the His tag antibody, showing that all of the visible bands on the Coomassie-stained gel are also immunoreactive and therefore represent multiple forms of the His-SOD protein. (C and D) The activity of the recombinant protein was assayed following separation on either SDS-PAGE (panel C) or native polyacrylamide gels (panel D) in the absence of reducing agent, with the inhibition of NBT reduction technique of Beauchamp and Fridovich (10). The clear zones on the gels indicate SOD activity.
Activity of the purified protein.
The activity of the purified His-SOD was assayed in two ways. The first simple assay makes use of the reduction of NBT to formazan and is done in situ in polyacrylamide gels (10). The resulting inhibition of the reduction of NBT by SODs results in a clear zone in a blue background, indicative of SOD activity. Figures 2C and D demonstrate the activity of the purified protein on both SDS and native PAGE gels under nonreducing conditions. Although this assay is usually performed on native gels, it has been demonstrated that the technique is also applicable to proteins resolved in SDS gels provided that the SDS is removed prior to assay for enzymatic activity (14). Separation of proteins on nonreducing SDS gels has the added benefit of making it possible to determine the size of the active protein. In Fig. 2C it is evident that the multiple forms of the protein observed on stained gels (Fig. 2A) are active. Together, the activity gels clearly demonstrate that the His-SOD recombinant protein is active as a superoxide dismutase.
We also assayed the His-SOD recombinant protein for activity with stopped-flow spectrophotometry to measure the SOD-catalyzed decay of superoxide. In this assay, the decay of superoxide is measured by change in absorbance at 250 nm. Initial velocities were calculated by fitting the first 10% of each curve to a second-order (uncatalyzed) and linear (catalyzed) process. No evidence of saturation was observed with increasing superoxide concentration (Fig. 3), consistent with previous results with bovine and human Cu-Zn SODs (22, 33). However, catalytic activity was not diffusion controlled, with kcat/Km values of 8.5 ± 0.8 × 107 M−1 s−1 at pH 8.0, an approximately 20-fold decrease in rate compared to the bovine Cu-Zn SOD, and a value of 4.1 ± 0.1 × 107 M−1 s−1 at pH 9.5, a 50-fold decrease from that of bovine SOD (33).
FIG. 3.

Activity of His-SOD determined with stopped-flow spectrophotometry. The initial velocities of the catalyzed disproportionation of superoxide anion at 20°C are shown. Final solutions contained 100 mM CHES, 1 mM CAPS, 1 mM EDTA, and 0.25 μM His-SOD at pH 9.5. The total enzyme concentration is based on the total Cu occupancy. The solid line is a least squares fit of the data to the Michaelis-Menten equation with kcat/Km = 4.1 ± 0.1 × 107 M−1 s−1.
Expression of the sod transcript.
Earlier analysis of the region upstream of AMV255 indicated some similarity to vaccinia virus late promoter sequences (9, 15), and thus AMVSOD was predicted to be a late protein. We confirmed that the mRNA is indeed expressed and that it is a late gene by Northern blot analysis (Fig. 4A). Three different oligonucleotides (Table 2) corresponding to different regions of the AMVsod gene were each used as probes in this analysis, and all three recognized a discrete transcript of 0.8 kb produced between 9 and 24 h postinfection. Only one blot is shown because all the blots gave the same result.
FIG. 4.

Time course of mRNA expression in wild-type AmEPV as determined by Northern blotting. Each blot contains 3 μg of total RNA per lane. The time points are 3, 6, 9, 12, 15, 18, 24, and 48 h postinfection (hpi). The rightmost three lanes of each blot contain RNA isolated from mock-infected cells at 12, 18, and 24 h, respectively. Each blot was hybridized with a single oligonucleotide to one AmEPV gene. The sequences of the oligonucleotide probes are given in Table 2. (A) AMV255 sod gene. The oligonucleotide probe is near the 5′ end of the gene. (B) AMV139 p4a. (C) AMV147 p4b. (D) AMV054 RAP94. (E) AMV016 thymidine kinase. (F) AMV199 DNA ligase.
To define the late and early expression profiles of genes in AmEPV, we performed a series of other hybridizations of Northern blots with AmEPV genes known to be early or late genes in vertebrate poxviruses. The vaccinia virus genes for the major structural proteins p4a (58) and p4b (46) as well as for the RNA polymerase-associated protein RAP94 (3) are expressed late during vaccinia virus infection. The AmEPV homologs of those genes, AMV139 (p4a), AMV147 (p4b), and AMV054 (RAP94), are expressed 9 to 24 h postinfection (Fig. 4B, C, and D). The profile of sod mRNA expression mimics that of the p4a, p4b, and RAP94 genes, suggesting that sod is expressed as a late gene. In contrast, our data shows that the early genes for thymidine kinase (AMV016) (Fig. 4E) (23) and DNA ligase (AMV199) (Fig. 4F) (51) are expressed from 3 to 9 h postinfection. A second unanticipated finding from this Northern analysis is the fact that all of the late AmEPV transcripts analyzed here are of unique size in contrast to those of the orthopoxviruses, which are polydisperse (41). Whether this is a general property of all late AmEPV transcripts as well as the mechanism by which unique-sized late AmEPV transcripts are generated remains to be determined.
AmEPV produces a functional SOD during infection.
To determine whether the AMVSOD protein was produced during infection, Ld652 cells infected with wild-type AmEPV were harvested 48 h after infection. In order to eliminate cellular iron and manganese SODs, cell extracts were chloroform-ethanol-extracted prior to analysis (59). This treatment does not affect the activity of Cu-Zn SODs and reduces the number of proteins in the sample. The chloroform-ethanol protein extracts were then resolved on nonreducing SDS-PAGE and either stained with Coomassie (Fig. 5A) or assayed in situ for SOD activity (Fig. 5B). Extracts from infected cells showed a pattern of two distinct bands of activity, one at approximately 50 kDa and one at 18 kDa (Fig. 5B). The upper band presumably corresponds to the host homodimeric Cu-Zn SOD because it is present in both infected and uninfected cells. The 18-kDa band is present only in the infected cell extracts and appears slightly larger than the predicted size of 16 kDa for the monomeric form of the AMVSOD. Clearly AMVSOD is expressed and is functional in the context of an AmEPV infection. The question remains whether the enzyme functions as a monomer or dimer but cannot be addressed with this method due to the presence of the cellular enzyme, which would obscure any AMVSOD dimer due to the similarity in size.
FIG. 5.

AmEPV SOD activity during infection. (A) Coomassie-stained gel of infected and uninfected cellular protein extracts. Extraction with chloroform-ethanol is indicated by + (lanes 3 and 4). Extracts were from Ld652 cells infected with wild-type AmEPV at a multiplicity of infection of 0.15 (lanes 2 and 4) or mock infected (lanes 1 and 3), and proteins were harvested at 48 h postinfection. Lane 5 contains purified His-SOD protein. (B) In situ gel activity assay. Protein extracts were separated by SDS-PAGE and then processed for activity as described in Materials and Methods. The arrow indicates the band of activity in the wild-type AmEPV extract (lane 2).
We produced a monoclonal antibody to the His-SOD protein (monoclonal antibody 2B8-1C9) in order to help identify the AMVSOD protein. Immunoblot experiments (see Fig. 7, lanes 1 and 2) confirmed that the AMVSOD protein is expressed in infected cells by the presence of a 16-kDa band not found in uninfected cells. Under these conditions, there is no indication of an AMVSOD dimer; however, a dimer may not withstand the SDS concentrations in the gel.
FIG. 7.
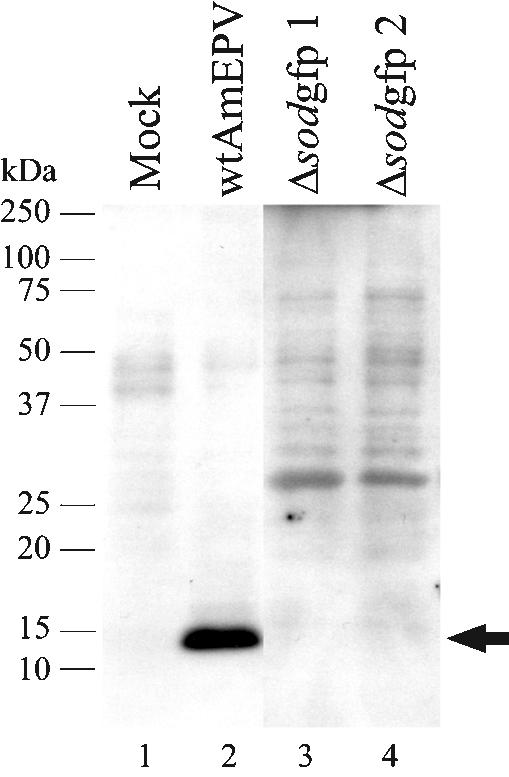
Inactivation of the sod gene in AmEPV. Immunoblot analysis with the SOD monoclonal antibody 2B8-1C9. Each lane contains 10 μg of denatured, unreduced protein. Lane 1 contains protein from mock-infected cells; lane 2 contains protein from wild-type AmEPV-infected cells. Δsodgfp refers to the two different recombinant viruses, each containing the GFP marker but in different orientations with respect to the transcription of the intact sod gene. The monoclonal antibody was used at a 1:1,000 dilution.
AMVSOD is a dimer in solution.
Since most active Cu-Zn SODs from other species are dimeric, we addressed the question of whether AMVSOD functions as a dimer or a monomer with the technique of gel filtration chromatography. A mixture of proteins from infected cells was separated with a calibrated Superdex 75 column (Fig. 6A). For comparison, bovine SOD was chromatographed separately on the column (Fig. 6B). The calibration curve for the column is shown in Fig. 6C. Upon separation, the fractions that were collected from the infected-cell extract were analyzed via Western blotting with monoclonal antibody 2B8-1C9 to determine which fractions contained AMVSOD. AMVSOD eluted from the column starting at 10 ml and continued to elute through 11 ml (Fig. 6D). Based on the corresponding Kav values, the size of the AMVSOD in solution is 32.4 ± 6.8 kDa. The predicted molecular mass of an AMVSOD dimer is 32.4 kDa, indicating that AMVSOD functions as a dimer during infection.
FIG. 6.

Size determination of AMVSOD from infected cells. (A) Chromatogram from a Superdex 75 column of a mixture of proteins from wild-type AmEPV-infected cells. Arrows at the top indicate the locations of peaks for each size standard. Elution volume is indicated on the x axis. (B) Chromatogram of bovine SOD for comparison. The calculated size is 34.6 kDa. (C) The standard curve for the column is derived from proteins of known size: albumin, 67 kDa; ovalbumin, 43 kDa; chymotrypsinogen A, 25 kDa; and RNase A, 13.7 kDa. B indicates the position of the blue dextran peak used to determine the void volume of the column. (D) Western blot of 0.5-ml fractions from the column with monoclonal antibody 2B8-1C9 to detect AMVSOD. Lane 1, total protein; lane 2, 0.25 μg of purified His-SOD, lanes 3 to 10 are labeled according to the starting elution volume of the fraction and correspond to fractions 19 to 26, respectively. AMVSOD elutes predominantly at 10 to 11 ml.
AMVsod gene is not essential for virus growth in cell culture.
We asked whether the AMVsod gene is essential for virus growth. The nonessentiality of a given gene is indicated by little or no effect following deletion or disruption of the gene in question. The open reading frame located downstream of AMV255, AMV256, is similar to the vaccinia virus G1L gene (9). This adjacent gene is transcribed from the opposite strand from sod, and the transcripts overlap at their 3′ ends. In order to leave this gene intact, we opted to generate viruses in which only a small portion of the sod gene was deleted and which concurrently had the gene disrupted by the insertion of a visible marker (GFP). In the plasmid that we constructed, 85 bp of AMV255 were deleted, eliminating two of the His residues involved in Cu binding as well as one of the Cys residues that participates in the key intrastrand disulfide bond formation. The GFP gene was inserted in either orientation. AMV255 disruption viruses were obtained from each of the two plasmids following transfection of infected cells and selection followed by purification of fluorescent plaques.
Western blotting with monoclonal antibody 2B8-1C9 (Fig. 7, lanes 3 and 4) showed that these viruses failed to produce any SOD protein. The genomic structure of the viruses was also confirmed by PCR (data not shown). The viruses that were deficient in SOD production could be grown to high titer and produced plaques similar in size to those of wild-type virus. Our ability to construct viruses deficient in SOD indicates that, as in vaccinia virus and myxoma virus, AMVsod is not an essential gene for growth in tissue culture despite the fact that the AMVSOD is catalytically active.
Comparative models suggest structural similarities between AMVSOD and active SODs.
We used comparative protein structure prediction methods to evaluate similarities and differences between AMVSOD and functional SOD enzymes with solved crystal structures.
BLAST2P (6) was used to search for suitable templates to generate a homology model of AMVSOD. Running pairwise alignments with the 152 residues of AMVSOD identified solved structures of active SODs that ranged in sequence identity from 57.9% to 61.5%. These structures provided a framework for an atomic model generated by ProModII (24). The loop regions were rebuilt based on a database of structural fragments derived from the Protein Data Bank, and the best-fitting fragments were used as the new loop. The side chain conformations were optimized, and energy minimization was conducted. To generate a homology model of AMVSOD in a dimeric form, we superimposed the homology model of AMVSOD on each subunit of the bovine SOD dimer (PDB 1E9O) and conducted conjugate gradient energy minimization. The model of dimeric AMVSOD is shown in red in Fig. 8, superimposed on the crystal structure of bovine dimeric SOD, shown in blue, and illustrates the predicted structural similarity.
FIG. 8.

Model of the AMVSOD dimer. The model shows an AMVSOD dimer (in red) superimposed on a bovine SOD dimer (PDB 1E9O) in blue. Disulfide bonds are shown in yellow. The model shows a high level of similarity between the two SOD proteins. The figure was generated with SETOR.
Overall, the predicted AMVSOD structure is extremely similar to that of bovine SOD, as indicated by average root mean squared deviations of 0.5 Å for Cα atoms. Conformational similarities between the AMVSOD homology model and bovine SOD crystal structures in the region close to the metal atoms are striking. These structural observations are consistent with functional data demonstrating that AMVSOD is an active enzyme.
DISCUSSION
In this study, we primarily addressed the question of whether the SOD from AmEPV is an active superoxide dismutase. As expected from the sequence analysis, the SOD from AmEPV is functional. The kcat/Km value obtained indicates that AMVSOD is active, although 20-fold less active than bovine SOD at pH 8. The need for supplementation of the medium with copper and zinc for maximum activity has been demonstrated previously and hypothesized to be necessary, in part, due to the low levels of intracellular copper (20, 28, 45). We clearly saw multiple forms of the purified protein on Coomassie-stained gels, and the lower activity level of His-SOD may be due in part to the different forms of the protein that are present in the purified preparation, each of which may have different levels of activity.
Incorrect placement of the metal ions is known to cause aberrant mobility of SODs on polyacrylamide gels as well as reduced activity (31, 56). Another possibility is that the presence of two Cys residues at positions 4 and 104 which are not normally involved in disulfide bond formation might contribute to abnormal configurations of the protein via additional disulfide bonds. The Cys residues at positions 53 and 143 in AMVSOD are conserved and would be expected to form the intrasubunit disulfide bond required for activity. It has been demonstrated that, in both bovine and human SOD, replacement of free cysteines within the protein increases the thermostability of the enzyme and that the decreased thermostability of wild-type SOD is due to the formation of oligomers and aggregates via atypical disulfide bond formation with the free Cys residues (27, 35, 40). Alternatively, the presence of the N-terminal His tag might reduce the activity of the protein.
One of the most fascinating and unanticipated findings in our study is that, unlike those of vertebrate poxviruses, many if not all of the AmEPV late transcripts are homogeneous in size rather being polydisperse. While the preponderance of late vertebrate orthopoxvirus gene transcripts are polydisperse, there are some exceptions, including the cowpoxvirus A-type inclusion gene (7). In the case of the A-type inclusion gene, the transcript is initially polydisperse, like other late orthopoxvirus genes, but is cleaved at the 3′ end to eliminate the polydispersity (7, 29). It is possible that, like those of orthopoxviruses, AmEPV late transcripts are also initially polydisperse but cleavage at the 3′ end is more global to render most if not all late transcripts unique in size. The implication of this model is that late transcripts contain either a signal or a structure recognized by a nuclease. A second model for the generation of unique-sized late transcripts is the presence of a 3′ transcription termination signal, similar to what has been observed for unique-sized poxvirus early transcripts (30). This model implies a late transcription termination signal and appropriate termination factor. Elucidation of the mechanism by which unique AmEPV transcripts are generated and the significance of this difference from orthopoxviruses remain to be determined.
AMVSOD is similar to most active Cu-Zn SODs in that it is dimeric in solution. This is not surprising given its similarity to both human and bovine SODs. The role of superoxide dismutase during infection is still unknown. One possibility that we have not yet explored is whether the AMVSOD is able to form a heterodimer with the copper chaperone and thus function in a fashion similar to the Shope fibroma virus SOD (55) to reduce the activity of the host SOD.
Despite its activity, AMVsod is clearly not essential for growth, since virus containing a disrupted sod gene can be grown to high titer in cell culture and plaques normally. Furthermore, experiments in which Lymantria dispar larvae were injected intrahemocoelically with either wild-type AmEPV or AmEPV lacking a functional SOD indicated little difference in pathogenesis or virulence (M. Grove, K. Hoover, M. N. Becker, and R. W. Moyer, unpublished results). It should be pointed out, however, that Lymantria dispar is not the native host for this virus, which was isolated from the red hairy caterpillar of India. The red hairy caterpillar is not available in culture to study. It is conceivable that the minimal effect of removing the SOD from AmEPV on virulence in Lymantria dispar larvae is reflective of this fact. It should also be noted that injection of nonoccluded virus is not the normal mechanism for infection in nature, and it might be that the requirement for an active SOD is only necessary for infection per os.
The finding that the SOD in AmEPV is active dispels the notion that all poxvirus SODs lack enzymatic activity and further heightens the question of what function vertebrate poxvirus SODs might have. It has been proposed that the nonactive SODs of the vertebrate poxviruses may operate by interacting with the relevant chaperones of the host cell to withhold critical copper residues and thereby reduce endogenous Cu-Zn SOD activity (55); however, deletions of sod from Shope fibroma virus and vaccinia virus have little effect in animal models (4, 13). On the other hand, the exact role that an active SOD plays during an infection also remains to be determined, and we propose several possibilities.
It may be that the usefulness of a functional SOD is related to the host organism for the virus, in this case a caterpillar. However, there is nothing intrinsic about the interaction of a poxvirus within an insect environment that dictates the need for a functional SOD, since the only other fully sequenced entomopoxvirus, Melanoplus sanguinipes, does not appear to encode an SOD at all (2). It might be that the diverse host species, moths versus grasshoppers, account for this discrepancy. This view is supported by the presence of genes with homology to those encoding SODs in many baculoviruses which infect members of lepidopteran family, as does AmEPV. An argument against this is the report that the well-studied baculovirus Autographa californica nuclear polyhedrovirus appears to encode a functional SOD, based on its sequence, but studies indicate that the enzyme is not active (57). At present, the role of an active SOD in infection is not clear. It is possible that examination of the pathology of infected insects will provide the clues needed to answer this question.
Acknowledgments
We thank Kelli Hoover and Mike Grove, Pennsylvania State University, for permission to use unpublished data. We are grateful to David N. Silverman, Department of Pharmacology, University of Florida, for advice and support of the enzyme activity assays. Jose A. Hernandez assisted with production of structural figures. Monoclonal antibodies were produced at the University of Florida Hybridoma Core Lab. We thank Danielle Aramburo and her mentor, Alison Bawden, for providing the original clones of AMVSOD.
This work was supported by NIH grant AI46479 to R.W.M. and by NIH grant GM54903 to D. N. Silverman.
REFERENCES
- 1.Abernethy, J. L., H. M. Steinman, and R. L. Hill. 1974. Bovine erythrocyte superoxide dismutase. Subunit structure and sequence location of the intrasubunit disulfide bond. J. Biol. Chem. 249:7339-7347. [PubMed] [Google Scholar]
- 2.Afonso, C. L., E. R. Tulman, Z. Lu, E. Oma, G. F. Kutish, and D. L. Rock. 1999. The genome of Melanoplus sanguinipes entomopoxvirus. J. Virol. 73:533-552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ahn, B. Y., and B. Moss. 1992. RNA polymerase-associated transcription specificity factor encoded by vaccinia virus. Proc. Natl. Acad. Sci. USA 89:3536-3540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Almazan, F., D. C. Tscharke, and G. L. Smith. 2001. The vaccinia virus superoxide dismutase-like protein (A45R) is a virion component that is nonessential for virus replication. J. Virol. 75:7018-7029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Alscher, R. G., N. Erturk, and L. S. Heath. 2002. Role of superoxide dismutases (SODs) in controlling oxidative stress in plants. J. Exp. Bot. 53:1331-1341. [PubMed] [Google Scholar]
- 6.Altschul, S. F., W. Gish, W. Miller, E. W. Myers, and D. J. Lipman. 1990. Basic local alignment search tool. J. Mol. Biol. 215:403-410. [DOI] [PubMed] [Google Scholar]
- 7.Antczak, J. B., D. D. Patel, C. A. Ray, B. S. Ink, and D. J. Pickup. 1992. Site-specific RNA cleavage generates the 3′ end of a poxvirus late mRNA. Proc. Natl. Acad. Sci. USA 89:12033-12037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Battistoni, A., S. Folcarelli, R. Gabbianelli, C. Capo, and G. Rotilio. 1996. The Cu,Zn superoxide dismutase from Escherichia coli retains monomeric structure at high protein concentration. Evidence for altered subunit interaction in all the bacteriocupreins. Biochem. J. 320:713-716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bawden, A. L., K. J. Glassberg, J. Diggans, R. Shaw, W. Farmerie, and R. W. Moyer. 2000. Complete genomic sequence of the Amsacta moorei entomopoxvirus: analysis and comparison with other poxviruses. Virology 274:120-139. [DOI] [PubMed] [Google Scholar]
- 10.Beauchamp, C., and I. Fridovich. 1971. Superoxide dismutase: improved assays and an assay applicable to acrylamide gels. Anal. Biochem. 44:276-287. [DOI] [PubMed] [Google Scholar]
- 11.Berweger, C. D., W. Thiel, and W. F. van Gunsteren. 2000. Molecular-dynamics simulation of the beta domain of metallothionein with a semi-empirical treatment of the metal core. Proteins 41:299-315. [DOI] [PubMed] [Google Scholar]
- 12.Cameron, C., S. Hota-Mitchell, L. Chen, J. Barrett, J. X. Cao, C. Macaulay, D. Willer, D. Evans, and G. McFadden. 1999. The complete DNA sequence of myxoma virus. Virology 264:298-318. [DOI] [PubMed] [Google Scholar]
- 13.Cao, J. X., M. L. Teoh, M. Moon, G. McFadden, and D. H. Evans. 2002. Leporipoxvirus Cu-Zn superoxide dismutase homologs inhibit cellular superoxide dismutase, but are not essential for virus replication or virulence. Virology 296:125-135. [DOI] [PubMed] [Google Scholar]
- 14.Chen, J. R., C. N. Weng, T. Y. Ho, I. C. Cheng, and S. S. Lai. 2000. Identification of the copper-zinc superoxide dismutase activity in Mycoplasma hyopneumoniae. Vet. Microbiol. 73:301-310. [DOI] [PubMed] [Google Scholar]
- 15.Davison, A. J., and B. Moss. 1989. Structure of vaccinia virus late promoters. J. Mol. Biol. 210:771-784. [DOI] [PubMed] [Google Scholar]
- 16.Deng, H. X., A. Hentati, J. A. Tainer, Z. Iqbal, A. Cayabyab, W. Y. Hung, E. D. Getzoff, P. Hu, B. Herzfeldt, R. P. Roos, C. Warner, G. Deng, E. Soriano, C. Smyth, H. E. Parge, A. Ahmed, A. D. Roses, R. A. Hallewell, M. A. Pericakvance, and T. Siddique. 1993. Amyotrophic-lateral-sclerosis and structural defects in Cu,Zn superoxide-dismutase. Science 261:1047-1051. [DOI] [PubMed] [Google Scholar]
- 17.Esposito, J. J., and F. Fenner. 2001. Poxviruses, p. 2885-2921. In D. M. Knipe and P.M. Howley (ed.), Fields virology. Lippincott Williams & Wilkins, Philadelphia, Pa.
- 18.Evans, S. V. 1993. SETOR: hardware-lighted three-dimensional solid model representations of macromolecules. J. Mol. Graph. 11:134-138. [DOI] [PubMed] [Google Scholar]
- 19.Fridovich, I. 1995. Superoxide radical and superoxide dismutases. Annu. Rev. Biochem. 64:97-112. [DOI] [PubMed] [Google Scholar]
- 20.Fujii, J., T. Myint, H. G. Seo, Y. Kayanoki, Y. Ikeda, and N. Taniguchi. 1995. Characterization of wild-type and amyotrophic lateral sclerosis-related mutant Cu,Zn-superoxide dismutases overproduced in baculovirus-infected insect cells. J. Neurochem. 64:1456-1461. [DOI] [PubMed] [Google Scholar]
- 21.Goodwin, R. H., J. R. Adams, and M. Shapiro. 1990. Replication of the entomopoxvirus from Amsacta moorei in serum-free cultures of a gypsy-moth cell line. J. Invertebr. Pathol. 56:190-205. [Google Scholar]
- 22.Goto, J. J., H. Zhu, R. J. Sanchez, A. Nersissian, E. B. Gralla, J. S. Valentine, and D. E. Cabelli. 2000. Loss of in vitro metal ion binding specificity in mutant copper-zinc superoxide dismutases associated with familial amyotrophic lateral sclerosis. J. Biol. Chem. 275:1007-1014. [DOI] [PubMed] [Google Scholar]
- 23.Gruidl, M. E., R. L. Hall, and R. W. Moyer. 1992. Mapping and molecular characterization of a functional thymidine kinase from Amsacta moorei entomopoxvirus. Virology 186:507-516. [DOI] [PubMed] [Google Scholar]
- 24.Guex, N., A. Diemand, and M. C. Peitsch. 1999. Protein modelling for all. Trends Biochem. Sci. 24:364-367. [DOI] [PubMed] [Google Scholar]
- 25.Hall, R. L., Y. Li, J. Feller, and R. W. Moyer. 1996. The Amsacta moorei entomopoxvirus spheroidin gene is improperly transcribed in vertebrate poxviruses. Virology 224:427-436. [DOI] [PubMed] [Google Scholar]
- 26.Hall, R. L., and R. W. Moyer. 1991. Identification, cloning, and sequencing of a fragment of Amsacta moorei entomopoxvirus DNA containing the spheroidin gene and three vaccinia virus-related open reading frames. J. Virol. 65:6516-6527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hallewell, R. A., K. C. Imlay, P. Lee, N. M. Fong, C. Gallegos, E. D. Getzoff, J. A. Tainer, D. E. Cabelli, P. Tekampolson, G. T. Mullenbach, and L. S. Cousens. 1991. Thermostabilization of recombinant human and bovine CuZn superoxide dismutases by replacement of free cysteines. Biochem. Biophys. Res. Comm. 181:474-480. [DOI] [PubMed] [Google Scholar]
- 28.Hartman, J. R., T. Geller, Z. Yavin, D. Bartfeld, D. Kanner, H. Aviv, and M. Gorecki. 1986. High-level expression of enzymatically active human Cu-Zn superoxide dismutase in Escherichia coli. Proc. Natl. Acad. Sci. USA 83:7142-7146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Howard, S. T., C. A. Ray, D. D. Patel, J. B. Antczak, and D. J. Pickup. 1999. A 43-nucleotide RNA cis-acting element governs the site-specific formation of the 3′ end of a poxvirus late mRNA. Virology 255:190-204. [DOI] [PubMed] [Google Scholar]
- 30.Ink, B. S., and D. J. Pickup. 1989. Transcription of a poxvirus early gene is regulated both by a short promoter element and by a transcriptional termination signal controlling transcriptional interference. J. Virol. 63:4632-4644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jewett, S. L., G. S. Latrenta, and C. M. Beck. 1982. Metal-deficient copper-zinc superoxide dismutases. Arch. Biochem. Biophys. 215:116-128. [DOI] [PubMed] [Google Scholar]
- 32.Kinnula, V. L., and J. D. Crapo. 2003. Superoxide dismutases in the lung and human lung diseases. Am. J. Respir. Crit Care Med. 167:1600-1619. [DOI] [PubMed] [Google Scholar]
- 33.Klug, D., I. Fridovich, and J. Rabani. 1972. Direct demonstration of catalytic action of superoxide dismutase through use of pulse radiolysis. J. Biol. Chem. 247:4839-4842. [PubMed] [Google Scholar]
- 34.Laemmli, U. K. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680-685. [DOI] [PubMed] [Google Scholar]
- 35.Lepock, J. R., H. E. Frey, and R. A. Hallewell. 1990. Contribution of conformational stability and reversibility of unfolding to the increased thermostability of human and bovine superoxide dismutase mutated at free cysteines. J. Biol. Chem. 265:21612-21618. [PubMed] [Google Scholar]
- 36.Maier, C. M., and P. H. Chan. 2002. Role of superoxide dismutases in oxidative damage and neurodegenerative disorders. Neuroscientist 8:323-334. [DOI] [PubMed] [Google Scholar]
- 37.McClune, G. J., and J. A. Fee. 1978. A simple system for mixing miscible organic solvents with water in 10-20 ms for the study of superoxide chemistry by stopped-flow methods. Biophys. J. 24:65-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McCord, J. M. 2002. Superoxide dismutase in aging and disease: an overview. Methods Enzymol. 349:331-341. [DOI] [PubMed] [Google Scholar]
- 39.McCord, J. M., and I. Fridovich. 1969. Superoxide dismutase. An enzymic function for erythrocuprein (hemocuprein). J. Biol. Chem. 244:6049-6055. [PubMed] [Google Scholar]
- 40.McRee, D. E., S. M. Redford, E. D. Getzoff, J. R. Lepock, R. A. Hallewell, and J. A. Tainer. 1990. Changes in crystallographic structure and thermostability of a Cu,Zn superoxide dismutase mutant resulting from the removal of a buried cysteine. J. Biol. Chem. 265:14234-14241. [DOI] [PubMed] [Google Scholar]
- 41.Moss, B. 2001. Poxviridae: the viruses and their replication, p. 2849-2883. In D. M. Knipe and P. M. Howley (ed.), Fields' virology. Lippincott Williams & Wilkins, Philadelphia, Pa.
- 42.Peitsch, M. C., T. Schwede, and N. Guex. 2000. Automated protein modelling—the proteome in 3D. Pharmacogenomics 1:257-266. [DOI] [PubMed] [Google Scholar]
- 43.Potterton, E., S. McNicholas, E. Krissinel, K. Cowtan, and M. Noble. 2002. The CCP4 molecular-graphics project. Acta Crystallogr. D Biol. Crystallogr. 58:1955-1957. [DOI] [PubMed] [Google Scholar]
- 44.Rabani, J., and S. O. Nielsen. 1969. Absorption spectrum and decay kinetics of O2− and HO2 in aqueous solutions by pulse radiolysis. J. Phys. Chem. 73:3736-3744. [Google Scholar]
- 45.Rae, T. D., P. J. Schmidt, R. A. Pufahl, V. C. Culotta, and T. V. O'Halloran. 1999. Undetectable intracellular free copper: the requirement of a copper chaperone for superoxide dismutase. Science 284:805-808. [DOI] [PubMed] [Google Scholar]
- 46.Rosel, J., and B. Moss. 1985. Transcriptional and translational mapping and nucleotide sequence analysis of a vaccinia virus gene encoding the precursor of the major core polypeptide 4b. J. Virol. 56:830-838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rosen, D. R., T. Siddique, D. Patterson, D. A. Figlewicz, P. Sapp, A. Hentati, D. Donaldson, J. Goto, J. P. O'Regan, H. X. Deng, Z. Rahmani, A. Drizus, D. McKenna-Yasek, A. Cayabyab, S. M. Gaston, R. Berger, R. E. Tanzi, J. J. Halperin, B. Herzfeldt, R. Van Den Bergh, W. Y. Hung, T. Bird, G. Deng, D. W. Mulder, C. Smyth, N. G. Lainge, E. Soriano, M. A. Pericak-Vance, J. Haines, G. A. Rouleau, J. S. Gusella, H. R. Horvitz, and R. H. Brown. 1993. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 362:59-62. [DOI] [PubMed] [Google Scholar]
- 48.Schwede, T., J. Kopp, N. Guex, and M. C. Peitsch. 2003. SWISS-MODEL: An automated protein homology-modeling server. Nucleic Acids Res. 31:3381-3385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Seet, B. T., J. B. Johnston, C. R. Brunetti, J. W. Barrett, H. Everett, C. Cameron, J. Sypula, S. H. Nazarian, A. Lucas, and G. McFadden. 2003. Poxviruses and immune evasion. Annu. Rev. Immunol. 21:377-423. [DOI] [PubMed] [Google Scholar]
- 50.Smith, G. L., Y. S. Chan, and S. T. Howard. 1991. Nucleotide sequence of 42 kbp of vaccinia virus strain WR from near the right inverted terminal repeat. J. Gen. Virol. 72:1349-1376. [DOI] [PubMed] [Google Scholar]
- 51.Sriskanda, V., R. W. Moyer, and S. Shuman. 2001. NAD+-dependent DNA ligase encoded by a eukaryotic virus. J. Biol. Chem. 276:36100-36109. [DOI] [PubMed] [Google Scholar]
- 52.Steinman, H. M., V. R. Naik, J. L. Abernethy, and R. L. Hill. 1974. Bovine erythrocyte superoxide dismutase. Complete amino acid sequence. J. Biol. Chem. 249:7326-7338. [PubMed] [Google Scholar]
- 53.Stocker, U., and W. F. van Gunsteren. 2000. Molecular dynamics simulation of hen egg white lysozyme: a test of the GROMOS96 force field against nuclear magnetic resonance data. Proteins 40:145-153. [PubMed] [Google Scholar]
- 54.Stroppolo, M. E., M. Sette, P. O'Neill, F. Polizio, M. T. Cambria, and A. Desideri. 1998. Cu,Zn superoxide dismutase from Photobacterium leiognathi is an hyperefficient enzyme. Biochemistry 37:12287-12292. [DOI] [PubMed] [Google Scholar]
- 55.Teoh, M. L. T., P. J. Walasek, and D. H. Evans. 2003. Leporipoxvirus Cu,Zn-superoxide dismutase (SOD) homologs are catalytically inert decoy proteins that bind copper chaperone for SOD. J. Biol. Chem. 278:33175-33184. [DOI] [PubMed] [Google Scholar]
- 56.Tiwari, A., and L. J. Hayward. 2003. Familial amyotrophic lateral sclerosis mutants of copper/zinc superoxide dismutase are susceptible to disulfide reduction. J. Biol. Chem. 278:5984-5992. [DOI] [PubMed] [Google Scholar]
- 57.Tomalski, M. D., R. Eldridge, and L. K. Miller. 1991. A baculovirus homolog of a Cu/Zn superoxide dismutase gene. Virology 184:149-161. [DOI] [PubMed] [Google Scholar]
- 58.Vanslyke, J. K., S. S. Whitehead, E. M. Wilson, and D. E. Hruby. 1991. The multistep proteolytic maturation pathway utilized by vaccinia virus P4a protein: a degenerate conserved cleavage motif within core proteins. Virology 183:467-478. [DOI] [PubMed] [Google Scholar]
- 59.Weisiger, R. A., and I. Fridovich. 1973. Superoxide dismutase. Organelle specificity. J. Biol. Chem. 248:3582-3592. [PubMed] [Google Scholar]
- 60.Zelko, I. N., T. J. Mariani, and R. J. Folz. 2002. Superoxide dismutase multigene family: a comparison of the CuZn-SOD (SOD1), Mn-SOD (SOD2), and extracellular SOD (SOD3) gene structures, evolution, and expression. Free Radic. Biol. Med. 33:337-349. [DOI] [PubMed] [Google Scholar]
- 61.Zolotukhin, S., M. Potter, W. W. Hauswirth, J. Guy, and N. Muzyczka. 1996. A “humanized” green fluorescent protein cDNA adapted for high-level expression in mammalian cells. J. Virol. 70:4646-4654. [DOI] [PMC free article] [PubMed] [Google Scholar]