

Abstract
We introduce a simple intermembrane junction system in which to explore pattern and structure formation by membrane-bound proteins. The junction consists of a planar lipid bilayer to which one species of protein (an IgG antibody) is bound, forming a 2D, compressible fluid. Upon the adhesion of a second lipid bilayer, the formerly uniformly distributed proteins rapidly reorganize into patterns of dense and sparse zones. Using a combination of complementary imaging techniques (fluorescence microscopy, fluorescence interference contrast microscopy, and fluorescence resonance energy transfer), we reconstruct the 3D structure of these intermembrane patterns with nanometer-scale topographic resolution, revealing the orientation of the proteins. The patterns form as the rapid bilayer-bilayer adhesion, often radiating outward from an initial, circular contact site, pushes aside the antibodies, sweeping them into areas of high density and clearing low-density regions. Coarsening of these local features is energetically costly and therefore kinetically trapped; the patterns do not change over tens of minutes. These studies demonstrate that membrane mechanical forces alone, i.e., in the absence of specific biochemical interactions, can drive μm-scale organization of membrane proteins.
Cell membranes are complex 2D fluids in which the biochemical reactivity of mobile, membrane-associated proteins can be coupled to the mechanics of the membrane itself. Several natural systems make use of these features: protein pattern formation at intercellular junctions is an important aspect of intercellular communication (1–6). Disproportionately few studies (7–9), however, have attempted to examine the structure and dynamics of proteins in artificial intermembrane junctions. Such efforts are worthwhile both from the perspective of creating model systems that might eventually illuminate the mechanisms behind biological protein reorganization and from the more general perspective of creating new classes of self-assembled, biologically inspired, or biologically templated (10) materials.
We have constructed a simple experimental system, amenable to nanometer-scale imaging methods, in which to explore the self-organization of mobile, membrane-bound elements at intermembrane junctions. Lipid bilayers can be readily interfaced with solid substrates while retaining their natural 2D fluidity (11–13); half of our junction consists of such a supported bilayer, assembled on SiO2. A single species of protein (an IgG antibody) is bound to the supported bilayer, and a ruptured giant lipid vesicle (14, 15) provides the second lipid bilayer half of the intermembrane junction. The quasi-planar geometry allows us to image the intermembrane topography via several concurrent, complementary techniques: direct fluorescence microscopy, intermembrane fluorescence resonance energy transfer (FRET) microscopy (16–19), and fluorescence interference contrast (FLIC) microscopy (14–18, 20, 21).
We find that the initially uniformly distributed, mobile proteins reorganize themselves into patterns of dense and sparse zones upon the formation of the intermembrane junctions. In this article we first describe these patterns and the structural information they provide. Then, we explore the mechanics behind their formation, finding that membrane mechanics alone can drive μm-scale protein organization. Although our experimental system is highly abstracted from biological intercellular junctions, the lessons drawn from it should be of broad relevance, exposing general dynamics driving protein motions that may be superimposed on other, more specific, biochemical interactions.
Materials and Methods
A schematic illustration of an intermembrane junction, consisting of a supported lipid bilayer, antibodies bound to the supported bilayer, and an upper lipid bilayer formed by a ruptured giant vesicle, is sketched in Fig. 1 A–C.
Fig. 1.

Protein patterns at lipid bilayer junctions. (A–C) Schematic (side view) of the experimental setup. (A) A supported lipid bilayer, with ≈1% biotinylated lipid, self-assembles on a glass substrate. (B) Antibiotin antibodies bind to the supported bilayer, forming a layer of fluid, membrane-bound protein. (C) Giant lipid vesicles tens of micrometers in diameter (schematic not shown) are introduced to the supported bilayer/antibody system. The giant vesicles rupture, creating a bilayer-bilayer junction. Interbilayer adhesion leads to reorganization of the proteins into dense and sparse regions. (D) Fluorescence image (top view) of FITC-labeled antibiotin; the central region, corresponding to the bilayer-bilayer junction, is patterned into zones of high and low density. The protein patterns often exhibit considerable regularity; the 2D Fourier transform (2DFT) shows a broad ring corresponding to a spatial periodicity ≈1–2 μm in wavelength. (Inset) The squared magnitude of the 2DFT. (E) Fluorescence image (top view) of the Texas red-labeled upper bilayer; its finite extent defines the area of the intermembrane junction. The system is in an aqueous solution of 3 mM NaCl.
Lipids. The following lipids were purchased from Avanti Polar Lipids: dimyristoylphosphatidylcholine (DMPC), 1,2-dioleoylsn-glycero-3-phosphocholine (DOPC), l-α-phosphatidylinositol from soy plants (plant PI), 1,2-dimyristoyl-sn-glycero-3-[phospho-l-serine] (DMPS), dioleoyl-dimethylammonium propane (DODAP), 1,2-dioleoyl-3-trimethylammonium-propane (DOTAP), and 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine-N-(cap biotinyl). Monosialoganglioside GM1 was purchased from Matreya (Pleasant Gap, PA). The fluorescent conjugates of the lipid 1,2-dihexadecanoyl-sn-glycero-3-phosphoethanolamine (DHPE), Texas red DHPE and Marina blue DHPE, were purchased from Molecular Probes. The peak excitation and emission wavelengths are 583 and 601 nm, respectively, for Texas red DHPE and 365 and 460 nm, respectively, for Marina blue DHPE.
Supported Lipid Bilayers. First, supported lipid bilayers were formed on glass coverslips (or Si/SiO2 wafers, for experiments involving FLIC) by standard vesicle fusion techniques (19, 22) (Fig. 1 A). In brief, lipid solutions of the desired composition were mixed in chloroform, and then the chloroform was removed by evaporation. The dried lipid films were hydrated with distilled, deionized water (2 mg of lipid per ml) at 4°C for ≈12 h. Small unilamellar vesicles (SUVs) were formed by repeated extrusion of the lipid suspension at 50°C through 0.1-μm polycarbonate filters by using a Lipex extruder purchased from Northern Lipids (Vancouver, Canada). Supported bilayers form spontaneously via rupture and fusion of SUVs onto glass coverslips or SiO2/Si wafers, cleaned previously with piranha solution (3:1 concentrated sulfuric acid/30% hydrogen peroxide). Silicon wafers with a 100-nm thermal oxide layer were purchased from Silicon Quest International (Santa Clara, CA), and wafers with 60- and 125-nm oxide layers were obtained from the Microfabrication Laboratory at the University of California at Berkeley. In all experiments, the bilayer composition was primarily phosphatidylcholine lipids, with a few mole percent Marina blue DHPE fluorescent lipid probes and 0–2 mole percent lipids with biotinylated headgroups. The exact compositions of the bilayers shown in the figures are provided as Supporting Text, which is published as supporting information on the PNAS web site).
Antibodies and Streptavidin. Antibiotin antibodies (mouse monoclonal, as well as goat polyclonal, isotype IgG1) were purchased from Sigma, with and without FITC fluorescent labels. The supported bilayers were incubated in a 1–10 μg/ml solution of antibiotin antibodies for at least 20 min, sufficient to approach saturation of biotin binding sites (23). Unbound antibodies were washed away, leaving a monolayer of protein bound to the supported bilayer (Fig. 1B). The antibodies bound only to the biotinylated lipids; in their absence, nonspecific binding was at most ≈100 molecules per square μm. The excitation and emission wavelengths for FITC are 494 and 518 nm, respectively. Alexa Fluor 488-labeled streptavidin was purchased from Molecular Probes and bound to supported bilayers in the same manner as the antibodies.
Giant Lipid Vesicles and Intermembrane Junctions. Formation of giant lipid vesicles was similar to that described by Akashi et al. (24). Dried lipid films were made as described above, then hydrated with 0.5 M aqueous sucrose solution (0.5 mg lipid per ml), initially at 50°C, cooling to room temperature over ≈12 h. The membrane composition was primarily phosphatidylcholine lipids, with 0.5–1.0 mole percent Texas red DHPE fluorescent lipid probes; there was no biotinylated lipid in the giant vesicles. The exact compositions of the membranes shown in the figures are provided in Supporting Text. We determined from polarization studies that the excitation and emission transition dipole moments of the Texas red fluorophores lie in the bilayer plane, consistent with reports on similar fluorophores (25). Typically, a few microliters of giant vesicle suspension was dropped into the dish containing the supported bilayer and bound antibodies. The vesicles initially sank because of the higher density of the sucrose solution; then, strong electrostatic and van der Waals interactions drove the vesicles toward the supported bilayer, often causing their rupture, depositing an upper bilayer patch tens of micrometers in size (Fig. 1C). For the experiments illustrated in Figs. 1, 2, 3, 4, NaCl was added to the distilled, deionized aqueous environment, after the formation of the intermembrane junctions, to a concentration of 3–10 mM.
Fig. 2.

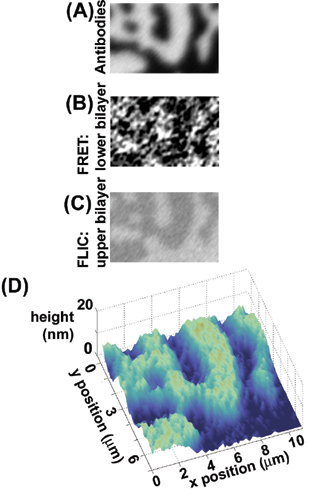
Topographic reconstruction via FLIC microscopy and FRET. (A) Schematic illustration. The supported bilayer sits on a 60-nm SiO2 layer grown on areflective silicon substrate, allowing FLIC; interference between the excitation and emission light of the fluorophores in the upper bilayer with their reflections from the Si surface leads to height-dependent fluorescence intensity. Images B–D are 10 μm wide. (B) Fluorescence image of FITC-labeled antibodies at an intermembrane junction, as in Fig. 1D.(C) Fluorescence image of the Texas red-labeled upper bilayer. Higher intensity corresponds to greater distance from the reflective Si surface. (D) Fluorescence image of the Marina blue-labeled supported bilayer. FRET between the Marina blue donors and the Texas red and FITC acceptors diminishes the blue fluorescence, with the protein/bilayer FRET dominating. Hence, areas of sparse antibodies, where there is no FITC/Marina blue FRET, appear brighter than dense areas. (E) Topographic reconstruction from the FLIC data of C, showing the upper bilayer patch draped over hills and valleys of antibodies ≈14 nm in height. The system is in an aqueous solution of 10 mM NaCl.
Fig. 3.

Reconstruction of the intermembrane structure using FLIC microscopy and FRET, with unlabeled proteins. As in Fig. 2, the supported bilayer sits on a 60-nm SiO2/Si substrate. Images A and B are 9 μm wide. (A) Fluorescence image of the Texas red-labeled upper bilayer. Higher intensity corresponds to greater distance from the Si surface. (B) Fluorescence intensity of the Marina blue-labeled supported bilayer. Energy transfer between the Marina blue donors and the Texas red acceptors is the only FRET mode possible. Hence, areas of sparse antibodies, where the upper and lower bilayers are in tight contact, appear darker than dense areas, in contrast to Fig. 2. (C) Topographic reconstruction from the FLIC data of A, showing the upper bilayer patch draped over hills and valleys of antibodies ≈14 nm in height. The system is in an aqueous solution of 5 mM NaCl.
Fig. 4.

Dynamics of intermembrane protein patterns. Images A–E are fluorescence images of FITC-labeled antibodies. (A and B) Patterns that include a nearly radially symmetric feature. (Bar = 10 μm.) (C) Images taken during the formation of an intermembrane junction. Initially (C1, time t = 0), the antibodies are uniformly distributed along the supported bilayer. A giant vesicle approaches, redistributing the proteins at a circular contact zone (C2, t = 0.44 s); the vesicle then ruptures, forming a larger intermembrane junction (C3, t = 0.88 s). At the junction edges, fingering of the spreading upper bilayer patch pushes antibodies outward (C3, t = 0.88 s; C4, t = 1.32 s); the spreading slows and soon stops (C5, t = 7.48 s). Fine patterns in the junction area are not visible at the low resolution and short exposure times at which this sequence was taken. (Bar = 5 μm.) (D) After their formation, the protein distribution is static and does not coarsen with time; shown are images of the same region, separated in time by 15 min. (Bar = 3 μm.) (E) Heating the intermembrane junctions from room temperature (23°C) to 40°C leaves the protein patterns unaffected; shown are images of the same junction, formed at 23°C, taken at 23°C and 36°C. (F) Schematic illustration: the strong bilayer-bilayer adhesion energy pushes antibodies into dense zones to maximize the area of tight bilayer-bilayer contact.
Microscopy. Images were obtained at room temperature (unless otherwise noted), typically by using a Nikon TE300 inverted fluorescence microscope with a Hamamatsu ORCA 2 (C4742–98) charge-coupled device camera (Hamamatsu, Tokyo) and simple pci (Compix, Cranberry Township, PA) acquisition software. Some images (including Fig. 4 C and D) were obtained by using a Nikon TE2000-E inverted fluorescence microscope with a Quantix 57 back-illuminated charge-coupled device camera (Roper Scientific, Tucson, AZ) and metamorph (Universal Imaging, Downingtown, PA) acquisition software. Illumination was provided by a mercury arc lamp. Filter cubes from Chroma Technology (Rockingham, VT) were used to examine fluorescence from Marina blue (excitation band 325–380 nm; emission band 435–485 nm), FITC (excitation band 455–500 nm; emission band 510–560 nm), and Texas red (excitation band 530–580 nm; emission band 605–675 nm). The fluorophores' spectra were sufficiently distinct to allow three-color imaging, while enough overlap existed between excitation and emission spectra of different fluorophores to allow FRET. For FLIC studies, interference contrast in the fluorescence of the Texas red probes in the upper bilayer patch was monitored, making use of Nikon objective lenses of fixed and adjustable numerical aperture (NA) (air immersion: ×60, NA = 0.7 and ×100, NA = 0.9; oil immersion: ×100, NA = 0.5–1.3).
Image Analysis. Images were analyzed by using photoshop (Adobe Systems, San Jose, CA), metamorph (Universal Imaging), and matlab 6.5 (Mathworks, Natick, MA) software. In the Marina blue (supported membrane) fluorescence images shown, as well as the FITC/antibiotin images of Figs. 1D and 4 D and E, a uniform background intensity has been subtracted to enhance the display contrast. For FITC/antibiotin images transformed for analysis into binary (bright and dark) images, the threshold intensity level for the transformation was chosen by using the graythresh function of matlab, which implements an algorithm by N. Otsu (26): the image intensity levels were sorted into two classes, and the optimal level was that which maximized the variance between classes. We verified that the level so chosen provided the best-fit (minimal mean-square deviation) binary version of the original image. The uncertainty in the threshold level, and in the resulting image properties, was estimated by subdividing each image into several parts, calculating the properties of each, and examining the standard deviation of the resulting set of values.
Results and Discussion
We observed that when sandwiched at the intermembrane junction formed between a ruptured giant vesicle and a supported lipid bilayer (sketched schematically in Fig. 1 A–C) initially homogeneously distributed, fluid, membrane-bound antibodies often reorganize themselves into highly nonuniform μm-scale patterns of dense and sparse regions (Fig. 1 C and D). By examining the structure of the junctions and the dynamics of adhesion, as described below, we concluded that this protein reorganization is driven by the mechanical properties of the membranes themselves: rather than maintain a fixed separation dictated by the antibody size, the membranes push the proteins into new arrangements that allow regions of close interbilayer contact. Although we concentrate here on patterns formed by antibiotin antibodies, similar patterns were also seen in experiments conducted with streptavidin, each molecule of which also binds to two biotinylated lipids in the supported membrane (see Fig. 6, which is published as supporting information on the PNAS web site).
Nanometer-Scale Imaging. The structure of these protein distributions can be independently determined by several means, which together confirm the diagram of Fig. 1C. First, the fluorescent labeling of the antibodies allows a direct map of antibody concentration (Fig. 1D). The fluorescently labeled upper and lower lipid bilayers remain uniform and continuous; in Fig. 1E we show the upper bilayer fluorescence image for the same junction whose antibodies are shown in Fig. 1D.
Second, FLIC microscopy, largely developed in the past decade by Peter Fromherz and colleagues (20, 21, 27), allows nanometer-scale reconstruction of the upper bilayer topography. In FLIC, the experiment is performed as described above, but with a reflective silicon substrate underlying a thin oxide layer (schematically sketched in Fig. 2A). Interference of the excitation and emission light of upper bilayer fluorophores with their reflections from the Si substrate leads to a height-dependent fluorescent intensity; intensity maps topography, with a height resolution of a few nanometers. We plot fluorescence images from the antibodies (Fig. 2B), upper lipid bilayer (Fig. 2C), and supported bilayer (Fig. 2D, discussed in the next paragraph) of an experiment like that of Fig. 1, but performed on a silicon chip with a 60-nm oxide layer. As in Fig. 1D, the antibody image shows spatial patterns. The Texas red fluorescence of the upper bilayer is not uniform, but shows features that correspond to the antibody distribution. Accounting for the imperfectly reflective Si surface, numerical aperture of the microscope optics, and the orientation of the fluorophores in the lipid bilayer (16), we reconstructed from the intensity profile the topography of the upper bilayer patch, its height as a function of lateral position (Fig. 2E). Peaks and valleys match the dense and sparse regions of antibodies. The heights of these features (14 ± 2 nm) match the crystallographically known size of IgG antibodies (28) plus a 1- to 2-nm hydration layer. (The feature heights can be read directly from the topography plot, or, more accurately, by determining the plateau height that, when convolved with the microscope's optical response function, best fits the observed topography. For our data, the two methods differ by only 2 nm.) The FLIC data show that the antibodies are oriented upright, with their Fc domains pointing away from the supported bilayer and covering an area of ≈30 nm2 on the membrane, rather than oriented flat, which would correspond to a height of ≈4 nm. For FLIC, labeling of the proteins in the intermembrane junction is not necessary, because only the upper lipid bilayer fluorescence is relevant. We show in Fig. 3 data from a similar experiment, but with unlabeled proteins. Again, the upper bilayer topography (fluorescence image in Fig. 3A and reconstructed surface in Fig. 3C) maps the structure of the (now invisible) protein structures. FLIC imaging of intermembrane junctions formed with streptavidin, rather than antibodies, bound to the lower membrane shows a protein height of 5.5 ± 2 nm, in agreement with the crystallographically determined protein height (4 nm) plus a hydration layer (Fig. 6).
Third, FRET helps confirm the molecular arrangement sketched in Fig. 1C. In FRET, overlap of the emission spectrum of the donor fluorophore with the excitation spectrum of the acceptor fluorophore leads to a quenching of the donor's fluorescence (16, 29, 30). Quenching by FRET is sensitive to the degree of spectral overlap and the spatial separation of the donor and acceptor, the decay of FRET efficiency being given by the fluorophores' Förster radius, typically a few nm. We make use of FRET between fluorescent probes on apposed lipid bilayers (16, 17, 19) and between the labeled proteins and fluorescent lipid probes. We show in Fig. 2D the fluorescence of a supported bilayer, labeled with Marina blue fluorophores. Brighter regions of Marina blue correspond to sparse zones of FITC (green)-labeled antibodies, the FRET between the adjacent Texas red labels in the upper bilayer patch and the Marina blue labels in the lower bilayer being weaker than the FITC/Marina blue FRET caused by the lower spectral overlap. In Fig. 3B we show fluorescence images of a supported lipid bilayer, again Marina blue labeled, from an experiment in which unlabeled antibiotin was bound. Here, there is no FITC/Marina blue FRET, and brighter regions of Marina blue correspond to dense regions of antibodies, where the upper bilayer is several nanometers from the lower bilayer, and the Texas red/Marina blue FRET is reduced. The FRET signal is also useful in the interpretation of the FLIC data. Interference contrast microscopies in general are sensitive to differences or ratios of heights. Because of its short spatial range (a few nm), FRET between bilayers indicates tight contact, providing a reference point from which topography can accurately be measured.
Dynamics. Before the introduction of the second bilayer, the membrane-bound antibodies are uniformly distributed and fluid, with diffusion coefficients of the same order of magnitude as the lipids (≈1 μm2/s), as determined by fluorescence recovery after photobleaching. Upon the adhesion of the upper bilayer, the proteins reorganize into dense and sparse regions, as discussed above. Several observations help identify the mechanism behind the protein patterning.
Often, the antibody distribution shows a ring, circle, or spoke pattern amid a larger field of “spots” (e.g., Fig. 4 A and B). About 60% of >300 antibody patterns examined show some such radially symmetric feature or circular zone of peculiar patterning. These structures are a consequence of the manner in which the intermembrane junctions form, as illustrated by the time series of protein fluorescence images shown in Fig. 4C: initially, an unruptured giant lipid vesicle in contact with the supported membrane and bound antibodies pushes the antibodies aside at the circular contact zone. Then the vesicle ruptures, forming the larger intermembrane junction surrounding the original spot. The time scale of the vesicle rupture and protein reorganization is faster than our temporal resolution of a few hundred milliseconds, prohibiting direct observation of the protein motions. We can, however, observe the slower dynamics of proteins at the edges of the junctions, where fingering instabilities of the upper bilayer patches push the antibodies along the supported membrane (Fig. 4C), initially with a velocity between 0.3 and 0.9 μm/s and then slowing within a few seconds as the bilayer spreading ceases.
After the intermembrane junction is formed, the antibody patterns are stable with respect to time as well as changes in ionic strength and temperature. There is no coarsening of the patterns (merging of dense or sparse regions) over tens of minutes, as illustrated by images of the same protein pattern separated in time by 15 min in Fig. 4D. We do not find, using FLIC, fluctuations of the upper membrane topography. Addition of 0.1–20 mM NaCl (corresponding to tuning the electrostatic screening length from 30 to 2 nm, spanning the protein size) does not affect the protein organization. The intermembrane junctions are typically formed at room temperature (23°C); subsequently raising the temperature to 40°C does not affect the protein organization, as illustrated in Fig. 4E. [At higher temperatures, IgG antibodies are known to denature and aggregate (31).] There is a large variety in the 2D appearance of the protein patterns formed at room temperature; intermembrane junctions formed at 40°C do not appear different from those formed at room temperature and show no morphologies not also seen in their room temperature counterparts (see Fig. 7, which is published as supporting information on the PNAS web site).
The lipid bilayers themselves are known to have a strong, attractive adhesion energy [mediated by electrostatic, van der Waals, and hydration forces (32)]. Surface-force-apparatus studies of interactions between phosphatidylcholine bilayers have revealed interaction energies of ≈0.1 mJ/m2, decaying with separation over a few nanometers and with a preferred intermembrane distance of ≈1–2 nm (19, 32, 33). Over the 30-nm2 area covered by an antibody molecule the adhesion energy gained by pushing aside the proteins (whose presence and upright orientation necessitate a separation of ≈14 nm) and bringing the bilayers into tight contact is ≈0.7 kBT, where kB is Boltzmann's constant and the temperature T = 296 K.
We propose, therefore, that the observed protein reorganization is driven by the large bilayer–bilayer adhesion energy: the bilayers push the proteins into dense zones to maximize their area of tight contact (sketched in Fig. 4F). While the global energetic minimum configuration would be that in which all proteins are expelled from the intermembrane area, reaching this configuration is not possible during the rapid adhesion time; only local rearrangements, creating local voids of protein, appear possible. The spacing of the zones of high and low density, we suggest, is seeded by topographic fluctuations during the giant vesicles' fast rupture and adhesion: studies of fluctuating membranes (14) reveal spatial length scales similar to those of the intermembrane protein patterns. Energetic considerations explain the observed lack of coarsening: collective rearrangements of collections of proteins would require separation of tightly adhered bilayers. Given the large adhesion energy, for any more than a few proteins the energetic cost would far exceed the ambient thermal energy. Motion of just a few proteins, however, is made difficult by the large bending energy of a membrane following the contour of a small number of antibodies: the bending modulus of a lipid bilayer is kc ≈25 kBT; draping the upper membrane over a single protein would require a curvature energy Ec ≈ 4πkc ≈ 300 kBT. (For a cluster of proteins, Ec is larger still, growing as the square root of the number of proteins in the cluster.) The stability of the patterns with respect to small changes in temperature (≈20°C) further corroborates the proposed mechanism of adhesion-driven organization: small changes in the ambient thermal energy are insignificant compared to the adhesive energy quoted above. Entropic concerns do not play a major role in stabilizing the protein patterns: because both the intermembrane adhesion energy and the entropic free energy of separating the proteins into zones of high- and low-density scale as the area of the zones, there can be no equilibrium balance between these two at any finite zone size, in contrast to our observations. The lack of coarsening of the protein patterns further suggests that the system is not sampling a range of conformations, but is trapped far from equilibrium, either caught in a deep, local energetic minimum of the accessible free energy landscape or kinetically arrested away from the free energy minimum.
Given this picture of bilayer-bilayer adhesion pushing the proteins into dense zones, we would expect the area fraction occupied by dense regions (φd) to increase as a function of the protein concentration initially uniformly bound to the supported membrane (c0), keeping the protein concentration in the dense zones (cmax) constant. (This finding is in contrast, for example, to mechanisms that might prefer a particular partitioning of the adhesive contact between membranes into phases of tight and weak adhesion, adjusting the protein concentration in the dense zones to keep φd fixed.) To test this, for each of three series of data we varied the antibiotin density while keeping the concentration of nonbiotinylated lipids, and therefore the intermembrane interaction energy, nearly constant. As above, we obtained fluorescence images of the resulting protein patterns. The edges of the dense (bright) and sparse (dark) pattern features are blurred by the diffraction-limited lateral resolution (≈300 nm); to extract structural information about the patterns we transformed each image into a binary image (as described in Materials and Methods). The resulting graph of φd versus c0 is plotted in Fig. 5. There is considerable variability in the size and structure of the antibody patterns. Moreover, there are likely to be systematic uncertainties associated with the determination of the dense-phase area fractions (arising from the binary image transformation), shifting the points of Fig. 5 upward or downward, and with the conversion from initial fluorescence intensity to protein density (c0), linearly rescaling the density axis of Fig. 5. (For the intensity to density conversion, we estimate an uncertainty of 20%.) Nonetheless, qualitatively, each set of data shows an upward trend of dense protein area fraction versus initial protein density, consistent with adhesion-driven protein reorganization. Quantitatively, we expect φd to rise linearly with c0, with a slope of 1/cmax. We note (cautiously, given the above-mentioned uncertainties) that the best-fit slope value averaged over the three data sets gives cmax = 17,000 ± 4,000 molecules per um2, of the same order of magnitude as the maximally dense packing, given the antibody orientation, of 33,000 molecules per μm2. The value of cmax being <33,000 molecules per μm2 raises the intriguing possibility that the driven proteins, like macroscopic spheres or colloidal glasses, might jam into a state that is less dense than the densest possible (crystalline) packing (34–36).
Fig. 5.

The area fraction occupied by dense protein regions (φd) increases as a function of the initial (uniform) protein density. Data were collected from three series of samples. Solid lines are linear fits, as discussed in the text. Despite the large amount of variability in the patterns, a similar upward trend is evident in each of the data sets.
Conclusions
Adhesion between simple lipid bilayers can lead to reorganization of bound proteins into complex patterns, trapped far from the uniform, equilibrium state. A variety of imaging techniques allow nanometer-scale reconstruction of the junction topography and determination of molecular orientations. Within the junctions, the dense and sparse protein zones are often remarkably uniform in size (e.g., Figs. 1D and 4 A and B), suggesting an underlying simplicity to their formation.
The proposed mechanism of pattern formation is different from the (equilibrium) aggregation of adhesion molecules in junctions involving intact lipid vesicles observed by Sackmann and coworkers (7–9), most likely because of stronger adhesion in our system. This sort of protein reorganization is also different from proposed mechanisms of T cell immunological synapse formation that rely on a dynamic equilibrium between molecular binding and unbinding (37–39). However, the mechanical effects uncovered here are quite general. The antibodies involved do not play any role in adhering opposing membranes and also do not have any intrinsic affinity for self-aggregation, and yet they find themselves organized into μm-scale patterns. Nearly a decade ago, the idea of size-dependent segregation of membrane proteins, in which adhesion mediated by short adhesion molecules leads to expulsion of large membrane proteins from the contact zone, was put forth in the context of immunological intercellular junctions (40). Although not at equilibrium, the protein rearrangement observed in our junctions is conceptually similar (with bilayer-bilayer adhesion replacing specific adhesive proteins), providing a simple experimental realization of size-dependent sorting. In conjunction with whatever other dynamics that may be occurring, coupling between intermembrane adhesion and the motions of membrane-bound molecules, as well as kinetic stabilization of protein structures far from their equilibrium configuration, may play a role in protein organization at natural intercellular junctions.
Supplementary Material
Acknowledgments
We thank Paul Cripe for help with experiments on membrane-bound streptavidin. R.P. gratefully acknowledges a Miller Postdoctoral Research Fellowship. This work was supported by National Institutes of Health Grant 1-R01-GM64900-01 and a Burroughs Wellcome Career Award in the Biomedical Sciences (to J.T.G.).
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: FRET, fluorescence resonance energy transfer; FLIC, fluorescence interference contrast; DHPE, 1,2-dihexadecanoyl-sn-glycero-3-phosphoethanolamine.
References
- 1.Dustin, M. L. & Colman, D. R. (2002) Science 29, 785–789. [DOI] [PubMed] [Google Scholar]
- 2.Monks, C. R. F., Freiberg, B. A., Kupfer, H., Sciaky, N. & Kupfer, A. (1998) Nature 395, 82–86. [DOI] [PubMed] [Google Scholar]
- 3.Grakoui, A., Bromley, S. K., Sumen, C., Davis, M. M., Shaw, A. S., Allen, P. M. & Dustin, M. L. (1999) Science 285, 221–227. [DOI] [PubMed] [Google Scholar]
- 4.Lee, K.-H., Dinner, A. R., Tu, C., Campi, G., Raychaudhuri, S., Varma, R., Sims, T. N., Burack, W. R., Wu, H., Wang, J., et al. (2003) Science 302, 1218–1222. [DOI] [PubMed] [Google Scholar]
- 5.Igakura, T., Stinchcombe, J. C., Goon, P. K. C, Taylor, G. P., Weber, J. N., Griffiths, G. M., Tanaka, Y., Osame, M. & Bangham, C. R. M. (2003) Science 299, 1713–1716. [DOI] [PubMed] [Google Scholar]
- 6.Singer, S. J. (1992) Science 255, 1671–1677. [DOI] [PubMed] [Google Scholar]
- 7.Bruinsma, R., Behrisch, A. & Sackmann, E. (2000) Phys. Rev. E 61, 4253–4267. [DOI] [PubMed] [Google Scholar]
- 8.Kloboucek, A., Behrisch, A., Faix, J. & Sackmann, E. (1999) Biophys. J. 77, 2311–2328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Albersdörfer, A., Feder, T. & Sackmann, E. (1997) Biophys. J. 73, 245–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mao, C., Solis, D. J., Reiss, B. D., Kottmann, S. T., Sweeney, R. Y., Hayhurst, A., Georgiou, G., Iverson, B. & Belcher, A. M. (2004) Science 303, 213–217. [DOI] [PubMed] [Google Scholar]
- 11.Sackmann, E. (1996) Science 271, 43–48. [DOI] [PubMed] [Google Scholar]
- 12.Boxer, S. G. (2000) Curr. Opin. Chem. Biol. 4, 704–709. [DOI] [PubMed] [Google Scholar]
- 13.Groves, J. T. & Boxer, S. G. (2002) Acc. Chem. Res. 35, 149–157. [DOI] [PubMed] [Google Scholar]
- 14.Kaizuka, Y. & Groves, J. T. (2004) Biophys. J. 86, 905–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Parthasarathy, R., Jackson, B. L., Lowery, T. J., Wong, A. P. & Groves, J. T. (2004) J. Phys. Chem. B 108, 649–657. [Google Scholar]
- 16.Parthasarathy, R. & Groves, J. T. (2004) Cell Biochem. Biophys., in press. [DOI] [PubMed]
- 17.Niles, W. D., Silvius, J. R. & Cohen, F. S. (1996) J. Gen. Physiol. 107, 329–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wong, A. P & Groves, J. T. (2001) J. Am. Chem. Soc. 123, 12414–12415. [DOI] [PubMed] [Google Scholar]
- 19.Wong, A. P. & Groves, J. T. (2002) Proc. Natl. Acad. Sci. USA 99, 14147–14152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lambacher, A. & Fromherz, P. (1996) Appl. Phys. A 63, 207–216. [Google Scholar]
- 21.Lambacher, A. & Fromherz, P. (2002) J. Opt. Soc. Am. B 19, 1435–1453. [Google Scholar]
- 22.Brian, A. A. & McConnell, H. M. (1984) Proc. Natl. Acad. Sci. USA 81, 6159–6163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Blake, R. C., Pavlov, A. R. & Blake, D. A. (1999) Anal. Biochem. 272, 123–134. [DOI] [PubMed] [Google Scholar]
- 24.Akashi, K., Hidetake, M., Hiroyasu, I. & Kinosita, K. (1996) Biophys. J. 71, 3242–3250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Medhage, B., Mukhtar, E., Kalman, B., Johansson, L. & Molotkovsky, J. G. (1992) J. Chem. Soc. Faraday Trans. 88, 2845–2841. [Google Scholar]
- 26.Otsu, N. (1979) IEEE Trans. Sys. Man. Cyb. 9, 62–66. [Google Scholar]
- 27.Braun, D. & Fromherz, P. (1997) Appl. Phys. A 65, 341–348. [Google Scholar]
- 28.Harris, L. J., Skaletsky, E. & McPherson, A. (1998) J. Mol. Biol. 275, 861–872. [DOI] [PubMed] [Google Scholar]
- 29.Clegg, R. M. (1995) Curr. Opin. Biotechnol. 6, 103–110. [DOI] [PubMed] [Google Scholar]
- 30.Selvin, P. R. (2000) Nat. Struct. Biol. 7, 730–734. [DOI] [PubMed] [Google Scholar]
- 31.Vermeer, A. W. P. & Norde, W. (2000) Biophys. J. 78, 394–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Israelachvili, J. (1991) Intermolecular and Surface Forces (Academic, New York), 2nd Ed.
- 33.Marra, J. & Israelachvili, J. (1985) Biochemistry 24, 4608–4618. [DOI] [PubMed] [Google Scholar]
- 34.Liu, A. J. & Nagel, S. R. (1998) Nature 396, 21–22. [Google Scholar]
- 35.Trappe, V., Prasad, V., Cipellitti, L., Segre, P. N. & Weitz, D. A. (2001) Nature 411, 772–775. [DOI] [PubMed] [Google Scholar]
- 36.Donev, A., Cisse, I., Sachs, D., Variano, E. A., Stillinger, F. H., Connelly, R., Torquato, S. & Chaikin, P. M. (2004) Science 303, 990–993. [DOI] [PubMed] [Google Scholar]
- 37.Lee, S. E., Hori, Y., Groves, J. T., Dustin, M. L. & Chakraborty, A. K. (2002) Trends Immunol. 23, 492–499. [DOI] [PubMed] [Google Scholar]
- 38.Weikl, T. R., Groves, J. T. & Lipowsky, R. (2002) Europhys. Lett. 59, 916–922. [Google Scholar]
- 39.Burroughs, N. J. & Wülfing, C. (2002) Biophys. J. 83, 1784–1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Davis, S. J. & van der Merwe, P. A. (1996) Immunol. Today 17, 177–187. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}