

Abstract
Efficient contractions of the left ventricle are ensured by the continuous transfer of adenosine triphosphate (ATP) from energy production sites, the mitochondria, to energy utilization sites, such as ionic pumps and the force-generating sarcomeres. To minimize the impact of intracellular ATP trafficking, sarcomeres and mitochondria are closely packed together and in proximity with other ultrastructures involved in excitation-contraction coupling, such as t-tubules and sarcoplasmic reticulum junctions. This complex microdomain has been referred to as the intracellular energetic unit. Here, we review the literature in support of the notion that cardiac homeostasis and disease are emergent properties of the hierarchical organization of these units. Specifically, we will focus on pathological alterations of this microdomain that result in cardiac diseases through energy imbalance and posttranslational modifications of the cytoskeletal proteins involved in mechanosensing and transduction.
1. Introduction
The dynamic range and durability of the heart are enabled by the hierarchical organization of sarcomeres, the organ's force-generating units [1]. Sarcomeres are highly ordered arrays of molecular motors developed and maintained through a finely regulated mechanotransductive mechanism, sarcomerogenesis [2]. A second mechanism, myofibrillogenesis, ensures that sarcomeres serially register along acting bundles, creating the parallel arrays of myofibrils responsible for the cardiomyocyte striated appearance [3]. Through dedicated cell-cell junctions, myofibrils register across multiple cardiomyocytes creating myocardial sheets that wrap themselves around the cardiac chambers ensuring efficient pumping of blood in the circulation [4, 5].
While the anisotropic organization of the contractile cytoskeleton ensures efficient organ-level contraction, large quantities of Ca2+ ions and adenosine-triphosphate (ATP) molecules are required at the subcellular level for sarcomere contraction. In fact, cardiomyocytes feature a network of dedicated calcium storages, known as sarcoplasmic reticulum, as well as a large number of mitochondria, the organelles responsible for synthesizing ATP molecules from a range of available energy substrates [6]. Specifically, while glycolytic mechanisms are sufficient to meet the organ's ATP requirements during development, an increased energy demand coupled with an abundance of energy dense fatty acids promotes a shift towards oxidative metabolism as the organism matures. Fatty acid oxidation requires a complex set of enzymes that cluster into mitochondria to effectively participate in the tricarboxylic acid cycle [7, 8].
To ensure rapid and efficient transfer of ATP molecules, mitochondria in cardiomyocytes localize in close proximity with sarcomeres, the sarcolemmal invaginations known as t-tubules, and the sarcoplasmic reticulum. This creates a functional microdomain, termed the intracellular energetic unit (ICEU), where rapid catabolism drives a chemical gradient of ATP from the mitochondria to the sarcoplasmic reticulum and sarcomeres [9, 10]. Here, we first review some recent results suggesting a link between the cell microenvironment, the contractile cytoskeleton, and metabolism, which we hypothesize to interact at the level of intracellular energetic units. We will then argue that pathological alterations of this microdomain, and not only of its components, result in cardiac diseases through both energy imbalance and the direct impairment of structural and contractile cytoskeleton components. Finally, we speculate that hiPS-derived cardiomyocytes and heart-on-a-chip platforms may be used to investigate the pathophysiology of cardiomyocyte intracellular energetic units.
2. Lessons Learned from Recent In Vitro Experiments
“How do cardiomyocytes build themselves?” [11] Following up on initial in vivo studies [12], in vitro and in silico assays were developed to characterize how the physicochemical characteristics of the cell microenvironment influence sarcomerogenesis and myofibrillogenesis [13–16]. In engineered isolated cardiomyocytes [17], cardiac microtissues [18], and laminar monolayers [19], microenvironmental cues constitute a chemomechanical signal that controls cell shape and the registration of the cell contractile apparatus. Intriguingly, recent results suggest that mechanotransduction may also affect cell metabolism [17, 20, 21] (Figure 1(a)) as we will describe in this section.
Figure 1.

Mechanosensitive control of cardiomyocytes metabolism. (a) Putative link between microenvironmental cues, sarcomere assembly, and mitochondria maturation. (b) Traction force microscopy experiments suggesting that in response to stiffening of their microenvironment cardiomyocytes may regulate their shape to maintain an optimal work output. (c) Metabolic flux analysis showing the cardiomyocytes cultured on soft substrates stiffness (5% gelatin, black) retained a greater respiratory capacity than on stiffer ones (polydimethylsiloxane, PDMS, red). (d) Qualitative and quantitative analysis of cardiomyocytes structure in a disease-on-a-chip study of Barth syndrome, an inherited cardiomyopathy caused by a mutation to the tafazzin gene. The analysis showed that hiPS-derived cardiomyocytes obtained from a Barth syndrome patient (BTH-H, nGFP) had significantly impaired contractile architecture with respect to cardiomyocytes obtained from a healthy individual (WT1, nGFP). Furthermore, introducing wild-type tafazzin (BTH-H, TAZ), but not restoring the basal ATP level by switching to the glycolytic pathway (BTH-H, Glucose), in diseased cells rescued the structural phenotype. Scale bar 10 μm. Together, these findings support the notion that mechanotransductive processes like sarcomerogenesis and myofibrillogenesis are linked not only to metabolism but also to mitochondria structure and function. Images were obtained with permission from the following sources: (b) McCain et al., 2014a [17]; (c) McCain et al., 2014b [20]; and (d) Wang et al., 2014 [21]. ∗,#Significant (p < 0.05) differences with respect to the nGFP-galactose and nGFP-glucose groups, respectively.
Hypertrophied and failing hearts have a higher fibronectin content and are less compliant than healthy organs, and their cardiomyocytes exhibit distinct morphological characteristics. In these pathological conditions, cardiomyocytes change not only in size, but also in aspect ratio—their length-to-width ratio changes from ~7 : 1 in healthy hearts to ~3 : 1 and ~11 : 1 in concentric and eccentric hypertrophy, respectively [22–24]. We hypothesized that cardiomyocytes in a diseased microenvironment remodel to preserve contractile function at the cellular level. To test this hypothesis, we developed a dedicated in vitro cell culture system. We engineered shape-controlled neonate rat cardiomyocytes on deformable gels of tunable stiffness. We then employed immunocytochemistry techniques, traction force microscopy, and a computational model of muscle mechanics to characterize the structure and function of the contractile apparatus and correlated them to the engineered cell shapes. We established that cardiomyocytes with normal length-to-width ratio exerted maximal contractile force on substrates with physiological, but not pathological, stiffness. On these diseased substrates, myocytes with a reduced length-to-width ratio (Figure 1(b)) exhibited better contractile performances. These results support the notion that cardiomyocytes remodel their shape, and therefore the relative amount of sarcomeres aligned in series or parallel to preserve their mechanical function in the presence of increasingly stiffer cell microenvironments [17]. In a separate study, we hypothesized that the fetal phenotype of stem cell derived cardiomyocytes could be improved by culturing these cells for an extended period of time in the presence of an adult-like engineered microenvironment [14, 25, 26]. We reasoned that heart development occurs over ~270 days in humans and therefore we hypothesized that long-term in vitro culture may coerce human stem cell derived cardiomyocytes towards a more mature phenotype. Unfortunately, traditional culture conditions utilize rigid materials from which stem cell derived cardiomyocytes tend to delaminate as they mature. We developed micromolded gelatin hydrogels to engineer rat primary and hiPS-derived cardiomyocytes into tissues that exhibit laminar architecture and mature and aligned sarcomeric structure and are functionally proficient. While the structural organization and functional proficiency of engineered neonatal rodent cardiomyocytes cultured on compliant gelatin hydrogels or stiffer fibronectin-coated polydimethylsiloxane (PDMS) substrates were similar [26, 27], we showed that myocytes had a greater spare respiratory capacity on soft than on stiff substrates (Figure 1(c)). These findings support the notion that microenvironmental cues, such as substrate stiffness and ECM coating, may impact cardiomyocyte metabolism independently from changes in cell structure or function [20].
Finally, we devised a disease-on-a-chip platform to study Barth syndrome using patient-specific hiPS-derived cardiomyocytes [21]. This disease is characterized by mutations of the gene encoding tafazzin, a protein involved in the maintenance of the mitochondrial inner membrane [28]. Patients develop a metabolic cardiomyopathy, but detailed molecular mechanisms and possible therapeutic strategies remain unknown. We hypothesized that impaired mitochondrial structure would cause mitochondrial dysfunction leading to disrupted sarcomeric structure and contractile function. To test this hypothesis, we cultured patient-specific hiPS-derived cardiomyocytes on our heart-on-a-chip platform, to support establishment of laminar tissue architecture and to enable direct measurement of contractile stress generation. Metabolic dysfunction was exacerbated by culturing tissues in a media containing galactose to prevent compensation via glycolytic pathways. In agreement with our hypothesis, engineered Barth cardiac tissues were characterized by depressed respiratory capacity, unorganized sarcomere structure, and decreased contractile twitch and peak systolic stress. Reintroduction of wild-type transcripts [29] for tafazzin was sufficient to rescue the disease phenotype structurally and functionally, confirming the causative role of mitochondrial dysfunction in this myopathy. Interestingly, when galactose was replaced with glucose, the basal whole-cell ATP level in diseased and healthy cardiomyocytes was rescued, but the structural impairment of the contractile cytoskeleton persisted, indicating that the ATP deficit was not the etiology of cardiac dysfunction in Barth syndrome (Figure 1(d)). Importantly, when we treated disease cells with ROS-scavengers we were able to rescue the disease phenotype, suggesting that the ROS buffering system in cardiomyocytes may be an overlooked target for pharmacological interventions in Barth syndrome. These results support the notion that mitochondria dynamics affect myofibrillogenesis not only by meeting the energy demand, but also through ROS-mediated mechanisms [21].
Taken together, these findings point to mechanosensitive feedback loop through which the cell microenvironment modulates the cross-talk between cardiomyocyte cytoskeletal architecture and metabolic profile, ultimately affecting the heart's ability to function (Figure 1(a)).
3. Intracellular Energetic Units in Healthy Hearts
We believe that the link between microenvironment, metabolism, and sarcomerogenesis resides in the mechanochemical signaling that occurs at the level of the intracellular energetic units, whose structural integrity is ensured by a variety of cytoskeletal elements previously implicated in mechanotransduction. Here we review the development of sarcomeres and mitochondria, as well as the establishment and maintenance of intracellular energetic units in cardiomyocytes (Figure 2).
Figure 2.
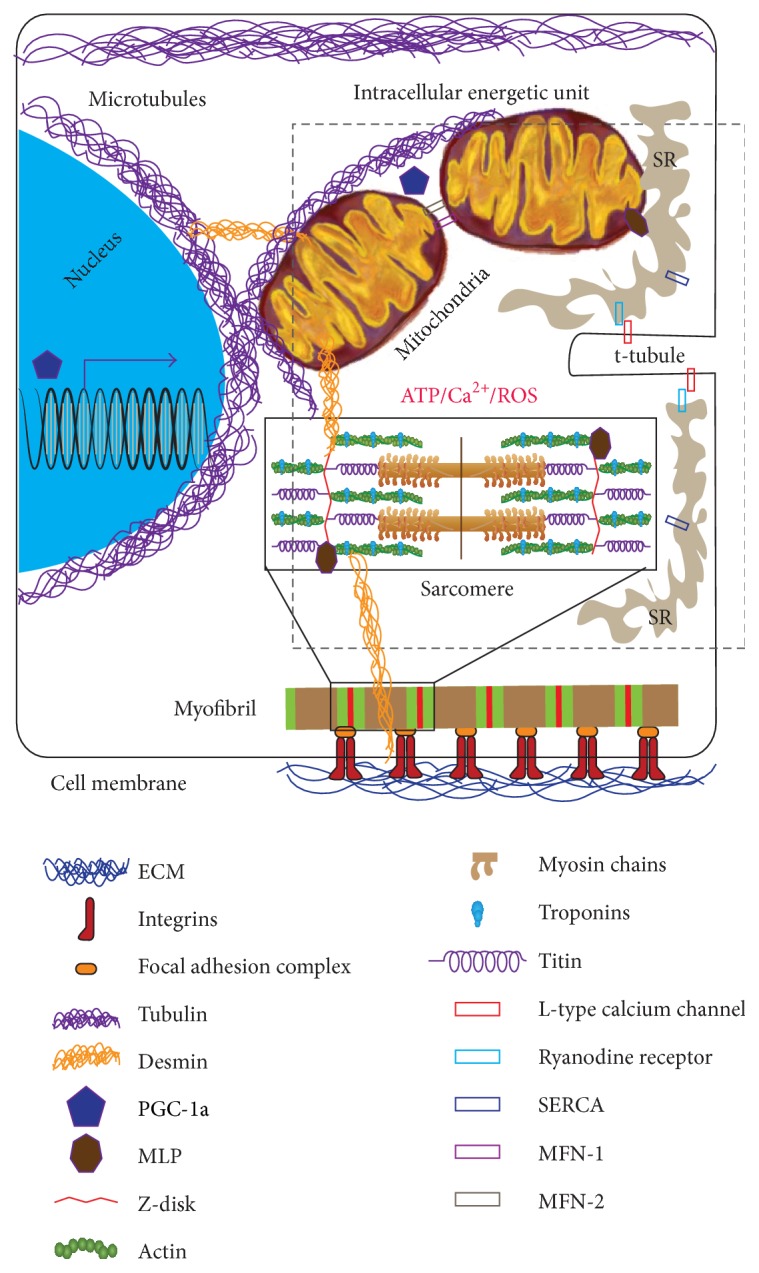
Schematic representation of the intracellular energetic unit. Mitochondria, sarcomeres t-tubules, and sarcoplasmic reticulum (SR) are closely packed together in a functional microdomain known as the intracellular energetic unit. Establishment and maintenance of these microdomains require tethering to a variety of cytoskeletal proteins that have been shown to participate in mechanotransduction. Furthermore, organelles and ultrastructures in the unit exchange through ATP, calcium ions, and reactive oxygen and nitrogen species.
3.1. Sarcomeres
In striated muscle cells, including cardiomyocytes, sarcomeres are serially arranged along parallel arrays of myofibrils. Their orchestrated activation is responsible for cell contraction and their physicochemical state determines how the organ contractile force changes in response to environmental challenges, such as pre/postload [30–32]. The correct spatial localization of sarcomeres, as well as their continuous renewal and remodeling along striated fibers, is ensured through the processes of sarcomerogenesis and myofibrillogenesis [33–35]. These processes start with the recruitment of integrins in focal adhesions near the cell membrane; these adhesions anchor the cell cytoskeleton to the extracellular matrix allowing actin filaments to extend outwards [22]. As the focal adhesions mature through the recruitment of additional integrins and signaling kinases, premyofibrils rich in Z-bodies (immature assemblies of contractile proteins) form near the spreading cell edge [36, 37]. Myosin, actin, tropomyosin, and various troponin isoforms polymerize and interweave to form sarcomeres via template proteins, nebulin and titin [37]. Thick and thin filaments are subsequently assembled [38] and terminated by actin capping proteins [12, 35, 37]. As the myofibrils mature and increase in width, linear arrays of Z-bodies fuse to form Z-bands [36]. These are further anchored by costameres and attachment sites that transmit force between the cell cytoskeleton and the extracellular matrix [39], thus closing the mechanotransductive feedback loop.
3.2. Mitochondria
Approximatively 35% of the volume of an adult cardiomyocyte is occupied by mitochondria, interspersed within the cell contractile apparatus [6]. To achieve the number and spatial distribution observed in adult cells and to limit the degradation of their DNA, mitochondria continuously merge together (fusion) and split apart (fission) in a process termed mitochondrial biogenesis [40]. Chief among the molecular mediators of this process is the coactivator PGC-1α (peroxisome proliferator-activated receptor γ—PPARγ—coactivator 1α) [41]. Downstream of PGC-1α, a family of large GTPases known as dynamin-related proteins (DRPs), further controls the fission and fusion cycles [42]. Mitochondrial fission requires helical DRPs known as Dnm1/Drp1 that wrap themselves around the mitochondrial membrane and, by hydrolyzing GTP, constrict the membranes together until they divide [43–45]. Mitochondrial fusion occurs in two phases: membrane tethering and lipid mixing [46]. These events are regulated by additional DRP such as mitofusins (Mfn-1 and Mfn-2) and OPA1/Mgm-1 [47–49].
During development, biogenesis affects structure, function, and tethering of mitochondria as it requires the remodeling of the organelle inner and outer membranes, as well as of the receptors and enzymes located in the cristae [50]. In immature cardiomyocytes, mitochondria begin as fragmented bodies with poorly developed cristae and are located primarily in the perinuclear regions of the cytoplasm of immature cells [51, 52]. As cardiomyocytes mature, mitochondria transform into organized tubular structures containing elongated cristae and display elevated mitochondrial membrane potential, increased oxygen consumption, and elevated ATP production relative to the fragmented structures observed in immature cells [51, 53]. Importantly, a study using embryonic mouse hearts demonstrated that the mitochondrial permeability pores are open during the early stages of heart development, and their closure correlates with cytoskeletal maturation [54]. Furthermore, the authors demonstrated that pore closure facilitated myofibrillogenesis completion by reducing the cytoplasmic levels of reactive oxygen species.
3.3. The Intracellular Energetic Unit
The interplay between sarcomeres and mitochondria is critical to myocardial function. During cell contraction, calcium ions and ATP molecules are consumed to ensure cross-bridge cycling, muscle shortening, and the development of contractile force [38, 55]. Efficient transfer from energy production sites to energy consumption sites has been evolutionarily achieved by creating a functional microdomain, the intracellular energetic unit [56–58]. That is, mitochondria, sarcomeres, t-tubules, and the sarcoplasmic reticulum are packed so close together that, during normal contraction, calcium ions and ATP molecules are released and uptaken in the intracellular energetic unit before they have the time to diffuse outside of it. There are at least two ways for this arrangement to impact cardiomyocyte mechanotransduction. First, mitochondrial function is necessary to ensure the energy demands of myofibrillogenesis and sarcomerogenesis are met [59, 60]. Secondly, tethering of the mitochondrial membrane to the cell cytoskeleton is required to facilitate nucleotide channeling [58] and the movement of metabolites involved in oxidative phosphorylation [57, 61]. For example, microtubules [62] and actin [63] have been implicated in long- and short-distance mitochondrial trafficking, respectively. Further, tethering to tubulin, actin [64], and intermediate filaments [65] is required for mitochondrion cristae to acquire their characteristics invaginated morphology. Tubulin tethering to mitochondria has been shown to play both structural and functional roles in striated muscle homeostasis [66] and disease [10]. Specifically, by physically and chemically interacting with the mitochondrial outer membrane, tubulin modulates the function of the permeability transition pore [67] and of voltage-dependent anion channels [68]. Mitochondrial tethering to cytoskeletal proteins involved in mechanosensing represents a mechanism through which the extracellular matrix may affect the contractile and metabolic apparatuses. In fact, integrins connect the cell cytoskeleton to the extracellular matrix at specific locations known as focal adhesions [22]. External mechanical forces activate the integrin-mediated response of protein-tyrosine kinases of the Src family and cytoplasmic form of focal adhesion kinases (FAK) and mitogen-activated protein kinases (MAPK) [69, 70]. Importantly, integrin signaling mediated by Rho family GTP-binding proteins was shown to directly affect mitochondria structure and function [71].
Finally, recent studies suggest that sarcomeres and mitochondria directly exchange internal forces through sarcomeric shortening and mitochondrial swelling [72, 73]. In fact, mitochondria located in between sarcomeres are squeezed during cell contraction, thus providing an intracellular load against which force must be generated [73]. At the same time, stimuli that lead to mitochondrial swelling cause these organelles to apply pressure against neighboring structures [72]. For example, through the microtubule network, swollen mitochondria severely deformed the nuclear shape, possibly initiating chromatin remodeling as well as epigenetic and transcriptional events [74].
Taken together these results suggest that the intracellular energetic unit itself can sense and respond to changes in the cell microenvironment through its cytoskeleton components.
4. Intracellular Energetic Units in Diseased Hearts
In the traditional view, the heart structure and function are ensured by the multiscale hierarchical organization of sarcomeres that are closely packed with mitochondria (Figures 3(a)-3(b)). Mutations to proteins involved in cell contraction and metabolism result in a variety of inherited cardiomyopathies, characterized by maladaptive remodeling at the organ, tissue, and cell level and by disorganized intracellular energetic units (Figures 3(c)–3(e)). If the intracellular energetic units are responsible for cardiac homeostasis, we reasoned that mechanotransductive alterations to their structure and function should result in similar disease phenotypes (Figure 3(f)). Here we first review animal and clinical studies relative to inherited diseases affecting the contractile and metabolic apparatuses, respectively. We then present a number of conditions that were linked to the dysfunctional interplay between sarcomeres, mitochondria, and the mechanosensitive constituents of the intracellular energetic unit. Finally, we discuss the possible pathological implications of excess reactive oxygen species in the intracellular energetic units.
Figure 3.

Multiscale disease mechanism in the heart. (a-b) Schematic representation of the multiscale hierarchical organization of sarcomeres in the healthy (a) and diseased (b) heart: the structure of the ventricular wall (i) is affected by the alignment of the myocytes within the myocardial layers (ii) that is modulated by the orientation of myofibrils within cardiomyocytes (iii), which in turn depends on the organization of the intracellular energetic units (iv, ICEUs). (c–f) Examples of organ (i), tissue (ii), cell (iii), and ICEU (iv) level structure in healthy control animals (c), as well as diseased animals with dysfunctional sarcomeres (d), mitochondria (e), and intracellular energetic units (f). Images were obtained with permission from the following sources: (c, d) Sussman et al., JCI, 1998 [85]; (e-i, ii) Krebs et al., PNAS, 2011 [126]; (e-iii) Zaglia et al., JCI, 2014 [93]; (e-iv) Perdomini et al., Nat Med, 2014 [127]; (f-i) Chen et al., Circ Res, 2012 [128]; (f-ii) Martin et al., Circ Res, 2014 [125]; (f-iii) Purevjav et al., JACC, 2010 [121]; and (f-iv) van den Bosch et al., Cardiovasc Res, 2005 [117]. Arrows and asterisks indicate pathologically disorganized sarcomeres, mitochondria, and ICEUs.
4.1. Sarcomerogenesis in Diseased Hearts
Many inherited cardiomyopathies have been linked to genetic defects in sarcomeric proteins such as myosin heavy chain (MHC), myosin light chain (MLC), cardiac troponin T (cTnT), and cardiac troponin I (cTnI) [75, 76]. Disease pathways are complex in that different mutations of the same sarcomeric protein can give rise to both hypertrophic and dilated cardiomyopathies [76]. In fact, abnormalities in sarcomeric proteins such as actin, titin, myosin and myosin-binding proteins, and cTnT impaired sarcomerogenesis and led to severe cardiac impairment, abnormal chamber morphology, and congenital heart disease [77–81].
Heart pathologies are often characterized by sarcomere disarray which leads to compromised contractile function. Titin is a major sarcomeric protein that acts as a template for myosin incorporation during sarcomerogenesis. The absence or mutation of the gene encoding for titin resulted in disruptions in sarcomerogenesis and dysfunctional sarcomeres which ultimately led to heart failure [82]. Myosin light chain kinases (MLCK) regulate sarcomerogenesis through the phosphorylation of ventricular MLCK2. Patients with dilated cardiomyopathy exhibited reduced ventricular MLCK2 expression, unstable myofilaments, and defective myosin thick filaments assembly [83]. Additionally, reduced cardiac MLCK levels in zebrafish embryos result in dilated ventricles with thinned walls and poor sarcomere assembly [84]. Finally, tropomodulin—a tropomyosin-binding protein—regulates actin thin film length and is essential for sarcomere maintenance [85]. Alterations in tropomodulin expression levels disrupt sarcomere structure [86], whereas overexpression can lead to the development of juvenile dilated cardiomyopathy due to myofibril disassembly [85].
Besides their function in contraction, specific myofibrillar and sarcomeric associated proteins act as sensors and transducers of internal strains. For example, cardiac troponin mutations disrupt sarcomere assembly [87] and calcium regulation resulting in arrhythmias, sudden death, and cardiomyopathies [88, 89]. Cardiac troponins' loss-of-function alters the regulatory role of the troponin complex leading to an increase in calcium sensitivity in hypertrophic cardiomyopathy and a decrease in sensitivity in dilated cardiomyopathy [90, 91]. Mutations in cTnI have also been linked to restrictive cardiomyopathy and result in greater calcium sensitivity of cardiac myofilaments [92].
Taken together, these results implicate sarcomere integrity and structural organization in heart function, as compromised sarcomere structure triggers organ-level disease development.
4.2. Bioenergetics and Biogenesis in Diseased Hearts
Heart function depends heavily on ATP generation; therefore impediments or alterations in metabolism inevitably lead to cardiomyocytes apoptosis through autophagy and contractile dysfunction [93–95]. The metabolic state of the heart can be gauged estimating the extent to which the organ relies on oxidative (mature) and glycolytic (fetal) pathways [96–98]. Alterations in energy substrate utilization and energy metabolism, including mitochondrial dysfunction and increased glucose utilization, are characteristic of the hypertrophied and failing myocardium [8, 99]. Following stress, that is, chronic pressure, volume overload, or infarction, the heart gradually loses the ability to generate a sufficient supply of ATP, prompting organ failure [100, 101]. In the early stages of heart failure, fatty acids are the predominant myocardial energy substrate. However, as the disease progresses, there are a concomitant increase in glycolysis and glucose oxidation, a decrease in fatty acid oxidation, and a reduction of respiratory chain activity which resembles the fetal metabolic profile [98, 102, 103]. Under pressure overload hypertrophy, the fetal metabolic profile is reactivated with an increase in glycolysis and a decrease in fatty acid oxidation in response to increased workload [104, 105]. Interestingly, the notion that these pathological changes were exclusively associated with lack of ATP was challenged by a study on mice completely lacking creatine—and therefore phosphocreatine—a key indicator of energy depletion in the heart [106]. While the role of creatine kinase pathways remains essential for myocardial homeostasis and disease [107], the creatine-deficient animals responded reasonably well to myocardial infarction and pressure overload suggesting that energy depletion may not be the dominant mechanism in heart failure. Instead, and in agreement with our own observations in vitro, accumulation of ROS [108] and other intermediate metabolites [109] may be responsible for pathological responses to hemodynamic overload and the onset of heart failure.
Abnormalities in cardiac energy metabolism have also been associated with the loss of mitochondrial structural integrity [110, 111], maintained through mitochondrial biogenesis [112]. For example, knocking out Mfn1 and Mfn2 in mice caused lethal heart failure, as it abolished mitochondrial tethering and fusion of the outer membrane, leading to fragmented, smaller, and poorly organized mitochondria [113]. Furthermore, mitochondria exhibited reduced size and a decline in structural integrity in the failing human myocardium [105]. Similarly, round mitochondria that lacked organization within the cytoskeleton were observed in dilated cardiomyopathy and hibernating myocardium [80, 114]. Pigs with hypertrophic cardiomyopathy were shown to have swollen cardiac mitochondria with disrupted cristae and disoriented myofibrils resulting from the internal forces exerted by mitochondrial growth [111]. Finally, mitochondria of failing hearts in rats are characterized by sparse and disorganized cristae [115]. These alterations in mitochondria morphology suggest interruptions in mitochondrial fusion and fission and suggest that mitochondria biogenesis is essential for structural organization.
Together, these studies suggest that maintenance of the correct mitochondrial structure and function is essential to cardiac homeostasis.
4.3. Intracellular Energetic Units in Diseased Hearts
Impairment of the structural interactions between components of the intracellular energetic unit also leads to cardiomyopathies. For example, muscle LIM protein (MLP), a mechanosensor in the Z-disc of myofilaments, has been shown to interact directly with metabolic enzymes [116]. In mice with MLP deficiency, mitochondria density around the myofilaments was reduced [117, 118]. Similarly, intracellular energetic unit disruption also led to impaired mitochondrial ATP channeling to the sarcoplasmic reticulum, due to the increased distance between mitochondria and the sarco/endoplasmic reticulum calcium-ATPase (SERCA) pumps [94, 118]. This suggests that cytoskeletal MLP may play a role in the sensing mechanisms that link mitochondria and the cell cytoskeleton [119, 120]. Similar studies with desmin-null mice hearts revealed that cytoskeletal disorganization led to looser packing of mitochondria within the myofibrils and sarcoplasmic reticulum [119] and impaired ATP transfer and myocardial dysfunction [10, 118]. Finally, cardiomyopathies caused by mutations in desmin and other intermediate filament related proteins are characterized by myofibril disruption [24, 94, 110, 121, 122].
Additionally, disruption of normal sarcomere function directly impairs metabolic function, an initial indication of disease. In a transgenic rat model overexpressing a mutated cTnT [123], rats exhibited normal contractile function and no hypertrophy compared to controls. After pretreatment with glucose-deprived media, transgenic rats showed decreased metabolic reserve resulting in reduced cardiac fractional shortening, contraction, and relaxation velocity. Similarly, in a group of patients diagnosed with familial hypertrophic cardiomyopathy due to mutations in β-myosin heavy chain (β-MHC), cTnT, or myosin-binding protein C (MyBPC) [124], altered cardiac metabolism was observed in both symptomatic and asymptomatic patients. The finding that altered cardiac metabolism occurred in patients with normal echocardiographs suggests that altered metabolism may be a cause, not an effect, of cardiac hypertrophy [125]. Additionally, disruption of normal mitochondrial function can also drive changes in cardiac structure and function, resulting in significant disease. For example, homozygous mutation to Med30zg, a mediator of gene expression, led to decreased transcription of genes requisite for oxidative phosphorylation and subsequently caused lethal cardiomyopathy in a mouse model [126]. Histological characterization of the myocardium indicated fibrosis, necrosis of cardiomyocytes, and dilation of both ventricles. Further, ultrastructural analysis showed loss of Z-disk organization demonstrating a critical role for metabolic regulation of cardiac structure over a range of spatial magnitudes. In a mouse model of Friedreich's ataxia [127], a mutation of the mitochondrial protein frataxin involved in oxidative phosphorylation resulted in the progressive development of hypertrophic cardiomyopathy.
Even when the structural integrity of the ICEU and its components are preserved, changes in calcium utilization patterns may lead to poorly functional microdomains. In fact, the sarcoplasmic reticulum is physically tethered to mitochondria through a variety of mechanisms [9], including Mfn-1 and Mfn-2 [128]. And this tethering may play a role in disease onset and progression; for example, Mfn-1/2 are downregulated in heart failure impairing mitochondrial fusion and SR function, which in turn lead to decreased SR calcium load and, over time, to decreased calcium release. Mitochondrial calcium uptake will also be affected at this point, as regeneration of pyridine nucleotides is impaired resulting in reduced antioxidative capacity [129] and increased oxidative stress-mediated myocardial remodeling [108, 130].
As a consequence of the critical coupling between sarcomere and mitochondria, new therapeutic strategies have emerged that target various aspects of metabolism in order to restore the mechanical function of the heart [131]. In a study of patients with nonobstructive hypertrophic cardiomyopathy [132], pharmacological treatment with perhexiline decreased fatty acid oxidation and increased glucose utilization as an energy substrate [131]. Patients experienced improved exercise capacity and overall decreased symptoms compared to placebo controls, suggesting that changes in metabolic substrate utilization may play an important role in disease progression. At the same time a number of clinical trials aimed at repurposing drugs that were approved for the treatment of metabolic disorders in other organs were not successful [133]. For example, Acadesine [134], a nonspecific agent targeting AMP-dependent protein kinase, L-arginine [135], an alternative energy substrate utilized in the treatment of other mitochondrial myopathies, and adenosine receptor agonists [136, 137] did not improve the conditions of myocardial infarction patients.
Together these studies demonstrate that impairment of the interaction between contractile and metabolic components in the intracellular energetic unit triggers disease development through both ATP dependent and ATP-independent mechanisms.
4.4. Evidence for a ROS-Mediated Disease Mechanism
In addition to ATP exchange, sarcomeres and mitochondria interact through calcium ions and reactive oxygen and nitrogen species (ROS for convenience). A number of reviews are available describing how mitochondria sequester calcium ions from the cytosol, directly competing with cardiac contractility, calcium handling, and biochemical signaling [8, 138–140]. Similarly, ROS are a byproduct of mitochondrial metabolism [141, 142] that can alter cardiac function [143, 144] and activate the cell apoptotic program [95]. Additionally, impairment of the ROS buffering system causes oxidative damage to ionic channels in the plasma membrane [145], sarcoplasmic reticulum [143, 146, 147], and alterations in signaling cascades involving calcium/calmodulin-dependent protein kinase II (CaMKII) [148], protein kinase-C [149], Rho-associated protein kinase (ROCK) [150] and nicotinamide adenine dinucleotide phosphate (NADPH) oxidase 2 (NOX2) [151]. Importantly, recent studies further point towards a direct, beat-to-beat dependence of mechanotransduction, metabolism, and contractility: as myocyte stretching [152] and increased afterload [153] induced NOX and NOS dependent ROS signaling that directly affected calcium release from the SR and contractility. Interestingly, computational models predict that these effects may be proarrhythmic causing action potential shortening [154] and altering automaticity [155]. Here we focus on posttranslational modifications of cytoskeletal proteins that can impact the formation and maintenance of competent intracellular energetic units [142, 156].
ROS damage to cytoskeletal proteins involved in cardiomyocytes contractility directly impacts cell and organ function [157]. For example, elevation of ROS depresses contractile function due to (i) reduced tropomodulin-actin binding in the thin filament and (ii) increased F-actin depolymerization leading to reduced cross-bridge sliding velocity [158, 159]. Furthermore, α-actinin polymerization kinetics is also depressed in the presence of elevated ROS levels, which destabilized the Z-disks and compromised longitudinal force transmission [156]. Moreover, excess ROS enhance the activity of matrix metalloproteinase-2 (MMP-2), which has been directly linked to increased degradation of myosin light chain isoforms [160, 161].
Additionally, damage to the proteins that maintain the structural integrity of the intracellular energetic unit may have pathological relevance. For example, titin oxidation decreases the extensibility of the N2B domain [162], which has been linked to diastolic dysfunction [163]. Similarly, ROS-mediated damage to the mitochondria-microtubules connection has been observed after ischemic damage [164] and in volume overload [165]. A tethering of mitochondria to the SR has been postulated [166], and ROS-mediated damage to the microtubule network has been associated with calcium sparks abrogation [167] and decreased velocity of calcium wave propagation [168].
Taken together, these results suggest that the inability of cardiomyocytes to buffer excessive ROS production may result in extensive damage to the architecture and therefore to the functional proficiency of the intracellular energetic units. This is consistent with our Barth syndrome-on-a-chip study, in which we observed that treating the cells with a ROS scavenger resulted in complete rescue of the structural and functional disease phenotype [21]. Furthermore, these findings are consistent with the observation that closure of the mitochondrial transition pore facilitates maturations of the contractile cytoskeleton in embryonic cardiomyocytes by lowering the cytoplasmic concentration of ROS [54].
5. Human Diseases-on-Chips and the Intracellular Energetic Units
If intracellular energetic units control cardiac homeostasis and disease, “How do these microdomains build themselves? How do they adapt to changes in the cell microenvironment?” Unfortunately, these questions are difficult to answer utilizing traditional animal models for a number of reasons: (i) humans and animals preferentially express distinct isoforms of proteins involved in the structure and function of intracellular energy units, such as α- and β-myosin heavy chains; (ii) the interaction between organelles and ultrastructural units within functional microdomains occur at spatial scales that are difficult to study with animal model; and (iii) altering the microenvironment of cardiomyocytes in vivo causes a systemic organ-level response that confounds the analysis. Here we review how patient-specific human induced pluripotent stem cells (hiPSCs) and hiPSC-derived cardiomyocytes may be utilized to study the molecular mechanisms responsible for the development and maintenance of intracellular energetic unit in humans. Moreover, engineered tissue-culture platforms, so called hearts-on-chips, can be adopted to recapitulate healthy and diseased microenvironmental conditions in vitro and measure how these variables affect structure and function of human induced pluripotent stem cell derived cardiomyocytes.
5.1. Human Induced Pluripotent Stem Cell Derived Cardiomyocytes
Technologies for reprograming somatic cells into pluripotent stem cells [169] and directing their differentiation towards the cardiomyogenic phenotype [170] enabled the creation of in vitro replicas of various human diseases including channelopathies, vasculopathies, and cardiomyopathies [171, 172]. For example, hiPS-derived cardiomyocytes carrying mutations to troponin T (TNTT2) and β-myosin heavy chain (MyH7) recapitulated in vitro dilated and hypertrophic cardiomyopathy, respectively [173, 174]. Interestingly, the intracellular energetic units were severely underdeveloped in these hiPSC-derived cardiomyocytes that displayed reduced myofibrillogenesis and immature, sparse mitochondria. Moreover, hiPSC-derived cardiomyocyte carrying mutations to plakoglobin [175] and plakophilin-2 [176–178] recapitulated in vitro arrhythmogenic right-ventricular dysplasia/cardiomyopathy (ARVD/C). This is a rare inherited disease characterized by pathological fat infiltration, fibrosis, and cardiomyocyte loss predominantly in the right ventricle resulting in right-ventricular dysfunction and ventricular arrhythmias [179]. The mutated plakophilin-2 coactivated both normal PPARα-dependent metabolism and abnormal PPARγ pathway ultimately resulting in exaggerated lipogenesis, apoptosis, and impaired calcium handling. Furthermore, a different set of mutations to plakophilin-2 were associated with a Brugada syndrome phenotype both in patients and in patient-specific hiPSC-derived cardiomyocytes [180]. Interestingly, at the ultrastructural level, these symptoms were associated with alterations to various components of the intracellular energetic unit, including intercalated disk and microtubules. In addition, hiPSC-derived cardiomyocytes carrying mutations to ryanodine receptors (RyR2) calcium/calmodulin-dependent protein kinase II (CaMKII) and calsequestrin (CLSQ) displayed calcium oversensitization, the hallmark of catecholaminergic polymorphic ventricular tachycardia (CPVT) [181–183]. Specifically, abnormalities in whole-cell calcium transients were observed, including proarrhythmic early/after depolarization events [181]. Interestingly, more abundant and longer local spontaneous releases of calcium were observed at spatial scale comparable to the intracellular energetic units [128, 183]. Computational studies suggest that this extra calcium may have functional implications for sarcomere and mitochondrial function and thus implicate the dysregulation of the intracellular energetic units in disease development [184].
Furthermore, mutations to either chromosomal or mitochondrial genes involved in cardiomyocyte metabolism also result in cardiomyopathies. For example, hiPSC-derived cardiomyocytes have been obtained for patients affected by Friedreich ataxia, a neurodegenerative disorder associated with hypertrophic cardiomyopathy [185]. In this model, deregulation of iron homeostasis leads to disorganized mitochondrial network and mitochondrial DNA depletion, which in turn results in energy deficiency that blunt the calcium cycling and contractile properties of cardiomyocytes. Similarly, in a recently proposed in vitro model for Pompe disease, hiPSC-derived cardiomyocytes exhibited reduced metabolic capacity associated with aberrant mitochondria morphology [186]. Together these studies suggest that hiPSC-derived cardiomyocytes can also be utilized to model dysregulation of the ATP local homeostasis in the intracellular energetic units responsible for the onset and development of various cardiomyopathies.
We previously argued that elevated ROS levels impair the mechanosensitive processes responsible for the genesis and maintenance of the intracellular energetic units. A number of hiPSC-based disease models appear to support this notion. For example, hiPSC-derived cardiomyocytes recapitulating genetically or environmentally caused diabetic cardiomyopathy exhibited deranged intracellular energetic units, with immature sarcomerogenesis and impaired mitochondrial biogenesis [187]. In this study, similarly to what we observed with hiPSC-based model of Barth syndrome [21], ROS-generation, and not a metabolic insufficiency, triggered the disease. Moreover, hiPSC lines were derived from patients exhibiting ROS-mediated damage to proteins of the intracellular energetic unit. For example, hiPSC-derived neurons obtained from patients affected by Parkinson's [188] and Huntington's [189] diseases exhibited deregulation of the peroxisome proliferator-activated receptor c coactivator 1 (PGC-1α) and dynamin-related protein 1 (DRP1) pathways, involved in mitochondrial fusion and fission, respectively. Similarly, mutations to parkin [190] and plakophilin-2 [180] have been shown to destabilize the microtubule network in hiPSC-derived neurons and cardiomyocytes, respectively.
Taken together this body of evidence suggests that patient-specific hiPSC lines that carry mutations to proteins of the intracellular energetic units, as well as genome editing techniques to engineer isogenetic hiPSC lines carrying arbitrary mutations [191], are promising tools to study the regulation of intracellular energetic units in a human genetic background.
5.2. Cardiac Tissue Engineering and Heart-on-a-Chip Platforms
The pathophysiological state of the heart results from the interaction between the genetic instructions cardiomyocytes carry from birth and the constantly evolving microenvironment in which they operate. While access to hiPSC lines that carry a variety of genetic mutations is a fundamental step in designing in vitro models to understand disease mechanisms mediated by intracellular energetic units, the physicochemical characteristics of healthy and diseased cell microenvironments must also be captured in vitro.
While we have not engineered a complete biomimicry of the native architecture of the myocardium that incorporates the transmurally changing alignment of the myofibrils [192], a number of two- and three-dimensional in vitro analogs have been developed. These platforms can be distinguished for the degree of control they offer over the cell microenvironment. For example, 3D collagen sponges [193] and biowires [194] of variable stiffness were utilized to improve the maturation of primary and stem cell derived cardiomyocytes. Alternatively, cardiac patches coated with various extracellular matrix proteins have been shown to increase the maturation of pluripotent stem cell derived cardiomyocytes and to permit contractility studies via traditional force-transducers [195]. One extremely sophisticated extension of these efforts was recently published that incorporates an engineered vascular network and can be surgically implanted into the native myocardium [196]. Among nonvascularized 2D and thin-3D platforms, microstructured chambers coated with extracellular matrix proteins, and dog-bone shaped thin gels promoted the alignment of hiPSC-derived cardiomyocytes. In these studies, cells were genetically engineered to express a calcium-sensitive fluorescent protein, enabling the assessment of contractility and calcium cycling parameters in vitro [197, 198]. Additionally, micropillars-based platforms were manufactured in a range of pathophysiological stiffness to study single cardiomyocytes [199] and tissues [200]. Finally, cardiomyocytes were also cultured on densely packed arrays of microposts, coated with various extracellular matrix proteins [201, 202].
In our laboratory, we developed a heart-on-a-chip platform based on the muscular thin film technology [203]. In open-well or microfluidic chip, cantilevers were precut from a thin layer of synthetic material and microcontact printed [27] or micromolded [20, 204] to support a two-dimensional laminar sheet of cardiomyocytes. These hearts-on-chips were utilized to model Barth syndrome [21] and heart failure [19] as well as to assess the proficiency of stem cell derived cardiomyocytes [14, 26]. Furthermore, we optimized a traction force microscopy approach where cardiomyocytes are cultured on deformable hydrogels embedding fluorescent nanospheres that, similarly to the posts, can be tracked optically during cell contraction. Specifically, we employed a range of fabrication and surface-chemistry techniques to independently control substrate stiffness, extracellular matrix composition, and topography. We utilized this technique to suggest that remodeling of cardiomyocyte shape in hypertrophic cardiomyopathies may be a compensatory effect aimed at maintaining optimal contractile output [17]; that is, the establishment of cell-matrix adhesion at the cell-cell interface is a characteristic of immature, diseased, or regenerating cardiac tissues [18, 205].
Together, these efforts suggest that advanced platforms for cardiac cell and tissue phenotyping can be utilized to study intracellular energetic units. One challenge will be the integration of superresolution microscopy techniques [206, 207] that can resolve many different proteins and organelles [208] (actin [209], tubulin [210], and mitochondria [211]) in intracellular energetic units whose spatial extent is below the resolution limit of traditional microscopy.
6. Conclusions
In conclusion, we believe that the interactions between sarcomeres and mitochondria not only affect cardiomyocyte excitation-contraction coupling directly, but also participate in mechanotransduction, through the cytoskeletal proteins involved in the development and maintenance of intracellular energetic units. Alterations in the structure and function of these microdomains, such as those generated by oxidative posttranslational modifications, represent a disease mechanism that can now be better investigated integrating recent advances in stem cell biology and tissue engineering.
Acknowledgments
The authors wish to thank Cristina Nardelli and Karaghen Hudson for their help with the illustrations. This work was funded by NCATS Grant UH3 TR000522 “Human Cardio-Pulmonary System on a Chip” and NHLBI Grant U01 HL100408 “Human Pluripotent Stem Cells and Progenitor Models of Cardiac and Blood Diseases.”
Competing Interests
The authors declare that there is no conflict of interests regarding the publication of this paper.
References
- 1.Buckberg G., Hoffman J. I. E., Mahajan A., Saleh S., Coghlan C. Cardiac mechanics revisited: the relationship of cardiac architecture to ventricular function. Circulation. 2008;118(24):2571–2587. doi: 10.1161/circulationaha.107.754424. [DOI] [PubMed] [Google Scholar]
- 2.Kontrogianni-Konstantopoulos A., Ackermann M. A., Bowman A. L., Yap S. V., Bloch R. J. Muscle giants: molecular scaffolds in sarcomerogenesis. Physiological Reviews. 2009;89(4):1217–1267. doi: 10.1152/physrev.00017.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ehler E., Perriard J.-C. Cardiomyocyte cytoskeleton and myofibrillogenesis in healthy and diseased heart. Heart Failure Reviews. 2000;5(3):259–269. doi: 10.1023/A:1009861504264. [DOI] [PubMed] [Google Scholar]
- 4.Lindsey S. E., Butcher J. T., Yalcin H. C. Mechanical regulation of cardiac development. Frontiers in Physiology. 2014;5, article no. 318 doi: 10.3389/fphys.2014.00318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Parker K. K., Ingber D. E. Extracellular matrix, mechanotransduction and structural hierarchies in heart tissue engineering. Philosophical Transactions of the Royal Society of London. Series B, Biological Sciences. 2007;362(1484):1267–1279. doi: 10.1098/rstb.2007.2114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dorn G. W., II Mitochondrial dynamics in heart disease. Biochimica et Biophysica Acta—Molecular Cell Research. 2013;1833(1):233–241. doi: 10.1016/j.bbamcr.2012.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Razeghi P., Young M. E., Alcorn J. L., Moravec C. S., Frazier O. H., Taegtmeyer H. Metabolic gene expression in fetal and failing human heart. Circulation. 2001;104(24):2923–2931. doi: 10.1161/hc4901.100526. [DOI] [PubMed] [Google Scholar]
- 8.Stanley W. C., Recchia F. A., Lopaschuk G. D. Myocardial substrate metabolism in the normal and failing heart. Physiological Reviews. 2005;85(3):1093–1129. doi: 10.1152/physrev.00006.2004. [DOI] [PubMed] [Google Scholar]
- 9.Kohlhaas M., Maack C. Calcium release microdomains and mitochondria. Cardiovascular Research. 2013;98(2):259–268. doi: 10.1093/cvr/cvt032. [DOI] [PubMed] [Google Scholar]
- 10.Anmann T., Varikmaa M., Timohhina N., et al. Formation of highly organized intracellular structure and energy metabolism in cardiac muscle cells during postnatal development of rat heart. Biochimica et Biophysica Acta—Bioenergetics. 2014;1837(8):1350–1361. doi: 10.1016/j.bbabio.2014.03.015. [DOI] [PubMed] [Google Scholar]
- 11.Chien K. R., Domian I. J., Parker K. K. Cardiogenesis and the complex biology of regenerative cardiovascular medicine. Science. 2008;322(5907):1494–1497. doi: 10.1126/science.1163267. [DOI] [PubMed] [Google Scholar]
- 12.Dabiri G. A., Turnacioglu K. K., Sanger J. M., Sanger J. W. Myofibrillogenesis visualized in living embryonic cardiomyocytes. Proceedings of the National Academy of Sciences of the United States of America. 1997;94(17):9493–9498. doi: 10.1073/pnas.94.17.9493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Grosberg A., Kuo P., Guo C., et al. Self-organization of muscle cell structure and function. PLoS Computational Biology. 2011;7(2) doi: 10.1371/journal.pcbi.1001088.e1001088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pasqualini F. S., Sheehy S. P., Agarwal A., Aratyn-Schaus Y., Parker K. K. Structural phenotyping of stem cell-derived cardiomyocytes. Stem Cell Reports. 2015;4(3):340–347. doi: 10.1016/j.stemcr.2015.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Parker K. K., Tan J., Chen C. S., Tung L. Myofibrillar architecture in engineered cardiac myocytes. Circulation Research. 2008;103(4):340–342. doi: 10.1161/CIRCRESAHA.108.182469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dasbiswas K., Majkut S., Discher D. E., Safran S. A. Substrate stiffness-modulated registry phase correlations in cardiomyocytes map structural order to coherent beating. Nature Communications. 2015;6, article 6085 doi: 10.1038/ncomms7085. [DOI] [PubMed] [Google Scholar]
- 17.McCain M. L., Yuan H., Pasqualini F. S., Campbell P. H., Parker K. K. Matrix elasticity regulates the optimal cardiac myocyte shape for contractility. American Journal of Physiology—Heart and Circulatory Physiology. 2014;306(11):H1525–H1539. doi: 10.1152/ajpheart.00799.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McCain M. L., Lee H., Aratyn-Schaus Y., Kléber A. G., Parker K. K. Cooperative coupling of cell-matrix and cell-cell adhesions in cardiac muscle. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(25):9881–9886. doi: 10.1073/pnas.1203007109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McCain M. L., Sheehy S. P., Grosberg A., Goss J. A., Parker K. K. Recapitulating maladaptive, multiscale remodeling of failing myocardium on a chip. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(24):9770–9775. doi: 10.1073/pnas.1304913110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McCain M. L., Agarwal A., Nesmith H. W., Nesmith A. P., Parker K. K. Micromolded gelatin hydrogels for extended culture of engineered cardiac tissues. Biomaterials. 2014;35(21):5462–5471. doi: 10.1016/j.biomaterials.2014.03.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang G., McCain M. L., Yang L., et al. Modeling the mitochondrial cardiomyopathy of Barth syndrome with induced pluripotent stem cell and heart-on-chip technologies. Nature Medicine. 2014;20(6):616–623. doi: 10.1038/nm.3545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McCain M. L., Parker K. K. Mechanotransduction: the role of mechanical stress, myocyte shape, and cytoskeletal architecture on cardiac function. Pflugers Archiv. 2011;462(1):89–104. doi: 10.1007/s00424-011-0951-4. [DOI] [PubMed] [Google Scholar]
- 23.Horton R. E., Yadid M., McCain M. L., et al. Angiotensin II induced cardiac dysfunction on a chip. PLoS One. 2016;11(1) doi: 10.1371/journal.pone.0146415.e0146415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gerdes A. M. Cardiac myocyte remodeling in hypertrophy and progression to failure. Journal of Cardiac Failure. 2002;8(6):S264–S268. doi: 10.1054/jcaf.2002.129280. [DOI] [PubMed] [Google Scholar]
- 25.Yang X., Pabon L., Murry C. E. Engineering adolescence: maturation of human pluripotent stem cell-derived cardiomyocytes. Circulation Research. 2014;114(3):511–523. doi: 10.1161/circresaha.114.300558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sheehy S. P., Pasqualini F., Grosberg A., Park S. J., Aratyn-Schaus Y., Parker K. K. Quality metrics for stem cell-derived cardiac myocytes. Stem Cell Reports. 2014;2(3):282–294. doi: 10.1016/j.stemcr.2014.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Agarwal A., Goss J. A., Cho A., McCain M. L., Parker K. K. Microfluidic heart on a chip for higher throughput pharmacological studies. Lab on a Chip. 2013;13(18):3599–3608. doi: 10.1039/c3lc50350j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schlame M., Ren M. Barth syndrome, a human disorder of cardiolipin metabolism. FEBS Letters. 2006;580(23):5450–5455. doi: 10.1016/j.febslet.2006.07.022. [DOI] [PubMed] [Google Scholar]
- 29.Zangi L., Lui K. O., Von Gise A., et al. Modified mRNA directs the fate of heart progenitor cells and induces vascular regeneration after myocardial infarction. Nature Biotechnology. 2013;31(10):898–907. doi: 10.1038/nbt.2682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Van Der Velden J., De Jong J. W., Owen V. J., Burton P. B. J., Stienen G. J. M. Effect of protein kinase a on calcium sensitivity of force and its sarcomere length dependence in human cardiomyocytes. Cardiovascular Research. 2000;46(3):487–495. doi: 10.1016/s0008-6363(00)00050-x. [DOI] [PubMed] [Google Scholar]
- 31.Allen D. G., Kentish J. C. The cellular basis of the length-tension relation in cardiac muscle. Journal of Molecular and Cellular Cardiology. 1985;17(9):821–840. doi: 10.1016/S0022-2828(85)80097-3. [DOI] [PubMed] [Google Scholar]
- 32.Hofmann P. A., Fuchs F. Bound calcium and force development in skinned cardiac muscle bundles: effect of sarcomere length. Journal of Molecular and Cellular Cardiology. 1988;20(8):667–677. doi: 10.1016/s0022-2828(88)80012-9. [DOI] [PubMed] [Google Scholar]
- 33.Du A., Sanger J. M., Sanger J. W. Cardiac myofibrillogenesis inside intact embryonic hearts. Developmental Biology. 2008;318(2):236–246. doi: 10.1016/j.ydbio.2008.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gregorio C. C., Antin P. B. To the heart of myofibril assembly. Trends in Cell Biology. 2000;10(9):355–362. doi: 10.1016/S0962-8924(00)01793-1. [DOI] [PubMed] [Google Scholar]
- 35.Huang W., Zhang R., Xu X. Myofibrillogenesis in the developing zebrafish heart: a functional study of tnnt2. Developmental Biology. 2009;331(2):237–249. doi: 10.1016/j.ydbio.2009.04.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rhee D., Sanger J. M., Sanger J. W. The premyofibril: evidence for its role in myofibrillogenesis. Cell Motility and the Cytoskeleton. 1994;28(1):1–24. doi: 10.1002/cm.970280102. [DOI] [PubMed] [Google Scholar]
- 37.Stout A. L., Wang J., Sanger J. M., Sanger J. W. Tracking changes in Z-band organization during myofibrillogenesis with FRET imaging. Cell Motility and the Cytoskeleton. 2008;65(5):353–367. doi: 10.1002/cm.20265. [DOI] [PubMed] [Google Scholar]
- 38.Ehler E., Horowits R., Zuppinger C., et al. Alterations at the intercalated disk associated with the absence of muscle LIM protein. Journal of Cell Biology. 2001;153(4):763–772. doi: 10.1083/jcb.153.4.763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Danowski B. A., Imanaka-Yoshida K., Sanger J. M., Sanger J. W. Costameres are sites of force transmission to the substratum in adult rat cardiomyocytes. The Journal of Cell Biology. 1992;118(6):1411–1420. doi: 10.1083/jcb.118.6.1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chan D. C. Fusion and fission: interlinked processes critical for mitochondrial health. Annual Review of Genetics. 2012;46(1):265–287. doi: 10.1146/annurev-genet-110410-132529. [DOI] [PubMed] [Google Scholar]
- 41.Ryan J. J., Marsboom G., Fang Y.-H., et al. PGC1α-mediated mitofusin-2 deficiency in female rats and humans with pulmonary arterial hypertension. American Journal of Respiratory and Critical Care Medicine. 2013;187(8):865–878. doi: 10.1164/rccm.201209-1687OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lackner L. L. Shaping the dynamic mitochondrial network. BMC Biology. 2014;12, article 35 doi: 10.1186/1741-7007-12-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bleazard W., McCaffery J. M., King E. J., et al. The dynamin-related GTPase Dnm1 regulates mitochondrial fission in yeast. Nature Cell Biology. 1999;1(5):298–304. doi: 10.1038/13014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ingerman E., Perkins E. M., Marino M., et al. Dnm1 forms spirals that are structurally tailored to fit mitochondria. Journal of Cell Biology. 2005;170(7):1021–1027. doi: 10.1083/jcb.200506078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mears J. A., Lackner L. L., Fang S., Ingerman E., Nunnari J., Hinshaw J. E. Conformational changes in Dnm1 support a contractile mechanism for mitochondrial fission. Nature Structural and Molecular Biology. 2011;18(1):20–26. doi: 10.1038/nsmb.1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Meeusen S., McCaffery J. M., Nunnari J. Mitochondrial fusion intermediates revealed in vitro. Science. 2004;305(5691):1747–1752. doi: 10.1126/science.1100612. [DOI] [PubMed] [Google Scholar]
- 47.Chen H., Detmer S. A., Ewald A. J., Griffin E. E., Fraser S. E., Chan D. C. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. Journal of Cell Biology. 2003;160(2):189–200. doi: 10.1083/jcb.200211046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cipolat S., De Brito O. M., Dal Zilio B., Scorrano L. OPA1 requires mitofusin 1 to promote mitochondrial fusion. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(45):15927–15932. doi: 10.1073/pnas.0407043101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Meeusen S., DeVay R., Block J., et al. Mitochondrial inner-membrane fusion and crista maintenance requires the dynamin-related GTPase Mgm1. Cell. 2006;127(2):383–395. doi: 10.1016/j.cell.2006.09.021. [DOI] [PubMed] [Google Scholar]
- 50.Nunnari J., Suomalainen A. Mitochondria: in sickness and in health. Cell. 2012;148(6):1145–1159. doi: 10.1016/j.cell.2012.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cho Y. M., Kwon S., Pak Y. K., et al. Dynamic changes in mitochondrial biogenesis and antioxidant enzymes during the spontaneous differentiation of human embryonic stem cells. Biochemical and Biophysical Research Communications. 2006;348(4):1472–1478. doi: 10.1016/j.bbrc.2006.08.020. [DOI] [PubMed] [Google Scholar]
- 52.Rimbaud S., Garnier A., Ventura-Clapier R. Mitochondrial biogenesis in cardiac pathophysiology. Pharmacological Reports. 2009;61(1):131–138. doi: 10.1016/S1734-1140(09)70015-5. [DOI] [PubMed] [Google Scholar]
- 53.Chung S., Dzeja P. P., Faustino R. S., Perez-Terzic C., Behfar A., Terzic A. Mitochondrial oxidative metabolism is required for the cardiac differentiation of stem cells. Nature Clinical Practice Cardiovascular Medicine. 2007;4(supplement 4):S60–S67. doi: 10.1038/ncpcardio0766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hom J. R., Quintanilla R. A., Hoffman D. L., et al. The permeability transition pore controls cardiac mitochondrial maturation and myocyte differentiation. Developmental Cell. 2011;21(3):469–478. doi: 10.1016/j.devcel.2011.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Clark K. A., McElhinny A. S., Beckerle M. C., Gregorio C. C. Striated muscle cytoarchitecture: an intricate web of form and function. Annual Review of Cell and Developmental Biology. 2002;18:637–706. doi: 10.1146/annurev.cellbio.18.012502.105840. [DOI] [PubMed] [Google Scholar]
- 56.Vendelin M., Béraud N., Guerrero K., et al. Mitochondrial regular arrangement in muscle cells: a ‘crystal- like’ pattern. American Journal of Physiology—Cell Physiology. 2005;288(3):C757–C767. doi: 10.1152/ajpcell.00281.2004. [DOI] [PubMed] [Google Scholar]
- 57.Appaix F., Kuznetsov A. V., Usson Y., et al. Possible role of cytoskeleton in intracellular arrangement and regulation of mitochondria. Experimental Physiology. 2003;88(1):175–190. doi: 10.1113/eph8802511. [DOI] [PubMed] [Google Scholar]
- 58.Saks V. A., Kaambre T., Sikk P., et al. Intracellular energetic units in red muscle cells. Biochemical Journal. 2001;356(2):643–657. doi: 10.1042/0264-6021:3560643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Auerbach D., Rothen-Ruthishauser B., Bantle S., et al. Molecular mechanisms of myofibril assembly in heart. Cell Structure and Function. 1997;22(1):139–146. doi: 10.1247/csf.22.139. [DOI] [PubMed] [Google Scholar]
- 60.Breckler J. L., Winters R. B. Myosin ATPase activity during avian cardiac and skeletal muscle development. Mechanisms of Ageing and Development. 1988;46(1–3):47–58. doi: 10.1016/0047-6374(88)90113-3. [DOI] [PubMed] [Google Scholar]
- 61.Anmann T., Guzun R., Beraud N., et al. Different kinetics of the regulation of respiration in permeabilized cardiomyocytes and in HL-1 cardiac cells: importance of cell structure/organization for respiration regulation. Biochimica et Biophysica Acta (BBA)—Bioenergetics. 2006;1757(12):1597–1606. doi: 10.1016/j.bbabio.2006.09.008. [DOI] [PubMed] [Google Scholar]
- 62.Elhanany-Tamir H., Yu Y. V., Shnayder M., Jain A., Welte M., Volk T. Organelle positioning in muscles requires cooperation between two KASH proteins and microtubules. Journal of Cell Biology. 2012;198(5):833–846. doi: 10.1083/jcb.201204102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Boldogh I. R., Pon L. A. Interactions of mitochondria with the actin cytoskeleton. Biochimica et Biophysica Acta—Molecular Cell Research. 2006;1763(5-6):450–462. doi: 10.1016/j.bbamcr.2006.02.014. [DOI] [PubMed] [Google Scholar]
- 64.Voeltz G. K., Prinz W. A. Sheets, ribbons and tubules—how organelles get their shape. Nature Reviews Molecular Cell Biology. 2007;8(3):258–264. doi: 10.1038/nrm2119. [DOI] [PubMed] [Google Scholar]
- 65.Tang H. L., Lung H. L., Wu K. C., Le A.-H. P., Tang H. M., Fung M. C. Vimentin supports mitochondrial morphology and organization. Biochemical Journal. 2008;410(1):141–146. doi: 10.1042/bj20071072. [DOI] [PubMed] [Google Scholar]
- 66.Varikmaa M., Bagur R., Kaambre T., et al. Role of mitochondria-cytoskeleton interactions in respiration regulation and mitochondrial organization in striated muscles. Biochimica et Biophysica Acta (BBA)—Bioenergetics. 2014;1837(2):232–245. doi: 10.1016/j.bbabio.2013.10.011. [DOI] [PubMed] [Google Scholar]
- 67.Kumazawa A., Katoh H., Nonaka D., et al. Microtubule disorganization affects the mitochondrial permeability transition pore in cardiac myocytes. Circulation Journal. 2014;78(5):1206–1215. doi: 10.1253/circj.CJ-13-1298. [DOI] [PubMed] [Google Scholar]
- 68.Rostovtseva T. K., Sheldon K. L., Hassanzadeh E., et al. Tubulin binding blocks mitochondrial voltage-dependent anion channel and regulates respiration. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(48):18746–18751. doi: 10.1073/pnas.0806303105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.de Cavanagh E. M., Ferder M., Inserra F., Ferder L. Angiotensin II, mitochondria, cytoskeletal, and extracellular matrix connections: an integrating viewpoint. American Journal of Physiology—Heart and Circulatory Physiology. 2009;296(3):H550–H558. doi: 10.1152/ajpheart.01176.2008. [DOI] [PubMed] [Google Scholar]
- 70.Schlaepfer D. D., Hunter T. Signal transduction from the extracellular matrix—a role for the focal adhesion protein-tyrosine kinase FAK. Cell Structure and Function. 1996;21(5):445–450. doi: 10.1247/csf.21.445. [DOI] [PubMed] [Google Scholar]
- 71.Werner E., Werb Z. Integrins engage mitochondrial function for signal transduction by a mechanism dependent on Rho GTPases. Journal of Cell Biology. 2002;158(2):357–368. doi: 10.1083/jcb.200111028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kaasik A., Kuum M., Joubert F., Wilding J., Ventura-Clapier R., Veksler V. Mitochondria as a source of mechanical signals in cardiomyocytes. Cardiovascular Research. 2010;87(1):83–91. doi: 10.1093/cvr/cvq039. [DOI] [PubMed] [Google Scholar]
- 73.Yaniv Y., Juhaszova M., Wang S., Fishbein K. W., Zorov D. B., Sollott S. J. Analysis of mitochondrial 3D-deformation in cardiomyocytes during active contraction reveals passive structural anisotropy of orthogonal short axes. PLoS ONE. 2011;6(7) doi: 10.1371/journal.pone.0021985.e21985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ramdas N. M., Shivashankar G. V. Cytoskeletal control of nuclear morphology and chromatin organization. Journal of Molecular Biology. 2015;427(3):695–706. doi: 10.1016/j.jmb.2014.09.008. [DOI] [PubMed] [Google Scholar]
- 75.Hamdani N., Kooij V., Van Dijk S., et al. Sarcomeric dysfunction in heart failure. Cardiovascular Research. 2008;77(4):649–658. doi: 10.1093/cvr/cvm079. [DOI] [PubMed] [Google Scholar]
- 76.Lakdawala N. K., Thune J. J., Colan S. D., et al. Subtle abnormalities in contractile function are an early manifestation of sarcomere mutations in dilated cardiomyopathy. Circulation: Cardiovascular Genetics. 2012;5(5):503–510. doi: 10.1161/CIRCGENETICS.112.962761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kamisago M., Sharma S. D., DePalma S. R., et al. Mutations in sarcomere protein genes as a cause of dilated cardiomyopathy. The New England Journal of Medicine. 2000;343(23):1688–1696. doi: 10.1056/nejm200012073432304. [DOI] [PubMed] [Google Scholar]
- 78.Olson T. M., Michels V. V., Thibodeau S. N., Tai Y.-S., Keating M. T. Actin mutations in dilated cardiomyopathy, a heritable form of heart failure. Science. 1998;280(5364):750–752. doi: 10.1126/science.280.5364.750. [DOI] [PubMed] [Google Scholar]
- 79.Watkins H., Conner D., Thierfelder L., et al. Mutations in the cardiac myosin binding protein-C gene on chromosome 11 cause familial hypertrophic cardiomyopathy. Nature Genetics. 1995;11(4):434–437. doi: 10.1038/ng1295-434. [DOI] [PubMed] [Google Scholar]
- 80.Schaper J., Froede R., Hein S., et al. Impairment of the myocardial ultrastructure and changes of the cytoskeleton in dilated cardiomyopathy. Circulation. 1991;83(2):504–514. doi: 10.1161/01.CIR.83.2.504. [DOI] [PubMed] [Google Scholar]
- 81.Hein S., Kostin S., Heling A., Maeno Y., Schaper J. The role of the cytoskeleton in heart failure. Cardiovascular Research. 2000;45(2):273–278. doi: 10.1016/S0008-6363(99)00268-0. [DOI] [PubMed] [Google Scholar]
- 82.Person V., Kostin S., Suzuki K., Labeit S., Schaper J. Antisense oligonucleotide experiments elucidate the essential role of titin in sarcomerogenesis in adult rat cardiomyocytes in long-term culture. Journal of Cell Science. 2000;113(21):3851–3859. doi: 10.1242/jcs.113.21.3851. [DOI] [PubMed] [Google Scholar]
- 83.Seguchi O., Takashima S., Yamazaki S., et al. A cardiac myosin light chain kinase regulates sarcomere assembly in the vertebrate heart. The Journal of Clinical Investigation. 2007;117(10):2812–2824. doi: 10.1172/jci30804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Rottbauer W., Wessels G., Dahme T., et al. Cardiac myosin light chain-2: a novel essential component of thick-myofilament assembly and contractility of the heart. Circulation Research. 2006;99(3):323–331. doi: 10.1161/01.res.0000234807.16034.fe. [DOI] [PubMed] [Google Scholar]
- 85.Sussman M. A., Welch S., Cambon N., et al. Myofibril degeneration caused by tropomodulin overexpression leads to dilated cardiomyopathy in juvenile mice. Journal of Clinical Investigation. 1998;101(1):51–61. doi: 10.1172/JCI1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Sussman M. A., Baqué S., Uhm C.-S., et al. Altered expression of tropomodulin in cardiomyocytes disrupts the sarcomeric structure of myofibrils. Circulation Research. 1998;82(1):94–105. doi: 10.1161/01.RES.82.1.94. [DOI] [PubMed] [Google Scholar]
- 87.Sehnert A. J., Huq A., Weinstein B. M., Walker C., Fishman M., Stainier D. Y. R. Cardiac troponin T is essential in sarcomere assembly and cardiac contractility. Nature Genetics. 2002;31(1):106–110. doi: 10.1038/ng875. [DOI] [PubMed] [Google Scholar]
- 88.Moolman J. C., Corfield V. A., Posen B., et al. Sudden death due to troponin T mutations. Journal of the American College of Cardiology. 1997;29(3):549–555. doi: 10.1016/S0735-1097(96)00530-X. [DOI] [PubMed] [Google Scholar]
- 89.Baudenbacher F., Schober T., Pinto J. R., et al. Myofilament Ca2+ sensitization causes susceptibility to cardiac arrhythmia in mice. The Journal of Clinical Investigation. 2008;118(12):3893–3903. doi: 10.1172/jci36642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Mogensen J., Kubo T., Duque M., et al. Idiopathic restrictive cardiomyopathy is part of the clinical expression of cardiac troponin I mutations. Journal of Clinical Investigation. 2003;111(2):209–216. doi: 10.1172/JCI200316336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.McDonough J. L., Arrell D. K., Van Eyk J. E. Troponin I degradation and covalent complex formation accompanies myocardial ischemia/reperfusion injury. Circulation Research. 1999;84(1):9–20. doi: 10.1161/01.RES.84.1.9. [DOI] [PubMed] [Google Scholar]
- 92.Yumoto F., Lu Q.-W., Morimoto S., et al. Drastic Ca2+ sensitization of myofilament associated with a small structural change in troponin I in inherited restrictive cardiomyopathy. Biochemical and Biophysical Research Communications. 2005;338(3):1519–1526. doi: 10.1016/j.bbrc.2005.10.116. [DOI] [PubMed] [Google Scholar]
- 93.Zaglia T., Milan G., Ruhs A., et al. Atrogin-1 deficiency promotes cardiomyopathy and premature death via impaired autophagy. Journal of Clinical Investigation. 2014;124(6):2410–2424. doi: 10.1172/JCI66339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Maloyan A., Sanbe A., Osinska H., et al. Mitochondrial dysfunction and apoptosis underlie the pathogenic process in α-B-crystallin desmin-related cardiomyopathy. Circulation. 2005;112(22):3451–3461. doi: 10.1161/circulationaha.105.572552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wallace D. C. Mitochondrial diseases in man and mouse. Science. 1999;283(5407):1482–1488. doi: 10.1126/science.283.5407.1482. [DOI] [PubMed] [Google Scholar]
- 96.Barger P. M., Kelly D. P. Fatty acid utilization in the hypertrophied and failing heart: molecular regulatory mechanisms. The American Journal of the Medical Sciences. 1999;318(1):36–42. doi: 10.1016/s0002-9629(15)40570-1. [DOI] [PubMed] [Google Scholar]
- 97.Makinde A.-O., Kantor P. F., Lopaschuk G. D. Maturation of fatty acid and carbohydrate metabolism in the newborn heart. Molecular and Cellular Biochemistry. 1998;188(1-2):49–56. doi: 10.1023/A:1006860104840. [DOI] [PubMed] [Google Scholar]
- 98.Sack M. N., Rader T. A., Park S., Bastin J., McCune S. A., Kelly D. P. Fatty acid oxidation enzyme gene expression is downregulated in the failing heart. Circulation. 1996;94(11):2837–2842. doi: 10.1161/01.CIR.94.11.2837. [DOI] [PubMed] [Google Scholar]
- 99.Ventura-Clapier R., Garnier A., Veksler V. Energy metabolism in heart failure. The Journal of Physiology. 2004;555(1):1–13. doi: 10.1113/jphysiol.2003.055095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Van Bilsen M., Smeets P. J. H., Gilde A. J., Van Der Vusse G. J. Metabolic remodelling of the failing heart: the cardiac burn-out syndrome? Cardiovascular Research. 2004;61(2):218–226. doi: 10.1016/j.cardiores.2003.11.014. [DOI] [PubMed] [Google Scholar]
- 101.Ingwall J. S., Weiss R. G. Is the failing heart energy starved? On using chemical energy to support cardiac function. Circulation Research. 2004;95(2):135–145. doi: 10.1161/01.RES.0000137170.41939.d9. [DOI] [PubMed] [Google Scholar]
- 102.Lopaschuk G. D., Ussher J. R., Folmes C. D. L., Jaswal J. S., Stanley W. C. Myocardial fatty acid metabolism in health and disease. Physiological Reviews. 2010;90(1):207–258. doi: 10.1152/physrev.00015.2009. [DOI] [PubMed] [Google Scholar]
- 103.Sanbe A., Tanonaka K., Kobayashi R., Takeo S. Effects of long-term therapy with ACE inhibitors, captopril, enalapril and trandolpril, on myocardial energy metabolism in rats with heart failure following myocardial infarction. Journal of Molecular and Cellular Cardiology. 1995;27(10):2209–2222. doi: 10.1016/S0022-2828(95)91551-6. [DOI] [PubMed] [Google Scholar]
- 104.Depre C., Shipley G. L., Chen W., et al. Unloaded heart in vivo replicates fetal gene expression of cardiac hypertrophy. Nature Medicine. 1998;4(11):1269–1275. doi: 10.1038/3253. [DOI] [PubMed] [Google Scholar]
- 105.Sharov V. G., Todor A. V., Silverman N., Goldstein S., Sabbah H. N. Abnormal mitochondrial respiration in failed human myocardium. Journal of Molecular and Cellular Cardiology. 2000;32(12):2361–2367. doi: 10.1006/jmcc.2000.1266. [DOI] [PubMed] [Google Scholar]
- 106.Lygate C. A., Aksentijevic D., Dawson D., et al. Living without creatine: unchanged exercise capacity and response to chronic myocardial infarction in creatine-deficient mice. Circulation Research. 2013;112(6):945–955. doi: 10.1161/circresaha.112.300725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Saks V., Schlattner U., Tokarska-Schlattner M., et al. Systems level regulation of cardiac energy fluxes via metabolic cycles: role of creatine, phosphotransfer pathways, and AMPK signaling. In: Aon M. A., Saks V., Schlattner U., editors. Systems Biology of Metabolic and Signaling Networks: Energy, Mass and Information Transfer. Vol. 16. Berlin, Germany: Springer; 2014. pp. 261–320. (Springer Series in Biophysics). [DOI] [Google Scholar]
- 108.Nickel A. G., von Hardenberg A., Hohl M., et al. Reversal of mitochondrial transhydrogenase causes oxidative stress in heart failure. Cell Metabolism. 2015;22(3):472–484. doi: 10.1016/j.cmet.2015.07.008. [DOI] [PubMed] [Google Scholar]
- 109.Nickel A., Löffler J., Maack C. Myocardial energetics in heart failure. Basic Research in Cardiology. 2013;108(4, article 358) doi: 10.1007/s00395-013-0358-9. [DOI] [PubMed] [Google Scholar]
- 110.Lichter J. G. Remodeling of the sarcomeric cytoskeleton in cardiac ventricular myocytes during heart failure and after cardiac resynchronization therapy. Journal of molecular and cellular cardiology. 2014;72:186–195. doi: 10.1016/j.yjmcc.2014.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Lin C. S., Sun Y. L., Liu C. Y. Structural and biochemical evidence of mitochondrial depletion in pigs with hypertrophic cardiomyopathy. Research in Veterinary Science. 2003;74(3):219–226. doi: 10.1016/S0034-5288(02)00189-3. [DOI] [PubMed] [Google Scholar]
- 112.Makino A., Suarez J., Gawlowski T., et al. Regulation of mitochondrial morphology and function by O-GlcNAcylation in neonatal cardiac myocytes. American Journal of Physiology—Regulatory Integrative and Comparative Physiology. 2011;300(6):1296–1302. doi: 10.1152/ajpregu.00437.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Chen Y., Liu Y., Dorn G. W., II Mitochondrial fusion is essential for organelle function and cardiac homeostasis. Circulation Research. 2011;109(12):1327–1331. doi: 10.1161/CIRCRESAHA.111.258723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kalra D. K., Zoghbi W. A. Myocardial hibernation in coronary artery disease. Current Atherosclerosis Reports. 2002;4(2):149–155. doi: 10.1007/s11883-002-0039-x. [DOI] [PubMed] [Google Scholar]
- 115.Bugger H., Schwarzer M., Chen D., et al. Proteomic remodelling of mitochondrial oxidative pathways in pressure overload-induced heart failure. Cardiovascular Research. 2010;85(2):376–384. doi: 10.1093/cvr/cvp344. [DOI] [PubMed] [Google Scholar]
- 116.Boateng S. Y., Belin R. J., Geenen D. L., et al. Cardiac dysfunction and heart failure are associated with abnormalities in the subcellular distribution and amounts of oligomeric muscle LIM protein. American Journal of Physiology - Heart and Circulatory Physiology. 2007;292(1):H259–H269. doi: 10.1152/ajpheart.00766.2006. [DOI] [PubMed] [Google Scholar]
- 117.van den Bosch B. J. C., van den Burg C. M. M., Schoonderwoerd K., et al. Regional absence of mitochondria causing energy depletion in the myocardium of muscle LIM protein knockout mice. Cardiovascular Research. 2005;65(2):411–418. doi: 10.1016/j.cardiores.2004.10.025. [DOI] [PubMed] [Google Scholar]
- 118.Wilding J. R., Joubert F., De Araujo C., et al. Altered energy transfer from mitochondria to sarcoplasmic reticulum after cytoarchitectural perturbations in mice hearts. Journal of Physiology. 2006;575(1):191–200. doi: 10.1113/jphysiol.2006.114116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Capetanaki Y. Desmin cytoskeleton: a potential regulator of muscle mitochondrial behavior and function. Trends in Cardiovascular Medicine. 2002;12(8):339–348. doi: 10.1016/s1050-1738(02)00184-6. [DOI] [PubMed] [Google Scholar]
- 120.Heggeness M. H., Simon M., Singer S. J. Association of mitochondria with microtubules in cultured cells. Proceedings of the National Academy of Sciences of the United States of America. 1978;75(8):3863–3866. doi: 10.1073/pnas.75.8.3863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Purevjav E., Varela J., Morgado M., et al. Nebulette mutations are associated with dilated cardiomyopathy and endocardial fibroelastosis. Journal of the American College of Cardiology. 2010;56(18):1493–1502. doi: 10.1016/j.jacc.2010.05.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Jane-Lise S., Corda S., Chassagne C., Rappaport L. The extracellular matrix and the cytoskeleton in heart hypertrophy and failure. Heart Failure Reviews. 2000;5(3):239–250. doi: 10.1023/A:1009857403356. [DOI] [PubMed] [Google Scholar]
- 123.Luedde M., Flögel U., Knorr M., et al. Decreased contractility due to energy deprivation in a transgenic rat model of hypertrophic cardiomyopathy. Journal of Molecular Medicine. 2009;87(4):411–422. doi: 10.1007/s00109-008-0436-x. [DOI] [PubMed] [Google Scholar]
- 124.Crilley J. G., Boehm E. A., Blair E., et al. Hypertrophic cardiomyopathy due to sarcomeric gene mutations is characterized by impaired energy metabolism irrespective of the degree of hypertrophy. Journal of the American College of Cardiology. 2003;41(10):1776–1782. doi: 10.1016/S0735-1097(02)03009-7. [DOI] [PubMed] [Google Scholar]
- 125.Martin O. J., Lai L., Soundarapandian M. M., et al. A role for peroxisome proliferator-activated receptor γ coactivator-1 in the control of mitochondrial dynamics during postnatal cardiac growth. Circulation Research. 2014;114(4):626–636. doi: 10.1161/circresaha.114.302562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Krebs P., Fan W., Chen Y.-H., et al. Lethal mitochondrial cardiomyopathy in a hypomorphic Med30 mouse mutant is ameliorated by ketogenic diet. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(49):19678–19682. doi: 10.1073/pnas.1117835108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Perdomini M., Belbellaa B., Monassier L., et al. Prevention and reversal of severe mitochondrial cardiomyopathy by gene therapy in a mouse model of Friedreich's ataxia. Nature medicine. 2014;20(5):542–547. doi: 10.1038/nm.3510. [DOI] [PubMed] [Google Scholar]
- 128.Chen Y., Csordás G., Jowdy C., et al. Mitofusin 2-containing mitochondrial-reticular microdomains direct rapid cardiomyocyte bioenergetic responses via interorganelle Ca2+ crosstalk. Circulation Research. 2012;111(7):863–875. doi: 10.1161/circresaha.112.266585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Kohlhaas M., Liu T., Knopp A., et al. Elevated cytosolic Na+ increases mitochondrial formation of reactive oxygen species in failing cardiac myocytes. Circulation. 2010;121(14):1606–1613. doi: 10.1161/circulationaha.109.914911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Nickel A., Kohlhaas M., Maack C. Mitochondrial reactive oxygen species production and elimination. Journal of Molecular and Cellular Cardiology. 2014;73:26–33. doi: 10.1016/j.yjmcc.2014.03.011. [DOI] [PubMed] [Google Scholar]
- 131.Ashrafian H., McKenna W. J., Watkins H. Disease pathways and novel therapeutic targets in hypertrophic cardiomyopathy. Circulation Research. 2011;109(1):86–96. doi: 10.1161/CIRCRESAHA.111.242974. [DOI] [PubMed] [Google Scholar]
- 132.Abozguia K., Elliott P., McKenna W., et al. Metabolic modulator perhexiline corrects energy deficiency and improves exercise capacity in symptomatic hypertrophic cardiomyopathy. Circulation. 2010;122(16):1562–1569. doi: 10.1161/CIRCULATIONAHA.109.934059. [DOI] [PubMed] [Google Scholar]
- 133.Walters A. M., Porter G. A., Brookes P. S. Mitochondria as a drug target in ischemic heart disease and cardiomyopathy. Circulation Research. 2012;111(9):1222–1236. doi: 10.1161/CIRCRESAHA.112.265660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Mangano D. T., Miao Y., Tudor I. C., Dietzel C. Post-reperfusion myocardial infarction: long-term survival improvement using adenosine regulation with acadesine. Journal of the American College of Cardiology. 2006;48(1):206–214. doi: 10.1016/j.jacc.2006.04.044. [DOI] [PubMed] [Google Scholar]
- 135.Arakawa K., Kudo T., Ikawa M., et al. Abnormal myocardial energy-production state in mitochondrial cardiomyopathy and acute response to L-arginine infusion. C-11 acetate kinetics revealed by positron emission tomography. Circulation Journal. 2010;74(12):2702–2711. doi: 10.1253/circj.cj-10-0044. [DOI] [PubMed] [Google Scholar]
- 136.Kopecky S. L., Aviles R. J., Bell M. R., et al. A randomized, double-blinded, placebo-controlled, dose-ranging study measuring the effect of an adenosine agonist on infarct size reduction in patients undergoing primary percutaneous transluminal coronary angioplasty: The ADMIRE (AMP579 Delivery for Myocardial Infarction REduction) study. American Heart Journal. 2003;146(1):146–152. doi: 10.1016/s0002-8703(03)00172-8. [DOI] [PubMed] [Google Scholar]
- 137.Ross A. M., Gibbons R. J., Stone G. W., Kloner R. A., Alexander R. W. A randomized, double-blinded, placebo-controlled multicenter trial of adenosine as an adjunct to reperfusion in the treatment of acute myocardial infarction (AMISTAD-II) Journal of the American College of Cardiology. 2005;45(11):1775–1780. doi: 10.1016/j.jacc.2005.02.061. [DOI] [PubMed] [Google Scholar]
- 138.Bers D. M. Calcium cycling and signaling in cardiac myocytes. Annual Review of Physiology. 2008;70:23–49. doi: 10.1146/annurev.physiol.70.113006.100455. [DOI] [PubMed] [Google Scholar]
- 139.Maack C., O'Rourke B. Excitation-contraction coupling and mitochondrial energetics. Basic Research in Cardiology. 2007;102(5):369–392. doi: 10.1007/s00395-007-0666-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Eisner V., Csordás G., Hajnóczky G. Interactions between sarco-endoplasmic reticulum and mitochondria in cardiac and skeletal muscle-pivotal roles in Ca2+ and reactive oxygen species signaling. Journal of Cell Science. 2013;126(14):2965–2978. doi: 10.1242/jcs.093609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Murphy E., Steenbergen C. Preconditioning: the mitochondrial connection. Annual Review of Physiology. 2007;69:51–67. doi: 10.1146/annurev.physiol.69.031905.163645. [DOI] [PubMed] [Google Scholar]
- 142.Steinberg S. F. Oxidative stress and sarcomeric proteins. Circulation Research. 2013;112(2):393–405. doi: 10.1161/CIRCRESAHA.111.300496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Barouch L. A., Harrison R. W., Skaf M. W., et al. Nitric oxide regulates the heart by spatial confinement of nitric oxide synthase isoforms. Nature. 2002;416(6878):337–340. doi: 10.1038/416005a. [DOI] [PubMed] [Google Scholar]
- 144.Saavedra W. F., Paolocci N., John M. E. St., et al. Imbalance between xanthine oxidase and nitric oxide synthase signaling pathways underlies mechanoenergetic uncoupling in the failing heart. Circulation Research. 2002;90(3):297–304. doi: 10.1161/hh0302.104531. [DOI] [PubMed] [Google Scholar]
- 145.Zima A. V., Blatter L. A. Redox regulation of cardiac calcium channels and transporters. Cardiovascular Research. 2006;71(2):310–321. doi: 10.1016/j.cardiores.2006.02.019. [DOI] [PubMed] [Google Scholar]
- 146.Damy T., Ratajczak P., Shah A. M., et al. Increased neuronal nitric oxide synthase-derived NO production in the failing human heart. The Lancet. 2004;363(9418):1365–1367. doi: 10.1016/s0140-6736(04)16048-0. [DOI] [PubMed] [Google Scholar]
- 147.Lancel S., Zhang J., Evangelista A., et al. Nitroxyl activates SERCA in cardiac myocytes via glutathiolation of cysteine 674. Circulation Research. 2009;104(6):720–723. doi: 10.1161/CIRCRESAHA.108.188441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Dries E., Bito V., Lenaerts I., Antoons G., Sipido K. R., Macquaide N. Selective modulation of coupled ryanodine receptors during microdomain activation of calcium/calmodulin-dependent kinase II in the dyadic cleft. Circulation Research. 2013;113(11):1242–1252. doi: 10.1161/CIRCRESAHA.113.301896. [DOI] [PubMed] [Google Scholar]
- 149.Avner B. S., Hinken A. C., Yuan C., Solaro R. J. H2O2 alters rat cardiac sarcomere function and protein phosphorylation through redox signaling. American Journal of Physiology—Heart and Circulatory Physiology. 2010;299(3):H723–H730. doi: 10.1152/ajpheart.00050.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Soliman H., Gador A., Lu Y.-H., Lin G., Bankar G., MacLeod K. M. Diabetes-induced increased oxidative stress in cardiomyocytes is sustained by a positive feedback loop involving Rho kinase and PKCβ 2 . American Journal of Physiology—Heart and Circulatory Physiology. 2012;303(8):H989–H1000. doi: 10.1152/ajpheart.00416.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Münzel T., Gori T., Keaney J. F., Maack C., Daiber A. Pathophysiological role of oxidative stress in systolic and diastolic heart failure and its therapeutic implications. European Heart Journal. 2015;36(38):2555–2564. doi: 10.1093/eurheartj/ehv305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Prosser B. L., Ward C. W., Lederer W. J. X-ROS signaling: rapid mechano-chemo transduction in heart. Science. 2011;333(6048):1440–1445. doi: 10.1126/science.1202768. [DOI] [PubMed] [Google Scholar]
- 153.Jian Z., Han H., Zhang T., et al. Mechanochemotransduction during cardiomyocyte contraction is mediated by localized nitric oxide signaling. Science Signaling. 2014;7(317, article ra27) doi: 10.1126/scisignal.2005046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Zhou L., Cortassa S., Wei A.-C., Aon M. A., Winslow R. L., O'Rourke B. Modeling cardiac action potential shortening driven by oxidative stress-induced mitochondrial oscillations in guinea pig cardiomyocytes. Biophysical Journal. 2009;97(7):1843–1852. doi: 10.1016/j.bpj.2009.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Li Q., Su D., O'Rourke B., Pogwizd S. M., Zhou L. Mitochondria-derived ROS bursts disturb calcium cycling and induce abnormal automaticity in guinea pig cardiomyocyte: a theoretical study. American Journal of Physiology-Heart and Circulatory Physiology. 2015;308(6):H623–H636. doi: 10.1152/ajpheart.00493.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Passarelli C., Di Venere A., Piroddi N., et al. Susceptibility of isolated myofibrils to in vitro glutathionylation: potential relevance to muscle functions. Cytoskeleton. 2010;67(2):81–89. doi: 10.1002/cm.20425. [DOI] [PubMed] [Google Scholar]
- 157.Heineke J., Kempf T., Kraft T., et al. Downregulation of cytoskeletal muscle LIM protein by nitric oxide: impact on cardiac myocyte hypertrophy. Circulation. 2003;107(10):1424–1432. doi: 10.1161/01.cir.0000055319.94801.fc. [DOI] [PubMed] [Google Scholar]
- 158.Canton M., Menazza S., Sheeran F. L., Polverino De Laureto P., Di Lisa F., Pepe S. Oxidation of myofibrillar proteins in human heart failure. Journal of the American College of Cardiology. 2011;57(3):300–309. doi: 10.1016/j.jacc.2010.06.058. [DOI] [PubMed] [Google Scholar]
- 159.Snook J. H., Li J., Helmke B. P., Guilford W. H. Peroxynitrite inhibits myofibrillar protein function in an in vitro assay of motility. Free Radical Biology and Medicine. 2008;44(1):14–23. doi: 10.1016/j.freeradbiomed.2007.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Kanski J., Behring A., Pelling J., Schöneich C. Proteomic identification of 3-nitrotyrosine-containing rat cardiac proteins: effects of biological aging. American Journal of Physiology—Heart and Circulatory Physiology. 2005;288(1):H371–H381. doi: 10.1152/ajpheart.01030.2003. [DOI] [PubMed] [Google Scholar]
- 161.Doroszko A., Polewicz D., Cadete V. J. J., et al. Neonatal asphyxia induces the nitration of cardiac myosin light chain 2 that is associated with cardiac systolic dysfunction. Shock. 2010;34(6):592–600. doi: 10.1097/shk.0b013e3181e14f1d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Grützner A., Garcia-Manyes S., Kötter S., Badilla C. L., Fernandez J. M., Linke W. A. Modulation of titin-based stiffness by disulfide bonding in the cardiac titin N2-B unique sequence. Biophysical Journal. 2009;97(3):825–834. doi: 10.1016/j.bpj.2009.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.LeWinter M. M., Granzier H. Cardiac titin: a multifunctional giant. Circulation. 2010;121(19):2137–2145. doi: 10.1161/circulationaha.109.860171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Devillard L., Vandroux D., Tissier C., et al. Tubulin ligands suggest a microtubule-NADPH oxidase relationship in postischemic cardiomyocytes. European Journal of Pharmacology. 2006;548(1–3):64–73. doi: 10.1016/j.ejphar.2006.08.004. [DOI] [PubMed] [Google Scholar]
- 165.Yancey D. M., Guichard J. L., Ahmed M. I., et al. Cardiomyocyte mitochondrial oxidative stress and cytoskeletal breakdown in the heart with a primary volume overload. American Journal of Physiology—Heart and Circulatory Physiology. 2015;308(6):H651–H663. doi: 10.1152/ajpheart.00638.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Santos C. X. C., Anilkumar N., Zhang M., Brewer A. C., Shah A. M. Redox signaling in cardiac myocytes. Free Radical Biology and Medicine. 2011;50(7):777–793. doi: 10.1016/j.freeradbiomed.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Iribe G., Ward C. W., Camelliti P., et al. Axial stretch of rat single ventricular cardiomyocytes causes an acute and transient increase in Ca2+ spark rate. Circulation Research. 2009;104(6):787–795. doi: 10.1161/circresaha.108.193334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Miura M., Murai N., Hattori T., Nagano T., Stuyvers B. D., Shindoh C. Role of reactive oxygen species and Ca2+ dissociation from the myofilaments in determination of Ca2+ wave propagation in rat cardiac muscle. Journal of Molecular and Cellular Cardiology. 2013;56(1):97–105. doi: 10.1016/j.yjmcc.2012.12.011. [DOI] [PubMed] [Google Scholar]
- 169.Sánchez Alvarado A., Yamanaka S. Rethinking differentiation: stem cells, regeneration, and plasticity. Cell. 2014;157(1):110–119. doi: 10.1016/j.cell.2014.02.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Zhang J., Wilson G. F., Soerens A. G., et al. Functional cardiomyocytes derived from human induced pluripotent stem cells. Circulation Research. 2009;104(4):e30–e41. doi: 10.1161/CIRCRESAHA.108.192237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Moretti A., Laugwitz K.-L., Dorn T., Sinnecker D., Mummery C. Pluripotent stem cell models of human heart disease. Cold Spring Harbor Perspectives in Medicine. 2013;3(11) doi: 10.1101/cshperspect.a014027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Matsa E., Burridge P. W., Wu J. C. Human stem cells for modeling heart disease and for drug discovery. Science Translational Medicine. 2014;6(239) doi: 10.1126/scitranslmed.3008921.239ps6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Sun N., Yazawa M., Liu J., et al. Patient-specific induced pluripotent stem cells as a model for familial dilated cardiomyopathy. Science Translational Medicine. 2012;4(130) doi: 10.1126/scitranslmed.3003552.130ra47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Lan F., Lee A. S., Liang P., et al. Abnormal calcium handling properties underlie familial hypertrophic cardiomyopathy pathology in patient-specific induced pluripotent stem cells. Cell Stem Cell. 2013;12(1):101–113. doi: 10.1016/j.stem.2012.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Asimaki A., Kapoor S., Plovie E., et al. Identification of a new modulator of the intercalated disc in a zebrafish model of arrhythmogenic cardiomyopathy. Science Translational Medicine. 2014;6(240) doi: 10.1126/scitranslmed.3008008.240ra74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Ma D., Wei H., Lu J., et al. Generation of patient-specific induced pluripotent stem cell-derived cardiomyocytes as a cellular model of arrhythmogenic right ventricular cardiomyopathy. European Heart Journal. 2013;34(15):1122–1133. doi: 10.1093/eurheartj/ehs226. [DOI] [PubMed] [Google Scholar]
- 177.Caspi O., Huber I., Gepstein A., et al. Modeling of arrhythmogenic right ventricular cardiomyopathy with human induced pluripotent stem cells. Circulation: Cardiovascular Genetics. 2013;6(6):557–568. doi: 10.1161/CIRCGENETICS.113.000188. [DOI] [PubMed] [Google Scholar]
- 178.Kim C., Wong J., Wen J., et al. Studying arrhythmogenic right ventricular dysplasia with patient-specific iPSCs. Nature. 2013;494(7435):105–110. doi: 10.1038/nature11799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Calkins H. Arrhythmogenic right-ventricular dysplasia/cardiomyopathy. Current Opinion in Cardiology. 2006;21(1):55–63. doi: 10.1097/01.hco.0000198984.70884.4d. [DOI] [PubMed] [Google Scholar]
- 180.Cerrone M., Lin X., Zhang M., et al. Missense mutations in plakophilin-2 cause sodium current deficit and associate with a brugada syndrome phenotype. Circulation. 2014;129(10):1092–1103. doi: 10.1161/circulationaha.113.003077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Di Pasquale E., Lodola F., Miragoli M., et al. CaMKII inhibition rectifies arrhythmic phenotype in a patient-specific model of catecholaminergic polymorphic ventricular tachycardia. Cell Death and Disease. 2013;4, article e843 doi: 10.1038/cddis.2013.369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Itzhaki I., Maizels L., Huber I., et al. Modelling the long QT syndrome with induced pluripotent stem cells. Nature. 2011;471(7337):225–230. doi: 10.1038/nature09747. [DOI] [PubMed] [Google Scholar]
- 183.Fatima A., Xu G., Shao K., et al. In vitro modeling of ryanodine receptor 2 dysfunction using human induced pluripotent stem cells. Cellular Physiology and Biochemistry. 2011;28(4):579–592. doi: 10.1159/000335753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Hake J., Edwards A. G., Yu Z., et al. Modelling cardiac calcium sparks in a three-dimensional reconstruction of a calcium release unit. The Journal of Physiology. 2012;590(18):4403–4422. doi: 10.1113/jphysiol.2012.227926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Lee Y.-K., Ho P. W.-L., Schick R., et al. Modeling of Friedreich ataxia-related iron overloading cardiomyopathy using patient-specific-induced pluripotent stem cells. Pflugers Archiv. 2014;466(9):1831–1844. doi: 10.1007/s00424-013-1414-x. [DOI] [PubMed] [Google Scholar]
- 186.Huang H.-P., Chen P.-H., Hwu W.-L., et al. Human Pompe disease-induced pluripotent stem cells for pathogenesis modeling, drug testing and disease marker identification. Human Molecular Genetics. 2011;20(24):4851–4864. doi: 10.1093/hmg/ddr424. [DOI] [PubMed] [Google Scholar]
- 187.Drawnel F. M., Boccardo S., Prummer M., et al. Disease modeling and phenotypic drug screening for diabetic cardiomyopathy using human induced pluripotent stem cells. Cell Reports. 2014;9(3):810–820. doi: 10.1016/j.celrep.2014.09.055. [DOI] [PubMed] [Google Scholar]
- 188.Ryan S. D., Dolatabadi N., Chan S. F., et al. Isogenic human iPSC Parkinson's model shows nitrosative stress-induced dysfunction in MEF2-PGC1α transcription. Cell. 2013;155(6):1351–1364. doi: 10.1016/j.cell.2013.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Guo X., Disatnik M.-H., Monbureau M., Shamloo M., Mochly-Rosen D., Qi X. Inhibition of mitochondrial fragmentation diminishes Huntington's disease-associated neurodegeneration. Journal of Clinical Investigation. 2013;123(12):5371–5388. doi: 10.1172/JCI70911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Ren Y., Jiang H., Hu Z., et al. Parkin mutations reduce the complexity of neuronal processes in iPSC-derived human neurons. STEM CELLS. 2015;33(1):68–78. doi: 10.1002/stem.1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Byrne S. M., Mali P., Church G. M. Genome editing in human stem cells. Methods in Enzymology. 2014;546:119–138. doi: 10.1016/B978-0-12-801185-0.00006-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Sosnovik D. E., Mekkaoui C., Huang S., et al. Microstructural impact of ischemia and bone marrow-derived cell therapy revealed with diffusion tensor magnetic resonance imaging tractography of the heart in vivo. Circulation. 2014;129(17):1731–1741. doi: 10.1161/CIRCULATIONAHA.113.005841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Radisic M., Park H., Shing H., et al. Functional assembly of engineered myocardium by electrical stimulation of cardiac myocytes cultured on scaffolds. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(52):18129–18134. doi: 10.1073/pnas.0407817101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Nunes S. S., Miklas J. W., Liu J., et al. Biowire: a platform for maturation of human pluripotent stem cell-derived cardiomyocytes. Nature Methods. 2013;10(8):781–787. doi: 10.1038/nmeth.2524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Liau B., Christoforou N., Leong K. W., Bursac N. Pluripotent stem cell-derived cardiac tissue patch with advanced structure and function. Biomaterials. 2011;32(35):9180–9187. doi: 10.1016/j.biomaterials.2011.08.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Zhang B., Montgomery M., Chamberlain M. D., et al. Biodegradable scaffold with built-in vasculature for organ-on-a-chip engineering and direct surgical anastomosis. Nature Materials. 2016;15(6):669–678. doi: 10.1038/nmat4570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Huebsch N., Loskill P., Mandegar M. A., et al. Automated video-based analysis of contractility and calcium flux in human-induced pluripotent stem cell-derived cardiomyocytes cultured over different spatial scales. Tissue Engineering Part C: Methods. 2015;21(5):467–479. doi: 10.1089/ten.tec.2014.0283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Huebsch N., Loskill P., Deveshwar N., et al. Miniaturized iPS-cell-derived cardiac muscles for physiologically relevant drug response analyses. Scientific Reports. 2016;6 doi: 10.1038/srep24726.24726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Kajzar A., Cesa C. M., Kirchgeßner N., Hoffmann B., Merkel R. Toward physiological conditions for cell analyses: forces of heart muscle cells suspended between elastic micropillars. Biophysical Journal. 2008;94(5):1854–1866. doi: 10.1529/biophysj.107.115766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Thavandiran N., Dubois N., Mikryukov A., et al. Design and formulation of functional pluripotent stem cell-derived cardiac microtissues. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(49):E4698–E4707. doi: 10.1073/pnas.1311120110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Rodriguez M. L., Graham B. T., Pabon L. M., Han S. J., Murry C. E., Sniadecki N. J. Measuring the contractile forces of human induced pluripotent stem cell-derived cardiomyocytes with arrays of microposts. Journal of Biomechanical Engineering. 2014;136(5) doi: 10.1115/1.4027145.051005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Yang X., Rodriguez M., Pabon L., et al. Tri-iodo-l-thyronine promotes the maturation of human cardiomyocytes-derived from induced pluripotent stem cells. Journal of Molecular and Cellular Cardiology. 2014;72:296–304. doi: 10.1016/j.yjmcc.2014.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Feinberg A. W., Feigel A., Shevkoplyas S. S., Sheehy S., Whitesides G. M., Parker K. K. Muscular thin films for building actuators and powering devices. Science. 2007;317(5843):1366–1370. doi: 10.1126/science.1146885. [DOI] [PubMed] [Google Scholar]
- 204.Kujala V. J., Pasqualini F. S., Goss J. A., Nawroth J. C., Parker K. K. Laminar ventricular myocardium on a microelectrode array-based chip. Journal of Materials Chemistry B. 2016;4(20):3534–3543. doi: 10.1039/c6tb00324a. [DOI] [PubMed] [Google Scholar]
- 205.Aratyn-Schaus Y., Pasqualini F. S., Yuan H., et al. Coupling primary and stem cell-derived cardiomyocytes in an in vitro model of cardiac cell therapy. The Journal of Cell Biology. 2016;212(4):389–397. doi: 10.1083/jcb.201508026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Huang B., Bates M., Zhuang X. Super-resolution fluorescence microscopy. Annual Review of Biochemistry. 2009;78:993–1016. doi: 10.1146/annurev.biochem.77.061906.092014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Chen F., Tillberg P. W., Boyden E. S. Optical imaging. Expansion microscopy. Science. 2015;347(6221):543–548. doi: 10.1126/science.1260088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Jungmann R., Avendaño M. S., Woehrstein J. B., Dai M., Shih W. M., Yin P. Multiplexed 3D cellular super-resolution imaging with DNA-PAINT and exchange-PAINT. Nature Methods. 2014;11(3):313–318. doi: 10.1038/nmeth.2835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Xu K., Zhong G., Zhuang X. Actin, spectrin, and associated proteins form a periodic cytoskeletal structure in axons. Science. 2013;339(6118):452–456. doi: 10.1126/science.1232251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Prahl L. S., Castle B. T., Gardner M. K., Odde D. J. Quantitative analysis of microtubule self-assembly kinetics and tip structure. Methods in Enzymology. 2014;540:35–52. doi: 10.1016/B978-0-12-397924-7.00003-0. [DOI] [PubMed] [Google Scholar]
- 211.Rosenbloom A. B., Lee S.-H., To M., Lee A., Shin J. Y., Bustamante C. Optimized two-color super resolution imaging of Drp1 during mitochondrial fission with a slow-switching Dronpa variant. Proceedings of the National Academy of Sciences of the United States of America. 2014;111(36):13093–13098. doi: 10.1073/pnas.1320044111. [DOI] [PMC free article] [PubMed] [Google Scholar]