

Abstract
Nuclear pore complexes (NPCs) mediate bidirectional transport of proteins, RNAs, and ribonucleoprotein complexes across the double-membrane nuclear envelope. In vitro studies with purified transport cofactors have revealed a general scheme of cofactor-dependent transport energetically driven by the G protein Ran. However, the size and complexity of NPCs have made it difficult to clearly define the loci and kinetics of the cofactor–NPC interactions required for transport. We now report the use of single-molecule fluorescence microscopy to directly monitor a model protein substrate undergoing transport through NPCs in permeabilized cells. This substrate, NLS-2xGFP, interacts with NPCs for an average of 10 ± 1 ms during transport. However, because the maximum nuclear accumulation rate of NLS-2xGFP was measured to be at least ≈103 molecules per NPC per s, NPCs must be capable of transporting at least ≈10 substrate molecules simultaneously. Molecular tracking reveals that substrate molecules spend most of their transit time randomly moving in the central pore of the NPC and that the rate-limiting step is escape from the central pore.
Macromolecular traffic between the nucleus and cytoplasm is enabled by nuclear pore complexes (NPCs), which span the double-membrane nuclear envelope (NE). Proteins destined for nuclear functions, such as nucleic acid polymerases, histones, and splicing and transcription factors, must transit into the nucleus after synthesis on cytoplasmic ribosomes. In addition, the major cellular RNAs generated by transcription in the nucleus (mRNAs, tRNAs, and rRNAs) are exported to the cytoplasm. Whereas small molecules (less than ≈20–40 kDa) can transit NPCs essentially unhindered (passive diffusion), larger molecules and molecular complexes (up to ≈25–50 MDa) transit through carrier-mediated, signal-dependent processes (facilitated translocation) (1–3).
NPCs are large (≈50–100 MDa), octagonal structures comprised of at least ≈30 different proteins [nucleoporins (Nups)], each present in an integer multiple of eight copies (2, 4). The pore itself is ≈90 nm in length with a minimum internal diameter of ≈50 nm. Flexible filaments extend ≈50 nm into the cytoplasm, and a basket structure extends ≈75 nm into the nucleoplasm. Thus, a transiting substrate can potentially interact continuously with NPC proteins over a distance spanning ≈200 nm (2, 5). Substrates of signal-dependent nucleocytoplasmic transport bind to importins (for nuclear import) or exportins (for export), soluble accessory proteins that mediate transient interactions with phenylalanine-glycine repeat domains (FG repeats) found on a number of Nups. Import complexes (ICs), consisting of transport substrate and importin(s), are dissociated by RanGTP after transit through the NPC. The Ran GTPase cycle, the location of FG–Nups within the pore, the strength of the importin/exportin–FG–Nup interactions, and how these interactions promote directional transport have been researched intensely (1–3, 6, 7), but the sequence of binding interactions between transport complexes and the NPC is not well understood.
To gain further insight into the mechanism of nucleocytoplasmic transport, substrate molecules in the process of transit through the NPC were tracked by using single-molecule fluorescence (SMF) microscopy. By directly monitoring the binding and relative movement of ICs interacting with the NPC, we directly observed and characterized the principle kinetic steps of the import mechanism.
Methods
Proteins and Labeling. The green fluorescent protein (GFP) used is a modified “superglow” (8) variant (S65T, Q80R, V163A). The NLS-2xGFP protein and its nuclear localization sequence (NLS) mutant as shown in Fig. 1a were cloned into the pTrcHisB vector (Invitrogen); coding regions were confirmed by DNA sequencing. Proteins were expressed in Escherichia coli and purified by nickel-nitrilotriacetic acid Superflow (Qiagen, Valencia, CA), MonoQ, and Superdex 200 (Amersham Pharmacia) chromatography after uninduced overnight growth in JM109 at 30°C to yield a single band by Coomassie-stained polyacrylamide gel electrophoresis.
Fig. 1.

The NLS-2xGFP model substrate and its transport into permeabilized HeLa cells. (a) Schematic diagram of the substrate. Two identical GFP domains, represented here by structural models (PDB ID code 1C4F), have N- and C-terminal extensions and are linked by the peptide shown. The identified cysteines are each labeled with a molecule of Alexa-555 maleimide. The N-terminal extension contains an NLS (red) that targets the protein for Im α/Im β-dependent transport. Control experiments used protein with the indicated K → T NLS mutation, which blocks recognition by Im α (16). (b–e) Extent of nuclear accumulation of NLS-2xGFP in permeabilized HeLa cells measured by epifluorescence. Substrate (0.44 μM) was incubated with cells for 10 min and then washed away. (Scale bar, 10 μm.) (b) Normal nuclear accumulation in the presence of GTP, Ran, NTF2, Im α and Im β. Successful nuclear transport is detected as bright fluorescence (red) in oval nuclei. (c) Control experiment with NTF2, Im α, and Im β but without Ran and GTP. Substrate is localized at the NE. Under these conditions, the substrate is expected to remain bound to the NPCs as an Im α/β–substrate complex, because there is no RanGTP available on the nucleoplasmic side of the NE to dissociate this complex (20). (d) Control experiment with WGA, an inhibitor of nucleocytoplasmic transport (18). When the cells are preincubated with WGA before addition of substrate and cofactors as described for b, no transport occurs because IC binding is blocked by WGA. (e) Control experiment with substrate containing a mutant NLS. The K → T NLS mutant substrate is not transported under the conditions of b, presumably because the mutant NLS is not recognized by Im α. (f) Dependence of initial nuclear accumulation rates determined by confocal microscopy on substrate concentration. Initial transport rates in μM/s were converted to molecules per NPC per s by assuming 3,000 NPCs per nucleus (17, 32). (Error bars, SDs over five nuclei.)
The intrinsic GFP fluorescence of the model proteins was unsuitable for single-molecule detection in our NPC transport experiments because of blinking, rapid photobleaching, and the green autofluorescence of permeabilized cells. Therefore, proteins were labeled with excess Alexa-555 maleimide (Molecular Probes) for 2 h at room temperature in 50 mM sodium phosphate/150 mM NaCl, pH 7.5. Reactions were quenched with 2-mercaptoethanol, and the products were dialyzed to remove the free dye. The two cysteines within each GFP domain are not reactive: the labeling reaction is complete with a 10-fold molar excess of dye to give two Alexa-555 molecules per NLS-2xGFP molecule, and no additional labeling is observed with up to a 160-fold molar excess of dye.
Cell Culture and Transport Conditions. Freshly split HeLa cells (American Type Culture Collection) were grown overnight on coverslips (9). For microscopy, flow cells were constructed by adding a top coverslip together with two lines of silicone grease as spacers. Cells were washed with import buffer (20 mM Hepes/110 mM KOAc/5 mM NaOAc/2 mM MgOAc/1 mM EGTA, pH 7.3), permeabilized for 2 min with 40 μg/ml digitonin in import buffer, and washed again with import buffer supplemented with 1.5% polyvinylpyrrolidone (360 kDa). Polyvinylpyrrolidone was included in all import buffer solutions after digitonin treatment to prevent osmotic swelling of nuclei. Import cofactors were isolated by literature protocols (10–13). Import reactions contained 1 mM GTP, 0.5 μM importin α (Im α), 0.5 μM importin β (Im β), 2 μM Ran, and 1 μM NTF2 unless otherwise indicated. For ensemble experiments (Fig. 1 b–e), external substrate was washed away after the transport incubation period. Fixation was unnecessary, because the substrate was retained by the nuclei for minutes with minimal loss of signal. Where indicated, cells were preincubated with 0.1 mg/ml wheat germ agglutinin (WGA) for 10 min. For the single-molecule experiments, the substrate concentration was reduced to ≈100 pM, and the cofactor concentrations were unchanged from the ensemble transport reactions. At these substrate and cofactor concentrations, >99% of the substrate is complexed with Im α and Im β (14).
Confocal Experiments. Im α and Im β concentrations matched the substrate concentration to ensure that all substrate could simultaneously form a 1:1:1 complex. Calculations using known nanomolar binding constants (14) predict that for 10 μM initial concentrations of Im α, Im β, and substrate, <2% of these proteins are uncomplexed. Thus, it is expected that the observed rates are dominated by transport of complexed substrate. The fluorescence intensities from cytoplasmic substrate at known concentrations were used to calculate concentrations of transported substrate from nuclear fluorescence intensities.
SMF Instrumentation. The SMF microscope includes a Zeiss Axiovert 200M, 300-mW, 532-nm solid-state laser (Coherent Radiation, Palo Alto, CA), intensified charge-coupled device (CCD) camera (I-Pentamax, Roper Scientific, Duluth, GA), and 1.45-numerical-aperture ×100 oil-immersion objective (Zeiss). A Dual-View imager with I-mask (Optical Insights, Santa Fe, NM) was used to mask a portion of the intensified CCD and position the illumination area to allow high frame rates in virtual chip mode. winview (Roper Scientific) and metamorph (Universal Imaging, Media, PA) software packages were used for data acquisition and processing. A CoolSnap CCD camera (Roper Scientific) was used to obtain high-resolution bright-field images. CCD and intensified CCD camera images were aligned with 100-nm fluorescent microspheres (Molecular Probes).
Narrow-Field Epifluorescence Microscopy. To improve the signal-to-noise (S/N) ratio of SMF images, a 400-μm pinhole was placed in the excitation path within a specimen-conjugate plane to restrict the specimen illumination area to an 8-μm diameter. By using the virtual chip mode of the intensified CCD camera, 56 × 56-pixel frames at 333 frames per s were typically collected for 500 frames with a single-molecule S/N ratio of >6 at a power density of 9 kW/cm2 measured at the specimen. Stage drift was 0.20 ± 0.04 nm/s and therefore was not significant on the time scale of our single-molecule transport experiments. Immediately before beginning the SMF experiments, autofluorescence in the illumination area was photobleached and the NE was localized by bright-field imaging. Coverslip-immobilized substrate molecules remained detectable 100 nm, but not 200 nm, away from optimum focus, indicating a focal-plane thickness of ≈300 nm.
Data Analysis. The position of fluorescent spots was determined by fitting to a two-dimensional Gaussian (15). The S/N ratio was operationally defined as 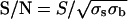 , where S is the integrated intensity above background, and σs and σb are the SDs of the signal and background noise, respectively. The range of motion of the IC during its interaction with the NE was computed as
, where S is the integrated intensity above background, and σs and σb are the SDs of the signal and background noise, respectively. The range of motion of the IC during its interaction with the NE was computed as 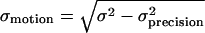 , where σ is the mean SD of the positions of the ICs, and σprecision is the mean SD of the positions of immobilized molecules measured in control experiments. Confidence intervals of σmotion were calculated by error propagation using Monte Carlo methods. Experimental measurements are reported as mean ± SE unless otherwise noted.
, where σ is the mean SD of the positions of the ICs, and σprecision is the mean SD of the positions of immobilized molecules measured in control experiments. Confidence intervals of σmotion were calculated by error propagation using Monte Carlo methods. Experimental measurements are reported as mean ± SE unless otherwise noted.
NE Localization. The position of the NE was determined by bright-field microscopy. The pixel intensities within a row or column approximately perpendicular to the NE were fit to a Gaussian. The peak position of the Gaussian for a particular set of pixel intensities was considered the NE position for that row or column. The peak positions of a series of such Gaussians then were fit with a second-degree polynomial, yielding the path of the NE within the entire image. In single-molecule transport experiments, only substrate fluorescence within ±200 nm of the measured position of the NE was selected for inclusion in the analysis. This distance is sufficiently large to include substrate bound at the periphery of the NPCs (cytoplasmic filaments and nuclear basket).
Results
Bulk Transport of the Model Substrate. We constructed a model import substrate, NLS-2xGFP, that consists of the NLS of the simian virus 40 large T antigen (16) followed by two GFP domains. This substrate was labeled at two cysteines with the fluorescent dye Alexa-555 for single-molecule visualization (Fig. 1a). As expected, in vitro transport assays indicated that the NLS-2xGFP model substrate was recognized by the Im α/Im β cofactor system and was transported through NPCs into the nuclei of permeabilized HeLa cells in a RanGTP-dependent manner (Fig. 1 b–e). Furthermore, the initial transport rate of the model substrate in the presence of GTP and the protein transport cofactors NTF2, Ran, Im α, and Im β was ≈10-fold larger than in their absence (data not shown). Thus, the facilitated transport mechanism predominated. By using confocal microscopy, it was determined that the maximum transport velocity for NLS-2xGFP is at least ≈103 molecules per NPC per s (Fig. 1f), in agreement with previous similar measurements for the transport of transportin (17).
Single-Molecule Transport of the Model Substrate. Having established that NLS-2xGFP is transported efficiently by the facilitated transport mechanism, we attempted detection of individual molecules undergoing transport. The conditions of the bulk transport experiments (Fig. 1b) were retained except that the substrate was diluted ≈4,000-fold to permit detection of single molecules by SMF. The NE at the nuclear equator of a permeabilized HeLa cell nucleus was examined by using “narrow-field” epifluorescence microscopy (see Methods). In the cytoplasmic compartment, transient spots of fluorescence emission from substrate molecules were occasionally observed. However, such events were rare (0.0074 ± 0.0018 events per s per μm2), as is expected from molecules that are free in solution and thus diffuse micrometer-scale distances on the time scale of a single video frame (3 ms). In contrast, transient, localized emission spots appeared far more frequently (1.1 ± 0.1 events per s per μm2) at the NE (Fig. 2a; see also Movie 1, which is published as supporting information on the PNAS web site). More than one such interaction event was often observed at the same location on the NE (Fig. 2b; see also Movie 2, which is published as supporting information on the PNAS web site). The average residence time of these NLS-2xGFP molecules on the NE under transport conditions was tave = 10 ± 1 ms. The distribution of residence times is approximately exponential (Fig. 2c), as would be expected for rate-limiting dissociation from a single class of binding sites.
Fig. 2.

Binding and release of single-substrate molecules at the NE visualized by SMF microscopy. (a) Consecutive video frames (3-ms duration) showing the appearance and disappearance of an Alexa-555-labeled NLS-2xGFP molecule at the NE. The position of the NE (dashed line) was determined by bright-field microscopy (see Methods). The nuclear (N) and cytoplasmic (C) compartments are indicated. Numbers denote time (ms). (Scale bar, 2 μm.) (b) Time course of the fluorescence intensity at the position of the NE-localized spot shown in a. Intensity of a 2 × 2-pixel area was quantified over the entire video sequence. Two NE-interaction events are observed, the first of which is shown in a.(Insets) Expanded views of the interaction events. (c) Histogram of NE residence times for 231 NE-interaction events (3-ms bins). The exponential fit yields a time constant of τ = 8.8 ± 0.6 ms. (d) Time course of the fluorescence intensity for substrate molecules immobilized on a glass surface. For comparison, the time-dependent fluorescence intensity observed for two isolated substrate molecules is shown under the same conditions and on the same intensity scale as described for b. The blue trace shows a single-step photobleach event and thus is a substrate molecule with one Alexa-555-dye label. The red trace shows two-step photobleaching, indicating a doubly labeled substrate molecule. Photobleaching (tave = 820 ± 30 ms) occurs on a time scale much longer than the NE-interaction time.
Although we could not unambiguously determine from which aqueous compartment (cytoplasm or nucleoplasm) single substrates came to the NE and to which compartment they went after release, several observations indicate that the transient fluorescent spots observed at the NE were single-substrate molecules undergoing signal-dependent transport through NPCs. (i) Even at the low substrate conditions required for the single-molecule experiments, an increase in nuclear fluorescence was observed within a few minutes, demonstrating that transport occurred under these conditions. (ii) The fluorescence intensities of spots at the NE correspond to that of a doubly labeled substrate molecule (Fig. 2d). Thus, the observed interaction events arose from single fluorescent molecules, not from aggregates. (iii) After addition of WGA, the frequency of observed NE spots decreased by >90% (Table 1). Because WGA binds to glycosylated Nups and thereby blocks binding of ICs (18), these data are consistent with the hypothesis that the observed NE-interaction events are a consequence of fluorescent substrate molecules interacting with NPCs. (iv) After GTP depletion (19), the NE residence time (tave) increased to 45 ± 5 ms (Table 1). These data indicate that GTP is required for rapid release from the NE, as is expected for signal-dependent transport (20). (v) A point mutation in the NLS that renders the substrate transport-incompetent reduced the NE-interaction event frequency by >95% (Table 1). Thus, the interaction of fluorescent substrate molecules with the NE is specific in that it requires a functional NLS.
Table 1. Kinetics of single-substrate molecule interaction events at the NE.
| NLS-2xGFP
|
Mutant NLS
|
|||
|---|---|---|---|---|
| Experimental conditions | Normal | -GTP, -Ran | +WGA | Normal |
| Mean residence time, tave, ms | 10 ± 1 | 45 ± 5 | 13 ± 3 | 150 ± 50 |
| Event frequency, events per s per μm2 | 1.1 ± 0.1 | 0.89 ± 0.07 | 0.081 ± 0.018 | 0.042 ± 0.015 |
| Total video examined, s | 82.5 | 75.0 | 75.0 | 75.0 |
Each set of experimental conditions (analogous to Fig. 1 b—e except that here the substrate is diluted for the single-molecule measurements) included lm α, lm β, NTF2, and unless otherwise indicated, Ran and GTP. For each set of conditions, 15-30 s of video was analyzed from each of four nuclei. +WGA, the nuclei were preincubated with WGA; mutant NLS, the K → T NLS mutant of NLS-2xGFP (Fig. 1a). Other conditions are as described in Methods.
Spacing of Interaction Sites. As shown in Fig. 2b, multiple interaction events were often observed at the same location on the NE. In one single-molecule experiment, we identified 11 such NE locations at each of which between 2 and 13 substrate molecules were observed to interact individually within a 33-s time window (Fig. 3a). Because NPCs do not diffuse extensively within the NE (21), we hypothesize that the successive events at a single location were from different substrate molecules binding to the same NPC. The median nearest-neighbor spacing between such NE binding locations was 0.56 μm (Fig. 3b). Because the focal-plane thickness for detection of single fluorescent molecules was ≈300 nm (see Methods), this linear separation agrees with previous surface-density estimates of approximately four to five NPCs per μm2 determined in structural studies by electron and fluorescence microscopy (21–23).
Fig. 3.

NE-interaction sites. (a) Distribution of substrate-interaction sites (red dots) on the NE detected under the conditions described for Fig. 2a in 33 s of video. Nuclear (N) and cytoplasmic (C) compartments are indicated. Green, illumination area; blue, NE position (see Methods). (Scale bar, 1 μm.) (Inset) Lower-magnification view showing the entire nucleus; the area of the main image is enclosed by the red box. (b) Distribution of nearest-neighbor spacings between interaction sites (n = 21).
Tracking of ICs Interacting with NPCs. The relative immobility of the NPCs in the NE, their large size, and previous successes with nanometer-scale tracking of single dye-labeled molecules within cells (24, 25) suggested that it might be possible to track the movement of substrate molecules relative to the NPC as they transit the pore. We first repeatedly measured the positions of substrate molecules immobilized on a glass surface (see Methods). The SDs of such position measurements, a measure of tracking precision, varied inversely with the S/N ratio of the fluorescence spots (Fig. 4a). For example, repeated position determinations for three immobilized substrate molecules with comparatively high S/N ratios of 7.8–10.3 at 3 ms per frame yielded a mean SD of 30.4 ± 1.8 nm (Fig. 4b), demonstrating that such substrate molecules could be localized with a precision considerably better than the ≈200-nm overall size of the NPC. The same tracking procedure was then applied to the high S/N ratio (>7) substrate-interaction events observed on the NE to characterize the locations and movements of substrate molecules undergoing NPC-mediated transport. Individual single-molecule trajectories (Fig. 4 c and d; see also Fig. 5, which is published as supporting information on the PNAS web site) were compact and centered close to the NE midplane. The sizes and positions of the trajectories strongly suggest that the observed substrate interactions are largely or completely confined to the central pore of the NPC. In control experiments without RanGTP, the variations in substrate position measured both parallel and perpendicular to the plane of the NE (Table 2, σ∥ and σ⊥) were not significantly greater than those observed in the control measurements on surface-immobilized molecules, indicating that the molecules are closely confined or immobilized by binding to the NPC under these conditions. Significant movement within the plane of the NE was also not observed for molecules under normal transport conditions. In contrast, molecules interacting with NPCs under transport conditions displayed a variation in position perpendicular to the NE significantly exceeding that of immobilized molecules (Table 2, σmotion,⊥), demonstrating substantial movement along the pore axis. The significantly smaller positional variance of the IC perpendicular to the NE in the absence of RanGTP than in its presence demonstrates increased movement of the IC within the pore in the presence of RanGTP. Significantly, net movement along the pore axis (Table 2, Δd⊥,ave) was negligible, indicating that the detected motion was largely random rather than directed. The average position coincided with the midplane of the NE (Table 2, d⊥,ave). Stepwise movement of substrate molecules through the NPC was not observed, indicating either that movement does not occur in discrete steps or that the steps are too small and/or rapid to be detected in these experiments.
Fig. 4.

Single-molecule tracking precision and transport trajectories. (a) Tracking precision measured for immobilized substrate molecules with different S/N ratios. The line is an exponential intended only to guide the eye. (b) Control: successive position measurements for three immobilized substrate molecules (S/N ratio = 7.8–10.3). A schematic (gray; to scale) of an NPC showing the cytoplasmic filaments (Upper), nuclear basket (Lower), and NE (horizontal lines; each line is a membrane bilayer) is underlaid for size comparison; the shape of the pore is adapted from figure 1 of Fahrenkrog and Aebi (2). Precisions in the vertical and horizontal directions are 32.0 ± 2.3 and 28.8 ± 1.2 nm, respectively. Acquisition conditions are as described for Fig. 2a.(c) Overlay of trajectories for three substrate molecules that interacted successively with the same NPC. The vertical position of the trajectories relative to the NE of the NPC schematic was determined by the NE-localization procedure (see Methods). The horizontal centroid from all the trajectories for this NPC was aligned with the horizontal center of the NPC schematic. For each trajectory, the points are numbered in sequence. (d) Superimposed plots of 17 trajectories (64 points) from 11 NPCs. The vertical position of the trajectories relative to the NPC schematic was aligned as described for b; the horizontal centroid from all the trajectories for each NPC was aligned with the horizontal center of the NPC schematic. By using the centroid of the trajectories for each NPC as reference points, the SDs of the position measurements in the plane of and perpendicular to the NE are 33 ± 4 and 50 ± 5 nm, respectively. The average position perpendicular to the NE is 0.2 ± 5.4 nm.
Table 2. Geometrical parameters (in nanometers) of single-molecule trajectories.
| Experimental conditions | σ∥ | σ⊥ | σmotion,∥ | σmotion,⊥ | Δd⊥,ave | d⊥,ave |
|---|---|---|---|---|---|---|
| Normal | 33 ± 4 | 50 ± 5 | 13 (0, 26) | 40 (28, 50) | 0 ± 24 | 0 ± 5 |
| -GTP, -Ran | 35 ± 6 | 31 ± 6 | 17 (0, 33) | 6 (0, 28) | 13 ± 17 | 2 ± 5 |
“Normal” and “-GTP, -Ran” experimental conditions are as defined in the Table 1 legend. The parameters σ∥ and σ⊥ are the SDs of the position measured for ICs interacting with the NE in directions parallel and perpendicular to the NPC, respectively. The σmotion values were calculated from σ∥ and σ⊥ (see Methods). For the average perpendicular displacement between first and last points of the trajectories, Δd⊥,ave, and the average perpendicular position, d⊥,ave, the positive direction is defined as toward the nucleus with the origin at the NE midplane. Values derived from 17 normal (64 points) and 12 -GTP, -Ran (50 points) trajectories are reported as mean ± SE or mean (90% confidence interval).
Discussion
In these studies, SMF imaging was used to identify single ICs interacting with the NPC with 3-ms and 30-nm resolution. By characterizing the predominant kinetic features of the movement of an IC relative to the NPC during transport, this approach reveals several basic features of the transport mechanism. First, the tracking data demonstrate that the import substrate complex spends the majority of its ≈10-ms interaction time with the NPC moving within a comparatively small region corresponding to the NPC central pore. Second, tracking also reveals that, while in this central pore, the movement of the IC along the pore axis is bidirectional, rapid, and unbiased to the limit of experimental detection. Thus, it has the characteristics of a random walk. Third, the roughly exponential distribution of NE residence times indicates that the overall transport process is rate-limited by one or at most a few reaction step(s). Control experiments indicate that the transport rate is stimulated by RanGTP. Because movement within the pore is rapid, these data suggest that the rate-limiting step is substrate exit from the pore. Finally, the measured residence times predict a maximum transport rate of ≈100 molecules per NPC per s, assuming that only one substrate molecule at a time can interact with the NPC. This rate is significantly below the saturated transport rate of approximately ≥103 molecules per NPC per s directly measured in ensemble experiments with the same substrate and NE preparation. The most parsimonious explanation for this difference is that multiple substrate complexes are simultaneously transported by the same NPC. This hypothesis is consistent with data indicating that multiple colloidal gold particles decorated with transport substrates interact with single NPCs in fixed cells (22, 26). Simultaneous transport of multiple substrate molecules together with the random rapid movement of the molecules along the length of the central pore suggest that there are multiple parallel transit paths through the central pore rather than one single-file path.
Published proposals for the mechanism of facilitated nuclear transport (17, 27–31) in general are more concerned with the thermodynamic and structural features of the process than with its kinetics. Nonetheless, the predictions of the selective phase model (17, 31) are remarkably consistent with the kinetic studies reported here. The selective phase model postulates that transport cofactors decrease the energy cost for a transport substrate to enter a meshwork, or phase, of FG–Nups within the pore region of the NPC. Our observations establish that the dominant interactions of ICs with the NPC occur in the pore region. An IC undergoing transit could diffuse back and forth throughout the proposed FG–meshwork multiple times before exiting. Such random walk behavior is illustrated by the observed import trajectories. Although there are many approximately equivalent binding sites in this proposed FG–meshwork of the selective phase model, such a transport mechanism predicts that the rate-limiting step is exit from the pore, a first-order process. This prediction is confirmed by the observed distribution of transport substrate residence times and the RanGTP dependence of substrate dissociation from the NPC. Finally, the size and fluidity of the predicted FG-Nup meshwork intimate that it could accommodate simultaneous transit of multiple ICs. Our data suggest that multiple substrates transit simultaneously through multiple transit paths.
Unlike the selective phase model, some models of nucleocytoplasmic transport (27–29) place a greater importance on the interaction between substrate complexes and FG–Nups located throughout the nuclear basket and cytoplasmic filaments. Our data do not exclude the possibility that these interactions play some role in transport; however, they demonstrate that the most kinetically important interactions take place in the central pore and that these interactions have the characteristics of unbiased diffusion.
Supplementary Material
Acknowledgments
We thank L. Davis for extensive advice and discussion and for the GFP clone; M. J. Moore for the HeLa cells; J. Wang for DNA sequencing and software assistance; M. S. Moore for providing import cofactors in the early stages of the project and the Im α clone; D. Görlich for the Im β, Ran, and NTF2 clones; M. Summers and S. Williamson for access to their confocal microscope; and R. Rock, A. LiWang, H. Bayley, and M. J. Moore for critical comments on the manuscript. This project was supported by The Welch Foundation (BE-1541), the Mallinckrodt Foundation, and the National Institute of General Medical Sciences.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: NPC, nuclear pore complex; NE, nuclear envelope; Nup, nucleoporin; FG, phenylalanine–glycine; IC, import complex; SMF, single-molecule fluorescence; NLS, nuclear localization sequence; Im α/β, importin α/β; WGA, wheat germ agglutinin; CCD, charge-coupled device; S/N, signal-to-noise.
References
- 1.Weis, K. (2003) Cell 112, 441–451. [DOI] [PubMed] [Google Scholar]
- 2.Fahrenkrog, B. & Aebi, U. (2003) Nat. Rev. Mol. Cell Biol. 4, 757–766. [DOI] [PubMed] [Google Scholar]
- 3.Fried, H. & Kutay, U. (2003) Cell Mol. Life Sci. 60, 1659–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rout, M. P. & Aitchison, J. D. (2001) J. Biol. Chem. 276, 16593–16596. [DOI] [PubMed] [Google Scholar]
- 5.Stoffler, D., Feja, B., Fahrenkrog, B., Walz, J., Typke, D. & Aebi, U. (2003) J. Mol. Biol. 328, 119–130. [DOI] [PubMed] [Google Scholar]
- 6.Görlich, D. & Kutay, U. (1999) Annu. Rev. Cell Dev. Biol. 15, 607–660. [DOI] [PubMed] [Google Scholar]
- 7.Strawn, L. A., Shen, T., Shulga, N., Golldfarb, D. S. & Wente, S. R. (2004) Nat. Cell Biol. 6, 197–206. [DOI] [PubMed] [Google Scholar]
- 8.Kahana, J. A. & Silver, P. A. (1998) in Green Fluorescent Protein: Properties, Applications, and Protocols, eds. Chalfie, M. & Kain, S. (Wiley–Liss, New York), pp. 139–151.
- 9.Adam, S. A. & Gerace, L. (1991) Cell 66, 837–847. [DOI] [PubMed] [Google Scholar]
- 10.Ribbeck, K., Lipowsky, G., Kent, H. M., Stewart, M. & Gorlich, D. (1998) EMBO J. 17, 6587–6598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Görlich, D., Prehn, S., Laskey, R. A. & Hartmann, E. (1994) Cell 79, 767–778. [DOI] [PubMed] [Google Scholar]
- 12.Kutay, U., Izaurralde, E., Bischoff, F. R., Mattaj, I. W. & Gorlich, D. (1997) EMBO J. 16, 1151–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Weis, K., Ryder, U. & Lamond, A. I. (1996) EMBO J. 15, 1818–1825. [PMC free article] [PubMed] [Google Scholar]
- 14.Kobe, B. (2003) Acta Chim. Slov. 50, 547–562. [Google Scholar]
- 15.Kubitscheck, U., Kückmann, O., Kues, T. & Peters, R. (2000) Biophys. J. 78, 2170–2179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Corbett, A. H. & Silver, P. A. (1997) Microbiol. Mol. Biol. Rev. 61, 193–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ribbeck, K. & Gorlich, D. (2001) EMBO J. 20, 1320–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Akey, C. W. & Goldfarb, D. S. (1989) J. Cell Biol. 1989, 971–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schwoebel, E. D., Ho, T. H. & Moore, M. S. (2002) J. Cell Biol. 157, 963–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schwoebel, E. D., Talcott, B., Cushman, I. & Moore, M. S. (1998) J. Biol. Chem. 273, 35170–35175. [DOI] [PubMed] [Google Scholar]
- 21.Daigle, N., Beaudouin, J., Hartnell, L., Imreh, G., Hallberg, E., Lippincott-Schwartz, J. & Ellenberg, J. (2001) J. Cell Biol. 154, 71–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Feldherr, C. M., Kallenbach, E. & Schultz, N. (1984) J. Cell Biol. 99, 2216–2222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kubitscheck, U., Wedekind, P., Zeidler, O., Grote, M. & Peters, R. (1996) Biophys. J. 70, 2067–2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kues, T., Peters, R. & Kubitscheck, U. (2001) Biophys. J. 80, 2954–2967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lommerse, P. H., Blab, G. A., Cognet, L., Harms, G. S., Snaar-Jagalska, B. E., Spaink, H. P. & Schmidt, T. (2004) Biophys. J. 86, 609–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dworetzky, S. I. & Feldherr, C. M. (1988) J. Cell Biol. 106, 575–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ben-Efraim, I. & Gerace, L. (2001) J. Cell Biol. 152, 411–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pyhtila, B. & Rexach, M. (2003) J. Biol. Chem. 278, 42699–42709. [DOI] [PubMed] [Google Scholar]
- 29.Rout, M. P., Aitchison, J. D., Suprapto, A., Hjertaas, K., Zhao, Y. & Chait, B. T. (2000) J. Cell Biol. 148, 635–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Macara, I. G. (2001) Microbiol. Mol. Biol. Rev. 65, 570–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kustanovich, T. & Rabin, Y. (2004) Biophys. J. 86, 2008–2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Maul, G. G., Maul, H. M., Scogna, J. E., Lieberman, M. W., Stein, G. S., Hsu, B. Y. & Borun, T. W. (1972) J. Cell Biol. 55, 433–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}