

Abstract
The difluoromethyl-allo-threonyl hydroxamate-based compound LPC-058 is a potent inhibitor of UDP-3-O-(R-3-hydroxymyristoyl)-N-acetylglucosamine deacetylase (LpxC) in Gram-negative bacteria. A scalable synthesis of this compound is described. The key step in the synthetic sequence is a transition metal/base-catalyzed aldol reaction of methyl isocyanoacetate and difluoroacetone, giving rise to 4-(methoxycarbonyl)-5,5-disubstituted 2-oxazoline. A simple NMR-based determination of enantiomeric purity is also described.
Graphical abstract
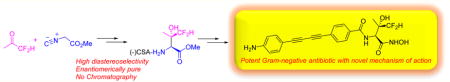
The alarming rise of antimicrobial resistance1–3 of Gram-negative pathogens has become a serious threat to public health and highlights the urgent need for antibiotics with novel modes of action. The UDP-3-O-(R-3-hydroxymyristoyl)-N-acetylglucosamine deacetylase (LpxC) is a novel, validated antibiotic target against Gram-negative pathogens.4 The enzyme catalyzes the committed step in the biosynthesis of lipid A, the membrane anchor of lipopolysaccharide and the predominant lipid component of the outer leaflet of the Gram-negative outer membrane. We have previously reported the design of potent LpxC inhibitors containing a difluoromethyl-allo-threonyl hydroxamate headgroup.4 This series of compounds, represented by LPC-058, displays broad-spectrum antibiotic activity against antibiotic resistant Gram-negative pathogens, thereby highlighting the therapeutic potential of this class of LpxC inhibitors. Herein, we describe a scalable and stereoselective synthesis of LPC-058, which sets the stage for preclinical in vivo studies of this novel series.4
The initial medicinal chemistry route for the synthesis of LPC-058 is shown in Scheme 1. The diacetylene carboxylic acid 4 was prepared in two steps from methyl-4-ethynyl benzoate (1). The first step employed modified Glaser reaction5,6 conditions under which an excess of 4-ethylnylbenzenamine 2 (5 equiv) and copper(II) acetate (2 equiv) in the presence of piperidine in dichloromethane (DCM) provided 3 in a low yield (35%).7 Subsequent alkaline hydrolysis of ester 3 provided the diacetylene carboxylic acid 4 in quantitative yield.7
Scheme 1.
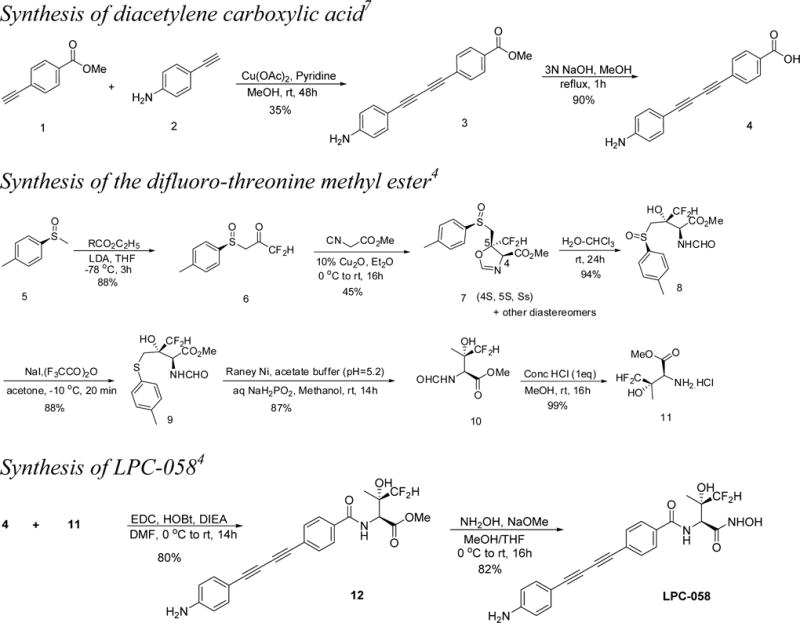
Initial Route to LPC-058
We recently published the medicinal chemistry route for (2S,3S)-3-difluoromethylthreonine methyl ester 11, which was generated in six steps starting from (S)-1-methyl-4-(methylsulfinyl)benzene (Scheme 1).4 Our synthetic strategy for amino ester 11 was adapted from a published procedure for the corresponding monofluoro analogue.8 The copper(I)-catalyzed [2+3] cycloaddition reaction between methyl isocyanoacetate and (S)-1,1-difluoro-3-(p-tolylsulfinyl)propan-2-one (6) provided oxazoline 7 as a mixture of diastereomers. The desired diastereoisomer of oxazoline 7 (i.e., 4S,5S,SS) is the less polar spot on TLC with Rf ~ 0.35 [EtOAc/hexanes (2:3)] and was isolated by flash chromatography [EtOAc/hexanes (0–40%)] as the major product in 45% yield. Oxazoline 7 was readily hydrolyzed in a chloroform/water mixture at room temperature to give the amido ester 8 in near quantitative yield. Sulfoxide 8 was reduced to corresponding sulfide 9 by using trifluoroacetic anhydride with sodium iodide in acetone at −40 °C.9 Desulfurization of 9 with a Raney nickel–sodium hypophosphite system provided the (2S,3S)-3-difluoromethyl-threonine derivative (10), which upon mild acidic hydrolysis afforded (2S,3S)-3-difluoromethylthreonine methyl ester 11.10
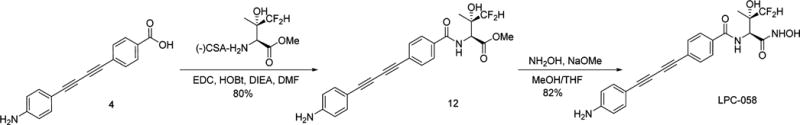
Amino ester 11 was reacted with diacetylene carboxylic acid 4 under standard coupling conditions to give amide ester 12. Treatment of 12 with hydroxylamine under alkaline conditions afforded desired hydroxamate LPC-058.4
A number of factors made this approach unsuitable for delivering substantial quantities (>100 g) of LPC-058. The major drawback of this strategy is the tedious chromatographic separation of oxazoline 7 from its diastereomers. Furthermore, chiral sulfoxide 5 is very expensive or has to be prepared.11 Finally, the copper-catalyzed cross-coupling of methyl 4-ethynylbenzoate 1 with excess 4-ethylnylbenzenamine 2 is complicated by the formation of homocoupled products that require tedious chromatographic separation. Therefore, we directed our attention to developing efficient and scalable routes to the key intermediates (2S,3S)-3-difluoromethylthreonine methyl ester 11 and diacetylene carboxylic acid 4.
Synthesis of unsymmetrical diynes has recently been the subject of extensive research. A survey of the literature indicates that diynes can be formed under various conditions, and many useful methods have been reported.12–16 In 2010, Balaraman et al. improved the synthesis of symmetrical and unsymmetrical 1,3-diynes by employing catalytic amounts (0.1 equiv) of copper(II) acetate in the presence of a stoichiometric amount of piperidine in 1,2-dichloroethane under aerobic conditions.12 With this method, the yield of unsymmetrical 1,3-diyne 3 was >60% (an improvement over that described above); however, unfortunately, it still involved 5 equiv of 4-ethylnylbenzenamine 2, and homocoupled byproducts remained a problem.
Efficient syntheses of unsymmetrical diynes using an alkynyl halide and a terminal alkyne have been reported.14,15 To reduce the need for excess 4-ethylnylbenzenamine 2, we replaced methyl 4-ethynylbenzoate 1 with methyl 4-(bromoethynyl)-benzoate 1617 as described in Scheme 2. Treatment of methyl 4-ethynylbenzoate 1 with N-bromosuccinimide in the presence of silver trifluoroacetate as a catalyst provided 4-(bromoethynyl)-benzoate 1617 in 83% yield. The reaction of 16 with a near-stoichiometric amount of 4-ethynylbenzenamine 2 (1.05 equiv) and copper(II) acetate (0.05 equiv) as a catalyst in the presence of piperidine proceeded efficiently to give diyne 3 (92% yield). In the scale-up process, crude intermediate 3 was hydrolyzed to give diacetylene acid 4 without purification.
Scheme 2.
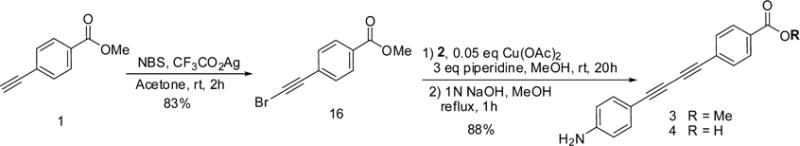
Synthesis of Diyne Carboxylic Acid 4
We next turned our attention to the large scale synthesis of (2S,3S)-3-difluoromethylthreonine methyl ester 11. The chiral sulfoxide-controlled stereoselective synthesis of optically pure (2S,3S)-3-difluoromethylthreonine methyl ester 11 (Scheme 1) is efficient and useful on a small scale. However, on a large scale, this approach has significant drawbacks, including (1) the high cost of (S)-1-methyl-4-(methylsulfinyl)benzene, (2) the fact that there was only a single crystalline intermediate in the sequence (i.e., chiral difluoroacetone 6), and (3) the low boiling point of trifluoroacetic anhydride that can be a safety concern when it is used in large scale reactions. Taken together, these concerns led us to re-evaluate our strategy. During the development of an alternative scalable route, our main objectives were (a) to address the safety issues, (b) to enhance the robustness of the synthetic route, and (c) to devise a sequence that had the potential for additional crystalline intermediates to facilitate purification by recrystallization. We took advantage of the well-documented transition metal/base-catalyzed aldol reaction to construct oxazolines in a stereo-selective manner.18 It is worth mentioning that the authors in that work have included entries of monofluoroacetone and trifluoroacetone reacting with methyl α-isocyanoacetate to yield corresponding oxazolines stereoselectively in ratios of 71:29 and 99:1, respectively. Inspired by their results, we had hoped to obtain stereoselectivity somewhere between the aforementioned ratios when reacting difluoroacetone with methyl α-isocyanoacetate as described in Scheme 3.
Scheme 3.
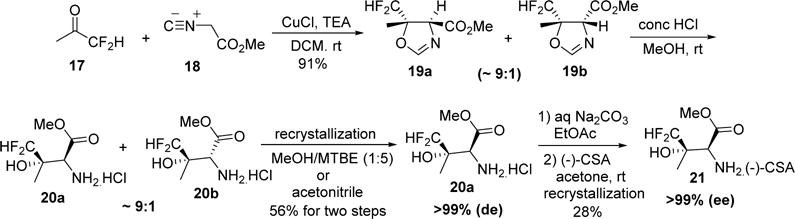
Synthesis of Amine
Difluoroacetone 17 was reacted with methyl α-isocyanoacetate 18 to give the diastereomeric mixture of oxazolines 19a and 19b. The reaction was performed in dry 1,1-dichloroethane (DCE) at ambient temperature in the presence of 4 mol % CuCl and 5 mol % TEA. The ratio of oxazoline diastereomers 19a and 19b is 87:13, which was determined by NMR chemical shifts of the crude reaction mixture after removal of the catalyst and solvent. Replacing DCE with DCM as the reaction solvent also provided similar results in terms of chemical yield and diastereoselectivity. Hydrolysis of the diastereomeric mixture of oxazolines 19a and 19b in methanolic HCl at room temperature afforded the corresponding amino acid hydrochloride salts 20a and 20b. Desired diastereomer 20a was obtained in >99% purity after recrystallizaion from a MeOH/MTBE solvent at room temperature with a chemical yield of 42%. Alternatively, recrystallization can be accomplished from hot acetonitrile to provide 20a in 56% yield and >99% purity. To obtain optically pure 21, amino acid hydrochloride salt 20a was neutralized by Na2CO3 and the racemic free base was reacted with equimolar amounts of R-(−)-camphorsulfonic acid [R-(−)-CSA] at room temperature in acetone. When the mixture was cooled, the desired amino ester 21 was obtained in 60% yield based on a single enantiomer with an optical purity of >99%. Optical purity was established using 1H NMR and 19F NMR as follows: 1H NMR and 19F NMR of racemic hydrochloride salt in the presence of a 10-fold excess of (+)-tartaric acid in CD3OD yielded a spectrum with well-resolved peaks. In particular, the α-CH, ester CH3, and 19F of the racemic methyl ester (Figure 1A,D) were well-resolved and readily identified when the spectra were recorded in the presence of excess tartaric acid (Figure 1B,E). It should be noted that 1H NMR and 19F NMR of a recrystallized optically pure sample of 21 in the presence of a 10-fold excess of (+)-tartaric acid in CD3OD displayed only one set of peaks corresponding to the desired enantiomer (Figure 1C,F). The modified route to chiral amino ester 21 was shorter and cheaper than that described earlier for the corresponding hydrochloride salt 11 and was deemed suitable for large scale synthesis.
Figure 1.
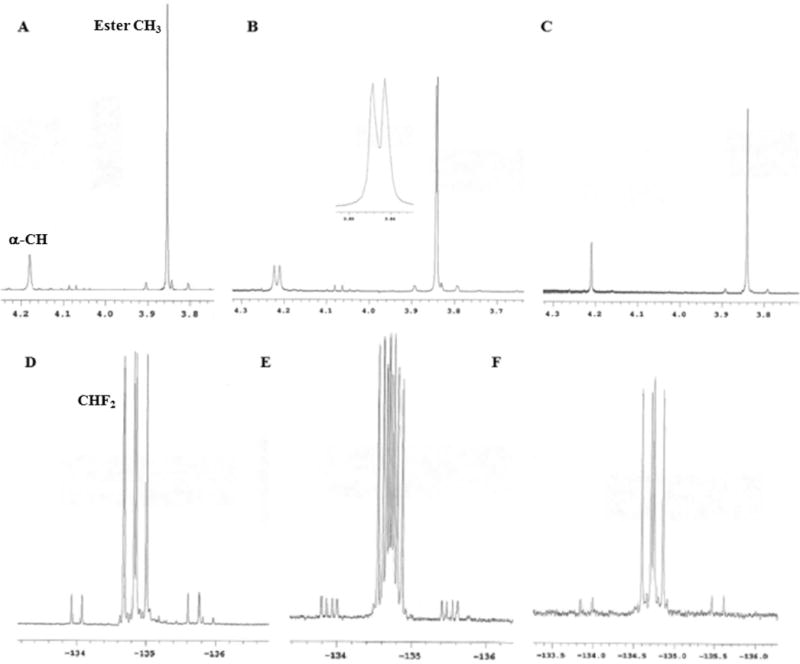
(A and D) Racemic hydrochloride salt 20a in CD3OD. (B and E) Racemic hydrochloride salt 20a in the presence of a 10-fold excess of (+)-tartaric acid in CD3OD. (C and F) Recrystallized optically pure sample of 21 in the presence of a 10-fold excess of (+)-tartaric acid in CD3OD. (A–C) 1H spectra and (D–F) 19F spectra.
With multigram quantities of amino ester 21 and diacetylene carboxylic acid 4 in hand, we proceeded to the large scale synthesis of LPC-058 (Scheme 4). Amide coupling under EDC/HOBt conditions provided diacetylene ester 12. Treatment of 12 with hydroxylamine and sodium methoxide at 0 °C provided hydroxamate LPC-058 in 67% yield over two steps. It is noted that no epimerization at the α-carbon was observed by LC–MS or NMR under these conditions.
Scheme 4.
Amide Coupling
To determine the absolute configuration of 11 and 21, they were crystallized in methanol/MTBE and acetone solvents, respectively. The absolute configuration was determined by the measurement of the Flack parameter, which is calculated during the structural refinement.19,20 If the value is near 0, the crystal is optically pure, in which case the absolute structure of the molecule can be determined with certainty by the X-ray data. In our study, the final refinement of the Cu Kα data of the crystal of 11 and 21 resulted in Flack parameters of 0.0(0) and −0.0(1), allowing an unambiguous assignment of the absolute structure as shown in Figure 2. The two chiral centers of 11, C-2 and C-3, and the four chiral centers of 21, C-2, C-3, C-8, and C-11, were thus determined to be S and S and S, S, R, and S, respectively. In summary, a short and scalable synthesis of hydroxamic acid derivative LPC-058 is described. The syntheses of key intermediates diacetylene carboxylic acid 4 and difluorothreonine methyl ester 21 were accomplished without any chromatography steps.
Figure 2.
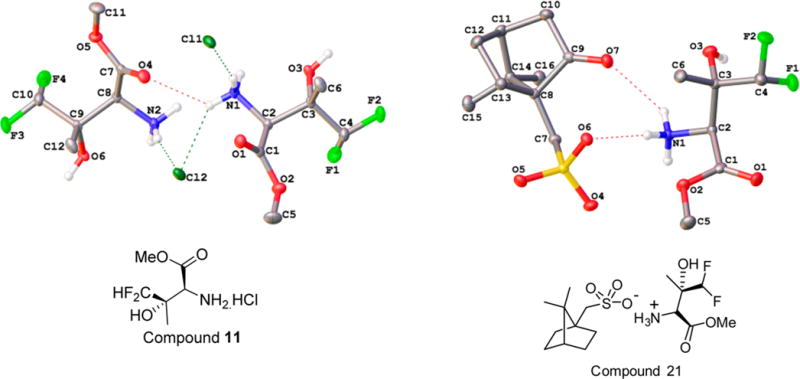
X-ray crystallographic structures of 11 and 21. The compounds are colored in the following atom colors: carbon, gray; nitrogen, blue; oxygen, red; sulfur, yellow; fluorine, green; chlorine, deep green.
EXPERIMENTAL SECTION
Methyl 4-ethynylbenzoate, 4-ethylnylbenzenamine, 1,1-difluoroacetone, and methyl α-isocyanoacetate were purchased from commercial vendors and used without further purification. LC–MS analysis was conducted on a HPLC instrument with a quadrapole mass analyzer and reverse phase C18 column (4.6 mm × 50 mm, 1.8 μm) with a water/acetonitrile [each with 0.2% (v/v) formic acid] gradient at a flow rate of 0.5 mL/min. The HRMS analyses were conducted on a TOF mass spectrometer using ESI mode. 1H, 13C, and 19F spectra were recorded on 300 (or 400) MHz, 75 (or 100) MHz, and 282.4 (or 376.5) MHz spectrometers, respectively. Column chromatography was conducted using either silica gel flash chromatography or prepacked silica gel cartridges. All moisture-sensitive reactions were conducted using dry solvents under a slight pressure of ultrapure quality argon. Glassware was dried in an oven at 140 °C for at least 12 h prior to being used and then assembled quickly while hot, sealed with rubber septa, and allowed to cool under a stream of argon. Reaction mixtures were stirred magnetically using Teflon-coated magnetic stirring bars. Commercially available disposable syringes were used for transferring reagents and solvents.
Methyl 4-(Bromoethynyl)benzoate (16).17
To a solution of methyl 4-ethynylbenzoate 1 (10.00 g, 62.5 mmol, 1.0 equiv) in acetone (100 mL) were added NBS (12.24 g, 68.8 mmol, 1.05 equiv) and CF3CO2Ag (0.53 g, 0.31 mmol, 0.05 equiv) at room temperature under argon. The reaction mixture was stirred at room temperature for 2 h. The resulting solution was concentrated to remove acetone. The residue was diluted with water (100 mL) and extracted with EtOAc (3 × 80 mL). The combined organic layers were washed with water (2 × 60 mL) and brine (60 mL) and dried over anhydrous Na2SO4. Evaporation of the solvent afforded the crude product that was purified by silica gel chromatography (eluting with EtOAc in hexane, 0–10%) to give 16 as a white solid (12.4 g, 83% yield). 1H NMR (300 MHz, CDCl3): δ 3.9 (s, 3H), 7.50 (d, J = 8.1 Hz, 2H), 7.97 (d, J = 6.9 Hz, 2H). 13C NMR (75 MHz, CDCl3): δ 52.3, 53.4, 79.4, 127.3, 129.5, 129.9, 131.9, 166.3. MS (LC/MS, ESI): m/z 240 [M + H]+.
Methyl 4-[(4-Aminophenyl)buta-1,3-diyn-1-yl]benzoate (3).7
Copper(II) acetate (0.45 g, 0.50 mmol, 0.05 equiv) was added at room temperature under a stream of argon to a stirred solution of methyl (4-bromoethylnyl)benzoate (12.0 g, 50.0 mmol, 1.00 equiv), 4-ethynylaniline 2 (6.20 g, 52.5 mmol, 1.05 equiv), and piperidine (12.77 g, 150.0 mmoL, 3.00 equiv) in MeOH (200 mL, degassed with argon). The reaction mixture was stirred at room temperature for 20 h. The resulting suspension was diluted with water (400 mL) and stirred for 30 min at room temperature. The mixture was filtered, and the filtered solid was washed with water (2 × 200 mL). The solid obtained was dried to afford crude 3 (13.5 g) as a yellow solid that was carried on to the next step without further purification. 1H NMR (300 MHz, DMSO-d6): δ 3.83 (s, 3H), 5.85 (s, 2H), 6.54 (d, J = 8.1 Hz, 2H), 7.25 (d, J = 8.4 Hz, 2H), 7.64 (d, J = 8.1 Hz, 2H), 7.92 (d, J = 8.1 Hz, 2H). 13C NMR (75 MHz, DMSO-d6): δ 53.0, 71.8, 78.3, 80.5, 87.0, 105.7, 114.3, 126.7, 130.1, 130.3, 133.0, 134.8, 151.6, 166.2. MS (LC/MS, ESI): m/z 276 [M + H]+.
4-[(4-Aminophenyl)buta-1,3-diyn-1-yl]benzoic Acid (4).7
To a solution of crude compound 3 (13.5 g, 50.0 mmol, 1.00 equiv) in methanol (100 mL) was added 1 N NaOH (100 mL, 100.0 mmol, 2.00 equiv) at room temperature under a stream of argon. The reaction mixture was heated to reflux for 1 h. The solution was diluted with water (100 mL) and acidified with concentrated HCl to pH ~3. The precipitate was filtered, washed with water, and dried under vacuum to give crude acid 4 (11.5 g, two steps, 88% yield for two steps), which was carried to the next step without purification. 1H NMR (300 MHz, DMSO-d6): δ 6.95 (d, J = 8.4 Hz, 2H), 7.45 (d, J = 8.4 Hz, 2H), 7.66 (d, J = 8.4 Hz, 2H), 7.93 (d, J = 8.4 Hz, 2H). 13C NMR (75 MHz, DMSO-d6): δ 73.0, 77.2, 81.2, 85.0, 112.0, 118.8, 125.8, 130.2, 131.9, 133.1, 134.6, 144.2, 167.2. MS (LC/MS, ESI): m/z 262 [M + H]+.
(4S,5S)-Methyl 5-(Difluoromethyl)-5-methyl-4,5-dihydrooxazole-4-carboxylate (19a)
Methyl 2-isocyanoacetate (20.00 g, 201.8 mmol, 1.00 equiv) was added slowly to an ice-cold suspension of 1,1-difluoroacetone (22.78 g, 242.20 mmol, 1.20 equiv), CuCl (0.80 g, 8.10 mmol, 0.04 equiv), and TEA (1.41 mL, 10.10 mmol, 0.05 equiv) in anhydrous DCM (330 mL) under argon. The reaction mixture was stirred at 0 °C and gradually warmed to room temperature overnight (20 h). The resulting solution was diluted with DCM (150 mL). The mixture was washed with 10% aqueous ammonia (3 × 100 mL), water (100 mL), and brine (100 mL) and dried (anhydrous Na2SO4). Evaporation of the solvent afforded a crude mixture (86:14) of diastereomers 19a and 19b (brown liquid, 35.5 g, 91% yield), which was carried to the next step without further purification. 1H NMR (400 MHz, CDCl3): δ 1.35 (s, 3H), 1.54 (s, 0.5H), 3.67 (s, 0.5H), 3.74 (s, 3H), 4.47 (s, 0.17H), 4.78 (s, 1H), 5.69 (t, JHF = 56 Hz, 1H), 5.88 (t, JHF = 56 Hz, 0.17H), 6.94 (s, 1H). 13C NMR (100 MHz, CDCl3): δ 15.4, 43.4, 52.5, 114.2 (t, JCF = 494 Hz), 168.7. 19F NMR (376.5 MHz, CDCl3): δ −132.83 (d, JHF = 56 Hz, major), −131.40 (ABq, d, 2F, JHF = 56 Hz, JFF = 300 Hz, minor). MS (LC/MS, ESI): m/z 194 [M + H]+.
Methyl 2-Amino-4,4-difluoro-3-hydroxy-3-methylbutanoate Hydrochloride (racemic 20a)
To a stirred solution of oxazoline 19a and 19b (35.30 g, 182.70 mmol, 1.00 equiv) in methanol (180 mL) kept at room temperature in a water bath under argon was added dropwise concentrated HCl (36 mL). The reaction mixture was stirred at room temperature for 14 h. The resulting solution was concentrated to dryness. The residue was diluted with MTBE (400 mL) and stirred vigorously for 2 h. The suspension was concentrated to afford crude 20a and 20b as a light brown solid. 1H NMR (400 MHz, CD3OD): δ 1.30 (s, 3H), 1.48 (s, 0.5H), 3.85 (s, 0.5H), 3.86 (s, 3H), 4.15 (s, 0.17H), 4.16 (s, 1H), 5.95 (t, JHF = 56 Hz, 0.17H), 5.99 (t, JHF = 56 Hz, 1H). 19F NMR (376.5 MHz, CD3OD): δ −134.25, −135.15 (ABq, d, 2F, JHF = 56 Hz, JFF = 301 Hz). 13C NMR (100 MHz, CD3OD): δ 15.9, 52.6, 55.9, 71.2 (t, 2JCF = 42.5 Hz), 115.4 (t, JCF = 491 Hz), 166.7. MS (LC/MS, ESI): m/z 184 [M + H]+. The crude product was dissolved to ~55 mL of methanol and MTBE (∼270 mL) at room temperature. Then the solution was left at −20 °C for 2 days to give the racemic 20a (17.00 g, 42% yield) as an off-white solid. Alternatively, a crude mixture of 20a and 20b can be recrystallized from hot acetonitrile (∼200 mL) to provide racemic 20a in 56% yield. Mp: 142 °C. 1H NMR (400 MHz, CD3OD): δ 1.30 (s, 3H), 3.87 (s, 3H), 4.16 (s, 1H), 5.99 (t, JHF = 56 Hz, 1H). 19F NMR (376.5 MHz, CD3OD): δ −134.25, −135.15 (ABq, d, 2F, JHF = 56 Hz, JFF = 301 Hz). 13C NMR (100 MHz, CD3OD): δ 15.9, 52.6, 56.0, 71.2 (t, 2JCF = 42.5 Hz), 115.4 (t, JCF = 490 Hz). 19F NMR (376 MHz, CDCl3): δ −134.70. HRMS (ESI/TOF): m/z [M + H]+ calcd for C6H12F2NO3 184.0785, found 184.0776.
(2S,3S)-Methyl 2-Amino-4,4-difluoro-3-hydroxy-3-methylbutanoate (−)-CSA (21)
To an ice bath-cooled solution of racemic 20 (16.70 g, 76.00 mmol, 1.00 equiv) in water (100 mL) was added Na2CO3 (24.17 g, 228.00 mmol, 3.00 equiv) under argon. The reaction mixture was stirred at room temperature for 1 h. Then the resulting solution was extracted with EtOAc (6 × 100 mL). The combined organic layers were dried over anhydrous Na2SO4. Evaporation of the solvent afforded the free amine (14.0 g) as a light brown liquid. The free amine (13.50 g, 73.7 mmol, 1.00 equiv) was dissolved in acetone (100 mL), and (1R)-(−)-10-camphorsulfonic acid (17.12 g, 73.70 mmol, 1.00 equiv) was added. The mixture was stirred at room temperature overnight. The white solid was filtered and washed with acetone (20 mL) and dried to give (−)-CSA salt 21 (8.40 g, 28% yield). Mp: 160 °C. [α]D25 = −24.1 (c = 0.4; MeOH). 1H NMR (400 MHz, CD3OD): δ 0.83 (s, 3H), 1.09 (s, 3H), 1.30 (s, 3H), 1.37–1.42 (m, 1H), 1.57–1.64 (m, 1H), 1.88 (d, J = 18.4 Hz, 1H), 2.00–2.04 (m, 2H), 2.29–2.35 (m, 1H), 2.57–2.61 (m, 1H), 2.76 (d, J = 14.8 Hz, 1H), 3.86 (s, 3H), 4.21 (s, 1H), 5.98 (t, JHF = 56 Hz, 1H). 19F NMR (376.5 MHz, CD3OD): δ −134.23, −135.13 (ABq, d, 2F, JHF = 56 Hz, JFF = 301 Hz). 13C NMR (100 MHz, CD3OD): δ 5.8, 18.7, 19.0, 24.3, 26.4, 42.2, 42.6, 46.8, 52.6, 56.0, 58.2, 71.2 (t, 2JCF = 42.5 Hz), 115.4 (t, JCF = 491 Hz), 166.7, 218.1. 19F NMR (376 MHz, CDCl3): δ −134.68 (t, JHF = 56 Hz). HRMS (ESI/TOF): m/z [M + H]+ calcd for C6H12F2NO3 184.0785, found 184.0781.
(2S,3S)-Methyl 2-{4-[(4-Aminophenyl)buta-1,3-diyn-1-yl]-benzamido}-4,4-difluoro-3-hydroxy-3-methylbutanoate (12).4
To a solution of diacetylene carboxylic acid 4 (3.00 g, 12.49 mmol) in anhydrous DMF (25 mL) were added amino ester 21 (5.40 g, 13.11 mmol, 1.05 equiv), EDC-HCl (2.64 g, 13.80 mmol, 1.2 equiv), and HOBt (1.87 g, 13.80 mmol, 1.2 equiv) at room temperature under argon. The mixture was cooled to 0 °C, and DIEA (8.1 mL, 45.90 mmol, 4.00 equiv) was added. The reaction mixture was stirred at 0 °C for 2 h and then allowed to warm to room temperature for 14 h. The yellow solution was then concentrated to dryness. The residue was treated with water (100 mL) and extracted with EtOAc (3 × 80 mL). The combined extracts were washed with water (80 mL) and brine (80 mL) and dried (anhydrous Na2SO4). The crude product was purified by silica gel chromatography (eluting with MeOH in DCM, 0–2.5%) to give 12 as a yellow solid (4.23 g, 80%). Mp: 167–168 °C dec. [α]D25 = +28.2 (c = 0.2; MeOH). 1H NMR (300 MHz, CD3OD): δ 1.37 (d, J = 2.1 Hz, 3H), 3,76 (s, 3H), 4.267 (dd, J = 9.3, 19.5 Hz, 1H), 4.42 (dd, J = 9.3, 19.8 Hz, 1H), 4.81 (s, 1H), 5.85 (t, JHF = 56 Hz, 1H), 6.61 (d, J = 8.4 Hz, 2H), 7.24 (d, J = 8.4 Hz, 2H), 7.56 (d, J = 8.1 Hz, 2H), 7.82 (d, J = 8.1 Hz, 2H). 19F NMR (400 MHz, CD3OD): δ −131.06, −137.61 (ABq, d, 2F, JHF = 56 Hz, JFF = 300 Hz). 13C NMR (75 MHz, CD3OD): δ 20.5, 51.7, 70.9, (71.9, 72.2), 76.9, 79.1, 84.7, (85.5, 87.8), 108.3, 114.3, 126.2, 127.2, 127.6, 132.2, 133.6, 133.9, 150.2, 168.2, 170.6. MS (LC/MS, ESI): m/z 427 [M + H]+. HRMS (ESI/TOF): m/z [M]+ calcd for C23H20F2N2O4 426.1391, found 426.1392.
4-[(4-Aminophenyl)buta-1,3-diyn-1-yl]-N-[(2S,3S)-4,4-difluoro-3-hydroxy-1-(hydroxyamino)-3-methyl-1-oxobutan-2-yl]benzamide (LPC-058).4
To an ice-cold solution of 12 (4.00 g, 9.4 mmol) dissolved in anhydrous MeOH (15 mL) and THF (15 mL) was added hydroxylamine hydrochloride (3.27 g, 46.9 mmol, 5.0 equiv) followed by 25% sodium methoxide in a methanol solution (16.0 mL, 70.50 mmol, 7.5 equiv). The reaction mixture was stirred under argon at 0 °C for 2 h and then allowed to warm to ambient temperature while stirring was continued overnight (14 h). The resulting yellow suspension was condensed to dryness with a rotary evaporator, and the residue obtained was treated with water (200 mL) and saturated NH4Cl (80 mL) and extracted with EtOAc (3 × 100 mL). The combined extracts were washed with water (80 mL) and brine (80 mL) and dried over anhydrous Na2SO4. Evaporation of the solvent afforded the crude product, which was purified by silica gel chromatography (eluting with 0–5% MeOH in DCM) to afford the title compound as a yellow solid (3.29 g, 82% yield). Mp: 155–157 °C dec. [α]D25 = +61.2 (c = 0.4; MeOH). 1H NMR (300 MHz, CD3OD): δ 1.36 (s, 3H), 4.73 (s, 1H), 5.80 (t, JHF = 56 Hz, 1H), 6.61 (d, J = 8.7 Hz, 2H), 7.24 (d, J = 8.7 Hz, 2H), 7.57 (d, J = 8.1 Hz, 2H), 7.82 (d, J = 8.4 Hz, 2H). 19F NMR (400 MHz, CD3OD): δ −130.00, −137.91 (ABq, d, 2F, JHF = 56 Hz, JFF = 300 Hz). 13C NMR (75 MHz, CD3OD): δ 16.5, 54.9, 70.8, (72.6, 72.9, 73.2), 76.8, 79.0, 84.7, 108.4, (112.7, 116.0, 119.2), 114.3, 126.3, 127.5, 132.2, 133.5, 133.8, 150.2, 166.5, 167.5. MS (LC/MS, ESI): m/z 428.2 [M + H]+. HRMS (ESI/TOF): m/z [M]+ calcd for C22H19F2N3O4 427.1344, found 427.1346.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health Grants AI055588, AI094475, and GM115355 awarded to P.Z. We thank Dr. Kalpathy Santhosh for experimental assistance.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.joc.6b00589.
Crystallographic data for 11 (CIF)
Crystallographic data for 21 (CIF)
1H, 13C, and wherever applicable 19F NMR spectra of all compounds synthesized (PDF)
Notes
The authors declare no competing financial interest.
References
- 1.Vergidis PI, Falagas ME. Curr Opin Invest Drugs. 2008;9:176. [PubMed] [Google Scholar]
- 2.Boucher HW, Talbot GH, Bradley JS, Edwards JE, Gilbert D, Rice LB, Scheld M, Spellberg B, Bartlett Clin Infect Dis. 2009;48:1. doi: 10.1086/595011. [DOI] [PubMed] [Google Scholar]
- 3.Cooper MA, Shlaes D. Nature. 2011;472:32. doi: 10.1038/472032a. [DOI] [PubMed] [Google Scholar]
- 4.Lee CJ, Liang XF, Wu QL, Najeeb J, Zhao J, Gopalaswamy R, Titecat M, Sebbane F, Lemaitre N, Toone EJ, Zhou P. Nat Commun. 2016;7:10638. doi: 10.1038/ncomms10638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Glaser C. Ber Dtsch Chem Ges. 1869;2:422. [Google Scholar]
- 6.Nicolaou KC, Zipkin RE, Petasis NA. J Am Chem Soc. 1982;104:5558. [Google Scholar]
- 7.Liang XF, Lee CJ, Chen X, Chung HS, Zeng DN, Raetz C, Li Y, Zhou P, Toone EJ. Bioorg Med Chem. 2011;19:852. doi: 10.1016/j.bmc.2010.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Arnone A, Gestmann D, Meille SV, Resnati, Sidoti G. Chem Commun. 1996;22:2569. [Google Scholar]
- 9.Drabowicz J, Oae S. Synthesis. 1977;1977:404. [Google Scholar]
- 10.Nishide K, Shigeta Y, Obata K, Inoue T, Node M. Tetrahedron Lett. 1996;37:2271. [Google Scholar]
- 11.Zhao SH, Samuel O, Kagan HB. Organic Syntheses. 1993;8:464. [Google Scholar]
- 12.Balaraman K, Kesavan V. Synthesis. 2010;2010:3461. [Google Scholar]
- 13.Nye SA, Potts KT. Synthesis. 1988;1988:375. [Google Scholar]
- 14.Weng Y, Cheng B, He C, Lei A. Angew Chem Int Ed. 2012;51:9547. doi: 10.1002/anie.201204112. [DOI] [PubMed] [Google Scholar]
- 15.Jiang HF, Wang AZ. Synthesis. 2007;2007:1649. [Google Scholar]
- 16.Paixao MW, Weber M, Braga AL, de Azeredo JB, Deobald AM, Stefani HA. Tetrahedron Lett. 2008;49:2366. [Google Scholar]
- 17.Karad SN, Liu RS. Angew Chem Int Ed. 2014;53:9072. doi: 10.1002/anie.201405312. [DOI] [PubMed] [Google Scholar]
- 18.Soloshonok VA, Kacharov AD, Avilov DV, Ishikawa K, Nagashima NJ, Hayashi T. J Org Chem. 1997;62:3470. [Google Scholar]
- 19.Flack HD, Bernardinelli G. Chirality. 2008;20:681. doi: 10.1002/chir.20473. [DOI] [PubMed] [Google Scholar]
- 20.Flack HD. Acta Crystallogr Sect A: Found Crystallogr. 1983;39:876. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.