

Abstract
Reactive oxygen species (ROS) and reactive nitrogen species (RNS) are produced, in part, from NADPH oxidase in response to host invasion and tissue injury. Defects in NADPH oxidase impair host defense; however, the role of ROS and RNS in the response to tissue injury is not known. We addressed this issue by subjecting leukocyte oxidase (Nox2)-deficient (Nox2-/-) mice to arterial injury. Femoral artery injury was associated with increased Nox2 expression, ROS/RNS production, and oxidative protein and lipid modification in wild-type mice. In Nox2-/- mice, RNS-mediated protein oxidation, as monitored by protein nitrotyrosine content, was significantly diminished. This was accompanied by reduced neointimal proliferation, as monitored by intimal thickness and intimal/medial ratio, in Nox2-/- compared to wild-type mice. In addition, Nox2 deficiency led to reduced cellular proliferation and leukocyte accumulation. These data indicate that Nox2-mediated oxidant production has a requisite role in the response to tissue injury.
Oxidative stress and production of both reactive oxygen species (ROS) and reactive nitrogen species (RNS) are thought to contribute to the pathophysiology of vascular diseases such as atherosclerosis (1, 2) and restenosis (3-9). ROS have been implicated in many aspects of vascular injury and neointimal formation, including endothelial cell dysfunction, inflammatory cell recruitment, foam cell formation, and smooth muscle cell (SMC) proliferation (reviewed in ref. 10). Although early studies of oxidant generation were typically limited to phagocytic cells that contain the prototypical NADPH oxidase, it is now well accepted that nonphagocytic cells, such as endothelial cells (11), vascular SMC (12), and fibroblasts (13), also express NADPH oxidase isoforms that participate in the generation of both ROS and RNS.
The NADPH oxidase found in phagocytic cells is a multisubunit complex consisting of membrane-bound and cytosolic components (14). The former consists of flavocytochrome b558, a heterodimer of gp91phox (now known as Nox2), and the smaller subunit, p22phox. Multiple cytoplasmic subunits (p47phox, p67phox, p40phox, Rac1, and Rac2) associate with the membrane component to provide complete enzymatic activity (15, 16)). Although less well characterized in nonphagocytic cells, NADPH oxidase subunits are present in vascular tissue. The cytosolic components and p22phox appear ubiquitously expressed, whereas the catalytic subunits (now known as Nox isoforms) vary among different vascular cell types. For example, vascular endothelium expresses predominantly Nox2 (11, 17) and Nox4. SMC express Nox1 (18-20), Nox4, and, to a lesser extent, Nox3 (20, 21). The expression of Nox2 and Nox4 has been demonstrated in the vascular adventitia (13). The precise molecular characterization of the Nox isoforms is incomplete, but available evidence suggests they have functional and structural similarity to Nox2 (22-24).
In vitro studies have established that ROS/RNS production by the NADPH oxidase complex serve as critical signals regulating gene transcription, cell growth, and apoptosis (25-27). In vivo studies indicate that vascular injury in atherosclerosis and restenosis is associated with markedly enhanced superoxide production and up-regulated expression of both membrane and cytosolic subunits of NADPH oxidase (reviewed in ref. 28). However, there is conflicting evidence concerning the role of NADPH oxidases in the biological responses to vascular injury (29-31). Thus, we sought to determine the role of NADPH oxidase activity in neointimal formation by using Nox2-deficient (Nox2-/-) mice exposed to arterial injury.
Methods
Femoral Artery Injury. Female wild-type and Nox2-/- C57BL/6J mice (>12 generations backcrossed; The Jackson Laboratory) aged 8-10 weeks were anesthetized on day 0 by using ketamine (80 mg/kg i.p.) and xylazine (5 mg/kg i.p.), and wire injury of the femoral artery was performed as described (32). All animals survived until the time of planned death without bleeding or infection. Animal care and procedures were reviewed and approved by the Harvard Medical School Standing Committee on Animals and were performed in accordance with the guidelines of the American Association for Accreditation of Laboratory Animal Care and the National Institutes of Health.
Tissue Harvesting and Analysis. Before (wild-type, n = 8; Nox2-/-, n = 8) and 7 d (wild-type, n = 7; Nox2-/-, n = 8) or 28 d after (wild-type, n = 15; Nox2-/-, n = 15) vascular injury, anesthesia was administered, the chest cavity opened, and the animals killed by right atrial exsanguination. A 22-gauge butterfly catheter was inserted into the left ventricle for in situ pressure perfusion at 100 mmHg (1 mmHg = 133 Pa) with 0.9% saline for 1 min followed by fixation with 4% paraformaldehyde in 0.1 M phosphate buffer, pH 7.3, for 10 min. The right and left femoral arteries were excised and immersed in buffered paraformaldehyde. Spleen and small intestine from three animals were harvested as control tissues for immunohistochemistry. All animals received BrdUrd, 50 mg/kg i.p., 18 and 1 h before death.
Femoral arteries were embedded, and two cross sections cut 1 mm apart were stained with hematoxylin/eosin and Verhoeff tissue elastin stain. A histologist blinded to the animal genotype measured the lumen, intimal, and medial areas of each cross-sectional plane using a microscope equipped with a charge-coupled device camera interfaced to a computer running nih image (version 1.60) software. Results for the two planes of each artery were averaged. For immunohistochemistry, standard avidin-biotin procedures for mouse CD45 (leukocyte common antigen, BD Biosciences), BrdUrd (Dako, Carpenteria, CA), SMC α-actin (Sigma), and Nox2 (Santa Cruz Biotechnology) were used. Apoptotic cells were detected by the terminal deoxynucleotidyltransferase-mediated dUTP nick end-labeling method with Apo Tag (Intergen, Purchase, NY). Immunostained sections were quantified as the number of immunostained positive cells per total number of nuclei.
Superoxide Detection. Frozen, enzymatically intact 30-μm-thick sections of sham-operated and injured femoral arteries were incubated at the same time with dihydroethidine (10 μmol/liter) in PBS for 30 min at 37°C in a humidified chamber protected from light. Vascular superoxide was estimated as ethidium bromide formation as described (33).
Quantification of Nitrotyrosine, Dityrosine, and Lipid Peroxidation Products. Protein-bound nitrotyrosine and dityrosine levels were determined by stable isotope dilution liquid chromatography-electrospray ionization tandem mass spectrometry-based methods by using an ion trap mass spectrometer (LCQ Deca, Thermo Finigann, San Jose, CA), as described (34). Before (wild-type, n = 9; Nox2-/-, n = 9) and 3 d after (wild-type, n = 19; Nox2-/-, n = 19) vascular injury, femoral arteries were harvested, rinsed, and submerged in PBS containing an antioxidant mixture composed of 100 μM diethylenetriaminepentaacetic acid and 100 μM butylated hydroxytoluene, covered with nitrogen, and snap frozen in liquid nitrogen before storage at -80°C. Synthetic 3-nitro-[13C6]tyrosine (2 pmol), o,o′-[13C12]dityrosine (2 pmol), and [13C915N1]tyrosine (2 nmol) were added to protein pellets both as internal standards and to simultaneously monitor nitrotyrosine, dityrosine, tyrosine, and potential artifactual formation of nitrotyrosine and dityrosine during analyses (34). Protein nitrotyrosine and dityrosine content in samples are expressed as the molar ratio between oxidized amino acid and the precursor amino acid tyrosine. Quantification of hydroxyoctadecadienoic acids (HODEs) and hydroxyeicosatetraenoic acids (HETEs) was performed by using stable isotope dilution on-line reverse-phase HPLC tandem mass spectrometry, as described (35). The content of lipid peroxidation products is expressed as the molar ratio between oxidized fatty acid and the precursor fatty acid.
Data Analysis. All data are presented as mean ± SD. Comparisons among groups used a nonpaired t test. P values of <0.05 were considered significant.
Results
Femoral Artery Injury, Nox2 Expression, and Superoxide Production. To determine whether Nox2 is causally related to neointimal formation, we subjected wild-type and Nox2-/- mice to transluminal wire injury of the femoral artery. This method of injury induces abundant intimal hyperplasia formation by 2 and 4 weeks and elicits the rapid accumulation of adhesion molecules and leukocytes on the endothelial denuded luminal surface (32). This model has been useful in demonstrating that inflammatory cell recruitment and function modulate neointimal formation (36, 37).
Immunohistochemical staining of femoral arteries from wild-type mice showed enhanced Nox2 expression within the media 7 d after injury that persisted 21 d after injury (Fig. 1 a-e). Expression of Nox2 was also evident in the neointima 21 d after injury (Fig. 1e). Double staining7dafter injury in wild-type mice for Nox2 and CD45 or SMC-specific α-actin shows both inflammatory and SMC expression of Nox2 (Fig. 1 f and g). This expression pattern is consistent with prior balloon injury studies in rat and rabbit carotid arteries showing enhanced expression of Nox2 within the media and neointima over time (20, 38).
Fig. 1.

Nox2 expression after carotid injury. Immunohistochemical staining of femoral artery 7 d after sham operation (a) or wire injury (b) showing enhanced Nox2 expression (reddish-brown color with hematoxylin counterstain delineating nuclei) within the media from wild-type mice (original magnification, ×150). Diminished staining is observed with nonimmune IgG (c) or in Nox2-/- vessel (d) 7 d after injury. Expression of Nox2 is also evident in the media and intima 21 d after injury in wild-type mice (e). Double staining without hematoxylin counterstaining 7 d after injury in wild-type mice for Nox2 (reddish-brown) and CD45 (blue, f) or smooth muscle cell α-actin (blue, g) shows both inflammatory cell and smooth muscle expression of Nox2, respectively. Arrow designates the internal elastic lamina; arrowheads designate double-positive cells.
Prior experimental studies indicate that leukocyte 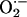 is reduced in Nox2-deficient mice (30) and that balloon injury leads to increased NADPH oxidase-dependent
is reduced in Nox2-deficient mice (30) and that balloon injury leads to increased NADPH oxidase-dependent 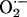 production (9, 39, 40). Therefore, we examined
production (9, 39, 40). Therefore, we examined 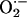 production in situ using dihydroethidium (DHE) fluorescence, as described (33). We observed low-intensity fluorescence in sham-operated vessels (Fig. 2). In contrast, wire injury of wild-type mice was associated with markedly increased DHE staining throughout the vessel wall that was particularly prominent in the adventitia, consistent with enhanced
production in situ using dihydroethidium (DHE) fluorescence, as described (33). We observed low-intensity fluorescence in sham-operated vessels (Fig. 2). In contrast, wire injury of wild-type mice was associated with markedly increased DHE staining throughout the vessel wall that was particularly prominent in the adventitia, consistent with enhanced 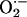 production. Enhanced DHE staining persisted 21 d after injury, with fluorescence evident in the media and the developing neointima. ROS levels, as monitored by DHE staining, increased in both wild-type and Nox2-/- mice after wire injury, although possibly to a lesser extent in Nox2-/- than in wild-type vessels at 7 d.
production. Enhanced DHE staining persisted 21 d after injury, with fluorescence evident in the media and the developing neointima. ROS levels, as monitored by DHE staining, increased in both wild-type and Nox2-/- mice after wire injury, although possibly to a lesser extent in Nox2-/- than in wild-type vessels at 7 d.
Fig. 2.

Superoxide production after wire injury. Superoxide production was detected in situ by staining femoral arteries with the superoxide-sensitive dye DHE (red fluorescence). (a) Sham-operated 7 d; (b) wild-type, wire injury 7 d; (c) Nox2-/-, wire injury 7 d; (d) sham-operated 21 d, (e) wild-type, wire injury 21 d; and (f) Nox2-/-, wire injury 21 d (original magnification, ×38). Each image is representative of results from three different animals.
To more quantitatively investigate oxidative events in response to wire injury, we measured femoral artery protein nitrotyrosine and dityrosine contents at baseline and after injury in wild-type and Nox2-/- vessels using stable isotope dilution liquid chromatography-electrospray ionization tandem mass spectrometry (34). Generation of NO-derived oxidants and tyrosyl radical, an intermediate in dityrosine formation, occurs through pathways that ultimately require 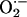 (34, 41). We hypothesized that Nox2 deficiency would result in diminished
(34, 41). We hypothesized that Nox2 deficiency would result in diminished 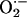 production and hence nitrotyrosine and dityrosine formation. Fig. 3 illustrates that vascular injury in wild-type mice is associated with a significant (80%) increase in femoral artery nitrotyrosine content (109 ± 81 to 197 ± 90 μmol of nitrotyrosine/mol of tyrosine, P = 0.019), and 50% increase in dityrosine (440 ± 170 to 674 ± 368 μmol of dityrosine/mol of tyrosine, P = 0.050) content. In marked contrast, nitrotyrosine levels did not change significantly in response to arterial injury in Nox2-/- mice (P = 0.77). Nitrotyrosine levels after wire injury were significantly higher in wild-type than in Nox2-/- mice (P = 0.02). Remarkably, dityrosine levels increased in both wild-type and Nox2-/- mice after injury and were unaffected by Nox2 status, suggesting that Nox2 participated in generation of NO-derived oxidants but not oxidative cross-linking of proteins through tyrosyl radical-generating pathways after vascular injury.
production and hence nitrotyrosine and dityrosine formation. Fig. 3 illustrates that vascular injury in wild-type mice is associated with a significant (80%) increase in femoral artery nitrotyrosine content (109 ± 81 to 197 ± 90 μmol of nitrotyrosine/mol of tyrosine, P = 0.019), and 50% increase in dityrosine (440 ± 170 to 674 ± 368 μmol of dityrosine/mol of tyrosine, P = 0.050) content. In marked contrast, nitrotyrosine levels did not change significantly in response to arterial injury in Nox2-/- mice (P = 0.77). Nitrotyrosine levels after wire injury were significantly higher in wild-type than in Nox2-/- mice (P = 0.02). Remarkably, dityrosine levels increased in both wild-type and Nox2-/- mice after injury and were unaffected by Nox2 status, suggesting that Nox2 participated in generation of NO-derived oxidants but not oxidative cross-linking of proteins through tyrosyl radical-generating pathways after vascular injury.
Fig. 3.

Femoral artery content of nitrotyrosine, dityrosine, and lipid peroxidation products. Protein-bound nitrotyrosine, dityrosine, and lipid peroxidation products, 8- and 15-HETE, were measured in femoral arteries before (n = 9 per group, open bars) and 3 d after (n = 19 per group, shaded bars) injury in wild-type and Nox2-/- mice by using electrospray ionization tandem mass spectrometry as described in Methods. Nitrotyrosine and dityrosine content in samples is expressed as the molar ratio between the oxidized tyrosine species and the precursor amino acid tyrosine; lipid peroxidation product content is expressed as the molar ratio between oxidized fatty acid and the precursor fatty acid arachidonate (mean ± SD).
The production of ROS/RNS from leukocytes has also been implicated in lipid peroxidation, and lipid-soluble antioxidants have been shown to limit the restenotic response after arterial injury (4, 42). Therefore, we quantified markers of lipid peroxidation in arterial samples before and after wire injury. Multiple specific oxidation products of both linoleic (9- and 13-HODE) and arachidonic (8-, 12-, 11-, and 15-HETE) acid increased as a function of wire injury in both control and Nox2-/- animals, with largest increases evident in products of 12-lipoxygenase (12-HETE and 13-HODE) (Table 2, which is published as supporting information on the PNAS web site). However, although the accumulation of the oxidized arachidonate derivatives 8- and 15-HETE was significantly reduced in Nox2-/- compared with wild-type mice (Fig. 3), no significant differences in tissue contents of other fatty acid oxidation products were observed (Table 2).
Deficiency of Nox2 Decreases Neointimal Thickening and Cellular Proliferation After Arterial Injury. In wild-type mice, intimal thickening was evident 7 d after injury and progressed significantly between 7 (2,928 ± 130 μm2) and 28 d (7,233 ± 3,678 μm2; Table 1). In Nox2-/- mice, intimal thickening was reduced by 34% at 28 d (P = 0.046) compared with wild-type mice (Table 1 and Fig. 4). Significant medial thickening was also observed from 0 to 28 d after injury and was similar in wild-type and Nox2-/- mice at 28 d (P = 0.24). In wild-type mice, the intima/media area ratio (I:M) increased from 0.24 ± 0.11 at 7 d to 0.97 ± 0.56 at 28 d. In Nox2-/- mice, I:M was reduced by 40% at 28 d (P = 0.036). Intimal and medial thickening was accompanied by vessel enlargement (i.e., positive remodeling), as determined by external elastic lamina (EEL) area measurements at 7d, that was comparable in wild-type and Nox2-/- mice. However, by 28 d, the EEL area was not significantly different from baseline in both wild-type and Nox2-/- mice.
Table 1. Quantitative morphometry and immunohistochemistry.
| Wild-type | Nox2−/− | P value | |
|---|---|---|---|
| Intimal area, μm2 | |||
| 7 d, n = 5 | 2,928±130 | 2,714±160 | 0.79 |
| 28 d, n = 15 | 7,233±3,678 | 4,800±2,563 | 0.046 |
| Medial area, μm2 | |||
| 0 d | 9,583±1,203 | 9,500±894 | 0.85 |
| 7 d | 12,286±2,812 | 12,427±2,992 | 0.928 |
| 28 d | 7,700±1,032 | 8,267±1,498 | 0.24 |
| I:M | |||
| 7 d | 0.24±0.11 | 0.21±0.09 | 0.58 |
| 28 d | 0.97±0.56 | 0.59±0.34 | 0.036 |
| EEL, μm2 | |||
| 0 d | 31,792±11,293 | 32,045±10,636 | 0.96 |
| 7 d | 49,393±16,133 | 47,071±6,445 | 0.73 |
| 28 d | 29,200±9,067 | 31,667±9,856 | 0.48 |
| BrdUnd-positive cells, % | |||
| Media, 7 d | 8.8±1.4 | 4.1±1.2 | 0.01 |
| Media, 28 d | 2.6±1.2 | 1.3±1.1 | 0.24 |
| CD45-positive cells, % | |||
| Intima, 7 d | 40.4±2.3 | 27.7±7.8 | 0.044 |
| Intima, 28 d | 13.2±7.9 | 8.8±1.1 | 0.44 |
| TUNEL-positive cells, % | |||
| Media, 7 d | 4.4±2.9 | 3.5±1.9 | 0.67 |
| Intima, 7 d | 3.5±1.5 | 3.3±1.5 | 0.83 |
EEL, external elastic lamina; I:M, intima/media area ratio; TUNEL, terminal deoxynucleotidyl-transferase-mediated dUTP nick end labeling.
Fig. 4.

Photomicrographs of mouse femoral arteries after injury. (a-d) VerHoeff elastin stain 28 d after injury: (a) wild-type; (b) Nox2-/- (original magnification, ×38); (c) wild-type; (d) Nox2-/- (×150). Neointima separates the internal elastic lamina (arrows) from the lumen. (e and f) Proliferating (BrdUrd-positive) cells 7 d after injury: (e) wild-type; (f) Nox2-/- (×150).
Because increasing evidence suggests that NADPH oxidases influence cellular proliferation (31, 43), we assessed cellular proliferation by quantifying incorporation of BrdUrd in wild-type and Nox2-/- vessels after injury. Substantial proliferation was observed 7 d after injury in wild-type arteries (8.8% of medial cells), and proliferation was still evident at 28 d (2.6% of intimal cells). The lack of Nox2 reduced medial proliferation at 7 d by 53% (P = 0.01) and at 28 d by 50% (P = 0.24; Table 1 and Fig. 4), providing direct experimental support for a role for Nox2 in the proliferative response to vascular injury.
Deficiency of Nox2 and Leukocyte Recruitment. Transluminal wire injury of the femoral artery is accompanied by prominent vascular inflammation, including alteration in surface expression of leukocyte adhesion molecules (32). A role for Nox2 in this process was implicated because altered leukocyte accumulation within vessels was observed in injured vessels from Nox2-/- compared to wild-type mice. Inf lammatory cells (CD45-positive) invading the intima at 7 d were reduced by 31% from 40.4 ± 2.3 to 27.7 ± 7.8% (P = 0.044) in wild-type compared to Nox2-/- mice.
Nox2 and Apoptosis. Because ROS/RNS activate both survival and proapoptotic signals (44), we investigated the effects of deficiency of Nox2 on apoptosis after injury. The number of apoptotic cells in the intima (P = 0.83) and media (0.67) at 7 d was comparable in wild-type and Nox2-/- arteries (Table 1).
Discussion
Our study provides direct evidence that deficiency of Nox2 reduces vascular oxidative stress after experimental angioplasty. Significant decreases in specific markers of protein and lipid oxidation were accompanied by reduced histological and biochemical indices of inflammation, cellular proliferation, and neointimal thickening after wire injury in the mouse. Thus, these results establish a role for Nox2 in modulating the biological response to vascular injury.
Although the response to arterial injury is characterized by an inflammatory response that is believed to involve the production of ROS, RNS, and, as a consequence, oxidative damage (45), evidence to support this contention was largely indirect. For example, injury of the rat aorta was associated with a modest (17%) increase in vessel thiobarbituric acid-reactive substances, an indirect marker of lipid peroxidation (6). Recently, direct measurement of lipid peroxidation byproducts in balloon-injured arteries has been reported (46), although the functional implications of this phenomenon were not known. The data presented herein provide direct evidence for induction of both lipid and protein oxidation in response to arterial injury (Fig. 3). We found that Nox2 contributes to the generation of NO-derived oxidant species (as monitored through nitrotyrosine formation) but could not establish a role for Nox2 in tyrosyl radical generation (as monitored by dityrosine production). Although the precise mechanism for this observation is not yet clear, the results strongly suggest that oxidation pathways alternative to Nox2-driven superoxide generation play a dominant role in formation of tyrosyl radical-dependent oxidative cross-linking and dityrosine formation in the femoral artery wire injury model used. Localized alternative enzymatic sources of superoxide generation in response to wire injury cannot be excluded, such as induction of an uncoupled inducible NOS (47), xanthine oxidase, or an alternative Nox isoform. Further investigation into this issue is warranted.
We did observe a significant role for Nox2 in the arterial content of some arachidonic acid oxidation products (i.e., 8- and 15-HETE) but no role with regard to the products of linoleic acid oxidation (HODEs). The mechanism(s) for this selectivity is not clear but likely derives from reduced free radical initiation of lipid peroxidation, as opposed to enzymatic- (i.e., lipoxygenase) driven reactions. In this regard, it is of interest to note that mice, unlike humans, lack a 15-lipoxygenase gene, and the murine 8-lipoxygenase appears localized predominantly in skin. Thus, the observed Nox2-dependent generation of 8- and 15-HETE suggests participation of a free radical-mediated process. Although the precise mechanisms for initiating lipid peroxidation in the injury response in this model are unclear, the generation of both nitrotyrosine and dityrosine in wild-type mice indicates generation of both NO-derived oxidants (e.g., peroxynitrite and/or nitrogen dioxide) and tyrosyl radical as likely contributors. Interestingly, we did observe significant inducible NOS staining in both wild-type and Nox2-/- mice (data not shown), suggesting a potential source of ROS/RNS that could facilitate lipid and protein oxidation. Finally, it should be noted that in all studies, generation of ROS/RNS by Nox2 was observed only in response to injury. No differences in basal levels of qualitative and quantitative indices of ROS/RNS were noted in any comparisons made between wild-type and Nox2-/- mice.
Despite the coexistence of oxidative stress with arterial injury, a more pressing question is whether oxidative events are necessary for intimal proliferation. Evidence addressing this question has primarily been restricted to studies examining antioxidant treatments and their effects on intimal thickening after arterial injury. Dietary probucol produced a 51% decrease in intima/media area ratio 5 weeks after carotid arterial injury in rabbits (48), and this effect has been replicated in both a number of animal models (3, 49) and human studies (42). Similar observations have been observed with other antioxidant treatments such as butylated hydroxytoluene (4) and a combination of vitamins C and E (39). The data presented herein provide direct experimental support for a causal role between oxidative events in the vessel wall and intimal thickening. In particular, we found that markers of oxidative stress were reduced in Nox2-/- mice concomitant with a reduction in both intimal thickening and cellular proliferation. Thus, our data are consistent with a requirement for oxidative stress in the proliferative response.
The precise mechanisms whereby oxidative stress contributes to the proliferative response remain a matter of question. Although many oxidized biomolecules can induce SMC proliferation, other mechanisms must be considered. For example, previous studies have implicated the respiratory burst oxidase in the adhesion of leukocytes to components of the extracellular matrix (50), and we observed a reduced accumulation of inflammatory cells within the neointima of Nox2-/- mice. These observations are likely to have consequences for neointimal growth, because targeting leukocyte recruitment and function are associated with reductions in neointimal thickening after vascular injury (51). A functional deficiency in Nox2 will also likely result in overall enhanced levels of NO. Modulation of the pleiotropic actions of NO through Nox2-generated 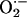 may indeed play a dominant role in the observed functional effects of Nox2 deficiency.
may indeed play a dominant role in the observed functional effects of Nox2 deficiency.
Numerous investigators have demonstrated that ROS and RNS have direct mitogenic effects on vascular cells (52) and serve as signaling intermediates for SMC mitogens, such as platelet-derived growth factor (PDGF) and thrombin (26, 53, 54). NADPH oxidase is a necessary component for SMC responses to PDGF, angiotensin II, and thrombin stimulation (18, 25, 30, 31, 55). Consistent with the notion that ROS/RNS are required for proliferation, we found that deficiency of Nox2 results in sustained reduction in cellular proliferation after vascular injury. It is important to realize, however, that Nox2 expression is not a prominent feature of SMC, suggesting that Nox2 from other cell types, such as fibroblasts (13, 56), may be involved in regulating SMC proliferation. Definitive answers concerning leukocyte vs. vascular cell Nox2 in arterial injury will require studies using bone marrow transplantation.
The reduction in neointimal thickening in Nox2 mice is statistically robust but somewhat modest. Intimal thickening and intima/media area ratio were reduced by 34% (P = 0.046) and 40% (P = 0.036), respectively, in Nox2-/- compared to wild-type mice. Because vascular injury is associated with the coordinated up-regulation of multiple NADPH oxidase subunits, including additional isoforms of Nox2 (i.e., Nox1 and -4) (20), it is highly likely that their contribution to ROS/RNS production may attenuate the effects of single gene deficiency on vascular injury and repair responses. In addition, xanthine oxidases, lipoxygenases, and peroxidases may serve as additional sources of ROS production.
Conclusion
Deficiency of Nox2 reduces vascular inflammation, cellular proliferation, and neointimal thickening after experimental angioplasty. These data provide convincing support for the hypothesis that NADPH oxidases have a requisite role in neointimal formation. They also provide a rationale for further studies to dissect the contributions of distinctive oxidative pathways and their generating enzymes to vascular lesion formation.
Supplementary Material
Acknowledgments
This work was supported in part by grants from the National Institutes of Health [DK55875 (to J.F.K. and D.I.S.); HL57506 and HL73852 (to D.I.S.); and HL076491, HL077692, and HL70621 (to S.L.H.)]. Mass spectrometry studies were supported in part by the Cleveland Clinic Foundation General Clinical Research Center (M01 RR018390).
Abbreviations: ROS, reactive oxygen species; RNS, reactive nitrogen species; SMC, smooth muscle cell; Nox2-/-, Nox2 deficient; HODE, hydroxyoctadecadienoic acids; HETE, hydroxyeicosatetraenoic acids; DHE, dihydroethidium.
References
- 1.Halliwell, B. (1989) Br. J. Exp. Pathol. 70, 737-757. [PMC free article] [PubMed] [Google Scholar]
- 2.Ohara, Y., Peterson, T. E. & Harrison, D. G. (1993) J. Clin. Invest. 91, 2546-2551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kisanuki, A., Asada, Y., Hatakeyama, K., Hayashi, T. & Sumiyoshi, A. (1992) Arterioscler. Thromb. 12, 1198-1205. [DOI] [PubMed] [Google Scholar]
- 4.Freyschuss, A., Stiko-Tahm, A., Swedenborg, J., Henriksson, P., Bjorkhem, I., Berglund, L. & Nilsson, J. (1993) J. Clin. Invest. 91, 1282-1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sundaresan, M., Yu, Z. X., Ferrans, V. J., Irani, K. & Finkel, T. (1995) Science 270, 296-299. [DOI] [PubMed] [Google Scholar]
- 6.Gong, K. W., Zhu, G. Y., Wang, L. H. & Tang, C. S. (1996) J. Vasc. Res. 33, 42-46. [DOI] [PubMed] [Google Scholar]
- 7.Nunes, G. L., Robinson, K., Kalynych, A., King, S. B., III, Sgoutas, D. S. & Berk, B. C. (1997) Circulation 96, 3593-3601. [DOI] [PubMed] [Google Scholar]
- 8.Pollman, M. J., Hall, J. L. & Gibbons, G. H. (1999) Circ. Res. 84, 113-121. [DOI] [PubMed] [Google Scholar]
- 9.Shi, Y., Niculescu, R., Wang, D., Patel, S., Davenpeck, K. L. & Zalewski, A. (2001) Arterioscler. Thromb. Vasc. Biol. 21, 739-745. [DOI] [PubMed] [Google Scholar]
- 10.Chen, K., Thomas, S. R. & Keaney, J. F., Jr. (2003) Free Radic. Biol. Med. 35, 117-132. [DOI] [PubMed] [Google Scholar]
- 11.Jones, S. A., O'Donnell, V. B., Wood, J. D., Broughton, J. P., Hughes, E. J. & Jones, O. T. (1996) Am. J. Physiol. 271, H1626-H1634. [DOI] [PubMed] [Google Scholar]
- 12.Griendling, K. K., Minieri, C. A., Ollerenshaw, J. D. & Alexander, R. W. (1994) Circ. Res. 74, 1141-1148. [DOI] [PubMed] [Google Scholar]
- 13.Pagano, P. J., Clark, J. K., Cifuentes-Pagano, M. E., Clark, S. M., Callis, G. M. & Quinn, M. T. (1997) Proc. Natl. Acad. Sci. USA 94, 14483-14488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Meier, B., Jesaitis, A. J., Emmendorffer, A., Roesler, J. & Quinn, M. T. (1993) Biochem. J. 289, 481-486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.De Leo, F. R., Ulman, K. V., Davis, A. R., Jutila, K. L. & Quinn, M. T. (1996) J. Biol. Chem. 271, 17013-17020. [DOI] [PubMed] [Google Scholar]
- 16.Dusi, S., Donini, M. & Rossi, F. (1996) Biochem. J. 314, 409-412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bayraktutan, U., Draper, N., Lang, D. & Shah, A. M. (1998) Cardiovasc. Res. 38, 256-262. [DOI] [PubMed] [Google Scholar]
- 18.Ushio-Fukai, M., Zafari, A. M., Fukui, T., Ishizaka, N. & Griendling, K. K. (1996) J. Biol. Chem. 271, 23317-23321. [DOI] [PubMed] [Google Scholar]
- 19.De Keulenaer, G. W., Alexander, R. W., Ushio-Fukai, M., Ishizaka, N. & Griendling, K. K. (1998) Biochem. J. 329, 653-657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Szocs, K., Lassegue, B., Sorescu, D., Hilenski, L. L., Valppu, L., Couse, T. L., Wilcox, J. N., Quinn, M. T., Lambeth, J. D. & Griendling, K. K. (2002) Arterioscler. Thromb. Vasc. Biol. 22, 21-27. [DOI] [PubMed] [Google Scholar]
- 21.Sorescu, D., Weiss, D., Lassegue, B., Clempus, R. E., Szocs, K., Sorescu, G. P., Valppu, L., Quinn, M. T., Lambeth, J. D., Vega, J. D., et al. (2002) Circulation 105, 1429-1435. [DOI] [PubMed] [Google Scholar]
- 22.Suh, Y. A., Arnold, R. S., Lassegue, B., Shi, J., Xu, X., Sorescu, D., Chung, A. B., Griendling, K. K. & Lambeth, J. D. (1999) Nature 401, 79-82. [DOI] [PubMed] [Google Scholar]
- 23.Banfi, B., Maturana, A., Jaconi, S., Arnaudeau, S., Laforge, T., Sinha, B., Ligeti, E., Demaurex, N. & Krause, K. H. (2000) Science 287, 138-142. [DOI] [PubMed] [Google Scholar]
- 24.Lambeth, J. D., Cheng, G., Arnold, R. S. & Edens, W. A. (2000) Trends Biochem. Sci. 25, 459-461. [DOI] [PubMed] [Google Scholar]
- 25.Marumo, T., Schini-Kerth, V. B., Fisslthaler, B. & Busse, R. (1997) Circulation 96, 2361-2367. [DOI] [PubMed] [Google Scholar]
- 26.Patterson, C., Ruef, J., Madamanchi, N. R., Barry-Lane, P., Hu, Z., Horaist, C., Ballinger, C. A., Brasier, A. R., Bode, C. & Runge, M. S. (1999) J. Biol. Chem. 274, 19814-19822. [DOI] [PubMed] [Google Scholar]
- 27.Lassegue, B. & Clempus, R. E. (2003) Am. J. Physiol. 285, R277-R297. [DOI] [PubMed] [Google Scholar]
- 28.Griendling, K. K. & Ushio-Fukai, M. (1998) J. Lab. Clin. Med. 132, 9-15. [DOI] [PubMed] [Google Scholar]
- 29.Hsich, E., Segal, B. H., Pagano, P. J., Rey, F. E., Paigen, B., Deleonardis, J., Hoyt, R. F., Holland, S. M. & Finkel, T. (2000) Circulation 101, 1234-1236. [DOI] [PubMed] [Google Scholar]
- 30.Kirk, E. A., Dinauer, M. C., Rosen, H., Chait, A., Heinecke, J. W. & LeBoeuf, R. C. (2000) Arterioscler. Thromb. Vasc. Biol. 20, 1529-1535. [DOI] [PubMed] [Google Scholar]
- 31.Barry-Lane, P. A., Patterson, C., van der Merwe, M., Hu, Z., Holland, S. M., Yeh, E. T. & Runge, M. S. (2001) J. Clin. Invest. 108, 1513-1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Roque, M., Fallon, J. T., Badimon, J. J., Zhang, W. X., Taubman, M. B. & Reis, E. D. (2000) Arterioscler. Thromb. Vasc. Biol. 20, 335-342. [DOI] [PubMed] [Google Scholar]
- 33.Miller, F. J., Jr., Gutterman, D. D., Rios, C. D., Heistad, D. D. & Davidson, B. L. (1998) Circ. Res. 82, 1298-1305. [DOI] [PubMed] [Google Scholar]
- 34.Brennan, M. L., Wu, W., Fu, X., Shen, Z., Song, W., Frost, H., Vadseth, C., Narine, L., Lenkiewicz, E., Borchers, M. T., et al. (2002) J. Biol. Chem. 277, 17415-17427. [DOI] [PubMed] [Google Scholar]
- 35.Zhang, R., Brennan, M. L., Shen, Z., MacPherson, J. C., Schmitt, D., Molenda, C. E. & Hazen, S. L. (2002) J. Biol. Chem. 277, 46116-46122. [DOI] [PubMed] [Google Scholar]
- 36.Smyth, S. S., Reis, E. D., Zhang, W., Fallon, J. T., Gordon, R. E. & Coller, B. S. (2001) Circulation 103, 2501-2507. [DOI] [PubMed] [Google Scholar]
- 37.Roque, M., Kim, W. J., Gazdoin, M., Malik, A., Reis, E. D., Fallon, J. T., Badimon, J. J., Charo, I. F. & Taubman, M. B. (2002) Arterioscler. Thromb. Vasc. Biol. 22, 554-559. [DOI] [PubMed] [Google Scholar]
- 38.Paravicini, T. M., Gulluyan, L. M., Dusting, G. J. & Drummond, G. R. (2002) Circ. Res. 91, 54-61. [DOI] [PubMed] [Google Scholar]
- 39.Nunes, G. L., Sgoutas, D. S., Redden, R. A., Sigman, S. R., Gravanis, M. B., King, S. B., III, & Berk, B. C. (1995) Arterioscler. Thromb. Vasc. Biol. 15, 156-165. [DOI] [PubMed] [Google Scholar]
- 40.Souza, H. P., Laurindo, F. R., Ziegelstein, R. C., Berlowitz, C. O. & Zweier, J. L. (2001) Am. J. Physiol. 280, H658-H667. [DOI] [PubMed] [Google Scholar]
- 41.Beckman, J. S., Beckman, T. W., Chen, J., Marshall, P. A. & Freeman, B. A. (1990) Proc. Natl. Acad. Sci. USA 87, 1620-1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tardif, J.-C., Cote, G., Lesperance, J., Bourassa, M., Lambert, J., Doucet, S., Bilodeau, L., Nattel, S. & De Guise, P. (1997) N. Engl. J. Med. 337, 365-372. [DOI] [PubMed] [Google Scholar]
- 43.Lassegue, B., Sorescu, D., Szocs, K., Yin, Q., Akers, M., Zhang, Y., Grant, S. L., Lambeth, J. D. & Griendling, K. K. (2001) Circ. Res. 88, 888-894. [DOI] [PubMed] [Google Scholar]
- 44.Guyton, K. Z., Liu, Y., Gorospe, M., Xu, Q. & Holbrook, N. J. (1996) J. Biol. Chem. 271, 4138-4142. [DOI] [PubMed] [Google Scholar]
- 45.Serrano, C. V., Jr., Ramires, J. A., Venturinelli, M., Arie, S., D'Amico, E., Zweier, J. L., Pileggi, F. & da Luz, P. L. (1997) J. Am. Coll. Cardiol. 29, 1276-1283. [DOI] [PubMed] [Google Scholar]
- 46.Upston, J. M., Witting, P. K., Brown, A. J., Stocker, R. & Keaney, J. F., Jr. (2001) Free Radic. Biol. Med. 31, 1245-1253. [DOI] [PubMed] [Google Scholar]
- 47.Xia, Y., Roman, L., Masters, B. & Zweier, J. (1998) J. Biol. Chem. 273, 22635-22639. [DOI] [PubMed] [Google Scholar]
- 48.Ferns, G. A., Forster, L., Stewart-Lee, A., Konneh, M., Nourooz-Zadeh, J. & Anggard, E. E. (1992) Proc. Natl. Acad. Sci. USA 89, 11312-11316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ishizaka, N., Kurokawa, K., Taguchi, J., Miki, K. & Ohno, M. (1995) Atherosclerosis 118, 53-56. [DOI] [PubMed] [Google Scholar]
- 50.Yan, S. R. & Berton, G. (1998) J. Leukocyte Biol. 64, 401-408. [DOI] [PubMed] [Google Scholar]
- 51.Simon, D. I., Chen, Z., Seifert, P., Edelman, E. R., Ballantyne, C. M. & Rogers, C. (2000) J. Clin. Invest. 105, 293-300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rao, G. N. & Berk, B. C. (1992) Circ. Res. 70, 593-599. [DOI] [PubMed] [Google Scholar]
- 53.Callsen, D., Sandau, K. B. & Brune, B. (1999) Free Radic. Biol. Med. 26, 1544-1553. [DOI] [PubMed] [Google Scholar]
- 54.Brar, S. S., Kennedy, T. P., Whorton, A. R., Murphy, T. M., Chitano, P. & Hoidal, J. R. (1999) J. Biol. Chem. 274, 20017-20026. [DOI] [PubMed] [Google Scholar]
- 55.Rajagopalan, S., Kurz, S., Munzel, T., Tarpey, M., Freeman, B. A., Griendling, K. K. & Harrison, D. G. (1996) J. Clin. Invest. 97, 1916-1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rey, F. E. & Pagano, P. J. (2002) Arterioscler. Thromb. Vasc. Biol. 22, 1962-1971. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}