

ABSTRACT
Specialized secretion systems of bacteria evolved for selective advantage, either killing microbial competitors or implementing effector functions during parasitism. Earlier work characterized the ESAT-6 secretion system (ESS) of Staphylococcus aureus and demonstrated its contribution to persistent staphylococcal infection of vertebrate hosts. Here, we identify a novel secreted effector of the ESS pathway, EssD, that functions as a nuclease and cleaves DNA but not RNA. EssI, a protein of the DUF600 family, binds EssD to block its nuclease activity in the staphylococcal cytoplasm. An essD knockout mutant or a variant lacking nuclease activity, essDL546P, elicited a diminished interleukin-12 (IL-12) cytokine response following bloodstream infection of mice, suggesting that the effector function of EssD stimulates immune signaling to support the pathogenesis of S. aureus infections.
IMPORTANCE Bacterial type VII or ESAT-6-like secretion systems (ESS) may have evolved to modulate host immune responses during infection, thereby contributing to the pathogenesis of important diseases such as tuberculosis and methicillin-resistant S. aureus (MRSA) infection. The molecular mechanisms whereby type VII secretion systems achieve their goals are not fully elucidated as secreted effectors with biochemical functions have heretofore not been identified. We show here that MRSA infection relies on the secretion of a nuclease effector that cleaves DNA and contributes to the stimulation of IL-12 signaling during infection. These results identify a biological mechanism for the contribution of the ESS pathway toward the establishment of MRSA disease.
KEYWORDS: EssD, nuclease, MRSA, IL-12 signaling, ESS secretion, effector, DUF600, IL-12
INTRODUCTION
The ESAT-6-like secretion system (ESS) of Staphylococcus aureus is conserved in the phylum Firmicutes and bears similarities with the well-characterized type 7 secretion system (T7SS) of Actinobacteria, including Mycobacterium tuberculosis, Mycobacterium smegmatis, and Mycobacterium marinum (1–3). As with many other specialized secretion systems, genes encoding ESS and the T7SS are present within clusters (1). Genetic analyses have been used to identify the genes that specify the machinery for substrate secretion while secreted factors have been identified as immune reactive species in the extracellular medium of bacterial cultures (4–9). Candidate factors for secretion machines share little to no sequence similarity between ESS and T7SS clusters (1). However, both ESS and T7SS clusters encode secreted and conserved substrates that belong to the protein family pfam06013 (WXG100) within the EsxAB clan CL0352 (pfam.janelia.org/clan/EsxAB) (1, 10). In the Actinobacteria phylum but not Firmicutes, the EsxAB clan CL0352 also includes the protein families pfam00934 (PE), pfam00823 (PPE), and pfam10824 (Esp) (11, 12). Sequence identity between protein members of the pfam06013 group (WXG100) is low, often less than 15%, but all of these proteins share a small domain of approximately 100 amino acids with the distinctive WXG motif in the center (10). The atomic structures of several pfam06013 proteins revealed a conserved fold with two side-by-side α-helices linked with a hairpin bend formed by the WXG motif (13–16). S. aureus secretes EsxA and EsxB, two canonical WXG100 proteins (6). EsxA forms a dimer with itself and with EsxC, whereas EsxB forms a dimer with EsxD (9). While EsxC and EsxD associate tightly with WXG100 proteins, they do not share the typical sequence features of the pfam06013 family or any other member of the EsxAB clan CL0352 (9).
In virulent strains of M. tuberculosis, EsxA (Mt-EsxA) is secreted by intracellular bacteria and has been proposed to form discrete pores that allow mixing of vacuolar and cytosolic contents during infection (17). In a model involving bacterial DNA release, the accessibility to cytosolic molecules in turn leads to AIM2 and cyclic GMP-AMP synthase (cGAS) activation (18–20). Cytosolic DNA sensing by AIM2 leads to activation of the inflammasome interleukin-1β (IL-1β) pathway (21). Cyclic GMP-AMP (cGAMP) signaling is mediated by STING, the mammalian sensor for cytoplasmic DNA and bacterial cyclic dinucleotides, and drives type I interferon (IFN) activation (22). Thus, ESX-1 activity accounts for both the stimulation of IFN and IL-1β responses that contribute to disease progression in tuberculosis (TB) (18, 23, 24).
In S. aureus, the ESS pathway contributes to the stimulation of IL-12 (p40) and IL-12 (p35) production and secretion during mouse infection (25). This immunomodulatory activity is thought to contribute to S. aureus virulence and persistence within deep-seated abscess lesions (7, 26). Here, we demonstrate that S. aureus ESS secretes EssD, which functions as a nuclease to cleave DNA. EssD nuclease activity in the bacterial cytoplasm is blocked by EssI, a small cytoplasmic protein that binds to the nuclease domain of EssD and whose gene is located adjacent to essD. Because the EssD-EssI module is reminiscent of toxin-antitoxin modules, we examine whether ESS may contribute a competitive advantage over other staphylococcal species and skin commensals but find no evidence of such antimicrobial activity in vitro. Instead, we observe reduced IL-12 production in mice infected with S. aureus essD mutants. We therefore propose that EssD represents an effector of the staphylococcal ESS pathway.
RESULTS
essI suppresses essD induced toxicity.
essD encodes a large polypeptide with mosaic structure (Fig. 1A), reminiscent of effectors in specialized secretion systems (27). Initial attempts to express essD in Escherichia coli resulted in the selection of mutants with single nucleotide transversions and transitions in the 3′ portion of the gene, causing either codon substitutions (Asp544Gly, Leu546Pro, and Trp563Arg) or nonsense mutations (Lys584*). Plasmid-borne expression of essD in cells that also expressed essI1, a gene of unknown function located immediately downstream on the staphylococcal chromosome (Fig. 1B), alleviated the selection for mutational lesions. Of note, the essI1 (SAUSA300_0289) product is a member of the DUF600 protein family (domain of unknown function 600); nine genes (essI2 to essI10) encoding additional DUF600 family members are located downstream of essD (Fig. 1B). DUF600 proteins vary in length from 128 to 166 amino acids and share 66 to 93% sequence identity (Fig. 1C).
FIG 1.

EssD and EssI proteins of S. aureus USA300. (A) Domain organization of EssD. EssD carries the COG5444 domain (blue) and a predicted nuclease domain (endonuclease_NS_2; red). The hydrophobic segment was identified with the TMHMM server, version 2.0. Numbers indicate positions of amino acid residues in the linear polypeptide. L546 indicates the position of Leu546, a residue essential for cytotoxicity. (B) Diagram of the genomic region carrying the essD and essI genes in S. aureus subsp. aureus USA300_FPR3757 as annotated in NCBI RefSeq NC_007793.1. essI genes with DUF600 domains are shown in green. (C) Alignment of the 10 open reading frames bearing DUF600 in S. aureus subsp. aureus USA300_FPR3757 (NCBI RefSeq accession number NC_007793.1) using Clustal W. The symbols denote identical residues in all sequences (*), highly conserved columns (:), and weakly conserved columns (.).
Expression of essD from the isopropyl-β-d-thiogalactopyranoside (IPTG)-inducible lac promoter (pSRK-Kanr vector) and essI1 or essI6 to essI10 (essI6-10) from the arabinose-inducible promoter (pBAD-Ampr vector) in E. coli (Fig. 1C) revealed that growth of the strain carrying plasmids expressing Plac-essD and Para-essI1 in Luria broth supplemented with IPTG was strictly dependent on arabinose induction of essI1 (Fig. 2B). As a control, E. coli Plac-essD Para-essI1 growth in medium lacking IPTG was not dependent on arabinose (data not shown), whereas E. coli Plac-essD Para-essI6-10 also displayed a requirement for arabinose induction of DUF600 proteins for growth in the presence of IPTG (Fig. 2A and B). Growth inhibition in the presence of IPTG is caused by essD expression as E. coli Plac-Para-essI1, lacking the essD gene, did not display a growth impediment in the presence of IPTG (Fig. 2B).
FIG 2.

Cytotoxicity of EssD in E. coli. (A) Diagram depicting plasmid constructs with ara and lac promoters. (B to D) Growth and viability of E. coli variants harboring plasmids carrying the essD and essI genes. Overnight cultures were normalized to an A600 of 5, diluted 1:100 into fresh medium with (+) or without (−) arabinose (ara) and grown at 37°C. IPTG was added 3 h after dilution. Growth was monitored as increased absorbance (A600) over 6 h (B). At timed intervals (0, 1, and 2 h) after IPTG addition (T0, T1, and T2), cultures of the Plac-essD Para-essI6-10 strain grown with or without arabinose and with IPTG were examined for integrity of genetic content (C) and for viability (D). Cells were stained with DAPI and visualized with an Olympus AX70 microscope and UV filter. Pseudocolor was used for image display in panel C. Cultures were serially diluted (0 to −5), and 10-μl aliquots of each dilution were spotted on agar medium containing ampicillin, kanamycin, and arabinose. An image of the plate after overnight incubation at 30°C is shown in panel D.
E. coli Plac-essD Para-essI6-10 cultures were grown for 3 h, followed by induction with IPTG. Culture aliquots were withdrawn at 0 (T0), 1 (T1), and 2 (T2) h following addition of IPTG to the growth medium and examined with fluorescence microscopy of 4′,6′-diamidino-2-phenylindole (DAPI)-stained samples (Fig. 2B and C). Samples were also diluted and plated on agar with arabinose as well as kanamycin and ampicillin for selection of E. coli carrying Plac-essD (Kanr) and Para-essI6-10 (Ampr) (Fig. 2D). Following IPTG induction, E. coli Plac-essD Para-essI6-10 cells lost DAPI fluorescence staining of chromosomal DNA in cultures that had not received the arabinose inducer of DUF600 proteins (Fig. 2C). CFU counts of E. coli Plac-essD Para-essI6-10 were diminished at T1 and T2 after IPTG addition but not at T0 when IPTG was just added to E. coli Plac-essD Para-essI6-10 cultures without arabinose supplement (Fig. 2D). These data support a model whereby essD expression is detrimental for E. coli growth and causes loss of cell viability, presumably due to the degradation of chromosomal DNA. Coexpression of essI prevents the bactericidal activity of EssD.
Association of EssD and EssI.
Expression of wild-type essD along with essI1 from the constitutive hrpK promoter of plasmid pWWW412 (pessD-essI1) did not affect growth of the S. aureus essD mutant (essD::erm strain). Further, pWWW412 expression of essD* (pessD*) in E. coli or S. aureus did not impair bacterial growth, suggesting that the Leu546Pro substitution abolishes the toxicity of the essD product. We generated pessD*HIS, which carries the coding sequence for 10 histidyl residues inserted immediately upstream of the stop codon. Bacteria from 8-liter cultures of S. aureus USA300 essD::erm harboring plasmid pessD-essI1 or pessD*HIS were lysed, and cleared lysates were subjected to immobilized metal affinity chromatography (IMAC) on Ni-nitrilotriacetic acid (Ni-NTA)–Sepharose (Fig. 3). Eluates were analyzed by immunoblotting with rabbit antiserum raised against purified recombinant EssDN (residues 1 to 200; anti-EssDN), EssDC (residues 434 to 617; anti-EssDC), or EssI1 (anti-EssI). The data demonstrate that EssD*HIS (predicted Mr of 70,073) elutes as a 72-kDa anti-EssDN immune-reactive species following IMAC (Fig. 3, lanes 2) whereas untagged EssD does not (Fig. 3, lanes 1). Immunoblotting with anti-EssDC and Coomassie-stained SDS-PAGE analysis demonstrated that EssD*HIS elutes predominantly as C-terminal cleavage products of the full-length polypeptide (Fig. 3). Immunoblotting with anti-EssI revealed the coelution of EssI1 and EssD*HIS during IMAC (Fig. 3). Edman degradation identified SEGKH (EssD*HIS residues 434 to 438) and MTFEE (EssI1 residues 1 to 5) as phenyl-hydantoin-released residues in five subsequent Edman cycles. These data indicate that EssD*HIS (residues 434 to 617) and EssI (residues 1 to 163) were copurified from S. aureus USA300 essD::erm(pessD*HIS) during IMAC. Further, S. aureus cleaves EssD*HIS and wild-type EssD (see below) to generate C-terminal fragments that bind EssI1.
FIG 3.
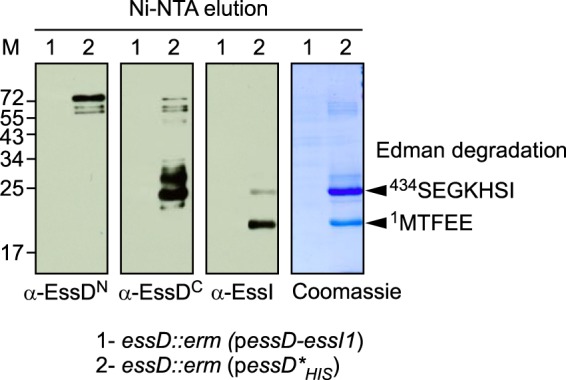
Purification of EssD from S. aureus. S. aureus strain USA300 essD::erm was transformed with plasmid pessD-essI1 or pessD*HIS to produce wild-type EssD or the nontoxic Leu546Pro variant with a C-terminal histidine tag (lanes 1 and 2, respectively). Both strains were grown at 37°C in TSB and chloramphenicol. Cultures were centrifuged, sedimented bacteria were lysed in buffer A, and cleared bacterial lysates were subjected to Ni-NTA chromatography. Beads were washed twice with 20 volumes of buffer A containing 30 and 50 mM imidazole, and bound proteins were eluted with 0.5 M imidazole. Eluted samples were separated by SDS-PAGE and either stained with Coomassie or transferred to PVDF membrane for immunoblot analyses with anti-EssDN, anti-EssDC, and anti-EssI polyclonal sera. Numbers to the left indicate the mobility of molecular mass markers (lane M). Arrowheads point to the two bands subjected to Edman degradation with the resulting amino acid sequences identified during this analysis.
EssD nuclease activity is inhibited by EssI.
EssD harbors a 133-residue domain (residues 475 to 607) with homology to pfam13930 (predicted DNA/RNA nonspecific nuclease from various bacterial species; E value, 6.3 × 10−48) (Fig. 1A). We hypothesized that EssD consisting of residues 475 to 607 (EssD475–607) may function as a nuclease and that the Leu546Pro substitution abolishes its nuclease activity (Fig. 1A). To test this, primer pairs were designed to amplify DNA fragments from essD and essD* encoding EssD434–617, EssD456–617, EssD476–617, EssD486–617, and EssD496–617 (Fig. 4A). essD alleles were cloned into the pSRK vector and transformed into E. coli Para-essI6-10, and transformants were plated on Amp/Kan agar supplemented with arabinose. Plasmids expressing Plac-essD434–617, Plac-essD456–617, Plac-essD476–617, and Plac-essD486–617 supported growth of E. coli Para-essI6-10 on Amp/Kan plates with arabinose, whereas the plasmid expressing Plac-essD496–617 did not (Fig. 4A). As a control, Plac-essD*434–617 and Plac-essD*496–617 supported growth of E. coli Para-essI6-10 plated on Amp/Kan agar supplemented with arabinose. These data suggest that EssI requires EssD residues 486 to 496 to neutralize the cytotoxicity associated with the nuclease activity encoded by Plac-essD496–617 (Fig. 4A). In agreement with this conjecture, growth of E. coli Plac-essD434–617/HIS Para-essI6-10 but not of E. coli Plac-essD*434–617/HIS Para-essI6-10 in LB medium supplemented with IPTG was dependent on arabinose supplement for the induced expression of DUF600 proteins (Fig. 4BC).
FIG 4.

Biochemical characterization of EssD434–617/HIS. (A) EssD sequence elements required for DNase activity and inhibition by EssI. Plasmids encoding N-terminal truncations of EssD were transformed in E. coli Para-essI6-10 for the selection of viable clones in the presence of kanamycin, ampicillin, and arabinose. (B) Diagram depicting plasmid constructs with lac and ara promoters. (C) Growth of E. coli variants carrying Para-essI6-10 with either Plac-essD434–617/HIS or Plac-essD*434–617/HIS plasmids was monitored as described in the legend to Fig. 2B. (D) Wild-type EssD434–617/HIS was purified from cleared lysate of E. coli Plac-essD434–617/HIS Para-essI6-10. Aliquots of samples prior to purification (Load), flowthrough, wash, and eluates were separated by SDS-PAGE and transferred to PVDF membrane for immunoblotting with anti-EssDC and anti-EssI polyclonal sera. Lanes 1 to 4 and 5 to 8 were loaded with sample aliquots of preparations purified in buffer A without urea (no denaturant) or buffer A containing 8 M urea (+ denaturant), respectively. Bound proteins were eluted in buffer A containing 0.5 M imidazole. Numbers to the left indicate the mobility of molecular mass markers. (E) Plasmid DNA, dsDNA, or RNA was incubated with proteins eluted in fraction 4 or 8 or buffer control. Following incubation, reaction products were separated on an agarose gel and visualized with a UV transilluminator for image acquisition. (F) Reaction products following incubation of dsDNA with sample 4, sample 8, or buffer control C were separated by reverse-phase chromatography, and absorbance was recorded at 260 nm (A260). AU, arbitrary units.
The cleared lysate of E. coli Plac-essD434–617/HIS Para-essI6-10 was subjected to affinity chromatography on Ni-NTA–Sepharose, and samples were analyzed by immunoblotting (Fig. 4D, lanes 1 to 4). EssI coeluted with EssD434–617/HIS when 0.5 M imidazole was flowed over Ni-NTA–Sepharose (Fig. 4D, lanes 1 to 4). When 8 M urea was flowed over Ni-NTA–Sepharose loaded with E. coli Plac-essD434–617/HIS Para-essI6-10 lysate, EssI was displaced from the column and did not elute with 0.5 M imidazole buffer, unlike EssD434–617/HIS (Fig. 4D, lanes 5 to 8). Purified, refolded EssD434–617/HIS and EssD434–617/HIS-EssI complex were added to plasmid DNA, to annealed oligonucleotides (double-stranded DNA [dsDNA]), or to RNA, and reaction products were analyzed by agarose gel electrophoresis (Fig. 4E). Both plasmid DNA and dsDNA, but not RNA, were degraded by EssD434–617/HIS (Fig. 4E). In contrast, EssD434–617/HIS-EssI did not degrade plasmid DNA, dsDNA, or RNA (Fig. 4E). Mock-treated (control) or EssD434–617/HIS- or EssD434–617/HIS-EssI-treated dsDNA was also analyzed by reverse-phase high-performance liquid chromatography (rpHPLC), and eluate was analyzed by absorbance at 260 nm (A260). Data in Fig. 4F demonstrate that EssD434–617/HIS degrades dsDNA, whereas EssD434–617/HIS-EssI did not significantly impact the integrity of dsDNA. Together, these data suggest that EssD434–617 functions as a nuclease that cleaves dsDNA and that this enzymatic activity is inhibited by binding of EssI. We think it is likely that other members of the DUF600 protein family of S. aureus USA300, i.e., EssI1 to EssI10, may also bind EssD and block its nuclease activity.
EssD is secreted in an ESS-dependent manner.
Earlier work characterized the ESS pathway in S. aureus strain Newman (6, 7) and reported that the N-terminal part of EssD is exposed on the bacterial surface (26). S. aureus Newman carries an L-to-A change at position 18 encoded by saeS (saeS-L18P), which encodes the sensory kinase of the two-component virulence regulator of staphylococci (28, 29). Because of the saeS-L18P mutation, the ESS pathway cannot be fully activated in S. aureus Newman (9, 30). To analyze the expression and subcellular localization of EssD in cultures of the American MRSA epidemic clone, USA300 harboring wild-type saeS (31), staphylococci were grown in serum-conditioned medium to an A600 of 1.0. Lysostaphin, a staphylococcal murein hydrolase (32), was added to lyse staphylococci in culture aliquots; proteins were precipitated with TCA and analyzed by immunoblotting with anti-EssDN, anti-EssDC, anti-EssI, and anti-SrtA (Fig. 5A). Wild-type USA300 cultures produced full-length 72-kDa EssD and its cleavage products of 45 (anti-EssDN), 66, 63, and 25 (anti-EssDC) kDa as well as EssI and SrtA, whereas the essD::erm mutant produced only EssI and SrtA (Fig. 5A). USA300 cultures were centrifuged to separate the extracellular medium (Fig. 5B, MD) from the staphylococcal sediment (Fig. 5B, Cell). Staphylococci were lysed with lysostaphin, and bacterial extracts were centrifuged at 108,000 × g to separate cytoplasmic proteins from membrane-associated proteins in the sediment (Fig. 5B, Sol and Insol, respectively). Full-length EssD and its cleavage products were secreted into the extracellular medium, and bacterial EssD was found predominantly associated with bacterial membranes (Fig. 5B). In wild-type USA300, EssI was detected as a soluble component of the bacterial cytoplasm and associated with the membrane sediment (Fig. 5B, α-EssI). Of note, membrane-associated EssI was not detected in essD::erm mutant staphylococci (Fig. 5B, α-EssI). These data suggest that EssI membrane association requires EssD. Although the abundance of EssI clearly exceeds that of EssD, EssI dissociates from EssD during its travels across the cell envelope as EssI is not found in the extracellular medium (Fig. 5B). As controls, α-hemolysin, a secreted product (anti-HLA), was found in the culture medium, whereas sortase A sedimented with the bacterial membranes (anti-SrtA) and ribosomal protein L6 remained in the cytoplasm (anti-L6).
FIG 5.

Subcellular localization of EssD and EssI proteins. S. aureus strain USA300 (lanes 1) and the isogenic essD::erm variant (lanes 2) were grown at 37°C in TSB supplemented with serum. Cultures were either incubated with lysostaphin yielding whole culture lysates (A) or fractionated into medium (MD), soluble (Sol), and insoluble (Insol) subcellular contents (B). Proteins were precipitated by the addition of TCA, suspended in sample buffer prior to separation by SDS-PAGE, and transferred to PVDF membranes for immunoblot analyses with anti-EssDN, anti-EssDC, anti-EssI, anti-SrtA, anti-L6, or anti-HLA polyclonal sera. Numbers to the left indicate the mobility of molecular mass markers. (C) EssD is secreted in an essB-dependent manner. S. aureus strain USA300 (lanes 1), the isogenic essD::erm strain (lanes 2), the ΔessB mutant (lanes 3), and ΔessB/pessB (lanes 4) complemented variant were grown at 37°C in TSB supplemented with serum and appropriate antibiotics. Cultures were centrifuged to separate medium (MD) from the bacterial sediment. Staphylococci were treated with lysostaphin for complete lysis (cell). Proteins were precipitated by the addition of TCA, suspended in sample buffer prior to separation by SDS-PAGE, and transferred to PVDF membranes for immunoblot analyses with anti-EssDC, anti-EssI, anti-EsxC, or anti-HLA polyclonal sera. Numbers to the left indicate the mobility of molecular mass markers.
To test whether EssD is secreted via the ESS pathway, we fractionated wild-type USA300 and essD::erm, ΔessB, and ΔessB/pessB cultures to separate the extracellular medium from bacterial lysates and analyzed samples by immunoblotting (Fig. 5C, MD and Cell, respectively). As a control, wild-type USA300 secreted EsxC into the extracellular medium, whereas essD and essB mutant strains did not (Fig. 5C). The EsxC secretion defect of the ΔessB mutant was complemented by plasmid-borne expression of wild-type essB (ΔessB/pessB strain) (Fig. 5C). Similarly, EssD was secreted by wild-type staphylococci and the pessB-complemented strain but not by the essD and essB mutants, suggesting that EssD is indeed a secretion substrate of the ESS pathway (Fig. 5C). As expected, all strains examined secreted α-hemolysin (HLA) into the medium and retained EssI in the bacterial cytoplasm (Fig. 5C).
The nuclease activity of EssD is not required for secretion.
To examine whether the nuclease activity of EssD contributes to ESS secretion, the essD-L546P allele was recombined on the chromosome of S. aureus USA300, yielding the essDL546P strain. As before, cultures of wild-type USA300 and isogenic essD::erm and essDL546P strains were centrifuged to separate the extracellular medium from bacterial lysates to analyze the fate of EssD, and EsxC by immunoblotting (Fig. 6A, MD and cell, respectively). Both EssD and EsxC were found in the extracellular medium of the USA300 and essDL546P cultures but failed to be secreted by the essD::erm mutant strain (Fig. 6A). All strains secreted α-hemolysin (HLA) into the medium and retained the ribosomal protein L6 in the bacterial cytoplasm (Fig. 6A). Culture supernatants of the USA300, essD::erm, essDL546P, ΔessB, and ΔessB/pessB strains were filtered, and nuclease activity was assessed by using plasmid DNA as a substrate (Fig. 6B). Degradation of DNA was observed only for culture filtrates of wild-type and complemented ΔessB/pessB strains, confirming that secreted essDL546P is not active.
FIG 6.
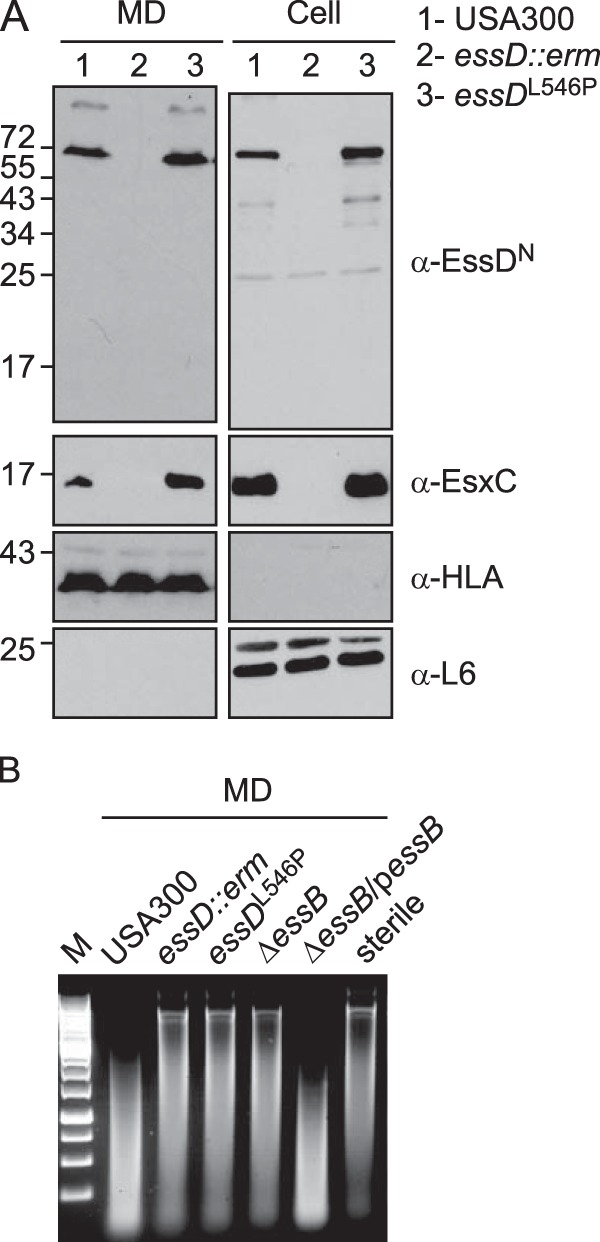
The nuclease activity of EssD is dispensable for secretion. (A) S. aureus strain USA300 (lanes 1) and the isogenic essD::erm (lanes 2) or essDL546P (lanes 3) variant were grown at 37°C in TSB supplemented with serum. Cultures were spun to separate the medium (MD) from intact cells that were subsequently lysed with lysostaphin to release all cellular content (cell). Proteins were TCA precipitated, suspended in sample buffer prior to separation by SDS-PAGE, and transferred to PVDF membranes for immunoblot analyses with anti-EssDN, anti-EsxC, anti-L6, or anti-HLA polyclonal sera. Numbers to the left indicate the mobility of molecular mass markers. (B) Cultures of S. aureus strain USA300 and the isogenic essD::erm, essDL546P, ΔessB, and ΔessB/pessB variants were grown at 37°C in TSB supplemented with serum and appropriate antibiotics. The supernatant of these cultures was used to assay for nuclease activity using plasmid DNA. Sterile TSB was used as a control (sterile). Following incubation, reaction products were separated on an agarose gel and visualized with a UV transilluminator for image acquisition. Lane M, molecular mass markers.
S. aureus ESS does not impact microbial competition.
The EssD-EssI module is reminiscent of pairs of effector toxins and their cognate immunity proteins identified in the type VI secretion systems, which endow Gram-negative bacteria with competitive advantages over other microbial species (33). We therefore wondered whether S. aureus ESS may also provide a competitive advantage during the bacteria's encounters with other microbes. We first assessed whether EssD production affects S. aureus replication. When overnight cultures of wild-type strain USA300 or the essD::erm mutant strain were diluted into fresh medium and bacterial replication was monitored at 600 nm (A600), the two strains multiplied at similar rates (Fig. 7A). Culture aliquots were collected at 3, 4, and 6 h following dilution of overnight cultures and plated on agar medium for colony formation and enumeration. No significant differences in plating efficiencies were observed (Fig. 7B). Next, we asked whether secreted EssD might confer a growth advantage for S. aureus over bacteria that lack essI-like genes. To test this possibility, wild-type USA300 or the isogenic essD::erm variant was grown in the presence of Staphylococcus simulans MK148 or Staphylococcus epidermidis 12228, isolates which do not harbor genes for ESS clusters or EssI (DUF600) immunity proteins. To assess competitive advantage during prolonged coincubation of microbes, bacteria were enumerated by plating on agar and enumeration of colonies (Fig. 7C). During S. simulans MK148 coincubation with S. aureus, replication of S. simulans MK148 was diminished by ∼1 log. However, this growth inhibition cannot be attributed to the ESS pathway or EssD secretion as deletion of essD in S. aureus USA300 did not alleviate the growth inhibition of S. simulans MK148 or S. epidermidis 12228 (Fig. 7C). Immunoblot analyses of lysates derived from S. simulans MK148 and S. epidermidis 12228 cultures confirmed that these isolates did not produce proteins immunoreactive with the anti-EssI polyclonal serum (Fig. 7D).
FIG 7.

Expression of essD does not confer any growth advantage and elicits an IL-12 response during infection. (A) Overnight cultures of wild-type USA300 and the isogenic essD::erm variant were normalized to an A600 of 5, diluted 1:100 into fresh medium, and grown with shaking at 37°C. Growth was monitored by recording the A600. (B) Aliquots of cultures shown in panel A were removed at 3, 4, and 6 h (T3, T4, and T6, respectively) and plated to assess viability. (C) S. epidermidis 12228 or S. simulans was cocultured with S. aureus USA300 or the isogenic essD::erm variant. Bacteria were mixed at a 1:1 ratio and plated on TSA. Immediately and 9 h following plating, cocultured bacteria were serially diluted and plated for enumeration of viable target cell counts. The data are presented as the average ± standard error of the mean from three independent competition experiments. (D) Identification of EssI in culture lysates of S. epidermidis (S. epi) 12228, S. simulans, and S. aureus USA300 using Western blotting. Samples were prepared and analyzed as described in the legend to Fig. 6A. (E) Cultures of P. acnes and P. granulosum grown to an A600 of 1.0 were plated on TSA, and the S. epidermidis 14.1.R1, USA300, essD::erm, or essDL546P strain was spotted on top of Propionibacterium lawns. Pictures of plates incubated at 37°C under anaerobic conditions for 72 h are shown. (F) Cohorts of C57BL/6 mice (n = 5) were inoculated intravenously with PBS (control) or with 5 × 107 CFU USA300 or with the essD::erm and essDL546P isogenic variants. Blood was sampled 12 h postinfection, and serum IL-12 (p70) was analyzed by enzyme-linked immunosorbent assay. Data were averaged, repeated for reproducibility, and analyzed by one-way ANOVA using Bonferroni's multiple-comparison test and with an unpaired t test in panel D (*, P < 0.05; ns, not significant).
Recently, Christensen et al. demonstrated that S. epidermidis 14.1.R1, a skin isolate, effectively killed Propionibacterium acnes and Propionibacterium granulosum, commensals of the human skin (34). Genome sequencing of S. epidermidis 14.1.R1 revealed an ESS cluster with DUF600 proteins similar but not identical to the four different variants identified in the genome sequences of S. aureus isolates (34, 35). It is, however, not clear whether the ESS cluster of S. epidermidis 14.1.R1 is indeed responsible for implementation of competitive growth advantages over P. acnes and P. granulosum isolates. To test whether the S. aureus ESS cluster affects microbial competition, P. acnes and P. granulosum were used as indicator strains on agar plates also inoculated with wild-type S. aureus USA300 and either its isogenic essD::erm or essDL546P variant (Fig. 7E). Compared to the positive control, S. epidermidis 14.1.R1, the S. aureus isolates did not impose growth inhibition on either P. acnes or P. granulosum (Fig. 7E). Further, S. epidermidis 14.1.R1 did not kill S. aureus USA300 or its essD mutants (data not shown).
S. aureus EssD nuclease activity affects IL-12 signaling during bloodstream infection.
Anderson et al. reported that the surge of IL-12 cytokine signaling associated with S. aureus bloodstream infection in mice requires EssE-mediated secretion of ESS pathway effectors (25). We wondered whether the EssD nuclease activity of S. aureus USA300 is also required for the induction of IL-12 signaling. To address this question, cohorts (n = 5) of C57BL/6 mice were infected by intravenous inoculation with 5 × 107 CFU of S. aureus USA300 wild-type or its essD::erm and essDL546P variants (Fig. 7F). IL-12 cytokine levels were determined in blood samples drawn 12 h after challenge and compared to those of mice mock infected with phosphate-buffered saline (PBS). As expected, S. aureus USA300 bloodstream infection caused a surge in IL-12 signaling (36), which was diminished during infection with the essD::erm and essDL546P variant strains (Fig. 7F). These data suggest that the nuclease activity of EssD exerts an effector function during S. aureus host infection, stimulating IL-12 cytokine signaling to modulate host immune responses.
DISCUSSION
EssD belongs to the cluster of orthologous group 5444 (COG5444), members of which are widely distributed in Gram-positive bacteria. COG5444 genes are associated with ESS gene clusters (8, 26). However, the C-terminal domain of this gene is highly variable. In S. aureus, the C-terminal domain of COG5444 (EssD) is annotated as endonuclease_NS_2 (protein family PF13930). In Listeria monocytogenes, COG5444 (Lmo0066) carries a VIP2 C-terminal domain, which is otherwise known to modify host actin via ADP-ribosylation (26). In Bacillus anthracis, COG5444 (EsxL) harbors a C-terminal domain of the nucleotide deaminase superfamily (8). The C-terminal domains have been grouped into the novel SUKH superfamily that encompasses the nuclease and nucleic acid deaminase families (37). As with the contact-dependent inhibition (CDI) systems (38) and colicin-like nuclease toxins of proteobacteria (39), SUKH superfamily members are found in close association with a distinct superfamily of proteins proposed to function as SUKH immunity factors (37, 40, 41). Holberger et al. provided biochemical validation for this model by demonstrating that the C-terminal domains of three COG5444-like proteins (YobL, YxiD, and YqcG) of B. subtilis 168 display cytotoxic RNase activities that are neutralized by the binding of cognate antitoxin (immunity) proteins (40). However, the biological significance of B. subtilis cytotoxic RNase activities has not yet been elucidated (40).
SUKH and immunity proteins appear to cluster with various known or predicted secretion systems and have been termed polymorphic toxin systems (37, 42). These secretion systems include filamentous surface proteins such as the rearrangement hot spot (Rhs) and related YD-peptide repeat proteins, contact-dependent inhibition (CDI), and T5SS, T6SS, and ESS secretion systems (37, 42). Similar to CDI and Rhs- and T6SS-mediated contact-dependent inhibition, toxins are injected into target cells presumably to help establish a niche and defend it against other bacteria (43, 44). We demonstrate here that EssD is a secreted effector of the ESS pathway in S. aureus with broad nuclease activity toward double-stranded DNA. Intracellular EssD is highly cytotoxic, and its DNase activity is inhibited by binding to EssI. However, the EssD-EssI module does not appear to confer on S. aureus a growth advantage over other commensals of the human skin and nares, including P. acnes, P. granulosum, S. epidermidis, and S. simulans.
Bacterial pathogens evolved with their hosts and manipulate the recognition of their pathogen-associated molecular patterns by pattern recognition receptors or usurp vertebrate immune signaling schemes to establish an environment conducive to their lifestyle. Escherichia coli, Shigella boydii, Haemophilus ducreyi, Actinobacillus actinomycetemcomitans, Helicobacter hepaticus, and Campylobacter jejuni produce cytolethal distending toxin (CDT) (45). CDT is comprised of three subunits (CdtA, CdtB, and CdtC), and its CdtB effector, a homologue of DNase I, cleaves double-stranded DNA in vitro and host cell chromatin in vivo (46). Association of CdtA and CdtC with CdtB is necessary for CDT binding and uptake into eukaryotic cells (47), where CdtB travels into the nucleus to generate double-strand breaks, arresting the cell cycle of intoxicated cells at G2M and activating DNA damage response pathways (45). Salmonella enterica serovar Typhi typhoid toxin is assembled as an A2B5 molecule with two A subunits, PltA, a homologue of Bordetella pertussis pertussis toxin with ADP-ribosyltransferase activity, and CdtB, a homologue of C. jejuni CdtB (48). When analyzed in a humanized mouse model, injection purified typhoid toxin (PltA-CdtB-PltB5) causes clinical signs of typhoid fever and eliminates bloodstream neutrophils in a manner that is dependent on CdtB nuclease but not PltA activity (48, 49). Thus, nuclease effectors represent a key virulence strategy for bacterial pathogenesis. Here, we add further support to this model by demonstrating that S. aureus EssD, a nuclease effector, contributes to the pathogenesis of bloodstream infections. S. Typhi produces typhoid toxin only during infection, when it is released into the extracellular environment by a unique transport mechanism involving vesicle carrier intermediates (50). Similarly, EssD secretion also occurs during infection, and we presume that the ESS pathway may direct EssD into the nucleus of host cells but do not yet know whether this involves intracellular or extracellular staphylococci.
Earlier work identified inflammatory myeloid cells as sources of IL-12 secretion during bacterial infection (51). In agreement with this conjecture, depletion of dendritic cells in mice abrogates IL-12 signaling during S. aureus infection (52). IL-12 production skews the immune system of infected hosts toward TH1 responses and the activation of cellular immunity by promoting the differentiation of naive CD4 T cells into gamma interferon-producing TH1 cells (51). As S. aureus abscess lesions require persistent supplies of immune cells for the destruction of host tissues and the drainage of purulent material with staphylococci, it seems plausible that staphylococci activate IL-12 signaling to accomplish these goals. In addition, IL-12 skewing of gamma interferon-producing TH1 cells also impedes TH2 polarization (53), suggesting that the ESS pathway also interferes with the host's ability to produce antibodies. Thus, ESS secretion of EssD may contribute to both virulence and immune evasive strategies of S. aureus.
MATERIALS AND METHODS
Bacterial growth conditions.
S. aureus, S. epidermidis, and S. simulans were grown in tryptic soy broth (TSB) or agar (TSA) at 37°C, supplemented with chloramphenicol (Cm; 20 μg/ml) for plasmid selection and 0.2% heat-inactivated horse serum (Gibco/Life Technologies) for induction of the ESS pathway (30). For coculture assays between staphylococcal species, bacteria were grown in TSB supplemented with 0.2% heat-inactivated horse serum at 37°C to an absorbance at 600 nm (A600) of 10.0. Ten microliters of bacterial suspensions was mixed in a 1:1 ratio and spotted on TSA supplemented with 0.2% heat-inactivated horse serum at 37°C for 9 h. Bacteria grown on these plates were transferred to PBS, serially diluted, and plated in triplicate for CFU enumeration on TSA containing chloramphenicol (Cm; 5 μg/ml) or kanamycin (Kan; 20 μg/ml) to enumerate S. simulans or S. epidermidis and S. aureus USA300. To assess whether USA300 displays a growth advantage over Propionibacterium strains, cultures of P. acnes and P. granulosum were incubated in reinforced clostridial medium (BD) at 37°C under anaerobic conditions (80% N2, 10% CO2, and 10% H2) for 48 to 72 h and diluted to an A600 of 1.0, and 500 μl was plated on TSA. Cultures (3 μl) of test bacteria were added on top of Propionibacterium lawns, and plates were incubated at 37°C under anaerobic conditions for another 72 h. E. coli was grown in Luria-Bertani (LB) broth or agar at 30°C or 37°C. Where necessary, ampicillin (Amp; 100 μg/ml), Kan (50 μg/ml), arabinose (Ara; 0.2%), and isopropyl β-d-1-thiogalactopyranoside (IPTG; 0.5 mM) were added to E. coli cultures. To examine growth of E. coli carrying various plasmids, overnight cultures grown at 30°C in LB broth containing Amp, Kan, and arabinose were diluted 1:100 in 50 ml of fresh broth supplemented with both antibiotics and in the presence or absence of arabinose. IPTG was added 3 h after dilution of cultures. Bacterial growth in cultures was monitored by recording absorbance (A600). To assess viability, culture aliquots were serially diluted, plated on agar in the presence of antibiotic and arabinose when needed, and grown overnight at 30°C.
Bacterial strains and plasmids.
S. aureus RN4220 was used to passage plasmid DNA. USA300 LAC, a clone of the American community-acquired methicillin-resistant S. aureus (CA-MRSA) epidemic strain (31), was used as the wild-type S. aureus strain for all other experiments. The essD::erm, ΔessB, and complemented ΔessB/pessB strains have been described earlier (26, 30). S. epidermidis ATCC 12228 (S. epidermidis 12228), P. acnes (ATCC 6919), and P. granulosum (ATCC 25564) were obtained from the American Type Culture Collection (ATCC.org). S. simulans MK148 (ATCC 27848) and S. epidermidis 14.1.R1 were gifts from Friedrich Götz and Holger Brüggemann, respectively. For the cloning of essD, three additional codons were included upstream of the start codon assigned in the referenced sequence (Staphylococcus aureus subsp. aureus USA300_FPR3757; NCBI RefSeq accession number NC_007793.1). Primers 1 (5′-ATAAGCTAGCAGGAGGTGCCAACATGACATTTG-3′) and 2 (5′-TGGCGGTACCTTATTCTTCTAGCTCTTTAATATATTGCTCGAT-3′) as well as primers 3 (5′-CGATGCTAGCTACTGAACCAAGCAGTGATGAA-3′) and 4 (5′-GCCGGTACCCTACTCTTGCTCTTTAACATACTTCTCTAC-3′) were used to amplify by PCR the gene SAUSA300_0289 or the last five contiguous DUF600-encoding genes, SAUSA300_0298 to SAUSA300_0302, using USA300 chromosomal DNA as the template. Inserts were cloned into the NheI and KpnI sites of the pBAD24 vector carrying the arabinose promoter and ampicillin resistance cassette (pBAD-Ampr). The resulting plasmids expressed Para-essI1 and Para-essI6-10, respectively. Primers 5 (5′-GGCAAACCATATGACAAAAGATATTGAATATCTAACAGCTG-3′) and 6 (5′-ATGTCTCGAGCTACTTATTTAATATTCTTCTAATATTTCTTTCACCATTAA-3′) or primers 7 (5′-GGCAGGTCATATGTACACCAAGGTTGAATTCGGAGAACACTAT-3′) and 8 (5′-ATATCTCGAGCTAATGGTGATGGTGATGGTGCTTATTTAATATTCTTCTAATATTTCTTTCACCATTAA-3′) were used to amplify full-length essD or the nuclease-encoding fragment with six appended histidine codons at the 3′ end, essD434–617/HIS, respectively. Wild-type template DNA was used for PCR. Cloning of essD also resulted in the isolation of the essD Leu546Pro allele where leucine at position 546 within the nuclease domain was changed to proline (essD*). This plasmid was used as a template for amplification using primers 7 and 8. All essD-bearing fragments were cloned into the NdeI and XhoI sites of the E. coli pSRK vector carrying the lactose promoter and resistance marker for kanamycin (pSRK-Kanr). The three resulting plasmids were named Plac-essD, Plac-essD434–617/HIS, and Plac-essD*434–617/HIS.
For the cloning of fragments encompassing the last 162, 142, 132, and 122 amino acids of EssD, the following primers were used for 5′ amplification: 9 (5′-GGCAGGTCATATGTACACCAAGGTTGAATTCGGAGAACACTAT-3′), 10 (5′-GGCACCGCATATGAATATTGAATACACAACACCTACTGGTCAC-3′), 11 (5′-GGCAGGGCATATGATATATCGAACCGATCATAAAGGTCGCATA-3′), and 12 (5′-CCCCGGCCATATGAAAGAAGTTTATGTAGACAATCTCTCTCT-3′). Cloning of these fragments was performed as described for wild-type essD using primer 6 for 3′ amplification. For the purification of recombinant proteins used to generate antibodies, primers 13 (5′-ATCGGATCCATGCATGACATGACAAAAGATATTGAATATC-3′) and 14 (5′-GCAGAATTCCTAAGCTACTGTCATGTAATCACAAGAA-3′) or 15 (5′-ATCGGATCCAGTGAAGGCAAACATAGTATAAGTAGC-3′) and 16 (5′-TACCCGGGCTACTTATTTAATATTCTTCTAATATTTCTTTCACCATTAA-3′) were used to amplify the coding region for the first 200 amino acids or last 185 amino acids of EssD, respectively, using template DNA from the Leu546Pro variants. Primers 17 (5′-AAGGATCCATGACATTTGAAGAGAAG-3′) and 18 (5′-AAGAATTCTTATTCTTCTAGCTCTTTA-3′) were used to amplify the coding region of essI1, and the insert was subsequently cloned into BamHI and EcoRI or BamHI and SmaI sites of vector pGEX-2T, respectively. For complementation studies in S. aureus, primers 19 (5′-GGCAGGCCATATGCATGACATGACAAAAGATATTGAATATCTAACAGCTG-3′) and 20 (5′-ACGGGATCCTTATTCTTCTAGCTCTTTAATATATTGCTCGAT-3′) were used to amplify essD-essI1 from wild-type DNA and for cloning into the NdeI and BamHI sites of pWWW412, resulting in the plasmid pessD-essI1. For cloning of essI1 into pSRK, primers 21 (5′-GGCAAACCATATGACATTTGAAGAGAAGCTTAGCAAAATATACAAT-3′) and 22 (5′-ATGTCTCGAGTTATTCTTCTAGCTCTTTAATATATTGCTCGAT-3′) were used. Primers 5 and 8 were used to amplify DNA from the template bearing the mutant allele for the Leu546Pro substitution. The insert was also cloned in pWWW412, resulting in plasmid pessD*HIS. To construct the isogenic essDL546P variant recombined on the chromosome of strain USA300, primer 21 (5′-GGGGACAAGTTTGTACAAAAAAGCAGGCTATGACAAAAGATATTGAATATCTAACAGCTG-3′) and 22 (5′-GGTTATCAATGTCTTTTGAACCACCAAACATTCTAGCGAT-3′) and primers 23 (5′-CTGTGGCACAAAGTAAATTTATCAACCGTCCATTTAAGGA-3′) and 24 (5′-GGGGACCACTTTGTACAAGAAAGCTGGGTTTATTCTTCTAGCTCTTTAATATATTGCTC-3′) were used to amplify two DNA fragments that were ligated together and reamplified using primers 21 and 24. The new product was cloned into the allelic replacement vector, pKOR1, as described previously (54). Whole-genome sequencing of the wild-type USA300 parent and two essDL546P recombined isolates was performed using Truseq DNA-seq Library Preparation and Illumina MiSeq technology by the Next Generation Sequencing Core at Argonne National Laboratory. Raw sequence data were analyzed using the bioinformatics software Geneious.
Microscopy.
Aliquots of culture samples were fixed with 4% formaldehyde for 10 min, washed three times with PBS, and transferred onto a microscope slide. One drop of SlowFade Gold solution with 4′,6-diamidino-2-phenylindole (DAPI) (Life Technologies) was added to each slide. Specimens were overlaid with a coverslip and visualized with an Olympus AX70 microscope using a 100× objective and a UV filter, and images were captured with a digital camera.
Purification of proteins and biochemical assays.
For the purification of glutathione S-transferase (GST)- or histidine-tagged proteins from E. coli, strains were grown at 37°C in LB broth with appropriate antibiotics and inducers in a 4-liter volume until they reached an A600 of 1.0. Spent culture medium was discarded following centrifugation at 9,000 × g for 10 min. Bacteria in cell pellets were suspended in 20 ml of buffer A (50 mM Tris-HCl, pH 7.5, 300 mM NaCl) and lysed by passage through a French pressure cell (twice at 15,000 lb/in2). Lysates were cleared by centrifugation at 100,000 × g for 2 h at 4°C, and the supernatants were loaded by gravity flow onto glutathione-Sepharose beads (GE Healthcare) or Ni-NTA beads (Qiagen) preequilibrated in buffer A. Beads were washed with 20 volumes of buffer A, and bound proteins were eluted with either 20 mM reduced glutathione or increasing concentrations of imidazole (up to 0.5 M). Eluted proteins were dialyzed into PBS or buffer B (see below), quantified by bicinchoninic acid assay (Pierce), and kept at 4°C for immediate use or stored frozen at −80°C. Proteins dialyzed in PBS were used for the generation of rabbit polyclonal antibodies as described earlier (55). For purification of proteins from S. aureus, 8-liter cultures were grown in TSB with appropriate antibiotics at 37°C with shaking until they reached an A600 of 2.0. Samples were processed as described for the purification of histidine-tagged proteins from E. coli except that bacterial suspensions were first incubated with lysostaphin (100 μg/ml) at 37°C for 1 h prior to passage through the French pressure cell. To assess nuclease activity, proteins (5 μl; ∼ 0.05 μg) or 5 μl of supernatant of cultures grown to an A600 of 3.0 was incubated with either 500 ng of pGEX-2T DNA or 100 ng of annealed complementary 48-nucleotide-long oligomers (double-stranded DNA substrate; dsDNA) or 100 ng of total RNA from E. coli purchased from Life Technologies. Reactions were performed in a 20-μl volume in buffer B (50 mM potassium acetate, 20 mM Tris-acetate, 10 mM magnesium acetate, and 1 mM dithiothreitol [DTT], pH 7.8), and tubes were incubated at 37°C for 3 h. DNA loading buffer (6×; 30% [vol/vol] glycerol, 0.25% [wt/vol] bromophenol blue, 0.25% [wt/vol] xylene cyanol) was added to each tube prior to separation of samples on a 1% or 2% agarose gel. DNA in the gel was visualized with a UV transilluminator for image acquisition. For HPLC analysis, 7 μg of dsDNA was incubated with ∼0.7 μg of proteins or buffer control in a total reaction volume of 700 μl in buffer B for 20 h at 37°C. Samples were heated at 95°C for 10 min, centrifuged at 17,000 ×g for 10 min, and loaded onto a C18 column (250 mm by 3 mm; 5-μm particle size [BDS Hypersil C18; Thermo Fisher Scientific]) maintained at 42°C and run at a flow rate of 0.5 ml/min. Buffer C (65 mM potassium phosphate, pH 6.0) was used as the mobile phase for 10 min, followed by a 50-min-long gradient of 1 to 100% buffer D (65 mM potassium phosphate, 25% methanol, pH 6.0) and a 5-min wash with 100% buffer D.
Protein secretion and subcellular fractionation.
To examine the production and subcellular localization of EssD in S. aureus, overnight cultures were diluted 1:100 in 50 ml of TSB supplemented with appropriate antibiotics and heat-inactivated horse serum. Cultures were grown with shaking at 37°C until an A600 of 1.0. Lysostaphin was added to the whole culture (20 μg/ml) at 37°C for 1 h, yielding samples labeled as culture lysate. To identify proteins secreted in the medium, cultures were subjected to centrifugation (9,000 × g for 10 min), and the spent medium was filtered with a 0.22-μm-pore-size Millex-GP unit and transferred to a fresh tube (medium fraction [MD]). Cells in the pellet were washed and lysed with lysostaphin (20 μg/ml) at 37°C for 1 h, and cellular lysates were further subjected to centrifugation at 100,000 × g for 2 h at 4°C to separate soluble (Sol) from insoluble (Insol) proteins associated with the membrane. Proteins in all fractions were precipitated by adding 100% trichloroacetic acid (TCA) to a final concentration of 10% and left on ice for 1 h. Precipitated materials were sedimented by centrifugation (13,000 × g for 10 min at 4°C) and washed with cold acetone, and pellets were air dried before suspension in 160 μl of 0.5 M Tris-HCl, pH 8.0.
SDS-PAGE and immunoblotting.
Bacterial and protein extracts were mixed with 1/5 volume of sample buffer (0.1 M Tris-HCl [pH 8.0], 4% SDS, 20% glycerol, 2 mM β-mercaptoethanol, 0.04% bromophenol blue), heated at 90°C for 10 min, and subjected to 12% SDS-PAGE. Proteins were visualized either by direct staining of gels using Coomassie brilliant blue R-250 (Aldrich-Sigma) or by immunoblotting following electrotransfer to polyvinylidene difluoride (PVDF) membranes. For immunoblotting, membranes were blocked by incubation in PBS containing 0.1% Tween-20 (PBST) and 5% milk for 1 h at room temperature. To prevent binding of primary antibodies to protein A, 0.8 mg of human IgG was added to 10 ml of blocking buffer. After incubation (1 h at room temperature), immune serum (primary polyclonal antibodies) was added, and the membranes were incubated for up to 16 h at 4°C. Membranes were washed four times for 10 min in PBST and incubated with secondary antibody (goat anti-rabbit HRP-linked antibody; Cell Signaling Technology) in PBST with 5% milk for 1 h. Membranes were washed four times with PBST, incubated with SuperSignal West Pico Chemiluminescent Substrate (Life Technologies), and developed on Amersham Hyperfilm ECL (GE Healthcare Life Sciences).
Antisera.
New Zealand White rabbits were purchased from Harlan Sprague Dawley and used to generate the polyclonal EssDN (first 200 amino acids of EssD), EssDC (last 185 amino acids of EssD), and EssI (EssI1 encoded by SAUSA300_0289) antisera. Purified antigens (100 μg) were emulsified with complete Freund's adjuvant (Difco), and emulsions were injected subcutaneously into the rabbits. At 21-day intervals, two booster immunizations with 100 μg of antigen emulsified with incomplete Freund's adjuvant were performed. Sera were obtained from blood samples, mixed with 0.02% sodium azide, aliquoted, and frozen for long-term storage at −80°C or stored at 4°C for reiterative use.
Animal infection.
Cohorts of five 5-week-old C57BL/6 mice (Jackson Laboratory) were anesthetized with 65 mg/ml ketamine and 6 mg/ml xylazine per kilogram of body weight via intraperitoneal injection. Animals were infected via retro-orbital injection with 5 × 107 CFU of bacteria grown to an A600 of 0.4, washed, and suspended in 100 μl of PBS. Control animals were inoculated with 100 μl of PBS alone. At 12 h postinfection, mice were euthanized via CO2 inhalation, and blood was collected by cardiac puncture for serum preparation and analysis. A Novex Mouse IL-12 Antibody Pair kit CMC0123 (Life Technologies) was used to measure IL-12 (p70) levels.
Ethics statement.
The preparation of rabbit antibodies and mouse challenge experiments were performed according to protocols that were reviewed, approved, and performed under the regulatory supervision of The University of Chicago's Institutional Animal Care and Use Committee (IACUC). Animal experiments were conducted in accordance with recommendations detailed in the Guide for the Care and Use of Laboratory Animals (56). Animal care was managed by The University of Chicago Animal Resource Center, which is accredited by the American Association for Accreditation of Laboratory Animal Care and acts in compliance with the NIH guidelines on laboratory animal care and use.
ACKNOWLEDGMENTS
We thank Holger Brüggemann and Friedrich Götz for providing strains S. epidermidis 14.1.R1 and S. simulans MK148, respectively. We are especially grateful to Khaled Aly, Yvonne Chan, Carla Emolo, Fabiana Falugi, Aretha Fiebig, Hwan Keun Kim, and Vilasack Thammavongsa for technical assistance and advice and Phoebe Rice, Sean Crosson, and members of the Schneewind and Missiakas laboratory for discussion.
M.A. was supported by a National Institute of Allergy and Infectious Diseases (NIAID) Biodefense Training Grant in Host-Pathogen Interactions at the University of Chicago (T32 AI065382) and was a recipient of an American Heart Association Award (11PRE7600117). R.J.O. was supported by a Molecular Cell Biology Training Grant at the University of Chicago (T32 GM007183). This work was supported by grants AI075258 and AI110937 from NIAID to D.M. Work in the laboratory of O.S. is supported by NIAID grants AI038897 and AI052474.
Footnotes
For a companion article on this topic, see https://doi.org/10.1128/JB.00527-16.
REFERENCES
- 1.Abdallah AM, Gey van Pittius NC, Champion PA, Cox J, Luirink J, Vandenbroucke-Grauls CM, Appelmelk BJ, Bitter W. 2007. Type VII secretion—mycobacteria show the way. Nat Rev Microbiol 5:883–891. doi: 10.1038/nrmicro1773. [DOI] [PubMed] [Google Scholar]
- 2.Bitter W, Houben EN, Bottai D, Brodin P, Brown EJ, Cox JS, Derbyshire K, Fortune SM, Gao LY, Liu J, Gey van Pittius NC, Pym AS, Rubin EJ, Sherman DR, Cole ST, Brosch R. 2009. Systematic genetic nomenclature for type VII secretion systems. PLoS Pathog 5:e1000507. doi: 10.1371/journal.ppat.1000507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stoop EJ, Bitter W, van der Sar AM. 2012. Tubercle bacilli rely on a type VII army for pathogenicity. Trends Microbiol 20:477–484. doi: 10.1016/j.tim.2012.07.001. [DOI] [PubMed] [Google Scholar]
- 4.Pym AS, Brodin P, Brosch R, Huerre M, Cole ST. 2002. Loss of RD1 contributed to the attenuation of the live tuberculosis vaccines Mycobacterium bovis BCG and Mycobacterium microti. Mol Microbiol 46:709–717. doi: 10.1046/j.1365-2958.2002.03237.x. [DOI] [PubMed] [Google Scholar]
- 5.Stanley SA, Raghavan S, Hwang WW, Cox JS. 2003. Acute infection and macrophage subversion by Mycobacterium tuberculosis require a specialized secretion system. Proc Natl Acad Sci U S A 100:13001–13006. doi: 10.1073/pnas.2235593100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Burts ML, Williams WA, DeBord K, Missiakas DM. 2005. EsxA and EsxB are secreted by an ESAT-6-like system that is required for the pathogenesis of Staphylococcus aureus infections. Proc Natl Acad Sci U S A 102:1169–1174. doi: 10.1073/pnas.0405620102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Burts ML, DeDent AC, Missiakas DM. 2008. EsaC substrate for the ESAT-6 secretion pathway and its role in persistent infections of Staphylococcus aureus. Mol Microbiol 69:736–746. doi: 10.1111/j.1365-2958.2008.06324.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Garufi G, Butler E, Missiakas D. 2008. ESAT-6-like protein secretion in Bacillus anthracis. J Bacteriol 190:7004–7011. doi: 10.1128/JB.00458-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Anderson M, Aly KA, Chen YH, Missiakas D. 2013. Secretion of atypical protein substrates by the ESAT-6 secretion system of Staphylococcus aureus. Mol Microbiol 90:734–743. doi: 10.1111/mmi.12395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pallen MJ. 2002. The ESAT-6/WXG100 superfamily—and a new Gram-positive secretion system? Trends Microbiol 10:209–212. doi: 10.1016/S0966-842X(02)02345-4. [DOI] [PubMed] [Google Scholar]
- 11.Cole ST, Brosch R, Parkhill J, Garnier T, Churcher C, Harris D, Gordon SV, Eiglmeier K, Gas S, Barry CE III, Tekaia F, Badcock K, Basham D, Brown D, Chillingworth T, Connor R, Davies R, Devlin K, Feltwell T, Gentles S, Hamlin N, Holroyd S, Hornsby T, Jagels K, Krogh A, McLean J, Moule S, Murphy L, Oliver K, Osborne J, Quail MA, Rajandream M-A, Rogers J, Rutter S, Seeger K, Skelton J, Squares R, Squares S, Sulston JE, Taylor K, Whitehead S, Barrell BG. 1998. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 393:537–544. doi: 10.1038/31159. [DOI] [PubMed] [Google Scholar]
- 12.Strong M, Sawaya MR, Wang S, Phillips M, Cascio D, Eisenberg D. 2006. Toward the structural genomics of complexes: crystal structure of a PE/PPE protein complex from Mycobacterium tuberculosis. Proc Natl Acad Sci U S A 103:8060–8065. doi: 10.1073/pnas.0602606103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Renshaw PS, Lightbody KL, Veverka V, Muskett FW, Kelly G, Frenkiel TA, Gordon SV, Hewinson RG, Burke B, Norman J, Williamson RA, Carr MD. 2005. Structure and function of the complex formed by the tuberculosis virulence factors CFP-10 and ESAT-6. EMBO J 24:2491–2498. doi: 10.1038/sj.emboj.7600732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sundaramoorthy R, Fyfe PK, Hunter WN. 2008. Structure of Staphylococcus aureus EsxA suggests a contribution to virulence by action as a transport chaperone and/or adaptor protein. J Mol Biol 383:603–614. doi: 10.1016/j.jmb.2008.08.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ilghari D, Lightbody KL, Veverka V, Waters LC, Muskett FW, Renshaw PS, Carr MD. 2011. Solution structure of the Mycobacterium tuberculosis EsxG·EsxH complex: functional implications and comparisons with other M. tuberculosis Esx family complexes. J Biol Chem 286:29993–30002. doi: 10.1074/jbc.M111.248732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Poulsen C, Panjikar S, Holton SJ, Wilmanns M, Song YH. 2014. WXG100 protein superfamily consists of three subfamilies and exhibits an alpha-helical C-terminal conserved residue pattern. PLoS One 9:e89313. doi: 10.1371/journal.pone.0089313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Houben D, Demangel C, van Ingen J, Perez J, Baldeón L, Abdallah AM, Caleechurn L, Bottai D, van Zon M, de Punder K, van der Laan T, Kant A, Bossers-de Vries R, Willemsen P, Bitter W, van Soolingen D, Brosch R, van der Wel N, Peters PJ. 2012. ESX-1-mediated translocation to the cytosol controls virulence of mycobacteria. Cell Microbiol 14:1287–1298. doi: 10.1111/j.1462-5822.2012.01799.x. [DOI] [PubMed] [Google Scholar]
- 18.Wassermann R, Gulen MF, Sala C, Perin SG, Lou Y, Rybniker J, Schmid-Burgk JL, Schmidt T, Hornung V, Cole ST, Ablasser A. 2015. Mycobacterium tuberculosis differentially activates cGAS- and inflammasome-dependent intracellular immune responses through ESX-1. Cell Host Microbe 17:799–810. doi: 10.1016/j.chom.2015.05.003. [DOI] [PubMed] [Google Scholar]
- 19.Watson RO, Manzanillo PS, Cox JS. 2012. Extracellular M. tuberculosis DNA targets bacteria for autophagy by activating the host DNA-sensing pathway. Cell 150:803–815. doi: 10.1016/j.cell.2012.06.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Manzanillo PS, Shiloh MU, Portnoy DA, Cox JS. 2012. Mycobacterium tuberculosis activates the DNA-dependent cytosolic surveillance pathway within macrophages. Cell Host Microbe 11:469–480. doi: 10.1016/j.chom.2012.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hornung V, Ablasser A, Charrel-Dennis M, Bauernfeind F, Horvath G, Caffrey DR, Latz E, Fitzgerald KA. 2009. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature 458:514–518. doi: 10.1038/nature07725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gao D, Wu J, Wu YT, Du F, Aroh C, Yan N, Sun L, Chen ZJ. 2013. Cyclic GMP-AMP synthase is an innate immune sensor of HIV and other retroviruses. Science 341:903–906. doi: 10.1126/science.1240933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Watson RO, Bell SL, MacDuff DA, Kimmey JM, Diner EJ, Olivas J, Vance RE, Stallings CL, Virgin HW, Cox JS. 2015. The cytosolic sensor cGAS detects Mycobacterium tuberculosis DNA to induce type I interferons and activate autophagy. Cell Host Microbe 17:811–819. doi: 10.1016/j.chom.2015.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dey B, Dey RJ, Cheung LS, Pokkali S, Guo H, Lee JH, Bishai WR. 2015. A bacterial cyclic dinucleotide activates the cytosolic surveillance pathway and mediates innate resistance to tuberculosis. Nat Med 21:401–406. doi: 10.1038/nm.3813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Anderson M, Ohr RJ, Aly KA, Nocadello S, Kim HK, Schneewind CE, Schneewind O, Missiakas DM. 2017. EssE promotes Staphylococcus aureus ESS-dependent protein secretion to modify host immune responses during infection. J Bacteriol 199:e00527-16. doi: 10.1128/JB.00527-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Anderson M, Chen YH, Butler EK, Missiakas DM. 2011. EsaD, a secretion factor for the Ess pathway in Staphylococcus aureus. J Bacteriol 193:1583–1589. doi: 10.1128/JB.01096-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Galán JE. 2009. Common themes in the design and function of bacterial effectors. Cell Host Microbe 5:571–579. doi: 10.1016/j.chom.2009.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schäfer D, Lâm TT, Geiger T, Mainiero M, Engelmann S, Hussain M, Bosserhoff A, Frosch M, Bischoff M, Wolz C, Reidl J, Sinha B. 2009. A point mutation in the sensor histidine kinase SaeS of Staphylococcus aureus strain Newman alters the response to biocide exposure. J Bacteriol 191:7306–7314. doi: 10.1128/JB.00630-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Steinhuber A, Goerke C, Bayer M, Doring G, Wolz C. 2003. Molecular architecture of the regulatory locus sae of Staphylococcus aureus and its impact on expression of virulence factors. J Bacteriol 185:6278–6286. doi: 10.1128/JB.185.21.6278-6286.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen YH, Anderson M, Hendrickx AP, Missiakas D. 2012. Characterization of EssB, a protein required for secretion of ESAT-6 like proteins in Staphylococcus aureus. BMC Microbiol 12:219. doi: 10.1186/1471-2180-12-219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Diep BA, Gill SR, Chang RF, Phan TH, Chen JH, Davidson MG, Lin F, Lin J, Carleton HA, Mongodin EF, Sensabaugh GF, Perdreau-Remington F. 2006. Complete genome sequence of USA300, an epidemic clone of community-acquired methicillin-resistant Staphylococcus aureus. Lancet 367:731–739. doi: 10.1016/S0140-6736(06)68231-7. [DOI] [PubMed] [Google Scholar]
- 32.Schindler CA, Schuhardt VT. 1964. Lysostaphin: a new bacteriolytic agent for the Staphylococcus. Proc Natl Acad Sci U S A 51:414–421. doi: 10.1073/pnas.51.3.414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Russell AB, Peterson SB, Mougous JD. 2014. Type VI secretion system effectors: poisons with a purpose. Nat Rev Microbiol 12:137–148. doi: 10.1038/nrmicro3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Christensen GJ, Scholz CF, Enghild J, Rohde H, Kilian M, Thurmer A, Brzuszkiewicz E, Lomholt HB, Bruggemann H. 2016. Antagonism between Staphylococcus epidermidis and Propionibacterium acnes and its genomic basis. BMC Genomics 17:152. doi: 10.1186/s12864-016-2489-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Warne B, Harkins CP, Harris SR, Vatsiou A, Stanley-Wall N, Parkhill J, Peacock SJ, Palmer T, Holden MT. 2016. The Ess/type VII secretion system of Staphylococcus aureus shows unexpected genetic diversity. BMC Genomics 17:222. doi: 10.1186/s12864-016-2426-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ziegler C, Goldmann O, Hobeika E, Geffers R, Peters G, Medina E. 2011. The dynamics of T cells during persistent Staphylococcus aureus infection: from antigen-reactivity to in vivo anergy. EMBO Mol Med 3:652–666. doi: 10.1002/emmm.201100173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang D, Iyer LM, Aravind L. 2011. A novel immunity system for bacterial Nucleic acid degrading toxins and its recruitment in various eukaryotic and DNA viral systems. Nucleic Acids Res 39:4532–4552. doi: 10.1093/nar/gkr036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Aoki SK, Diner EJ, de Roodenbeke CT, Burgess BR, Poole SJ, Braaten BA, Jones AM, Webb JS, Hayes CS, Cotter PA, Low DA. 2010. A widespread family of polymorphic contact-dependent toxin delivery systems in bacteria. Nature 468:439–442. doi: 10.1038/nature09490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cascales E, Buchanan SK, Duche D, Kleanthous C, Lloubes R, Postle K, Riley M, Slatin S, Cavard D. 2007. Colicin biology. Microbiol Mol Biol Rev 71:158–229. doi: 10.1128/MMBR.00036-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Holberger LE, Garza-Sanchez F, Lamoureux J, Low DA, Hayes CS. 2012. A novel family of toxin/antitoxin proteins in Bacillus species. FEBS Lett 586:132–136. doi: 10.1016/j.febslet.2011.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jamet A, Jousset AB, Euphrasie D, Mukorako P, Boucharlat A, Ducousso A, Charbit A, Nassif X. 2015. A new family of secreted toxins in pathogenic Neisseria species. PLoS Pathog 11:e1004592. doi: 10.1371/journal.ppat.1004592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang D, de Souza RF, Anantharaman V, Iyer LM, Aravind L. 2012. Polymorphic toxin systems: comprehensive characterization of trafficking modes, processing, mechanisms of action, immunity and ecology using comparative genomics. Biol Direct 7:18. doi: 10.1186/1745-6150-7-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Koskiniemi S, Lamoureux JG, Nikolakakis KC, t'Kint de Roodenbeke C, Kaplan MD, Low DA, Hayes CS. 2013. Rhs proteins from diverse bacteria mediate intercellular competition. Proc Natl Acad Sci U S A 110:7032–7037. doi: 10.1073/pnas.1300627110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Silverman JM, Brunet YR, Cascales E, Mougous JD. 2012. Structure and regulation of the type VI secretion system. Annu Rev Microbiol 66:453–472. doi: 10.1146/annurev-micro-121809-151619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lara-Tejero M, Galán JE. 2000. A bacterial toxin that controls cell cycle progression as a deoxyribonuclease I-like protein. Science 290:354–357. doi: 10.1126/science.290.5490.354. [DOI] [PubMed] [Google Scholar]
- 46.Lara-Tejero M, Galán JE. 2002. Cytolethal distending toxin: limited damage as a strategy to modulate cellular functions. Trends Microbiol 10:147–152. doi: 10.1016/S0966-842X(02)02316-8. [DOI] [PubMed] [Google Scholar]
- 47.Lara-Tejero M, Galán JE. 2001. CdtA, CdtB, and CdtC form a tripartite complex that is required for cytolethal distending toxin activity. Infect Immun 69:4358–4365. doi: 10.1128/IAI.69.7.4358-4365.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Song J, Gao X, Galán JE. 2013. Structure and function of the Salmonella Typhi chimaeric A2B5 typhoid toxin. Nature 499:350–354. doi: 10.1038/nature12377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Song J, Willinger T, Rongvaux A, Eynon E, Stevens S, Manz MG, Flavell RA, Galán JE. 2010. A mouse model for the human pathogen Salmonella typhi. Cell Host Microbe 8:369–376. doi: 10.1016/j.chom.2010.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Spanò S, Ugalde JE, Galán JE. 2008. Delivery of a Salmonella Typhi exotoxin from a host intracellular compartment. Cell Host Microbe 3:30–38. doi: 10.1016/j.chom.2007.11.001. [DOI] [PubMed] [Google Scholar]
- 51.Gately MK, Renzetti LM, Magram J, Stern AS, Adorini L, Gubler U, Presky DH. 1998. The interleukin-12/interleukin-12-receptor system: role in normal and pathologic immune responses. Annu Rev Immunol 16:495–521. doi: 10.1146/annurev.immunol.16.1.495. [DOI] [PubMed] [Google Scholar]
- 52.Schindler D, Gutierrez MG, Beineke A, Rauter Y, Rohde M, Foster S, Goldmann O, Medina E. 2012. Dendritic cells are central coordinators of the host immune response to Staphylococcus aureus bloodstream infection. Am J Pathol 181:1327–1337. doi: 10.1016/j.ajpath.2012.06.039. [DOI] [PubMed] [Google Scholar]
- 53.Zundler S, Neurath MF. 2015. Interleukin-12: functional activities and implications for disease. Cytokine Growth Factor Rev 26:559–568. doi: 10.1016/j.cytogfr.2015.07.003. [DOI] [PubMed] [Google Scholar]
- 54.Bae T, Schneewind O. 2006. Allelic replacement in Staphylococcus aureus with inducible counter-selection. Plasmid 55:58–63. doi: 10.1016/j.plasmid.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 55.Chan YG, Kim HK, Schneewind O, Missiakas D. 2014. The capsular polysaccharide of Staphylococcus aureus is attached to peptidoglycan by the LytR-CpsA-Psr (LCP) family of enzymes. J Biol Chem 289:15680–15690. doi: 10.1074/jbc.M114.567669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.National Research Council Committee for the Update of the Guide for the Care and Use of Laboratory Animals. 2011. Guide for the care and use of laboratory animals, 8th ed National Academies Press, Washington, DC. [Google Scholar]