

Abstract
Although oxidatively damaged lipoproteins are implicated in vascular injury, there is little information regarding the role of high-density lipoprotein (HDL) oxidation in atherogenesis. One potential pathway involves hypochlorous acid (HOCl) produced by myeloperoxidase (MPO), a heme protein secreted by phagocytes. We previously showed that 3-chlorotyrosine is a specific product of HOCl. Therefore, to explore the role of oxidized HDL in the pathogenesis of vascular disease, we used MS to quantify 3-chlorotyrosine in HDL isolated from plasma and atherosclerotic tissue. HDL from human aortic atherosclerotic intima had an 8-fold higher level of 3-chlorotyrosine than plasma HDL. Tandem MS analysis identified MPO as a component of lesion HDL, suggesting that the two interact in the artery wall. Moreover, immunohistochemical studies found that specific epitopes derived from HOCl colocalized with apolipoprotein A-I, the major protein of HDL. These observations strongly support the hypothesis that MPO promotes HDL oxidation in the human artery wall. Levels of 3-chlorotyrosine were elevated in HDL isolated from the blood of humans with established coronary artery disease, suggesting that circulating levels of oxidized HDL represent a unique marker for clinically significant atherosclerosis. HDL or lipid-free apolipoprotein A-I exposed to HOCl was less able to remove cholesterol from cultured cells by a pathway requiring the cell membrane transporter ATP-binding cassette transporter A1. The detection of 3-chlorotyrosine in HDL isolated from vascular lesions raises the possibility that MPO, by virtue of its ability to form HOCl, may promote atherogenesis by counteracting the established antiatherogenic effects of HDL and the ATP-binding cassette transporter A1 pathway.
Many lines of evidence indicate that high-density lipoprotein (HDL) protects the artery wall against the development of atherosclerosis (reviewed in refs. 1 and 2). This atheroprotective effect is attributed mainly to the ability of HDL to mobilize excess cholesterol from arterial macrophages. Cell culture experiments have uncovered several mechanisms that enable components of HDL to remove cellular cholesterol (3, 4). For example, phospholipids in HDL absorb cholesterol that diffuses from the plasma membrane, a passive process facilitated by the interaction of HDL particles with scavenger receptor B1. In contrast, HDL apolipoproteins remove cellular cholesterol and phospholipids by a cholesterol-inducible active transport process mediated by a cell membrane protein called ATP-binding cassette transporter A1 (ABCA1) (5-8).
The most abundant protein in HDL is apolipoprotein A-I (apoA-I), which accounts for ≈70% of the total protein content of HDL. Lipid-poor apoA-I promotes efflux of cellular cholesterol and phospholipids exclusively by the ABCA1 pathway (5-8). This process appears to involve the amphipathic α-helical domains in apoA-I (9). Studies of synthetic peptides and deletion mutants of apoA-I suggest that the terminal helices of apoA-I penetrate into the phospholipid bilayer of membranes, promoting cooperative interactions between other α-helical segments and lipids to create an apo/lipid structure that dissociates from membranes (10).
This atheroprotective process may be inhibited by oxidative damage, which is implicated in the pathogenesis of atherosclerosis, a chronic inflammatory disease (11). Moreover, phagocytes, which congregate at sites of inflammation, might be an important source of oxidants that create such damage. One pathway involves myeloperoxidase (MPO), a heme protein released by phagocytes (12-14). MPO uses H2O2 and chloride to generate the powerful oxidant hypochlorous acid (HOCl):
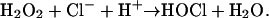 |
The importance of this reaction is underlined by the presence of enzymatically active MPO in human atherosclerotic lesions (15). Although most oxidation products generated by HOCl are either nonspecific or yield uninformative compounds, in vitro studies demonstrate that MPO converts tyrosine into 3-chlorotyrosine, a stable product (16). Studies of model systems and MPO-deficient mice have demonstrated that 3-chlorotyrosine is a molecular fingerprint that implicates the MPO pathway in oxidative damage (17, 18).
Chlorination of the phenolic ring of tyrosine may have physiological relevance because elevated levels of 3-chlorotyrosine and other products characteristic of MPO have been detected in low-density lipoprotein isolated from human atherosclerotic lesions (19-21). Moreover, methionine and phenylalanine residues in apoA-I are oxidized by reactive intermediates (22-25), and tyrosine residues are converted to o,o′-dityrosine by tyrosyl radical (26). We recently showed that HOCl selectively targets tyrosine residues in apoA-I that are suitably juxtaposed to primary amino groups (27). This mechanism might enable phagocytes to efficiently damage proteins during inflammation. However, remarkably little is known about the vulnerability of HDL to oxidation in the artery wall.
In the current study, we used MS to demonstrate that 3-chlorotyrosine levels are elevated in HDL isolated from human atherosclerotic tissue. Moreover, both HDL and lipid-free apoA-I exposed to physiologically plausible concentrations of HOCl became less able to promote cholesterol efflux from cultured cells by a pathway requiring ABCA1. Our observations raise the possibility that MPO promotes human atherogenesis by impairing the ability of HDL apolipoproteins to remove cholesterol from macrophages in the artery wall.
Experimental Procedures
Materials. Cambridge Isotope Laboratories (Andover, MA) supplied 13C-labeled amino acids. Carotid endarterectomy tissue resected at surgery and immediately frozen at -80°C was supplied by the Division of Vascular Surgery, Bowman Gray School of Medicine (Winston-Salem, NC).
Protein Oxidation Reactions. Reactions were carried out at 37°C in PBS (10 mM sodium phosphate/138 mM NaCl/2.7 mM KCl, pH 7.4) supplemented with 1 mg/ml HDL protein. Reactions were initiated by adding oxidant and terminated by adding a 10- to 50-fold mol excess of L-methionine. Concentrations of HOCl and H2O2 were determined spectrophotometrically (ε292 = 350 M-1·cm-1 and ε240 = 39.4 M-1·cm-1) (29, 30). Protein was determined by using the Lowry assay (Bio-Rad) with albumin as the standard.
Isolation of HDL. The Human Studies Committees at University of Washington School of Medicine and Wake Forest University School of Medicine approved all protocols involving human material. Blood collected from healthy adults and patients with documented coronary artery disease who had fasted overnight was anticoagulated with EDTA to obtain plasma. HDL (d = 1.125-1.210 g/ml) was prepared by sequential ultracentrifugation and was depleted of apoE and apoB-100 by heparin-agarose chromatography (31).
Lesion HDL was isolated from carotid endarterectomy specimens that had been snap frozen. Lesions from a single individual (≈0.5 g wet weight) were frozen in dry ice and pulverized with a stainless steel mortar and pestle. All subsequent procedures were carried out at 4°C. Tissue powder was suspended in 2 ml of buffer A [0.15 M NaCl/100 μM diethylenetriaminepentaacetic acid/100 μM butylated hydroxyl toluene, protease inhibitor mixture (Roche Diagnostics)/10 mM sodium phosphate, pH 7.4] in a 2-ml centrifuge tube and rocked gently overnight. Tissue was removed by centrifugation; the supernatant was collected, and the pellet was extracted a second time with buffer A for 1 h. The pooled supernatants were centrifuged at 100,000 × g for 30 min, and the pellet and uppermost lipemic layer were discarded. HDL was isolated from the tissue extract by sequential density ultracentrifugation (d = 1.063-1.210 g/ml; ref. 31). Diethylenetriaminepentaacetic acid and butylated hydroxyl toluene (both 100 μM) were included in all solutions used for lipoprotein isolation. Lesion HDL was equilibrated with buffer B (0.1 mM diethylenetriaminepentaacetic acid/50 mM sodium phosphate, pH 7.4) by using a 100-kDa cutoff filter device (Millipore). ApoA-I in lesion HDL was immunodetected by using polyclonal rabbit anti- (human apoA-I) IgG followed by a horseradish peroxidase-conjugated goat anti-rabbit IgG and enhanced chemiluminescence detection.
Immunohistochemical Studies. Human coronary artery segments were obtained from hearts excised at the time of cardiac transplantation and then fixed in neutral buffered formalin and embedded in paraffin (32). Atherosclerotic plaques were identified by morphological criteria in 6-μm sections stained with Movat's pentachrome stains. Macrophages, MPO, HOCl-modified proteins, and apoA-I were, respectively, identified with mAb HAM-56 (1:10 dilution, DAKO), rabbit polyclonal antisera (1:300 dilution; DAKO), hybridoma cell culture supernatant (HOP-1), and goat polyclonal antiserum (1:750 dilution). HOP-1 (clone 2D10G9) was provided by E. Malle (Medical University of Graz, Graz, Austria). Single-label immunohistochemistry was performed by using described techniques (32). Nova red (Vector Laboratories), which yields a red reaction product, was used as the peroxidase substrate, and cell nuclei were counterstained with hematoxylin.
MS Analysis. HDL protein was precipitated with ice-cold trichloroacetic acid (10% vol/vol), collected by centrifugation, washed with 10% trichloroacetic acid, and delipidated twice with water/methanol/water-washed diethyl ether (1:3:7 vol/vol) (33). Isotopically labeled internal standards were added, and samples were hydrolyzed at 110°C for 12 h under argon with 4 N methane sulfonic acid (Sigma) supplemented with 1% benzoic acid and 1% phenol. Amino acids were isolated from the acid hydrolysate with sequential solid-phase extractions by using C18 and Chrom P columns (Supelco, Bellefonte, PA) (18, 28). The t-butyl dimethylsilyl derivatives of amino acids were quantified by selected ion monitoring by using isotope dilution negative-ion chemical ionization GC/MS (28) performed on a Hewlett-Packard 6890 gas chromatograph equipped with a 15 m DB-5 capillary column (0.25 mm i.d., 0.33 μm film thickness, J & W Scientific, Folsom, CA) and interfaced with a Hewlett-Packard 5973 mass detector. Under these chromatography conditions, authentic compounds and isotopically labeled standards were baseline separated and exhibited retention times identical to those of analytes derived from tissue samples. The limit of detection (signal/noise >10) was <1 fmol for all of the amino acids. Authentic 3-chlorotyrosine was stable to acid hydrolysis, and recovery of the amino acid from the solid-phase extraction columns was >80%.
All samples were manually injected by using an on column injector. The level of chlorotyrosine was quantified by using the ratio between the ion of m/z 489 derived from 3-chlorotyrosine ([M-Cl-t-butyl-dimethylsilyl]-) and the ion of m/z 495 derived from 3-chloro[13C6]chlorotyrosine. Potential artifact formation was monitored as the appearance of ions at m/z 499 derived from l[13C9,15N]tyrosine added before sample work up. Under our experimental conditions, artifact formation was <5% of total 3-chlorotyrosine. The l-tyrosine and l-[13C6]tyrosine were quantified by using the ions ([M-COO-t-butyl-dimethylsilyl]-) at m/z 407 and m/z 413, respectively.
2D Liquid Chromatography-Tandem MS Analysis. Liquid chromatography-electrospray ionization-tandem MS (MS/MS) analyses were performed in the positive ion mode with a Finnigan LCQ ProteomeX ion trap instrument (San Jose, CA) coupled to a Surveyor (Thermo-Electron) quaternary HPLC pump, which in turn was interfaced with a strong cation exchange resin and a reverse-phase column (34). A fully automated 11-cycle chromatographic run was carried out on each sample. The sequest algorithm was used to interpret MS/MS spectra. Matches were visually assessed if unique peptides had highly significant sequest scores (34).
Cell Culture and Cholesterol Efflux. Baby hamster kidney (BHK) cells expressing mifepristone-inducible human ABCA1 were generated as described (35). Cellular cholesterol was labeled by adding 1 μCi/ml [3H]cholesterol (NEN) to the growth medium. Later (24 h), strong expression of ABCA1 was induced by incubating the cells for 20 h with DMEM containing 1 mg/ml BSA (DMEM/BSA) and 1 nM mifepristone (35). To measure cholesterol efflux, mock- or ABCA1-transfected cells were incubated with DMEM/BSA without or with HDL, apoA-I, or peptide. After 2-4 h, the medium and cells were assayed for [3H]cholesterol as described (35). Cholesterol efflux mediated by HDL, apoA-I, or peptide was calculated as the percentage of total [3H]cholesterol (medium plus cell) released into the medium after subtracting the value obtained with DMEM/BSA alone. BHK cells incubated with oxidized HDL or apoA-I for up to 24 h demonstrated no changes in morphology and cell protein or cholesterol content per well.
Statistical Analysis. Results represent means ± SEM. Differences between two groups were compared by using an unpaired Student's t test. Multiple comparisons were performed by using ANOVA. A P value < 0.05 was considered significant.
Results
ApoA-I Colocalizes with HOCl Epitopes in Human Atherosclerotic Tissue. To determine whether HOCl might modify HDL in vivo, we used Abs specific for apoA-I and HOCl-modified proteins to immunostain coronary arteries (n = 8) obtained from patients undergoing cardiac transplantation (32). ApoA-I colocalized with epitopes recognized by HOP-1, an Ab specific for proteins oxidized by HOCl (36), in the intima of atherosclerotic lesions (Fig. 1 A and B).
Fig. 1.

Immunohistochemical analysis of apoA-I, MPO, and proteins modified by HOCl in human atherosclerotic intima. Photomicrographs of adjacent sections of an atherosclerotic coronary artery demonstrating immunostaining for apoA-I (A), proteins modified by HOCl (B), macrophages (C), and MPO (D) are shown. Positive immunohistochemical staining is indicated by a red reaction product. HOCl-modified epitopes colocalize with extracellular apoA-I (A and B, arrows), whereas MPO staining is primarily associated with macrophages (C and D, arrowheads). Original magnification, ×100; hematoxylin counterstain.
We have demonstrated previously that MPO is present in atherosclerotic lesions, in both macrophage-associated and extracellular distributions (15). The vast majority of cell-associated MPO immunoreactivity was present in macrophages, and most of the extracellular MPO was juxtaposed with macrophages (Fig. 1 C and D). HOCl-modified proteins also colocalized with macrophages. However, the most robust staining for HOCl-modified proteins was extracellular and colocalized with apoA-I. These observations are consistent with the ability of HOCl to generate long-lived reactive intermediates such as chloramines, which can diffuse long distances to react with proteins. Indeed, we have shown that chloramines mediate tyrosine chlorination in apoA-I in vitro (27). The colocalization of HOCl-modified proteins with apoA-I suggests that HOCl oxidizes specific proteins in the human artery wall.
3-Chlorotyrosine Is Elevated in HDL Isolated from Human Vascular Lesions. To quantitatively assess whether MPO oxidizes proteins in the artery wall, we isolated HDL by sequential density gradient ultracentrifugation from human carotid atherosclerotic tissue recovered at surgery. Lesion HDL subjected to immunoblotting analysis with a rabbit polyclonal Ab monospecific for human apoA-I demonstrated a protein with the predicted molecular mass of apoA-I (Fig. 2A). Forms of immunoreactive apoA-I with higher molecular mass were also present. Apo A-I represented >50% of lesion HDL protein as assessed by Western blotting.
Fig. 2.

MS detection of 3-chlorotyrosine in HDL isolated from atherosclerotic human tissue harvested at surgery. Atherosclerotic tissue was obtained from subjects undergoing carotid endarterectomy. HDL was isolated from the supernatant of tissue powder by sequential ultracentrifugation. 13C-Labeled internal standards were added, and the protein was hydrolyzed with acid. (A) Western blot analysis of circulating HDL (1) and lesion HDL (2) with an Ab monospecific for apoA-I. Arrow, monomeric apoA-I. (B) Analysis of derivatized amino acids derived from HDL by isotope dilution negative-ion electron capture GC/MS with selected ion monitoring.
We used negative-ion chemical ionization GC/MS to determine whether 3-chlorotyrosine was present in HDL isolated from human atherosclerotic lesions. We detected a compound in the amino acid hydrolysate that exhibited major ions and retention time identical to those of authentic 3-chlorotyrosine. Selected ion monitoring showed that the ions derived from this amino acid co-eluted with those derived from 3-chloro[13C6]tyrosine (Fig. 2B). In contrast, there was little evidence for 3-chlorotyrosine formation during sample work-up and analysis (monitored as the appearance of 3-chloro[13C9,15N]tyrosine derived from exogenously added l-[13C9,15N]tyrosine). These results indicate that HDL isolated from human atherosclerotic lesions contains 3-chlorotyrosine, a specific marker of chlorination by MPO.
To determine how the level of 3-chlorotyrosine in lesion HDL compares to that in circulating HDL, we isolated HDL from human plasma and from human atherosclerotic aortic tissue. After delipidating and hydrolyzing the proteins, we quantified levels of the derivatized amino acid in acid hydrolysates with isotope dilution GC/MS (Fig. 3A). Remarkably, there was an 8-fold higher level of protein-bound 3-chlorotyrosine in lesion HDL (177 ± 27 μmol/mol Tyr; n = 10) than in circulating HDL (22 ± 7 μmol/mol Tyr; n = 17) isolated from humans (P < 0.0001).
Fig. 3.

MS quantification of 3-chlorotyrosine in HDL isolated from plasma and human atherosclerotic lesions. Plasma was obtained from healthy humans and humans with established coronary artery disease (CAD). Human atherosclerotic tissue was obtained at surgery from subjects undergoing carotid endarterectomy. HDL was isolated from plasma and tissue by sequential ultracentrifugation. Oxidized amino acids isolated from hydrolyzed HDL proteins were quantified by isotope dilution GC/MS with selected ion monitoring.
HDL Isolated from Human Atherosclerotic Lesions Contains Myeloperoxidase. Previous studies have shown that low-density lipoprotein binds MPO under physiologically relevant conditions (37). To determine whether HDL in the artery wall might behave similarly, we digested lesion HDL with trypsin and analyzed the resulting peptides with 2D liquid chromatography and electrospray ionization-MS. Four peptides in the digest were derived from MPO. We confirmed their origin by sequencing them with MS/MS (Fig. 4). This observation provides strong evidence that MPO is a component of HDL isolated by ultracentrifugation from atherosclerotic lesions and suggests that the enzyme has high affinity for HDL in the artery wall. Levels of 3-Chlorotyrosine Are Elevated in Plasma HDL from Humans with Coronary Artery Disease. To determine whether oxidized HDL might also be present in the circulation, we isolated HDL from plasma of healthy subjects (eight males, aged 34-63 years) and subjects with established coronary artery disease (seven males and two females, aged 33-67 years). The former had no known history of vascular disease or symptoms suggestive of angina, peripheral vascular disease, or cerebral vascular disease. The subjects with coronary artery disease had angiographically documented atherosclerosis.
Fig. 4.

Detection of MPO in lesion HDL by 2D liquid chromatography tandem MS analysis. (A) Sequence of MPO. (B) HDL isolated from human lesions was digested with trypsin and subjected to liquid chromatography-electrospray ionization-MS/MS analysis. Four peptides (underlined in A) unique to MPO were identified. The MS/MS spectrum of one peptide (WDGERLYQEARK) from the heavy chain of MPO is shown.
To determine whether levels of chlorinated lipoproteins were elevated in the subjects with coronary artery disease, we isolated HDL from their plasma and plasma of healthy subjects. After delipidating and hydrolyzing the proteins, we subjected the derivatized amino acid hydrolysate to isotope dilution GC/MS analysis (Fig. 3B). The level of protein-bound 3-chlorotyrosine was 13-times higher in circulating HDL from the patients (39 ± 7 μmol/mol Tyr; n = 9) than in circulating HDL from the healthy subjects (3 ± 2 μmol/mol Tyr; n = 8; P < 0.0001). This observation suggests that levels of chlorinated HDL (perhaps derived from vascular lesions) are elevated in the blood of humans suffering from clinically significant atherosclerosis.
Oxidation of HDL and ApoA-I Impairs Cholesterol Transport in Cultured Cells by ABCA1. The 10 amphipathic helices in apoA-I, the major protein of HDL, are thought to play essential roles in lipid binding, lipoprotein stability, and reverse cholesterol transport (38, 39). Five of the seven tyrosine residues in this protein lie in amphipathic helices, and we have previously shown that Tyr-192 in helix 8 is the major site of chlorination (27). We therefore hypothesized that HOCl might alter the ability of HDL and apoA-I to remove cholesterol from cells.
To test this idea, we exposed HDL or purified apoA-I to HOCl or H2O2 (80:1 or 25:1, mol/mol, oxidant/HDL particle or oxidant/apoA-I) in a physiological buffer (138 mM NaCl/2.7 mM KCl/10 mM sodium phosphate) at neutral pH for 120 min at 37°C, terminating the reaction with a 20-fold mol excess (relative to oxidant) of methionine. Because the average HDL3 particle contains 2 mol of apoA-I (seven tyrosine residues, 243 aa) and 1 mol of apoA-II (eight tyrosine residues, 154 aa), the ratio of oxidant to substrate (mol:mol) was ≈30:1 for apoA-I and apoA-II, 3:1 for tyrosine residues, and 1:8 for total amino acids. For lipid-free apoA-I, the ratio of oxidant to substrates was ≈30% greater than for apoA-I in HDL. We previously showed that ≈50% of Tyr-192 is chlorinated by HOCl under these conditions (27).
We next determined how oxidation affects the ability of HDL or apoA-I to promote cholesterol efflux from BHK cells that expressed very low or very high levels of ABCA1. With mock-transfected cells (low ABCA1), HDL promoted cholesterol efflux exclusively by diffusional mechanisms, and apoA-I had essentially no cholesterol efflux activity (Fig. 5A). Oxidation of HDL with HOCl or H2O2 (which oxidizes methionines) had no effect on or slightly increased HDL-mediated cholesterol efflux from these cells. When ABCA1 was overexpressed in transfected BHK cells, however, HDL-mediated cholesterol efflux increased and apoA-I became active (Fig. 5A). Whereas H2O2 oxidation had no effect, chlorination by HOCl was associated with a significant decrease in cholesterol efflux that was promoted by HDL or apoA-I (Fig. 5 A and B). These observations indicate that oxidation of HDL and apoA-I with HOCl selectively impairs their abilities to remove cholesterol from cells by a pathway requiring ABCA1.
Fig. 5.

Cholesterol efflux activities of native and HOCl-oxidized HDL, apoA-I, and peptide 18A. [3H]Cholesterol-labeled mock- (A and C) or ABCA1-transfected BHK cells were incubated for 4 h with native (Ctrl), H2O2-oxidized, HOCl-oxidized HDL (20 μg/ml), or apoA-I (5 μg/ml) (A), for 2 h with 5 μg/ml apoA-I oxidized with the indicated mol ratio of HOCl (B) or for 2 h with control (-) or HOCl-oxidized (+) peptide Ac-18A-NH2 (20 μg/ml) (C). At the end of the incubation, [3H]cholesterol efflux to the acceptor particle was measured. *, P < 0.01 compared with controls.
Oxidation of a Synthetic Peptide Containing Tyrosine Impairs Lipid Efflux Ability. Acetyl-18A-NH2 (18A), an 18-aa analog of the type of amphipathic α-helix found in apos, mimics apoA-I in promoting cholesterol efflux by the ABCA1 pathway (40, 41). 18A contains a single tyrosine residue in a KxxY motif (where K = lysine, Y = tyrosine, and x = an amino acid unreactive with HOCl), which juxtaposes the amino acid side chains of K and Y residues in an α-helical peptide (27). MS analysis revealed that ≈30% of the tyrosine residues in 18A were chlorinated when it was exposed to HOCl (5:1, oxidant/peptide, mol/mol).
We investigated the ability of native and oxidized 18A to promote cholesterol efflux from BHK cells. In contrast to apoA-I, 18A promoted cholesterol efflux from both mock- and ABCA1-transfected BHK cells, but to a much greater extent from the ABCA1-expressing cells. HOCl treatment significantly reduced the ability of 18A to remove cholesterol by both the ABCA1-independent and -dependent mechanisms (Fig. 5C). These studies suggest that site-specific oxidation of tyrosines in amphipathic α-helices can impair lipid transport activities.
Discussion
In this study, we determined whether MPO oxidizes HDL in vivo by analyzing the lipoprotein and its major protein, apoA-I, for 3-chlorotyrosine, a highly specific marker for the MPO-HOCl system. Remarkably, the level of 3-chlorotyrosine in HDL isolated from human atherosclerotic lesions was 8-fold higher than that in circulating HDL from human subjects. Moreover, the level of 3-chlorotyrosine was 13-fold higher in HDL isolated from plasma of subjects with coronary artery disease than in HDL from plasma of healthy subjects. These observations strongly support the hypothesis that HOCl derived from MPO contributes to HDL oxidation in the artery wall. They also suggest that elevated levels of 3-chlorotyrosine in circulating HDL might represent a unique marker for clinically significant atherosclerosis.
A key question is whether chlorination of HDL by MPO in the artery wall promotes the development of atherosclerotic plaque. One hypothesis to explain the ameliorative effects of HDL on atherogenesis is that HDL apos transfer excess sterol from macrophages into blood by a pathway requiring ABCA1 (9, 42). The observation that humans and mice lacking ABCA1 develop accelerated atherosclerosis (43, 44) is consistent with this proposal.
We found that HDL and lipid-free apoA-I oxidized by HOCl are less able to remove cholesterol from cells by the ABCA1 pathway than native HDL and apoA-I. Because HDL contains both phospholipids and apolipoproteins, it can remove cellular cholesterol by both ABCA1-independent and -dependent mechanisms. Treating HDL with HOCl did not inhibit cholesterol efflux by ABCA1-independent processes but significantly reduced efflux from ABCA1-expressing cells. Similarly, oxidizing lipid-free apoA-I (which removes cellular lipids exclusively by the ABCA1 pathway) with HOCl markedly reduced cholesterol efflux. This inhibitory effect was near maximal when HOCl had chlorinated ≈50% of the tyrosine residues in apoA-I. In contrast, treating HDL or apoA-I with hydrogen peroxide, which selectively oxidizes methionines in the absence of redox active metal ions, did not affect cholesterol efflux. Previous studies have shown that methionine oxidation fails to alter apoA-I-promoted cholesterol efflux from cultured cells (23). We also found that HOCl oxidation of an apolipoprotein-mimetic amphipathic α-helical peptide reduced its ability to remove cellular cholesterol. Thus, MPO-mediated chlorination of tyrosine residues in HDL apolipoproteins in the artery wall might impair cholesterol removal and enhances atherogenesis.
Our results showing relatively high levels of 3-chlorotyrosine in plasma HDL from coronary artery disease subjects might seem surprising, given that blood is richly endowed with anti-oxidants that scavenge reactive intermediates. If MPO interacted specifically with circulating HDL, however, it could promote HDL oxidation even in such an unfavorable environment by generating high local concentrations of reactive intermediates. Indeed, we found that MPO was associated with lesion HDL.
Another intriguing possibility is that the oxidized HDL found in plasma is generated elsewhere, in a microenvironment that is depleted of antioxidants and contains high concentrations of HOCl. Many lines of evidence indicate that atherosclerotic lesions represent such an environment (45). The levels of 3-chlorotyrosine in HDL isolated from human atherosclerotic lesions are ≈20% of those observed in bacteria that have been phagocytosed by neutrophils (46, 47). Phagocytosis triggers the production of superoxide and the secretion of MPO into the phagolysosome, where the microbe is bathed in high local concentrations of H2O2 and MPO-derived oxidants (48). Like the phagolysosome, the inflammatory milieu may provide all of the factors that MPO needs to chlorinate proteins. Thus, some of the chlorinated HDL found in plasma could leak into blood from atherosclerotic lesions or other sites of acute and chronic inflammation. Regardless of whether it originates in blood or lesions, however, 3-chlorotyrosine in plasma HDL might be a useful indicator of the risk of cardiovascular disease and the efficacy of antioxidant interventions.
We previously showed that the primary ε amino group of lysine facilitates the regioselective chlorination of tyrosine residues in the YxxK motif of apoA-I and synthetic peptides by a pathway involving a chloramine intermediate (27). Modeling and structural studies indicate that tyrosine and lysine residues separated by two amino acids are adjacent on the same face of an α-helix, suggesting that the YxxK motif could direct protein chlorination if it resided in an α-helix. Consistent with this proposal, we found that a single tyrosine residue in the 8th amphipathic α-helix of apoA-I was the major site of chlorination by HOCl and that this tyrosine resided in the YxxK motif (27). In future studies, it will be of interest to determine whether regiospecific chlorination of specific tyrosine residues can account for the impaired ability of oxidized apoA-I to promote cholesterol efflux from cells.
Conclusion
The observations made in this study have generated the following model. Oxidative species generated by phagocytes chlorinate specific tyrosine residues in apoA-I. Modification of these residues impairs the ability of the protein to promote cholesterol efflux from lipid-laden macrophages, contributing to the formation of atherosclerotic lesions. Because phagocytes store NADPH oxidase and MPO in their plasma membrane and secretory compartments, respectively, oxidation is likely to be tightly restricted in space by local changes in oxidant concentrations. It is important to note that apoA-I promotes cholesterol efflux from cells by interacting with ABCA1 at the plasma membrane of macrophages. Our findings suggest that local pericellular production of oxidants by phagocytes is a physiological mechanism for oxidizing apoA-I and inhibiting HDL function during atherogenesis. Moreover, 3-chlorotyrosine in HDL protein may serve as a molecular fingerprint for the pathway that mediates oxidative damage in patients suffering from coronary artery disease.
Acknowledgments
We thank Dr. Ernst Malle for generously providing HOP-1 antibody. This work was supported by grants from the National Institutes of Health and Environmental Health (AG021191, HL64344, HL18645, HL55362, HL34343, HL75381, HL073996, HL030086, ES07033, and DK02456) and from the Donald W. Reynolds Foundation. S.P. was supported by a Career Development Award from the Juvenile Diabetes Research Foundation. MS analyses were performed in the Mass Spectrometry Resource (Department of Medicine, University of Washington).
Abbreviations: apoA-I, apolipoprotein A-I; HDL, high-density lipoprotein; ABCA1, ATP-binding cassette transporter A1; MPO, myeloperoxidase; HOCl, hypochlorous acid; BHK, baby hamster kidney; MS/MS, tandem MS.
References
- 1.Miller, N. E., Thelle, D. S., Forde, O. H. & Mjos, O. D. (1977) Lancet 1, 965-968. [DOI] [PubMed] [Google Scholar]
- 2.Keys, A. (1980) Lancet 2, 603-606. [DOI] [PubMed] [Google Scholar]
- 3.Oram, J. F. & Yokoyama, S. (1996) J. Lipid Res. 37, 2473-2491. [PubMed] [Google Scholar]
- 4.Rothblat, G. H., de la Llera-Moya, M., Atger, V., Kellner-Weibel, G., Williams, D. L. & Phillips, M. C. (1999) J. Lipid Res. 40, 781-796. [PubMed] [Google Scholar]
- 5.Brooks-Wilson, A., Marcil, M., Clee, S. M., Zhang, L. H., Roomp, K., van Dam, M., Yu, L., Brewer, C., Collins, J. A., Molhuizen, H. O., et al. (1999) Nat. Genet. 22, 336-345. [DOI] [PubMed] [Google Scholar]
- 6.Bodzioch, M., Orso, E., Klucken, J., Langmann, T., Bottcher, A., Diederich, W., Drobnik, W., Barlage, S., Buchler, C., Porsch-Ozcurumez, M., et al. (1999) Nat. Genet. 22, 347-351. [DOI] [PubMed] [Google Scholar]
- 7.Rust, S., Rosier, M., Funke, H., Real, J., Amoura, Z., Piette, J. C., Deleuze, J. F., Brewer, H. B., Duverger, N., Denefle, P., et al. (1999) Nat. Genet. 22, 352-355. [DOI] [PubMed] [Google Scholar]
- 8.Lawn, R. M., Wade, D. P., Garvin, M. R., Wang, X., Schwartz, K., Porter, J. G., Seilhamer, J. J., Vaughan, A. M. & Oram, J. F. (1999) J. Clin. Invest. 104, R25-R31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Oram, J. F. (2003) Arterioscler. Thromb. Vasc. Biol. 23, 720-727. [DOI] [PubMed] [Google Scholar]
- 10.Gillotte, K. L., Zaiou, M., Lund-Katz, S., Anantharamaiah, G. M., Holvoet, P., Dhoest, A., Palgunachari, M. N., Segrest, J. P., Weisgraber, K. H., Rothblat, G. H., et al. (1999) J. Biol. Chem. 274, 2021-2028. [DOI] [PubMed] [Google Scholar]
- 11.Diaz, M. N., Frei, B., Vita, J. A. & Keaney, J. F., Jr. (1997) N. Engl. J. Med. 337, 408-416. [DOI] [PubMed] [Google Scholar]
- 12.Hurst, J. K. & Barrette, W. C., Jr. (1989) Crit. Rev. Biochem. Mol. Biol. 24, 271-328. [DOI] [PubMed] [Google Scholar]
- 13.Nauseef, W. M. (1988) Hematol. Oncol. Clin. North Am. 2, 135-158. [PubMed] [Google Scholar]
- 14.Klebanoff, S. J. (1980) Ann. Intern. Med. 93, 480-489. [DOI] [PubMed] [Google Scholar]
- 15.Daugherty, A., Dunn, J. L., Rateri, D. L. & Heinecke, J. W. (1994) J. Clin. Invest. 94, 437-444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Domigan, N. M., Charlton, T. S., Duncan, M. W., Winterbourn, C. C. & Kettle, A. J. (1995) J. Biol. Chem. 270, 16542-16548. [DOI] [PubMed] [Google Scholar]
- 17.Hazen, S. L., Hsu, F. F., Mueller, D. M., Crowley, J. R. & Heinecke, J. W. (1996) J. Clin. Invest. 98, 1283-1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gaut, J. P., Yeh, G. C., Tran, H. D., Byun, J., Henderson, J. P., Richter, G. M., Brennan, M. L., Lusis, A. J., Belaaouaj, A., Hotchkiss, R. S., et al. (2001) Proc. Natl. Acad. Sci. USA 98, 11961-11966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hazen, S. L. & Heinecke, J. W. (1997) J. Clin. Invest. 99, 2075-2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Leeuwenburgh, C., Rasmussen, J. E., Hsu, F. F., Mueller, D. M., Pennathur, S. & Heinecke, J. W. (1997) J. Biol. Chem. 272, 3520-3526. [DOI] [PubMed] [Google Scholar]
- 21.Heller, J. I., Crowley, J. R., Hazen, S. L., Salvay, D. M., Wagner, P., Pennathur, S. & Heinecke, J. W. (2000) J. Biol. Chem. 275, 9957-9962. [DOI] [PubMed] [Google Scholar]
- 22.Panzenboeck, U., Raitmayer, S., Reicher, H., Lindner, H., Glatter, O., Malle, E. & Sattler, W. (1997) J. Biol. Chem. 272, 29711-29720. [DOI] [PubMed] [Google Scholar]
- 23.Panzenbock, U., Kritharides, L., Raftery, M., Rye, K. A. & Stocker, R. (2000) J. Biol. Chem. 275, 19536-19544. [DOI] [PubMed] [Google Scholar]
- 24.Bergt, C., Oettl, K., Keller, W., Andreae, F., Leis, H. J., Malle, E. & Sattler, W. (2000) Biochem. J. 346, 345-354. [PMC free article] [PubMed] [Google Scholar]
- 25.Garner, B., Witting, P. K., Waldeck, A. R., Christison, J. K., Raftery, M. & Stocker, R. (1998) J. Biol. Chem. 273, 6080-6087. [DOI] [PubMed] [Google Scholar]
- 26.Francis, G. A., Mendez, A. J., Bierman, E. L. & Heinecke, J. W. (1993) Proc. Natl. Acad. Sci. USA 90, 6631-6635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bergt, C., Fu, X., Huq, N. P., Kao, J. & Heinecke, J. W. (2004) J. Biol. Chem. 279, 7856-7866. [DOI] [PubMed] [Google Scholar]
- 28.Gaut, J. P., Byun, J., Tran, H. D. & Heinecke, J. W. (2002) Anal. Biochem. 300, 252-259. [DOI] [PubMed] [Google Scholar]
- 29.Morris, J. C. (1966) J. Phys. Chem. 70, 3798-3805. [Google Scholar]
- 30.Nelson, D. P. & Kiesow, L. A. (1972) Anal. Biochem. 49, 474-478. [DOI] [PubMed] [Google Scholar]
- 31.Mendez, A. J., Oram, J. F. & Bierman, E. L. (1991) J. Biol. Chem. 266, 10104-10111. [PubMed] [Google Scholar]
- 32.O'Brien, K. D., Olin, K. L., Alpers, C. E., Chiu, W., Ferguson, M., Hudkins, K., Wight, T. N. & Chait, A. (1998) Circulation 98, 519-527. [DOI] [PubMed] [Google Scholar]
- 33.Pennathur, S., Wagner, J. D., Leeuwenburgh, C., Litwak, K. N. & Heinecke, J. W. (2001) J. Clin. Invest. 107, 853-860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McDonald, W. H. & Yates, J. R., III (2002) Dis. Markers 18, 99-105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vaughan, A. M. & Oram, J. F. (2003) J. Lipid Res. 44, 1373-1380. [DOI] [PubMed] [Google Scholar]
- 36.Hazell, L. J., Arnold, L., Flowers, D., Waeg, G., Malle, E. & Stocker, R. (1996) J. Clin. Invest. 97, 1535-1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Carr, A. C., Myzak, M. C., Stocker, R., McCall, M. R. & Frei, B. (2000) FEBS Lett. 487, 176-180. [DOI] [PubMed] [Google Scholar]
- 38.Segrest, J. P., Jones, M. K., De Loof, H., Brouillette, C. G., Venkatachalapathi, Y. V. & Anantharamaiah, G. M. (1992) J. Lipid Res. 33, 141-166. [PubMed] [Google Scholar]
- 39.Brouillette, C. G., Anantharamaiah, G. M., Engler, J. A. & Borhani, D. W. (2001) Biochim. Biophys. Acta 1531, 4-46. [DOI] [PubMed] [Google Scholar]
- 40.Mendez, A. J., Anantharamaiah, G. M., Segrest, J. P. & Oram, J. F. (1994) J. Clin. Invest. 94, 1698-1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Remaley, A. T., Thomas, F., Stonik, J. A., Demosky, S. J., Bark, S. E., Neufeld, E. B., Bocharov, A. V., Vishnyakova, T. G., Patterson, A. P., Eggerman, T. L., et al. (2003) J. Lipid Res. 44, 828-836. [DOI] [PubMed] [Google Scholar]
- 42.Wang, N. & Tall, A. R. (2003) Arterioscler. Thromb. Vasc. Biol. 23, 1178-1184. [DOI] [PubMed] [Google Scholar]
- 43.Joyce, C., Freeman, L., Brewer, H. B., Jr. & Santamarina-Fojo, S. (2003) Arterioscler. Thromb. Vasc. Biol. 23, 965-971. [DOI] [PubMed] [Google Scholar]
- 44.Aiello, R. J., Brees, D. & Francone, O. L. (2003) Arterioscler. Thromb. Vasc. Biol. 23, 972-980. [DOI] [PubMed] [Google Scholar]
- 45.Heinecke, J. W. (1998) Arterioscler. Thromb. 141, 1-15. [Google Scholar]
- 46.Rosen, H., Crowley, J. R. & Heinecke, J. W. (2002) J. Biol. Chem. 277, 30463-30468. [DOI] [PubMed] [Google Scholar]
- 47.Chapman, A. L., Hampton, M. B., Senthilmohan, R., Winterbourn, C. C. & Kettle, A. J. (2002) J. Biol. Chem. 277, 9757-9762. [DOI] [PubMed] [Google Scholar]
- 48.Weiss, S. J. (1989) N. Engl. J. Med. 320, 365-376. [DOI] [PubMed] [Google Scholar]