

ABSTRACT
The glycoprotein O (gO) is betaherpesvirus specific. Together with the viral glycoproteins H and L, gO forms a covalent trimeric complex that is part of the viral envelope. This trimer is crucial for cell-free infectivity of human cytomegalovirus (HCMV) but dispensable for cell-associated spread. We hypothesized that the amino acids that are conserved among gOs of different cytomegaloviruses are important for the formation of the trimeric complex and hence for efficient virus spread. In a mutational approach, nine peptide sites, containing all 13 highly conserved amino acids, were analyzed in the context of HCMV strain TB40-BAC4 with regard to infection efficiency and formation of the gH/gL/gO complex. Mutation of amino acids (aa) 181 to 186 or aa 193 to 198 resulted in the loss of the trimer and a complete small-plaque phenotype, whereas mutation of aa 108 or aa 249 to 254 caused an intermediate phenotype. While individual mutations of the five conserved cysteines had little impact, their relevance was revealed in a combined mutation, which abrogated both complex formation and cell-free infectivity. C343 was unique, as it was sufficient and necessary for covalent binding of gO to gH/gL. Remarkably, however, C218 together with C167 rescued infectivity in the absence of detectable covalent complex formation. We conclude that all highly conserved amino acids contribute to the function of gO to some extent but that aa 181 to 198 and cysteines 343, 218, and 167 are particularly relevant. Surprisingly, covalent binding of gO to gH/gL is required neither for its incorporation into virions nor for proper function in cell-free infection.
IMPORTANCE Like all herpesviruses, the widespread human pathogen HCMV depends on glycoproteins gB, gH, and gL for entry into target cells. Additionally, gH and gL have to bind gO in a trimeric complex for efficient cell-free infection. Homologs of gO are shared by all cytomegaloviruses, with 13 amino acids being highly conserved. In a mutational approach we analyzed these amino acids to elucidate their role in the function of gO. All conserved amino acids contributed either to formation of the trimeric complex or to cell-free infection. Notably, these two phenotypes were not inevitably linked as the mutation of a charged cluster in the center of gO abrogated cell-free infection while trimeric complexes were still being formed. Cysteine 343 was essential for covalent binding of gO to gH/gL; however, noncovalent complex formation in the absence of cysteine 343 also allowed for cell-free infectivity.
KEYWORDS: cell tropism, cytomegalovirus, glycoprotein O, glycoproteins, mutational studies
INTRODUCTION
Congenital human cytomegalovirus (HCMV) infection is the leading cause of sensorineural hearing loss and infectious brain damage in newborns (1–3). Furthermore, HCMV is a threatening complication for transplant recipients (4, 5). Development of an efficient vaccine relies greatly on understanding the role of the viral envelope proteins (6). Three of the viral glycoproteins are essential for fusion of herpesvirus envelopes with host cell membranes: the fusion protein gB and the glycoproteins gH and gL, presumably functioning as a trigger of gB fusion activity (7–9). In support of this model, Varnarsdall et al. demonstrated recently that HCMV gH/gL and gB interact within the virus envelope (10). In contrast to gH/gL of herpes simplex virus (HSV) or Kaposi's sarcoma-associated herpesvirus (KSHV), HCMV gH/gL is stably bound to accessory proteins which are crucial for infection of different host cells. Two alternative complexes are formed and incorporated into the virion envelope: the pentameric gH/gL complex with pUL128, pUL130, and pUL131A (11–13) and the trimeric gH/gL complex with pUL74 (gO) (14–16). The pentameric complex is indispensable for efficient infection of endothelial cells and epithelial cells but dispensable for infection of fibroblasts (13, 17–19), whereas the trimeric complex is necessary for efficient infection of fibroblasts, endothelial cells, and epithelial cells (20, 21). Deletion of gO in various HCMV strains severely reduced the infectivity of free virions, resulting in a small-plaque phenotype (20, 22–25). In the context of viruses containing both the pentamer and the trimer, dual deletion of the UL128 locus and gO was found to be lethal for HCMV (24, 26).
All betaherpesviruses express homologs of gO. In human herpesvirus 6 (HHV-6) and HHV-7, the homologous protein also forms a complex with gH and gL (27, 28), but to date no function has been assigned to this protein (29). Similarly to HCMV gO, murine and guinea pig cytomegalovirus (CMV) gO binds to gH/gL, and its deletion leads to a severely reduced cell-free infectivity (30, 31), indicating that the function of gO is conserved among the various CMVs. An essential role of the gH/gL/gO complex for pathogenesis of murine CMV (MCMV) was recently demonstrated in a mouse model: a gO-deficient MCMV mutant was unable to establish infection in mice (32), indicating that gO plays a pivotal role for host-to-host transmission of CMV. Immunization of mice with gH/gL/gO leads to full protection from otherwise lethal MCMV infection (33). For HCMV it was recently demonstrated that gH/gL/gO interacts with the cellular receptor platelet-derived growth factor receptor alpha (PDGFR-α) (34). Taken together, these findings highlight that gO is a promising target for future vaccination strategies.
In HCMV, gO is encoded by the open reading frame UL74. A thorough analysis of various HCMV strains with a panel of antibodies clarified that all strains contain the trimeric gH/gL/gO complex in the virion envelope (35). With most HCMV strains tested, the majority of gH/gL is incorporated into the virion as part of the trimer rather than the pentamer, and deletion of gO hence not only abrogates trimer formation but also greatly reduces the overall incorporation of gH/gL into virions (20, 35). gO is one of the most polymorphic HCMV proteins (36–38), and among gOs of different cytomegaloviruses, we found only few peptide sites to be conserved. We hypothesized that those parts of the protein that are conserved among all homologs contribute to the function that is common for all of them: the formation and incorporation of the trimeric complex. In silico alignment of gO sequences from different CMVs revealed 13 highly conserved amino acids. In this study, we present a comprehensive mutational analysis of all highly conserved sites within gO of HCMV in the context of a replicating virus. We demonstrate that (i) a central domain of gO plays a key role for the infectivity of gO, (ii) cysteine 343 of strain TB40-BAC4 is the site of covalent interaction with gH/gL, but (iii) covalent binding of gO is not necessary for infectivity.
RESULTS
Generation of a UL74stop mutant as a control virus for mutational analyses of pUL74.
While TB40-BAC4 served as a negative control, we generated a UL74stop virus as a positive control for the maximal effect of changes in UL74. We have previously reported a pUL74 null mutant in the background of HCMV strain TB40-BAC4 that was created by deletion of the N-terminal 37 amino acids (aa) of pUL74 (yielding ΔUL74nt1–37) (24). Meanwhile, a comprehensive analysis of the HCMV transcriptome indicated that this deletion might interfere with a transcript of the essential glycoprotein N (UL73) for which a splice acceptor site is located adjacent to the UL74 ATG (39). Hence, we suspected that the phenotype of the ΔUL74nt1–37 mutant could partially be due to an effect on this transcript of the neighboring gene. To avoid ambiguity and provide a more reliable control for our mutational analysis of pUL74, we constructed a new pUL74 null mutant by introducing two stop codons at aa 7 and aa 12 (UL74stop) via seamless mutagenesis of TB40-BAC4 (Table 1) (40, 41). For reconstitution of the gO stop mutant, bacterial artificial chromosome (BAC) DNA was transfected into fibroblasts.
TABLE 1.
Mutations of highly conserved peptide sites
| Designation | Wild-type sequencea | Mutated sequence | Phenotypic profileb |
||
|---|---|---|---|---|---|
| Focus size | Cell-free infectivity | Complex formation | |||
| UL74stop | MRSISK7–12 | *RSIS*c | — | — | — |
| aa108 | W108 | A | − | − | − |
| aa141 | C141 | A | + | − | − |
| aa146–149 | SMTC146–149 | AMTA | + | − | − |
| aa167–169 | CGN167–169 | AGA | + | + | + |
| aa181–186 | PRWNTK181–186 | AAANTA | — | — | — |
| aa193–198 | KVNVDS193–197 | AVNVAA | — | — | — |
| aa218 | C218 | A | + | + | + |
| aa249–254 | RKLKRK249–254 | AALAAA | + | — | + |
| aa343–349 | CKPDRNR343–349 | AAPAAAA | + | + | — |
Boldface highlights conservation among gO homologs. Subscript numbers indicate positions in the TB40-BAC4 sequence.
Relative to the wild type, as follows: —, severely reduced; −, moderately reduced; +, similar.
Asterisks indicate the stop codons introduced to construct the mutant.
The resulting HCMV-TB40-BAC4-UL74stop was analyzed regarding release of infectious progeny and focal growth in human foreskin fibroblasts (HFFs) and human endothelial cells (HECs). Both infection rates and focus size were determined by immunofluorescence detection of viral immediate early (IE) antigens. Expectedly, the UL74stop virus formed smaller foci than wild-type virus in HFFs. No change was observed in HECs, in which both viruses formed small plaques (Fig. 1A). Cell-free supernatants of the UL74stop virus were severely reduced in their infectivity both for fibroblasts and endothelial cells (Fig. 1B), which is in agreement with the notion that the cell-free route of infection depends on gO, irrespective of the cell type (20, 21). While cell-free infection of fibroblasts was almost completely abrogated with the UL74stop virus, a low level of infectivity in HECs was detectable. In order to further analyze this residual infectivity, freshly harvested, 60-fold-concentrated UL74stop virus was used for infection of HFFs and HECs. Using this approach, about 25% infection could be achieved in HECs, whereas infection rates in HFFs remained below 4% in all experiments. The difference between the 2,000-fold reduction of cell-free infectivity in HFFs and the 200-fold reduction in HECs indicates that infection of endothelial cells is less sensitive to disruption of gO.
FIG 1.

Growth characteristics of a novel TB40-BAC4-UL74stop mutant.(A) Focus formation was allowed for 5 days before infected cells were stained for viral IE antigens (magenta). Nuclei were stained with DAPI (blue). (B) Cell-free infectivity of wild-type and UL74stop in endothelial cells (HECs) and fibroblasts (HFFs). Supernatants of UL74stop were concentrated 60-fold (60×) by ultracentrifugation in order to analyze the tropism of the UL74stop virus. Infection rates were determined by calculating the ratio of IE-positive nuclei to the total number of nuclei.
As the detection of infectivity in cell-free supernatants suggested that mature virions are actually released from TB40-BAC4-UL74stop, we used this null mutant to revisit the effect of gO deletion on the secondary envelopment. The earlier UL74-mutant, TB40-BAC4-ΔUL74nt1–37, was defective in secondary envelopment, whereas an HCMV strain TR-based gO null mutant (TRΔgO) did not exhibit such a phenotype when investigated by transmission electron microscopy (TEM) (20, 24). HFFs infected with wild-type or UL74stop virus were cocultured with noninfected ones on carbon-coated sapphire discs, and focus formation was monitored. Comparable infection rates were reached on days 5 and 8 postcoculture for the wild-type and mutant cultures, respectively. At these time points, the cells were high-pressure frozen and freeze substituted. This technique allows observation of the ultrastructures closer to the living state (26, 42). Furthermore, it enhances the visibility of biological membranes to a level that the lipid bilayer leaflets are clearly distinguishable (43, 44). As an additional quality criterion to ensure comparability between the results, only cells with a clearly visible assembly complex and an accumulation of capsids in a kidney-shaped nucleus (45, 46) were analyzed. TEM images of the wild-type virus-infected cells showed accumulations of enveloped virus particles in the assembly complex (Fig. 2). The same phenotype with regard to secondary envelopment was also observed in the UL74stop virus-infected cells (Fig. 3). In order to confirm full envelopment of those virus particles and to rule out the possibility that they are rather stuck in a budding process into vesicles (i.e., still not fully enveloped), scanning transmission electron microscopy (STEM) tomography (47) of a UL74stop-infected cell was performed (see Movie S1 in the supplemental material). The tomogram reconstruction exhibits full envelopment of UL74stop virus particles in the cytoplasm.
FIG 2.

TEM images of TB40-BAC4. (A) An overview of an infected fibroblast showing both the nucleus and the cytoplasm. The boxed rectangle in the cytoplasm marks an area in the perinuclear assembly compartment which is magnified further in panel B, where various HCMV particles are visible. Panel C shows the boxed area of panel B at still higher magnification. Virion particles undergoing envelopment as well as completely enveloped particles in vesicles are visible.
FIG 3.

TEM images of TB40-BAC4-UL74stop. Similar to the images in Fig. 2, panel A shows an overview of a late-stage-infected fibroblast. The boxed area shows the perinuclear assembly compartment, which is shown at higher magnification in panel B. The four virus particles from this image are further magnified in panel C to allow discrimination of enveloped particles and particles in the process of envelopment.
In conclusion, HCMV-TB40-BAC4-UL74stop resembled previously published gO null viruses with regard to strongly reduced cell-free infectivity and a small-plaque phenotype (20, 22–25). With regard to secondary envelopment, the UL74stop virus differs from TB40-BAC4-ΔUL74nt1–37 but resembles TRΔgO as it can accumulate fully enveloped virus particles in the assembly complex. HCMV-TB40-BAC4-UL74stop was subsequently used for the mutational analysis of highly conserved peptide sites as a control representing the maximal effect of disruption in UL74.
Thirteen amino acids are conserved among gO homologs of different CMVs.
Under the assumption that strong conservation is associated with an involvement in common functions, such as the formation of the complex with gH and gL, we compared sequences of different CMV gOs. The alignment included sequences from CMVs infecting a broad range of species: chimpanzee, macaque, squirrel monkey, guinea pig, bat, mouse, tree shrew, and human. Among these gO homologs, only 13 amino acids (3% of HCMV gO) were found to be strictly conserved (Fig. 4). These 13 highly conserved amino acids were assigned to nine peptide sites which are spread over the central half of the protein. The largest accumulation of conserved amino acids was found from amino acids 140 to 280 (numbers according to TB40-BAC4). To confirm that these highly conserved amino acids are also conserved between different strains of HCMV, an alignment of eight HCMV strains was performed (Fig. 5). We found about 60% conservation among different HCMVs (similar to results reported in references 36 and 48). As with gOs of different CMVs, the highest degree of conservation was found in the center of the protein (aa 160 to 260). The N terminus is highly polymorphic among HCMV strains, and the C terminus is intermediate. As expected, all 13 amino acids that were found to be highly conserved among different CMVs were also conserved among different HCMV strains. In order to test whether these highly conserved sites are important for formation of the trimeric complex and virus growth, a set of nine peptide site mutants was designed (Table 1). To avoid secondary effects, all highly conserved amino acids were replaced with alanines. Under the assumption that protein-protein interactions are mediated by patches of the protein rather than by single amino acids, we also included neighboring amino acids if they were charged or potentially glycosylated (Table 1). The mutations were introduced seamlessly into the BAC-cloned HCMV strain TB40 (40, 41) and subsequently analyzed in terms of virus growth and complex formation.
FIG 4.

Alignment of pUL74 sequences from CMVs infecting different species. The percentage of conservation is indicated as a bar for each amino acid. Matching amino acids are displayed as dots. The red frames indicate the peptide sites that were chosen for mutation, including the 13 highly conserved amino acids.
FIG 5.

Alignment of pUL74 sequences from different HCMV strains. The percentage of conservation is indicated as a bar for each amino acid. Matching amino acids are displayed as dots. The red frames indicate the sites that were mutated.
Phenotypic screening of mutants lacking highly conserved amino acids.
Mutant viruses were reconstituted in HFFs along with the two control viruses, TB40-BAC4 wild type (unrestricted viral growth) and TB40-BAC4-UL74stop (complete small-plaque phenotype). Foci formed by the mutant viruses in HFFs differed in terms of both focus size and shape (Fig. 6A). The W108A (aa108) mutant grew with noticeably smaller foci than the wild type but had the typical comet shape which indicates cell-free transmission (49). In contrast, mutation of aa 249 to 254 (aa249–254 mutant) led to large but unusually confined foci. The strains with mutations of aa 181 to 186 and aa 193 to 198 (aa181–186 and aa193–198 mutants, respectively) (Table 1) displayed a complete small-plaque phenotype. To monitor the spreading capacity of the virus mutants in HFFs, aliquots of the cells were taken twice weekly when the transfection cultures were passaged and infection rates were determined by staining for viral IE antigens. Two of the nine mutants (aa181–186 and aa193–198) were strongly delayed regarding spread in HFF cultures (Fig. 6B). These virus mutants reached 50% infection only after 23 days, compared to 10 days in wild-type-transfected cultures. This initial experiment, which was performed with two independent clones of each virus mutant, suggested an important function of the peptide sites PRWNTK (aa 181 to 186) and KVNVDS (aa 193 to 198).
FIG 6.

Impact of highly conserved peptide sites within pUL74 on virus growth. (A) Representative images of foci formed in fibroblasts at 5 days posttransfection. Nuclei stained for the viral IE antigens are shown in magenta; DAPI-stained nuclei of noninfected cells are blue. (B) Growth of virus mutants was analyzed starting at day 7 posttransfection by measuring the infection rates within aliquots of transfected cultures. (C) For quantification of the spreading capacities of the virus mutants, infected fibroblasts (HFFs) were cocultured with noninfected HFFs or endothelial cells (HECs) for 5 days. Focus size was determined by staining for viral IE antigens and subsequent counting of the number of infected cells per focus. As not all nine peptide site mutants could be tested in both cell types at the same time, the results obtained in the different experiments were normalized to the wild-type values in the respective experiments. For each mutant the mean values of three independent experiments are shown. Error bars indicate standard errors of the means of the normalized values. (D) For comparison of the cell-free infectivity, supernatants of pUL74 mutants and controls (wild type and UL74stop) were used to infect HFFs and HECs in parallel. Infection rates were calculated from the number of IE-positive nuclei to the total number of nuclei (DAPI). The mean values of the infection rates in HFFs and HECs in at least three experiments were compared to those of wild-type virus in the respective experiments by an unpaired t test. (*, <0.05; **, <0.01; ***, <0.001). Error bars indicate standard errors of the means.
To validate these first findings, the spreading capacity of the mutants was further analyzed in focus expansion assays. In this series of experiments, infected cells were cocultured with more than a 100-fold excess of noninfected cells, and focus formation was allowed for 5 days. To control whether reduced growth of mutants was actually mediated by effects on UL74, human endothelial cells (HECs) were included in this analysis. In HECs, wild-type virus is also restricted to small plaques that are not further minimized by the complete disruption of gO expression due to a stop mutation (Fig. 1A). Hence, none of the UL74 mutations tested here was expected to affect focus expansion in HECs, and any reduction would be indicative of second site mutations affecting viral fitness in a more general manner. Infected cells were visualized by immunofluorescence staining for viral IE antigens (Fig. 6C). Quantification of the number of IE-positive cells per focus revealed that the mutation of aa 108 led to a 50% reduction of focus size in HFFs (115 infected cells per focus compared to 250 with wild-type virus), and this difference was significant (P = 0.02). The 80% reduction in focus size caused by mutation of the central peptide sites at aa 181 to 186 (P = 0.001) and aa 193 to 198 (P = 0.0004) resembled UL74stop (40, 41, and 40 infected cells per focus, respectively). Foci of all other mutants were indistinguishable from those formed by wild-type virus.
As expected, none of the mutations further reduced the focus size in HECs. This already argued against second site mutations generally affecting virus replication. To formally exclude the possibility that the phenotypes of our mutants were caused by second site mutations, the mutations of aa 108, aa 181 to 186, aa 193 to 198, and aa 249 to 254 were reversed. Focus sizes of the revertant viruses resembled the size of the wild type, demonstrating that the impaired growth in HFFs was actually caused by the mutations in UL74 (Fig. 7).
FIG 7.

Focal growth of virus revertants. Infected fibroblasts were cocultured with noninfected fibroblasts for 5 days. Focus size was determined by staining for viral IE antigens and subsequent counting of the number of infected cells per focus. Mean focus expansion values of three independent experiments are shown; error bars indicate standard errors of the means.
Wild-type virus forms large, comet-shaped foci, indicative of cell-free transmission (49). As foci formed by some UL74 mutants were rather small and/or confined, the hypothesis that these mutants are defective for cell-free infection was tested. Supernatants of freshly reconstituted virus mutants (harvested when cultures showed >90% cytopathic effect [CPE]) were used to infect HFFs and HECs in parallel. One day after infection, the fraction of infected cells was determined by immunofluorescence staining of viral IE antigens. Infection rates measured in at least three experiments in HFFs and HECs were compared between each mutant and the wild type to test for the ability of the mutants to infect via the cell-free route (Fig. 6D). Viruses with mutations of aa 167 to 169, aa 218, and aa 343 to 349 were similarly infectious as the wild type, whereas the mutations of aa 108, aa 141, and aa 146 to 149 reduced the cell-free infectivity for both cell-types by 50% compared to level of the wild type. The mutations of aa 181 to 186, aa 193 to 198, and aa 249 to 254 severely reduced cell-free infectivity, with infection rates remaining below 4% in all experiments. No infectivity for HFFs was observed with the mutants aa181–186 and aa193–198. These mutants resembled the gO null mutant not only regarding the almost complete reduction of cell-free infectivity in HFFs but also concerning the residual infectivity in HECs. Mutation of aa 249 to 254 was exceptional as it reduced cell-free infectivity equally strong for both cell types.
In order to test whether the reduced infectivity of the mutant viruses was associated with defects regarding accumulation of gO or decreased formation of the gH/gL/gO complex, Western blot analysis was performed under reducing and nonreducing conditions (Fig. 8). In cells infected with the aa108, aa141, aa146–149, aa167–169, aa218, and aa249–254 mutants (Table 1), signals were detected at the expected size of the gH/gL/gO complex (250 kDa). In contrast, no gH/gL/gO complex was detectable in lysates of mutants aa181–186, aa193–198, aa343–349, and UL74stop. Under reducing conditions, two subspecies of gO were detectable, a 130-kDa species and a faster-migrating species of 100 kDa. The 100-kDa species was detectable in all mutants except UL74stop, whereas the 130-kDa form, which might represent the fraction of gO that has acquired full glycosylation (15), was clearly detectable only in cells infected with the mutants aa141, aa146–149, aa167–169, aa218, and aa249–254. In lysates of the aa108, aa141, and aa146–149 mutants, additional gO-containing complexes were detectable. It is noteworthy that mutation of C141 as well as mutation of C149 induced the appearance of an additional gO-containing high-molecular-mass complex (>250 kDa), which might indicate that these mutations introduce a similar change to the protein. The lack of complex formation and accumulation of the 130-kDa gO by the mutants aa181–186 and aa193–198 can easily explain why these mutants resemble the UL74stop mutant regarding growth properties. In contrast, mutation of aa 249 to 254 severely reduced cell-free spread although the gH/gL/gO complex was formed as efficiently as in the wild type (Fig. 6 and 8). Notably, the phenotypes differed from those of mutants lacking the complex as mutant aa249–254 was not impaired regarding cell-associated spread. Finally, mutation of aa 343 to 349 was remarkable as it abrogated complex formation without causing severe phenotypic changes. As this peptide site includes C343, we hypothesized that C343 is the site of covalent binding to gH/gL.
FIG 8.

Importance of highly conserved peptide sites for formation of the gH/gL/gO complex and gO maturation. Infected fibroblasts were lysed when cultures showed >90% late-stage cytopathic effects. Lysates were analyzed either under reducing conditions to display gO accumulation in the cells or under nonreducing conditions to test for formation of the gH/gL/gO complex. Detection of gB was included to visualize the content of HCMV glycoproteins in each sample.
In summary, mutation of nine highly conserved peptide sites demonstrated that (i) mutations of aa 181 to 186 and aa 193 to 198 resemble a complete deletion of gO, (ii) aa 108, aa 141, and aa 146 to 149 participate in complex formation with gH/gL as indicated by reduced levels of gH/gL/gO in cell lysates and an intermediate reduction in growth, and (iii) aa 343 to 349 are dispensable for virus growth although they are necessary for covalent complex formation.
Cysteine 343 is the site of covalent interaction with gH/gL.
Surprisingly, none of the five mutants covering a cysteine had displayed a strong phenotype in the initial analysis concerning viral growth although each of them is highly conserved among gO proteins of different CMVs. Most remarkably, mutation of C343 in the context of the mutant aa343–349 abrogated complex formation without reducing infectivity. To further analyze the particular contribution of C343 to complex formation, combined mutations were performed (Table 2). As gO contains five cysteines, it seemed most likely that only one of them binds gH/gL, whereas the other four are involved in intramolecular bonds. To test whether one cysteine alone is sufficient for gH/gL/gO complex formation, we generated five virus mutants in each of which only one of the cysteines was left intact (mutants 4CtoA). In a maximal combination mutant, all five cysteines were replaced with alanines (mutant 5CtoA). Virus growth was monitored during reconstitution, and spreading efficiency was quantified in focus expansion assays (Fig. 9A and B). Not unexpectedly, the 5CtoA mutant was disabled regarding spread in HFFs to the same extent as the UL74stop virus, demonstrating that the cysteines are indispensable. Also, the mutants in which four of the cysteines were replaced by alanines displayed a complete growth reduction, with one exception, the mutant with C343 intact (4CtoA_C343intact) (Fig. 9A). Foci formed by this mutant were significantly larger than those of the UL74stop mutant (P = 0.036), whereas none of the other cysteines could significantly rescue virus growth (Fig. 9B). The rescue of virus growth by C343 was suggestive of the formation of the trimeric complex. To test this hypothesis, lysates of infected cells were analyzed under nonreducing conditions (Fig. 9C). Concordant with the growth phenotype of 4CtoA_C343intact, gH/gL/gO was detectable only in the mutant with an intact C343, whereas none of the other cysteines could individually rescue complex formation. Under reducing conditions, 4CtoA_C343intact was the only mutant for which the 130-kDa gO species remained detectable, albeit at greatly reduced levels compared with the wild-type level. These data demonstrate that C343 is the site of covalent interaction with gH/gL. It is remarkable that lack of C343 had no significant effect on virus growth when the other four cysteines were intact (Fig. 6, mutant aa343–349), suggesting that noncovalent complex formation might compensate for the loss of covalent binding to promote efficient virus growth. To address this, a virus mutant lacking only C343 (gO-C343A) was generated. The kinetics of virus growth after transfection resembled that of wild-type virus. As expected, no gH/gL/gO was detected in lysates of cells infected with this mutant. However, there were small amounts of the 130-kDa gO species (Fig. 10). Hence, the C343A mutant matches the aa343–349 mutant phenotypically. Purified virions of this mutant were analyzed with regard to incorporation of gO and gH/gL/gO (Fig. 11A). In immunoblotting analyses, a covalent gH/gL/gO complex was not detected with the C343A mutant under nonreducing conditions. Instead, a low-molecular-mass species (about 130 kDa) appeared when the membrane was probed with the anti-gO antibody. Furthermore, gO-C343A could be coimmunoprecipitated with an anti-gH antibody (Fig. 11B). Both findings together indicate that the mutant gO-C343A protein was incorporated into the virion envelope as part of the gH/gL/gO complex despite the lack of a covalent bond with gH/gL.
TABLE 2.
Mutations of highly conserved peptide sites
| Designation | Sequence at the indicated positiona |
Phenotypic profileb |
|||||
|---|---|---|---|---|---|---|---|
| 141 | 149 | 167 | 218 | 343 | Focus size | Complex formation | |
| Wild type | C | C | C | C | C | ||
| 5CtoA | A | A | A | A | A | — | — |
| 4CtoA_141intact | C | A | A | A | A | — | — |
| 4CtoA_149intact | A | C | A | A | A | — | — |
| 4CtoA_167intact | A | A | C | A | A | — | — |
| 4CtoA_218intact | A | A | A | C | A | — | — |
| 4CtoA_343intact | A | A | A | A | C | − | − |
| C167A C218A | C | C | A | A | C | + | + |
| C167A C343A | C | C | A | C | A | — | — |
| C218A C343A | C | C | C | A | A | — | — |
| C167A C218A C343A | C | C | A | A | A | — | — |
| C141A C149A | A | A | C | C | C | + | + |
| C141A C149A C343A | A | A | C | C | A | + | — |
Positions are according to pUL74 of TB40-BAC4.
Relative to the wild type, as follows: —, severely reduced; −, moderately reduced; +, similar.
FIG 9.

Cysteine 343 can rescue virus growth and complex formation. (A) Growth properties of mutants either lacking all five cysteines (5CtoA) or lacking four cysteines (4CtoA) were characterized during reconstitution. Infection rates were determined from aliquots of transfected cultures twice weekly, starting at day 2 posttransfection. Wild-type virus and UL74stop virus were included as references. (B) For analysis of the spreading capacities of the virus mutants, infected fibroblasts were cocultured with noninfected fibroblasts for 5 days. Focus size was quantified by staining for viral IE antigens and subsequent counting of the number of infected cells per focus. Mean focus expansion values of four independent experiments are shown. Error bars indicate standard errors of the means. A one-sided, paired t test was used to compare the different viruses with the UL74stop control (*, P < 0.5; **, P < 0.01). (C) In order to test for gO accumulation and complex formation, lysates of infected fibroblasts were subjected to SDS-PAGE under reducing and nonreducing conditions, respectively. Detection of actin served as a loading control. A staining for gB was included to control for the amount of HCMV glycoproteins in each sample.
FIG 10.
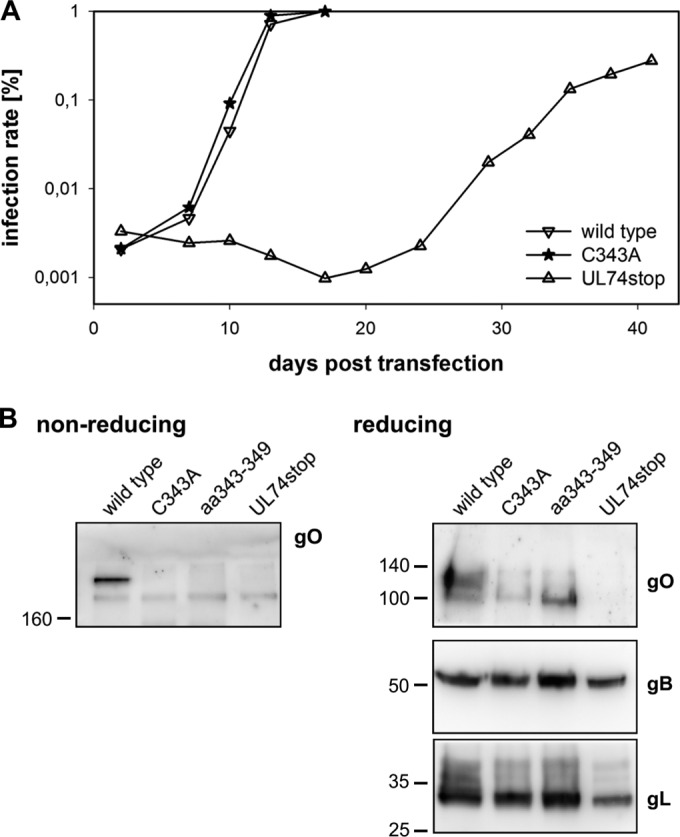
Characterization of the C343A mutant. (A) Virus growth was measured starting at day 2 posttransfection. The infection rates were determined over time in aliquots of the transfected cells by staining for the viral IE antigens. (B) To test whether the gH/gL/gO complex is also undetectable in a mutant lacking only C343, infected cells were subjected to SDS-PAGE under nonreducing conditions. The same lysates were also analyzed under reducing conditions to control for protein accumulation independent of complex formation.
FIG 11.
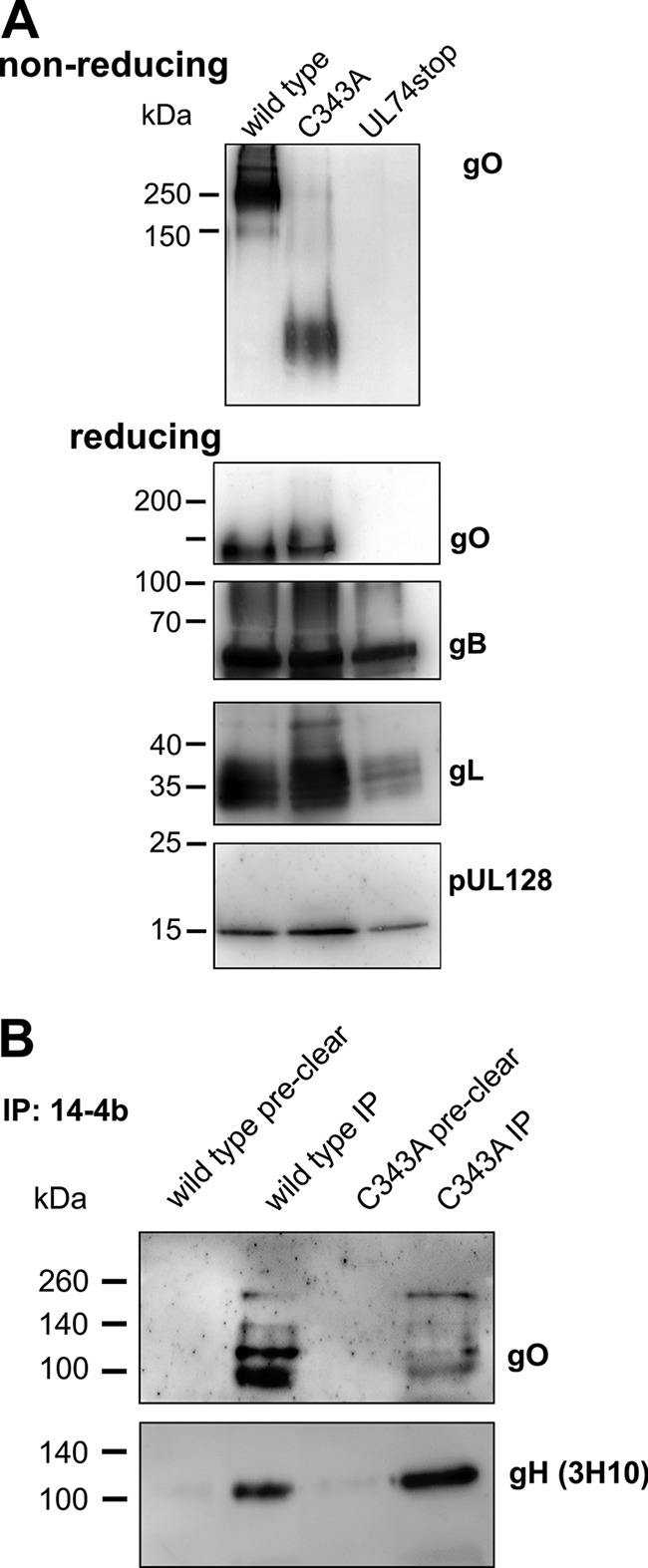
C343A virions contain large amounts of gO. (A) Gradient-purified virions were lysed and subjected to gel electrophoresis under nonreducing conditions to test whether the trimeric complex is detectable. Aliquots of the same virion preparation were analyzed in parallel under reducing conditions and probed against the viral proteins gO, gB, gL, and pUL128. (B) For coimmunoprecipitation, lysates of gradient-purified virions were either incubated with protein A-Sepharose alone (preclear) or with anti-gH antibody 14-4b and protein A-Sepharose (IP). The precipitated proteins were analyzed under reducing conditions. To control for input in each sample, gH was detected after stripping of the membrane.
The results obtained in the previous experiments served as a basis to further analyze the impact of the other four cysteines. The finding that mutations of C141 and C149 (as part of aa146–149) resulted in very similar phenotypes regarding virus growth and complex formation (Fig. 5 and 6), suggesting that these cysteines form a shared disulfide bond. Based on this, we assumed that C167 and C218 also bind to each other. As mutation of C343 abrogated covalent binding to gH/gL, the possibility that C167 and C218 represent additional sites of covalent binding to gH/gL could be excluded. In contrast, it seemed very likely that these two cysteines stabilize the structure of the large protein by formation of an intramolecular disulfide bond. Based on these considerations, mutants were generated that lacked either of the assumed intramolecular disulfide bonds alone or in combination with C343 (Table 2). Characterization during reconstitution (Fig. 12A) and focus expansion assays (Fig. 12B) revealed that a combined mutation of C141 and C149 had no significant impact on viral growth, irrespective of whether C343 was intact or mutated. A combined mutation of C167 and C218 was also well tolerated but resulted in a severe reduction of viral fitness when C343 was disrupted in addition. In line with these results, immunoblotting analyses showed that complex formation occurred with the combined C167A and C218A mutant but was disrupted in the triple mutant C167A C218A C343A (Fig. 12C). Combinations of C343A with either C167A or C218A caused a similarly strong impairment of viral growth, consistent with the assumption that C167 and C218 form a disulfide bond. Taken together, these data indicate that an intermolecular bond of C343 with gH/gL and an intramolecular bond between C167 and C218 are important for efficient virus growth.
FIG 12.
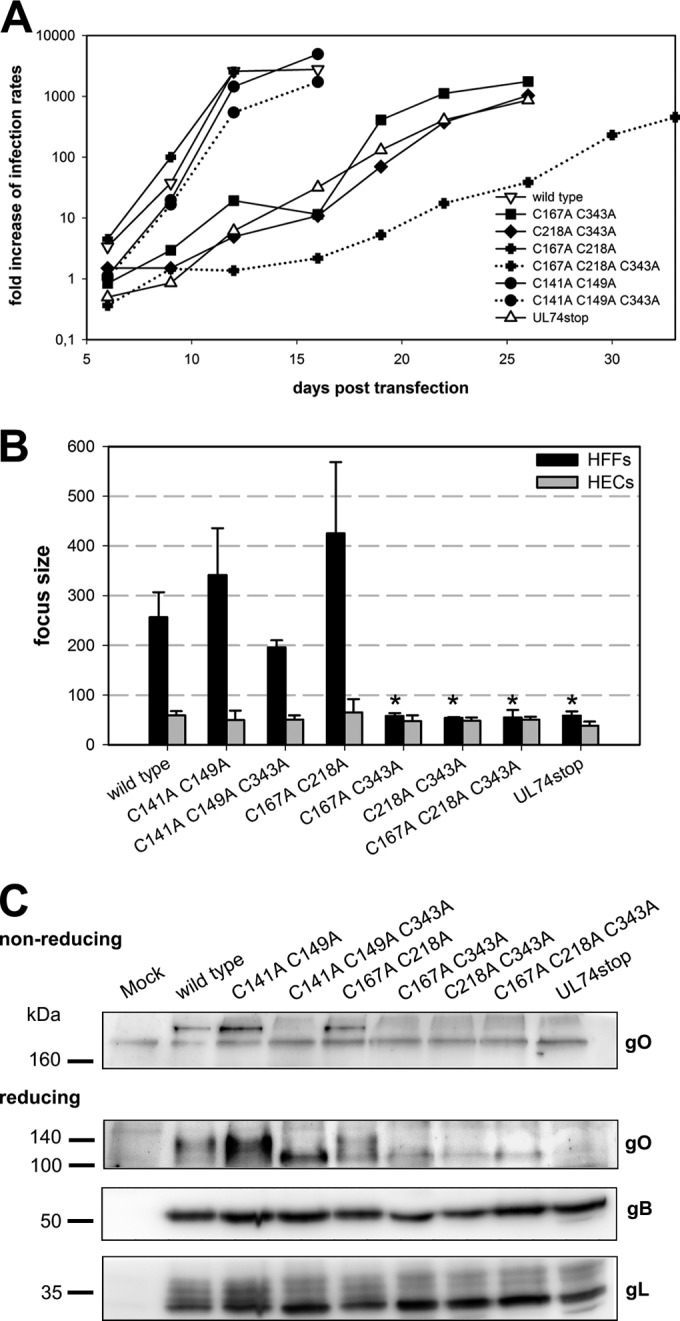
Either C343 or C167 and C218 are needed for efficient virus growth. (A) Growth of virus mutants was analyzed starting at day 6 posttransfection by measuring the infection rates within aliquots of transfected cultures. (B) Analysis of focal growth in fibroblasts (HFFs) and endothelial cells (HECs) over 5 days. Focus size was determined by counting the number of IE-positive nuclei per focus. The mean focus size of virus mutants in three independent experiments was compared to that of the wild type with a paired t test (*, P < 0.5). Error bars indicate standard errors of the means. (C) The virus mutants were analyzed regarding formation of the trimeric complex and gO accumulation by SDS-PAGE of cell lysates under nonreducing and reducing conditions, respectively. gB and gL served as internal controls for the content of viral glycoproteins in each sample.
DISCUSSION
By comparing sequences from different animal CMVs, we found 3% of the amino acids of gO to be highly conserved, and we hypothesized that the respective sites of the protein are important for the formation of the gH/gL/gO complex. This hypothesis was tested by exchanging these highly conserved amino acids and the neighboring charged or potentially glycosylated amino acids with alanines. Eight out of nine highly conserved peptide sites indeed had significant impact on the formation of the gH/gL/gO complex. Mutation of the peptide sites at aa 181 to 186, aa 193 to 198, and aa 343 to 349 abrogated covalent complex formation, with the former two mutations causing a complete loss of function similar to that of the UL74stop mutation. Mutation of aa 108, aa 141, and aa 146 to 149 reduced the amount of trimeric complex and induced formation of aberrant complexes. The peptide sites at aa 167 to 169 and aa 218 were dispensable for complex formation only in the presence of cysteine 343. The peptide site at aa 249 to 254 is dispensable for complex formation and focal growth but significantly influences cell-free infectivity for fibroblasts and endothelial cells. In conclusion, the data presented here demonstrate (i) that all highly conserved peptide sites within gO of HCMV contribute substantially to virus growth, (ii) that these conserved sites promote complex formation with gH/gL (except aa 249 to 254), and (iii) that the cysteine at position 343 is the site of covalent binding with gH/gL.
For the detailed analysis of highly conserved sites of gO, we first wanted to reevaluate the effect of gO deletion. Therefore, a UL74stop mutant was generated. This virus resembled other gO null viruses with respect to the small-plaque phenotype and severely reduced cell-free infectivity (20, 23, 24). When concentrated UL74stop virus was tested, residual infectivity for endothelial cells was detected. The same phenomenon was observed with two peptide site mutants (aa181–186 and aa193–198). In all experiments, gO-deficient virus mutants were less impaired in infection of HECs than of HFFs. This incomplete dependency of cell-free infection on gO could either be a characteristic of endothelial cells, e.g., via nonspecific endocytic uptake, or it could indicate that gO has two distinct functions in entry: a direct influence on fusion, which is important for entry into all cell types (21), and a fibroblast specific function, e.g., receptor binding, as suggested by a recent publication (34). A detailed analysis of HCMV-specific parts of gO will help to answer this question. In contrast to the phenotype of our previous pUL74 null mutant TB40-BAC4-ΔUL74nt1–37 (24), the novel TB40-BAC4-UL74stop mutant did not display an obvious envelopment defect. This is concordant with what was observed with a TR-based virus lacking gO (20). Indeed, STEM tomography of a late-stage UL74stop-infected cell confirmed the full envelopment of a fraction of the cytoplasmic particles, thus providing formal proof that the UL74stop virus can form fully enveloped virus particles. This difference between the TB40-BAC4-UL74stop mutant and the previously published ΔUL74nt1–37 virus might be due to a secondary effect of the deletion of nucleotides 1 to 37 on a previously unknown UL73 splice site (39).
Among the highly conserved amino acids, cysteine 343 was most peculiar as its mutation impaired covalent complex formation but had no significant effect on virus growth. In the context of a mutant lacking all other cysteines, C343 was sufficient to rescue virus growth to 50% of wild-type levels and to restore complex formation to some extent. These results strongly indicate that C343 is the site of covalent interaction with gH/gL. This is consistent with a recent report regarding transient expression of the respective proteins in HEK293 cells, where the disulfide bonds within the gH/gL complexes were analyzed by mass spectrometry (50). Those data demonstrated that the bond between gL and gO is most frequently formed by the cysteine at position 351 within pUL74 of strain Merlin, corresponding to C343 in TB40-BAC4. Ciferri et al. also detected an alternative binding between gL and gO C226 (corresponding to C218 in TB40 pUL74) (50). However, the data presented here argue against a relevant contribution of this alternative binding site in the context of replicating virus: (i) no covalent gH/gL/gO complex was detected by the mutant lacking C343, (ii) mutation of C218 alone had no significant influence on virus growth and complex formation, and (iii) only C343 but not C218 could rescue viral growth in the absence of the other four cysteines. This apparent discrepancy may be explained by the different experimental settings. Ciferri et al. analyzed the disulfide bonds formed in a transient expression system and used highly sensitive mass spectrometry for detection, whereas the data described here were obtained by mutational analysis in the viral background, with virus growth properties and Western blots being the main readouts. Hence, while not formally excluding a minor degree of C218 binding to gH/gL, our results clarify that this cysteine cannot functionally substitute for the loss of C343.
Surprisingly, mutation of C343 in the viral context revealed that covalent binding of gO to gH/gL is not necessary for complex formation. The fact that gO-C343A forms a noncovalent complex with gH/gL is demonstrated by the findings that the protein is incorporated into virions and is immunoprecipitated along with gH, yet a covalent complex is not detected in Western blotting under nonreducing conditions. The fact that the C343A mutation does not affect the functionality of the virus is demonstrated by their wild-type-like phenotype both in cell-free infection and focal spread. Regarding the molecular functions of gO for replication of HCMV, it has been reported that gH and gL alone are not sufficient for incorporation of functional gH/gL complexes into virions but need gO to form the trimer gH/gL/gO or the proteins of the UL128 locus to form the pentamer gH/gL/pUL128/pUL130/pUL131A (16, 18, 26). The trimer is necessary for cell-free infection irrespective of the cell type (21), most probably due to gO binding to the cellular receptor molecule PDGFR-α (34). Obviously, both of these essential functions (i.e., incorporation of trimer and promotion of cell-free infection) do not depend on a covalent binding of gO to gH/gL.
Most probably, in the absence of C343 these functions are maintained by gO that is noncovalently bound to gH/gL. An alternative explanation would be that gO-C343A acts as a chaperone promoting incorporation of gH/gL into virions (20) but then falls off and is no longer necessary for infection of target cells. Our finding that gO-C343A immunoprecipitates with gH in virion lysates argues against the assumption that gO-C343A falls off the complex after incorporation, and the finding that gO is the viral interaction partner of the cellular receptor PDGFR-α, thus promoting cell-free infection (34), argues against the idea that gH/gL alone could promote cell-free infection. Finally, the possibility that some of the gO-C343 that we detect in virions is located in the tegument rather than on the surface is not formally excluded by our data. However, given the current evidence regarding expression and maturation of gO, it is highly unlikely. Usually, tegument-associated proteins are expressed into the cytoplasm of infected cells, whereas envelope-associated proteins are expressed into the endoplasmic reticulum (ER) and subsequently matured in the Golgi compartment. gO is directed by an N-terminal signal peptide to the lumen of the ER where it binds to preassembled gH/gL. This premature trimeric complex is then transported to the Golgi compartment where complex glycosylation occurs (15, 43). The gO-C343A species that we detected in virions were all glycosylated forms considering their size, indicating that the mutant protein is localized in the secretory pathway. As gO itself does not contain a transmembrane domain (43), it would most probably be secreted unless bound to gH/gL or other membrane-anchored proteins. Hence, free uncomplexed gO would not be detectable in gradient-purified virions. Taken these observations together, the assumption of gO forming a functional noncovalent complex with gH/gL in the envelope of mutant virus is the most likely explanation of our findings. The question remains as to why C343 is highly conserved if it is not required for proper function of HCMV. It is noteworthy that gO levels in virions are reduced in C343 mutants, indicating that this cysteine might be necessary for highly efficient incorporation of trimer into the envelope. While we could not detect a reduction of infectivity in our assays compared to the wild-type level, the possibility remains of a more subtle phenotype that would become overt only under more restrictive conditions.
We analyzed the impact of the cysteines on complex formation also by combining various cysteine-to-alanine mutations. Mutation of C141 or C149 in addition to disruption of C343 had no significant effect on viral fitness, whereas mutation of C343 and either C167 or C218 decreased infectivity to UL74stop levels. As the outcomes were similar, irrespective of whether C167 or C218 was mutated in addition to C343, it can be assumed that these two sites bind to each other. Mutation of C343 alone was compensated by noncovalent complex formation allowing for unimpaired virus growth; however, when either C167 or C218 was mutated additionally, viral fitness was severely decreased. It is tempting to speculate that an intramolecular bond between C167 and C218 stabilizes the tertiary structure of gO and thereby promotes noncovalent complex formation in the absence of C343. Moreover, formation of a second intramolecular bond between C141 and C149 has a high likelihood. We found that mutants lacking either C141 or C149 showed the same abnormal patterns when analyzed in Western blotting under nonreducing conditions: a second signal appeared that was even larger than that of the 250-kDa gH/gL/gO complex (Fig. 8). Although separation is not linear in normal polyacrylamide gel electrophoresis for very large proteins, it can be speculated that this large gO-containing complex consists of gH/gL/gO homodimers. gH/gL was reported to form homodimers via C144 of gL when it was expressed in the absence of accessory proteins that would bind gL at C144 (50). Likewise, a free cysteine in gO (C141 in the absence of C149 and vice versa) might promote dimer formation of gH/gL/gO. Also, with regard to virus growth, disruption of C141 and C149 caused a similar decrease of cell-free infectivity. A disulfide bond at this site might be facilitated by the proline at position 145, e.g., by bending of the polypeptide backbone, which might further reduce the distance between the side chains of these two cysteines. However, these conjectures await validation by direct measurements, as for example by mass spectrometry.
Not only the highly conserved cysteines within gO but also the four other peptide sites, namely, W108, aa 181 to 186 (PRWNTK), aa 193 to 198 (KVNVDS), and aa 249 to 254 (RKLKRK), are relevant for HCMV infectivity. The peptide sites at aa 181 to 254 are located in the central part of the gO sequence and might form a core domain of the protein. Mutation of the peptide sites at aa 181 to 186 and aa 193 to 198 led to a deletion-like phenotype. Also, the single amino acid exchange of tryptophan 108 to alanine reduced viability to 50%, and complex formation was apparently reduced. In this context, it is noteworthy that tryptophan is very often found at protein contact sites (51). Another common feature of protein-protein interaction sites is the accumulation of charged amino acids; such a cluster can be found in the center of all gO homologs (in HCMV, RKLKRK). Mutation of all charged amino acids of this site led to a moderate impairment of focal spread (20%) but an unexpectedly severe reduction of cell-free infectivity (20-fold). Notably, in cultures of this mutant no comet-shaped foci, which are typical for cell-free transmission (49), were observed. This indicates that foci formed by this mutant depend mainly on cell-associated spread. The peptide site around aa 249 to 254 is predicted to form a helix with an asymmetrical distribution of basic amino acids (e.g., as predicted by HeliQuest [52]). Therefore, it is tempting to speculate that this site plays a role in attachment during cell-free infection.
The gH/gL/gO complex is thought to play a key role in entry of HCMV into host cells. The comprehensive mutational analysis of peptide sites conserved among gOs of cytomegaloviruses presented here sheds light on the characteristics of the gH/gL/gO complex with three main findings: (i) a core domain from aa 181 to 254 is crucial for complex formation and cell-free infectivity, (ii) cysteine 343 is necessary and sufficient for covalent interaction between gH/gL and gO, and (iii) noncovalent binding of gO to gH/gL is strong enough to promote its incorporation into the virion and to drive efficient infection.
MATERIALS AND METHODS
Cells.
Human foreskin fibroblasts (HFFs) were propagated in minimal essential medium (MEM) supplemented with GlutaMAX (Life Technologies) plus 5% fetal calf serum (FCS), 0.5 μg/liter basic fibroblast growth factor, and 0.1 g/liter gentamicin. During experiments, maintenance medium without growth factor was applied. Human endothelial cells (conditionally immortalized with large T antigen and telomerase; HEC-LTTs) (53, 54) were cultured in endothelial growth medium (EGM) (BulletKit; Lonza) in the presence of 2 mg/liter doxycycline. For experiments, doxycycline was omitted from the medium. Infection of endothelial cells was performed in MEM plus 5% FCS as EGM contains heparin at concentrations that would inhibit HCMV infection.
Mutagenesis.
All virus mutants were generated on the basis of HCMV strain TB40-BAC4 (40). Amino acid exchanges were introduced seamlessly as described by Tischer and colleagues (41). Overall integrity of the modified genomes was tested by restriction fragment comparison with at least two different restriction enzymes. Sequence integrity of UL74 was confirmed by sequencing of the DNA that was used for transfection and sequencing of the reconstituted virus. For all mutations of individual peptide sites, two independent clones were reconstituted and analyzed.
TEM.
The cells were seeded on 50-μm-thick carbon-coated sapphire discs, 3 mm in diameter, in a two- by nine-well μ-slide (Ibidi). A comparable degree of infection was reached at 5 days postcoculture of the wild-type-infected HFFs and at 8 days postcoculture of the mutant counterpart. The sapphire discs harboring the cells were then clamped between two aluminum planchettes (dipped in 1-hexadecene) whereby the cells were protected in the 100-μm-deep cavity as described previously (42, 45). These sealed specimen sandwiches were then high-pressure frozen using a Wohlwend HPF Compact 01 high-pressure freezer. The freeze substitution was performed as previously described (44) with minor changes, whereby the freeze substitution medium consisted of acetone with 0.2% osmium tetroxide, 0.1% uranyl acetate, and 5% water. The freeze substitution was done over a 17-h period with a gradual temperature increase from −90°C to 0°C. Afterwards, the samples were washed with acetone to get rid of the unbound uranyl acetate and osmium tetroxide. The samples were then gradually embedded in Epon and polymerized over 48 h at 60°C before ultrathin (80-nm-thick) sections were cut with a Leica Ultracut UCT ultramicrotome using a diamond knife. Ultrathin sections were then mounted on copper grids and observed with a Jeol 1400 transmission electron microscope (TEM), and the images were digitally acquired and processed using a Veleta camera.
Alignment of gO sequences.
In order to determine highly conserved peptide sites, seven amino acid sequences of gOs from different species were aligned using CLC Bio software (Qiagen). The following sequences (with NCBI accession numbers) were used for the alignment: pUL74 of HCMV strain TB40-BAC4 (ABV71596.1) (40), rhUL74 of macacine herpesvirus 3 (AAZ80605.1) (55), pUL74 of saimiriine herpesvirus 4 (NC_016448.1) (A. J. Davison, M. Holton, A. Dolan, D. J. Dargan, D. Gatherer, and G. S. Hayward, unpublished data), T74 of tupaiid herpesvirus 1 (NP_116424.1) (56), GP74 of caviid herpesvirus 2 (CDI95409.1) (M. R. Schleiss, N. Hernandez-Alvarado, T. Ramaraj, and J. A. Crow, unpublished data; A. Sundararajan, unpublished data), m74 of murine herpesvirus 1 (HE610452.1) (57), and B74 of bat beta herpesvirus (JQ805139.1) (58). Amino acids that were found to be conserved among all seven cytomegaloviruses were considered highly conserved. To test for conservation among HCMVs, an alignment of eight pUL74 sequences of different HCMV strains was performed with the following sequences (NCBI accession numbers): TB40-BAC4 (ABV71596.1) (40), VR1814 (ACZ79985.1) (59), Toledo (ACS93334.1) (60), AD169 (FJ527563.1) (61), TR (AGL96663.1) (62), Merlin (ACZ72817.1) (63), PH (AC146904.1) (37), and Towne (ACM48052.1) (61). The selection of HCMV strains reflects the different genotypes (36, 40).
Immunofluorescence.
Cells were fixed and permeabilized with 80% acetone for 5 min, followed by three washing steps in phosphate-buffered saline (PBS). Infection was assessed by staining for viral immediate early (IE) proteins pUL122/123 with a monoclonal mouse antibody (clone E13; Argene). A Cy3-conjugated goat anti mouse Ig secondary antibody (Jackson ImmunoResearch) was used for visualization. Nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI). For the calculation of infection rates, three images were taken from each well, and the number of IE-positive nuclei and DAPI signals was counted using Axio Vision software (Zeiss).
Characterization of virus growth during reconstitution.
For reconstitution of the mutated viruses, DNA was freshly prepared from Escherichia coli GS1783, and transfection was performed in duplicates with 2 μg of DNA per 300,000 HFFs. Wild-type and UL74stop viruses were always included as controls. At 2 days posttransfection, the cells were detached from the six-well plates and transferred to a 25-cm2 flask. In order to harvest reasonable amounts of the virus, the growth area for the cells was doubled once again at 7 days posttransfection. Afterwards, cells were reseeded on the same growth area twice weekly. In order to detect the increase of infection in the cultures, every time the cells were detached, three times 15,000 cells were seeded onto a 96-well plate and fixed after they were adherent again (4 h), followed by staining for the viral IE proteins. Infected cells and supernatants were harvested when the culture displayed >90% cytopathic effect. The cells were frozen in RPMI 1640 medium containing 12% glucose and 40% dimethyl sulfoxide. Dead cells and cell debris were removed from the supernatants by centrifugation at 3,220 × g for 10 min, and these cell-free virus preparations were stored at −80°C.
Analysis of focal growth.
Infected cultures with infection rates of 30 to 70% were cocultured with a 100-fold excess of indicator cells in quadruplicates. After 5 days, cells were fixed and stained for viral IE proteins. The number of infected cells in the largest focus of each replica was counted. The median of at least four replicas was calculated for each experiment.
Cell-free infection.
One day prior to infection, HFFs and HECs were seeded onto gelatin-coated 96-well plates at a density of 15,000 cells per well. The cells were preincubated with MEM supplemented with GlutaMAX plus 5% FCS for 30 min prior to infection. Supernatants harvested from freshly reconstituted virus were used for infection. Cell-free virus was incubated for 2 h with the cells before the cells were supplied with their respective maintenance medium and incubated for about 24 h until the experiment was stopped by fixation.
Purification of virions.
Supernatants of infected HFFs were collected when the cytopathic effect (CPE) was visible in more than 90% of the cells. Cells and large cell debris were removed by centrifugation at 3,220 × g for 10 min. For gradient purification of virions, cell-free supernatants were centrifuged at 70,000 × g for 70 min. Pellets were resuspended in 0.04 mol/liter sodium phosphate (pH 7.4) and carefully loaded onto glycerol-tartrate gradients (4 ml of 35% sodium tartrate and 5 ml of 15% sodium tartrate plus 30% glycerol) (64). After centrifugation at 70,000 × g for 45 min, the virion-containing band was extracted using a syringe and diluted in sodium phosphate buffer. The virions were pelleted via centrifugation at 100,000 × g for 70 min and subsequently lysed in radioimmunoprecipitation assay (RIPA) buffer (50 mM Tris, pH 7.4, 150 mM NaCl, 1 mM EDTA, 0.1% SDS, 0.5% deoxycholate, 1% NP-40, protease inhibitors [Roche]).
Immunoblotting.
Infected HFFs were lysed when they displayed >90% late-stage cytopathic effect. Lysis was conducted on ice for 30 min in RIPA buffer. Protein electrophoresis was performed in 8 to 12% polyacrylamide gels containing 0.1% SDS (SDS-PAGE). For nonreducing SDS-PAGE, no β-mercaptoethanol was added to the Laemmli sample buffer (126 mM Tris-HCl, 20% glycine, 4% SDS, 0,02% bromphenol blue), and a marker without reducing agents (SeeBlue Plus2 prestained protein ladder) was applied. Only samples intended for analysis under reducing conditions were boiled for 10 min at 95°C. Proteins were transferred onto polyvinylidene difluoride membranes in a Tris-glycine buffer (21.64 g/liter glycine, 4.58 g of Tris, and 15% methanol). The membranes were blocked overnight at 4°C in PBS–0.1% Tween with 5% dry milk powder. Antibodies were diluted in PBS–0.1% Tween with 0.5% dry milk powder. The rabbit anti-gO antiserum that was used for detection of the gH/gL/gO complex was kindly shared by B. J. Ryckman (35). Under reducing conditions, gO was detected using a monoclonal mouse antibody directed against full-length gO, which was kindly shared by B. Adler (26). D. C. Johnson generously provided the polyclonal rabbit anti-gL antiserum (12). The monoclonal mouse anti-pUL128 antibody was generously shared by G. Gerna (65). Glycoprotein B was detected using a monoclonal mouse antibody (clone 2F12; Abcam). Actin was stained with an anti-actin antibody produced in rabbit (Sigma). Proteins were visualized using horseradish peroxidase (HRP)-conjugated secondary antibodies (Santa Cruz), and Super Signal West Dura Extended Duration substrate was applied according to the manufacturer's instructions.
Immunoprecipitation.
Purified virions were lysed with a buffer containing 20 mM Tris-HCl, pH 8.0, 150 mM NaCl, 1% Triton X-100, and protease inhibitors. All steps of the immunoprecipitation were performed at 4°C. The lysates were precleared with 3 mg of protein A-Sepharose for 1 h. The beads were pelleted by centrifugation at 1,000 × g for 5 min, and the bound proteins were lysed with Laemmli sample buffer. The supernatants were precipitated with anti-gH antibody 14-4b overnight (66). Three milligrams of protein A-Sepharose was incubated with the mixture for 4 h before the precipitates were pelleted and lysed with Laemmli sample buffer. For detection of gH after immunoprecipitation, a recently isolated mouse monoclonal anti-gH antibody (3H10) was used (67).
Statistics.
In order to detect growth defects of virus mutants, we used one-sided t tests for the evaluation of all data. Cell-free infectivity of virus mutants was analyzed with an unpaired t test. For the analysis of focal growth, we used paired t tests to balance the influence of different preparations of indicator cells.
Supplementary Material
ACKNOWLEDGMENTS
We thank Guiseppe Gerna, David Johnson, and Barbara Adler for kindly providing antibodies against pUL128, gL, and gO. We also thank Brent Ryckman for generously sharing the gO antibody and for his invaluable suggestions to improve the project.
This work was supported by the Wilhelm-Sander-Foundation (Projekt 2013.002.1).
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JVI.01339-16.
REFERENCES
- 1.Kenneson A, Cannon MJ. 2007. Review and meta-analysis of the epidemiology of congenital cytomegalovirus (CMV) infection. Rev Med Virol 17:253–276. doi: 10.1002/rmv.535. [DOI] [PubMed] [Google Scholar]
- 2.Naing ZW, Scott GM, Shand A, Hamilton ST, van Zuylen WJ, Basha J, Hall B, Craig ME, Rawlinson WD. 2016. Congenital cytomegalovirus infection in pregnancy: a review of prevalence, clinical features, diagnosis and prevention. Aust N Z J Obstet Gynaecol 56:9–18. doi: 10.1111/ajo.12408. [DOI] [PubMed] [Google Scholar]
- 3.Pass RF. 1996. Immunization strategy for prevention of congenital cytomegalovirus infection. Infect Agents Dis 5:240–244. [PubMed] [Google Scholar]
- 4.Desai R, Collett D, Watson CJ, Johnson PJ, Moss P, Neuberger J. 2015. Impact of cytomegalovirus on long-term mortality and cancer risk after organ transplantation. Transplantation 99:1989–1994. doi: 10.1097/TP.0000000000000641. [DOI] [PubMed] [Google Scholar]
- 5.Limaye AP, Bakthavatsalam R, Kim HW, Randolph SE, Halldorson JB, Healey PJ, Kuhr CS, Levy AE, Perkins JD, Reyes JD, Boeckh M. 2006. Impact of cytomegalovirus in organ transplant recipients in the era of antiviral prophylaxis. Transplantation 81:1645–1652. doi: 10.1097/01.tp.0000226071.12562.1a. [DOI] [PubMed] [Google Scholar]
- 6.Plotkin S. 2015. The history of vaccination against cytomegalovirus. Med Microbiol Immunol 204:247–254. doi: 10.1007/s00430-015-0388-z. [DOI] [PubMed] [Google Scholar]
- 7.Connolly SA, Jackson JO, Jardetzky TS, Longnecker R. 2011. Fusing structure and function: a structural view of the herpesvirus entry machinery. Nat Rev Microbiol 9:369–381. doi: 10.1038/nrmicro2548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Eisenberg RJ, Atanasiu D, Cairns TM, Gallagher JR, Krummenacher C, Cohen GH. 2012. Herpes virus fusion and entry: a story with many characters. Viruses 4:800–832. doi: 10.3390/v4050800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Heldwein EE. 2016. gH/gL supercomplexes at early stages of herpesvirus entry. Curr Opin Virol 18:1–8. doi: 10.1016/j.coviro.2016.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vanarsdall AL, Howard PW, Wisner TW, Johnson DC. 2016. Human cytomegalovirus gH/gL forms a stable complex with the fusion protein gB in virions. PLoS Pathog 12:e1005564. doi: 10.1371/journal.ppat.1005564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Adler B, Scrivano L, Ruzcics Z, Rupp B, Sinzger C, Koszinowski U. 2006. Role of human cytomegalovirus UL131A in cell type-specific virus entry and release. J Gen Virol 87:2451–2460. doi: 10.1099/vir.0.81921-0. [DOI] [PubMed] [Google Scholar]
- 12.Ryckman BJ, Rainish BL, Chase MC, Borton JA, Nelson JA, Jarvis MA, Johnson DC. 2008. Characterization of the human cytomegalovirus gH/gL/UL128-131 complex that mediates entry into epithelial and endothelial cells. J Virol 82:60–70. doi: 10.1128/JVI.01910-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang D, Shenk T. 2005. Human cytomegalovirus virion protein complex required for epithelial and endothelial cell tropism. Proc Natl Acad Sci U S A 102:18153–18158. doi: 10.1073/pnas.0509201102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huber MT, Compton T. 1998. The human cytomegalovirus UL74 gene encodes the third component of the glycoprotein H-glycoprotein L-containing envelope complex. J Virol 72:8191–8197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huber MT, Compton T. 1999. Intracellular formation and processing of the heterotrimeric gH-gL-gO (gCIII) glycoprotein envelope complex of human cytomegalovirus. J Virol 73:3886–3892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li L, Nelson JA, Britt WJ. 1997. Glycoprotein H-related complexes of human cytomegalovirus: identification of a third protein in the gCIII complex. J Virol 71:3090–3097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Adler B, Sinzger C. 2013. Cytomegalovirus inter-strain variance in cell-type tropism, p 297–321. In MJ Reddehase. (ed), Cytomegaloviruses: from molecular pathogenesis to intervention, vol 1 Caister Academic Press, Norfolk, United Kingdom. [Google Scholar]
- 18.Hahn G, Revello MG, Patrone M, Percivalle E, Campanini G, Sarasini A, Wagner M, Gallina A, Milanesi G, Koszinowski U, Baldanti F, Gerna G. 2004. Human cytomegalovirus UL131-128 genes are indispensable for virus growth in endothelial cells and virus transfer to leukocytes. J Virol 78:10023–10033. doi: 10.1128/JVI.78.18.10023-10033.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ryckman BJ, Jarvis MA, Drummond DD, Nelson JA, Johnson DC. 2006. Human cytomegalovirus entry into epithelial and endothelial cells depends on genes UL128 to UL150 and occurs by endocytosis and low-pH fusion. J Virol 80:710–722. doi: 10.1128/JVI.80.2.710-722.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wille PT, Knoche AJ, Nelson JA, Jarvis MA, Johnson DC. 2010. A human cytomegalovirus gO-null mutant fails to incorporate gH/gL into the virion envelope and is unable to enter fibroblasts and epithelial and endothelial cells. J Virol 84:2585–2596. doi: 10.1128/JVI.02249-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhou M, Lanchy JM, Ryckman BJ. 2015. Human cytomegalovirus gH/gL/gO promotes the fusion step of entry into all cell types, whereas gH/gL/UL128-131 broadens virus tropism through a distinct mechanism. J Virol 89:8999–9009. doi: 10.1128/JVI.01325-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dunn W, Chou C, Li H, Hai R, Patterson D, Stolc V, Zhu H, Liu F. 2003. Functional profiling of a human cytomegalovirus genome. Proc Natl Acad Sci U S A 100:14223–14228. doi: 10.1073/pnas.2334032100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hobom U, Brune W, Messerle M, Hahn G, Koszinowski UH. 2000. Fast screening procedures for random transposon libraries of cloned herpesvirus genomes: mutational analysis of human cytomegalovirus envelope glycoprotein genes. J Virol 74:7720–7729. doi: 10.1128/JVI.74.17.7720-7729.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jiang XJ, Adler B, Sampaio KL, Digel M, Jahn G, Ettischer N, Stierhof YD, Scrivano L, Koszinowski U, Mach M, Sinzger C. 2008. UL74 of human cytomegalovirus contributes to virus release by promoting secondary envelopment of virions. J Virol 82:2802–2812. doi: 10.1128/JVI.01550-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yu D, Silva MC, Shenk T. 2003. Functional map of human cytomegalovirus AD169 defined by global mutational analysis. Proc Natl Acad Sci U S A 100:12396–12401. doi: 10.1073/pnas.1635160100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Laib Sampaio K, Stegmann C, Brizic I, Adler B, Stanton RJ, Sinzger C. 2016. The contribution of pUL74 to growth of human cytomegalovirus is masked in the presence of RL13 and UL128 expression. J Gen Virol 97:1917–1927. doi: 10.1099/jgv.0.000475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mori Y, Akkapaiboon P, Yonemoto S, Koike M, Takemoto M, Sadaoka T, Sasamoto Y, Konishi S, Uchiyama Y, Yamanishi K. 2004. Discovery of a second form of tripartite complex containing gH-gL of human herpesvirus 6 and observations on CD46. J Virol 78:4609–4616. doi: 10.1128/JVI.78.9.4609-4616.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sadaoka T, Yamanishi K, Mori Y. 2006. Human herpesvirus 7 U47 gene products are glycoproteins expressed in virions and associate with glycoprotein H. J Gen Virol 87:501–508. doi: 10.1099/vir.0.81374-0. [DOI] [PubMed] [Google Scholar]
- 29.Tang H, Mahmoud NF, Mori Y. 2015. Maturation of human herpesvirus 6A glycoprotein O requires coexpression of glycoprotein H and glycoprotein L. J Virol 89:5159–5163. doi: 10.1128/JVI.00140-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Coleman S, Hornig J, Maddux S, Choi KY, McGregor A. 2015. Viral glycoprotein complex formation, essential function and immunogenicity in the guinea pig model for cytomegalovirus. PLoS One 10:e0135567. doi: 10.1371/journal.pone.0135567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Scrivano L, Esterlechner J, Muhlbach H, Ettischer N, Hagen C, Grunewald K, Mohr CA, Ruzsics Z, Koszinowski U, Adler B. 2010. The m74 gene product of murine cytomegalovirus (MCMV) is a functional homolog of human CMV gO and determines the entry pathway of MCMV. J Virol 84:4469–4480. doi: 10.1128/JVI.02441-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lemmermann NA, Krmpotic A, Podlech J, Brizic I, Prager A, Adler H, Karbach A, Wu Y, Jonjic S, Reddehase MJ, Adler B. 2015. Non-redundant and redundant roles of cytomegalovirus gH/gL complexes in host organ entry and intra-tissue spread. PLoS Pathog 11:e1004640. doi: 10.1371/journal.ppat.1004640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang H, Huang C, Dong J, Yao Y, Xie Z, Liu X, Zhang W, Fang F, Chen Z. 2015. Complete protection of mice against lethal murine cytomegalovirus challenge by immunization with DNA vaccines encoding envelope glycoprotein complex III antigens gH, gL and gO. PLoS One 10:e0119964. doi: 10.1371/journal.pone.0119964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kabanova A, Marcandalli J, Zhou T, Bianchi S, Baxa U, Tsybovsky Y, Lilleri D, Silacci-Fregni C, Foglierini M, Fernandez-Rodriguez BM, Druz A, Zhang B, Geiger R, Pagani M, Sallusto F, Kwong PD, Corti D, Lanzavecchia A, Perez L. 2016. Platelet-derived growth factor-alpha receptor is the cellular receptor for human cytomegalovirus gHgLgO trimer. Nat Microbiol 1:16082. doi: 10.1038/nmicrobiol.2016.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhou M, Yu Q, Wechsler A, Ryckman BJ. 2013. Comparative analysis of gO isoforms reveals that strains of human cytomegalovirus differ in the ratio of gH/gL/gO and gH/gL/UL128-131 in the virion envelope. J Virol 87:9680–9690. doi: 10.1128/JVI.01167-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mattick C, Dewin D, Polley S, Sevilla-Reyes E, Pignatelli S, Rawlinson W, Wilkinson G, Dal Monte P, Gompels UA. 2004. Linkage of human cytomegalovirus glycoprotein gO variant groups identified from worldwide clinical isolates with gN genotypes, implications for disease associations and evidence for N-terminal sites of positive selection. Virology 318:582–597. doi: 10.1016/j.virol.2003.09.036. [DOI] [PubMed] [Google Scholar]
- 37.Murphy E, Yu D, Grimwood J, Schmutz J, Dickson M, Jarvis MA, Hahn G, Nelson JA, Myers RM, Shenk TE. 2003. Coding potential of laboratory and clinical strains of human cytomegalovirus. Proc Natl Acad Sci U S A 100:14976–14981. doi: 10.1073/pnas.2136652100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Paterson DA, Dyer AP, Milne RS, Sevilla-Reyes E, Gompels UA. 2002. A role for human cytomegalovirus glycoprotein O (gO) in cell fusion and a new hypervariable locus. Virology 293:281–294. doi: 10.1006/viro.2001.1274. [DOI] [PubMed] [Google Scholar]
- 39.Gatherer D, Seirafian S, Cunningham C, Holton M, Dargan DJ, Baluchova K, Hector RD, Galbraith J, Herzyk P, Wilkinson GW, Davison AJ. 2011. High-resolution human cytomegalovirus transcriptome. Proc Natl Acad Sci U S A 108:19755–19760. doi: 10.1073/pnas.1115861108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sinzger C, Hahn G, Digel M, Katona R, Sampaio KL, Messerle M, Hengel H, Koszinowski U, Brune W, Adler B. 2008. Cloning and sequencing of a highly productive, endotheliotropic virus strain derived from human cytomegalovirus TB40/E. J Gen Virol 89:359–368. doi: 10.1099/vir.0.83286-0. [DOI] [PubMed] [Google Scholar]
- 41.Tischer BK, Smith GA, Osterrieder N. 2010. En passant mutagenesis: a two step markerless red recombination system. Methods Mol Biol 634:421–430. doi: 10.1007/978-1-60761-652-8_30. [DOI] [PubMed] [Google Scholar]
- 42.Studer D, Michel M, Muller M. 1989. High pressure freezing comes of age. Scanning Microsc Suppl 3:253–269. [PubMed] [Google Scholar]
- 43.Theiler RN, Compton T. 2001. Characterization of the signal peptide processing and membrane association of human cytomegalovirus glycoprotein O. J Biol Chem 276:39226–39231. doi: 10.1074/jbc.M106300200. [DOI] [PubMed] [Google Scholar]
- 44.Walther P, Ziegler A. 2002. Freeze substitution of high-pressure frozen samples: the visibility of biological membranes is improved when the substitution medium contains water. J Microsc 208:3–10. doi: 10.1046/j.1365-2818.2002.01064.x. [DOI] [PubMed] [Google Scholar]
- 45.Buser C, Walther P, Mertens T, Michel D. 2007. Cytomegalovirus primary envelopment occurs at large infoldings of the inner nuclear membrane. J Virol 81:3042–3048. doi: 10.1128/JVI.01564-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Das S, Vasanji A, Pellett PE. 2007. Three-dimensional structure of the human cytomegalovirus cytoplasmic virion assembly complex includes a reoriented secretory apparatus. J Virol 81:11861–11869. doi: 10.1128/JVI.01077-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Villinger C, Schauflinger M, Gregorius H, Kranz C, Hohn K, Nafeey S, Walther P. 2014. Three-dimensional imaging of adherent cells using FIB/SEM and STEM. Methods Mol Biol 1117:617–638. doi: 10.1007/978-1-62703-776-1_27. [DOI] [PubMed] [Google Scholar]
- 48.Rasmussen L, Geissler A, Cowan C, Chase A, Winters M. 2002. The genes encoding the gCIII complex of human cytomegalovirus exist in highly diverse combinations in clinical isolates. J Virol 76:10841–10848. doi: 10.1128/JVI.76.21.10841-10848.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sinzger C, Schmidt K, Knapp J, Kahl M, Beck R, Waldman J, Hebart H, Einsele H, Jahn G. 1999. Modification of human cytomegalovirus tropism through propagation in vitro is associated with changes in the viral genome. J Gen Virol 80:2867–2877. doi: 10.1099/0022-1317-80-11-2867. [DOI] [PubMed] [Google Scholar]
- 50.Ciferri C, Chandramouli S, Donnarumma D, Nikitin PA, Cianfrocco MA, Gerrein R, Feire AL, Barnett SW, Lilja AE, Rappuoli R, Norais N, Settembre EC, Carfi A. 2015. Structural and biochemical studies of HCMV gH/gL/gO and pentamer reveal mutually exclusive cell entry complexes. Proc Natl Acad Sci U S A 112:1767–1772. doi: 10.1073/pnas.1424818112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Moreira IS, Fernandes PA, Ramos MJ. 2007. Hot spots–a review of the protein-protein interface determinant amino-acid residues. Proteins 68:803–812. doi: 10.1002/prot.21396. [DOI] [PubMed] [Google Scholar]
- 52.Gautier R, Douguet D, Antonny B, Drin G. 2008. HELIQUEST: a web server to screen sequences with specific alpha-helical properties. Bioinformatics 24:2101–2102. doi: 10.1093/bioinformatics/btn392. [DOI] [PubMed] [Google Scholar]
- 53.Lieber D, Hochdorfer D, Stoehr D, Schubert A, Lotfi R, May T, Wirth D, Sinzger C. 2015. A permanently growing human endothelial cell line supports productive infection with human cytomegalovirus under conditional cell growth arrest. Biotechniques 59:127–136. doi: 10.2144/000114326. [DOI] [PubMed] [Google Scholar]
- 54.May T, Butueva M, Bantner S, Markusic D, Seppen J, MacLeod RA, Weich H, Hauser H, Wirth D. 2010. Synthetic gene regulation circuits for control of cell expansion. Tissue Eng Part A 16:441–452. doi: 10.1089/ten.tea.2009.0184. [DOI] [PubMed] [Google Scholar]
- 55.Rivailler P, Kaur A, Johnson RP, Wang F. 2006. Genomic sequence of rhesus cytomegalovirus 180.92: insights into the coding potential of rhesus cytomegalovirus. J Virol 80:4179–4182. doi: 10.1128/JVI.80.8.4179-4182.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bahr U, Darai G. 2001. Analysis and characterization of the complete genome of tupaia (tree shrew) herpesvirus. J Virol 75:4854–4870. doi: 10.1128/JVI.75.10.4854-4870.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Smith LM, McWhorter AR, Shellam GR, Redwood AJ. 2013. The genome of murine cytomegalovirus is shaped by purifying selection and extensive recombination. Virology 435:258–268. doi: 10.1016/j.virol.2012.08.041. [DOI] [PubMed] [Google Scholar]
- 58.Zhang H, Todd S, Tachedjian M, Barr JA, Luo M, Yu M, Marsh GA, Crameri G, Wang LF. 2012. A novel bat herpesvirus encodes homologues of major histocompatibility complex classes I and II, C-type lectin, and a unique family of immune-related genes. J Virol 86:8014–8030. doi: 10.1128/JVI.00723-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dargan DJ, Douglas E, Cunningham C, Jamieson F, Stanton RJ, Baluchova K, McSharry BP, Tomasec P, Emery VC, Percivalle E, Sarasini A, Gerna G, Wilkinson GW, Davison AJ. 2010. Sequential mutations associated with adaptation of human cytomegalovirus to growth in cell culture. J Gen Virol 91:1535–1546. doi: 10.1099/vir.0.018994-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Davison AJ, Akter P, Cunningham C, Dolan A, Addison C, Dargan DJ, Hassan-Walker AF, Emery VC, Griffiths PD, Wilkinson GW. 2003. Homology between the human cytomegalovirus RL11 gene family and human adenovirus E3 genes. J Gen Virol 84:657–663. doi: 10.1099/vir.0.18856-0. [DOI] [PubMed] [Google Scholar]
- 61.Bradley AJ, Lurain NS, Ghazal P, Trivedi U, Cunningham C, Baluchova K, Gatherer D, Wilkinson GW, Dargan DJ, Davison AJ. 2009. High-throughput sequence analysis of variants of human cytomegalovirus strains Towne and AD169. J Gen Virol 90:2375–2380. doi: 10.1099/vir.0.013250-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Murrell I, Tomasec P, Wilkie GS, Dargan DJ, Davison AJ, Stanton RJ. 2013. Impact of sequence variation in the UL128 locus on production of human cytomegalovirus in fibroblast and epithelial cells. J Virol 87:10489–10500. doi: 10.1128/JVI.01546-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Stanton RJ, Baluchova K, Dargan DJ, Cunningham C, Sheehy O, Seirafian S, McSharry BP, Neale ML, Davies JA, Tomasec P, Davison AJ, Wilkinson GW. 2010. Reconstruction of the complete human cytomegalovirus genome in a BAC reveals RL13 to be a potent inhibitor of replication. J Clin Invest 120:3191–3208. doi: 10.1172/JCI42955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Talbot P, Almeida JD. 1977. Human cytomegalovirus: purification of enveloped virions and dense bodies. J Gen Virol 36:345–349. doi: 10.1099/0022-1317-36-2-345. [DOI] [PubMed] [Google Scholar]
- 65.Gerna G, Sarasini A, Patrone M, Percivalle E, Fiorina L, Campanini G, Gallina A, Baldanti F, Revello MG. 2008. Human cytomegalovirus serum neutralizing antibodies block virus infection of endothelial/epithelial cells, but not fibroblasts, early during primary infection. J Gen Virol 89:853–865. doi: 10.1099/vir.0.83523-0. [DOI] [PubMed] [Google Scholar]
- 66.Simpson JA, Chow JC, Baker J, Avdalovic N, Yuan S, Au D, Co MS, Vasquez M, Britt WJ, Coelingh KL. 1993. Neutralizing monoclonal antibodies that distinguish three antigenic sites on human cytomegalovirus glycoprotein H have conformationally distinct binding sites. J Virol 67:489–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Falk JJ, Laib Sampaio K, Stegmann C, Lieber D, Kropff B, Mach M, Sinzger C. 2016. Generation of a Gaussia luciferase-expressing endotheliotropic cytomegalovirus for screening approaches and mutant analyses. J Virol Methods 235:182–189. doi: 10.1016/j.jviromet.2016.06.008. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.