

ABSTRACT
Gammaherpesviruses are ubiquitous pathogens that establish lifelong infection in >95% of adults worldwide and are associated with a variety of malignancies. Coevolution of gammaherpesviruses with their hosts has resulted in an intricate relationship between the virus and the host immune system, and perturbation of the virus-host balance results in pathology. Interferon regulatory factor 1 (IRF-1) is a tumor suppressor that is also involved in the regulation of innate and adaptive immune responses. Here, we show that type I interferon (IFN) and IRF-1 cooperate to control acute gammaherpesvirus infection. Specifically, we demonstrate that a combination of IRF-1 and type I IFN signaling ensures host survival during acute gammaherpesvirus infection and supports IFN gamma-mediated suppression of viral replication. Thus, our studies reveal an intriguing cross talk between IRF-1 and type I and II IFNs in the induction of the antiviral state during acute gammaherpesvirus infection.
IMPORTANCE Gammaherpesviruses establish chronic infection in a majority of adults, and this long-term infection is associated with virus-driven development of a range of malignancies. In contrast, a brief period of active gammaherpesvirus replication during acute infection of a naive host is subclinical in most individuals. Here, we discovered that a combination of type I interferon (IFN) signaling and interferon regulatory factor 1 (IRF-1) expression is required to ensure survival of a gammaherpesvirus-infected host past the first 8 days of infection. Specifically, both type I IFN receptor and IRF-1 expression potentiated antiviral effects of type II IFN to restrict gammaherpesvirus replication in vivo, in the lungs, and in vitro, in primary macrophage cultures.
KEYWORDS: IRF-1, acute infection, gammaherpesvirus, interferon, viral replication
INTRODUCTION
Gammaherpesviruses are ubiquitous pathogens that establish lifelong infection in a majority of the adult population and are associated with cancer (1–3). Similar to replication of other viruses, replication of both human (Epstein-Barr virus [EBV] and Kaposi's sarcoma-associated herpesvirus [KSHV]) and murine (mouse gammaherpesvirus 68[MHV68]) gammaherpesviruses is suppressed by type I and type II interferons (IFNs), two partially overlapping yet distinct host networks that are critical for the control of gammaherpesvirus infection (4–10). In the case of MHV68, both acute and chronic MHV68 infection is attenuated by type I and type II IFNs (4, 6, 11, 12). While the antiviral role of IFNs in the context of gammaherpesvirus infection, including in vivo, is firmly established, the mechanism by which this restriction is imposed and the molecular players involved in this response are still being defined.
Interferon regulatory factor 1 (IRF-1) is a broadly antiviral transcription factor that restricts replication of diverse RNA and DNA viruses in cell culture via a poorly understood mechanism (13, 14). While initially IRF-1 was thought to induce type I interferon (IFN) expression (15), it soon became clear that IRF-1 functions downstream of IFN expression during viral infection in vitro and in vivo (16–19). We demonstrated (19) that one mechanism by which IRF-1 restricts gammaherpesvirus replication in primary macrophage cultures is via transcriptional induction of cholesterol-25-hydroxylase (CH25H), an enzyme whose product displays broad antiviral activities (20, 21). However, our studies also indicate that CH25H induction is not responsible for the entire spectrum of IRF-1 antiviral activity in vitro (19), suggesting that additional IRF-1-driven genes contribute to the antiviral activity.
In contrast to studies in vitro, few viruses have been tested in an IRF-1−/− host in vivo. Of those tested, IRF-1 restricted replication of West Nile virus (WNV) (16), vesicular stomatitis virus (VSV) (22), and murine norovirus (MNV) (18) during acute infection. Interestingly, IRF-1 contributes to IFN-γ-mediated control of MNV replication in primary macrophages in vitro and during acute infection in vivo, especially under conditions when type I IFN signaling is inhibited (18). However, expression of type I IFN receptor (IFNAR) is not required for IFN-γ-mediated control of MNV replication in primary macrophages in vitro (18). Further, recombinant type I IFN restricts MNV replication independent of IRF-1 expression, indicating that in the MNV system IRF-1 is specifically needed for type II- but not type I IFN-mediated antiviral activities (18). With respect to gammaherpesviruses, an early study by Dutia et al. demonstrated increased acute mortality of IRF-1−/− mice following a high-dose MHV68 infection (4 × 105 PFU) (6); however, neither the virus titers nor immune responses were measured. We showed that IRF-1 suppresses chronic MHV68 infection (23) via restriction of the germinal center reaction; however, the role of IRF-1 during acute MHV68 infection following a lower dose of viral inoculum remains unclear.
Here, we show that IRF-1 deficiency had minimal effects on the control of acute MHV68 replication following low-dose intranasal infection. In contrast, combined IRF-1 and type I IFN deficiency led to a significant increase in lung viral titers, decreased survival of the infected host, and inadequate expression of IFN-stimulated genes (ISGs) in the acutely infected lungs. Further, type II IFN-mediated restriction of MHV68 replication was significantly compromised in primary macrophages deficient for both type I IFN receptor and IRF-1. Thus, our study demonstrates that IRF-1 and type I and type II IFN cooperate in the control of acute gammaherpesvirus infection.
RESULTS
IRF-1 and type I IFN signaling independently and in cooperation attenuate MHV68 replication in the lungs at 7 days postinfection.
To investigate the role of IRF-1 in restricting MHV68 acute infection, mice were intranasally infected with a low dose of MHV68, and lung titers were measured at 7 days postinfection. IRF-1 deficiency resulted in a 6-fold increase in the lung titers compared to that in C57BL/6 (BL6) lungs (Fig. 1A).
FIG 1.
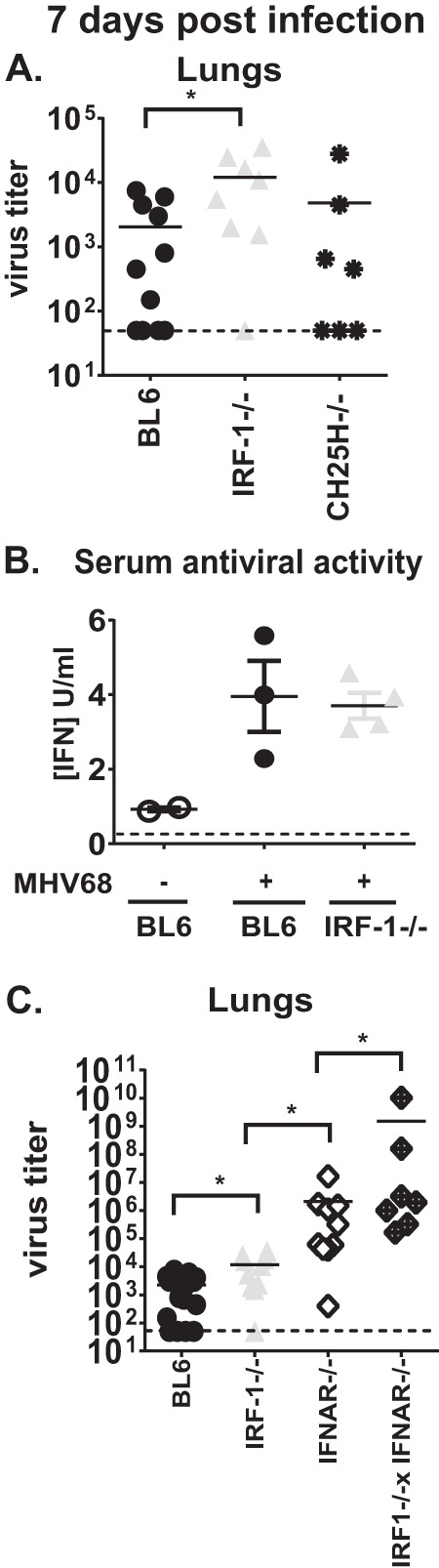
IRF-1 and type I IFN signaling independently and in cooperation attenuate MHV68 replication in the lungs at 7 days postinfection. Mice of indicated genotypes were intranasally inoculated with 500 PFU of MHV68. (A and C) At 7 days postinfection MHV68 titers were assessed in homogenized lungs. Each symbol represents an individual animal. Data were pooled from three to four independent experiments. Solid lines represent means, and dashed lines indicate limits of detection. (B) Antiviral activity was measured in serum collected at 7 days after mock or MHV68 infection. Data are representative of two independent experiments. *, P < 0.05.
Type I IFN plays an important role in limiting acute MHV68 infection. While IRF-1 was initially identified as a transcription factor that can associate with the IFN-β promoter, IRF-1 has not been found to be responsible for type I IFN expression in mice infected with WNV or Newcastle disease virus (16, 24). Because many cytokines engage type I IFN receptor, we have employed an encephalomyocarditis virus (EMCV) bioassay to comprehensively assess systemic antiviral activity in BL6 and IRF-1−/− MHV68-infected mice. Sera collected from mice of both genotypes had equivalent capacities to restrict replication of IFN-sensitive EMCV in fibroblast cultures (Fig. 1B), indicating that the increase in MHV68 lung titers observed in IRF-1−/− mice is not due to suboptimal expression of IFN in the absence of IRF-1.
IRF-1-mediated expression of CH25H is partially responsible for the antiviral effect of IRF-1 observed in MHV68-infected primary macrophage cultures (19). Thus, we examined the requirement for CH25H in the control of acute MHV68 infection. Interestingly, similar MHV68 titers were found in CH25H−/− and BL6 lungs at 7 days postinfection (Fig. 1A), suggesting that CH25H plays a lesser role in controlling acute MHV68 replication in the lungs than in vitro in macrophages.
Finally, we have previously shown that IRF-1 functions downstream of type I IFN to control MHV68 replication in primary macrophage cultures (19). To define the relationship between type I IFN and IRF-1 during acute MHV68 infection, MHV68 lung titers were measured at 7 days postinfection in mice with single or combined deficiency of IRF-1 and type I IFN receptor (IFNAR). As expected, IFNAR−/− mice had significantly elevated MHV68 lung titers compared to those of IRF-1−/− and BL6 mice (Fig. 1C), consistent with the role of type I IFN in controlling acute MHV68 replication (4, 6). In contrast to results observed in vitro (19), combined IRF-1 and IFNAR deficiency resulted in a further increase in MHV68 lung titers, indicating that IRF-1 independently and in cooperation with type I IFN restricts acute MHV68 replication at 7 days postinfection.
IRF-1 cooperates with type I IFN to ensure survival of the MHV68-infected host.
To investigate the extent to which IRF-1 contributes to control of MHV68 replication during later times in acute infection, MHV68 replication was assessed at 9 days postinfection. In contrast to titers observed at 7 days postinfection, MHV68 lung titers were similar in BL6 and IRF-1−/− mice (Fig. 2A), suggesting that increased MHV68 replication associated with IRF-1 deficiency is overcome by 9 days postinfection.
FIG 2.
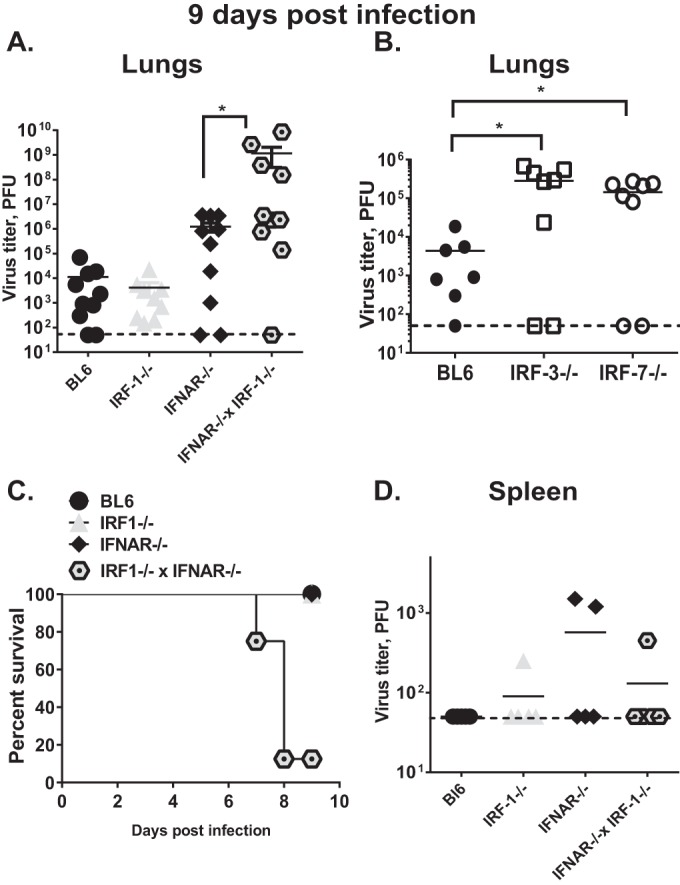
IRF-1 cooperates with type I IFN to ensure survival of MHV68-infected host. Mice were infected as described in the legend of Fig. 1. MHV68 titers were measured in the lungs (A and B) or spleens (D) at 9 days postinfection. Lung MHV68 titers for the IFNAR−/− IRF-1−/− mice were obtained using lung tissues from moribund animals at 7 to 8 days postinfection. Each symbol represents an individual animal; data were pooled from two to five independent experiments. The dashed line indicates the limit of detection; solid lines indicate means for each group. *, P < 0.05. (C) A survival curve was derived from the same cohorts as described in panels A and D.
IRF-3 and IRF-7 are transcription factors that are related to IRF-1 and represent major effectors of antiviral innate responses (25). We previously showed that IRF-3 restricts MHV68 replication in primary macrophages in vitro (26). In contrast to titers observed in IRF-1−/− animals, MHV68 titers were significantly elevated in IRF-3−/− and IRF-7−/− lungs compared to those in BL6 lungs (Fig. 2B), indicating that, unlike IRF-1, IRF-3 and IRF-7 play nonredundant functions in controlling lung MHV68 replication at 9 days postinfection.
Unexpectedly, we found that at around 8 days postinfection, a majority of MHV68-infected IFNAR−/− IRF-1−/− mice became moribund, in contrast to mice with individual IRF-1 or IFNAR deficiencies, which were uniformly surviving the infection (Fig. 2C). Further, average lung MHV68 titers of the moribund IFNAR−/− IRF-1−/− mice harvested at 8 to 9 days postinfection were 400-fold higher than those in IFNAR−/− mice and 70,000-fold higher than those in IRF-1−/− mice examined at 9 days postinfection (Fig. 2A).
The dramatic increase in lung virus titers observed in IFNAR−/− IRF-1−/− mice was restricted to lungs as MHV68 titers in IFNAR−/− IRF-1−/− livers and spleens were similar to those observed in IFNAR−/− mice (Fig. 2D; also data not shown), suggesting that the combined IRF-1/IFNAR deficiency did not further promote MHV68 spread from lungs to spleen and liver, in spite of greatly elevated viral titers in the lungs. In conclusion, both IRF-1 and type I IFN signaling were required to control lung MHV68 replication and ensure survival of acute gammaherpesvirus infection.
IRF-1 and type I IFN signaling are required to induce expression of antiviral ISGs in acutely infected lungs.
One plausible explanation for increased MHV68 lung titers in IFNAR−/− IRF-1−/− mice could be attenuated IFN-γ expression in the absence of IRF-1. CD4 T cells in IRF-1−/− mice are thought to be skewed toward a Th2 phenotype, with decreased expression of IFN-γ due to attenuated interleukin-12 (IL-12) expression (27). IFN-γ is induced during the late stages of acute MHV68 infection (28) and facilitates clearance of lytically replicating MHV68 in the lungs (29). Given the importance of IFN-γ in MHV68 clearance, serum IFN-γ levels were measured at 9 days postinfection. Surprisingly, IRF-1 deficiency did not attenuate systemic IFN-γ levels; instead, the highest serum levels were observed in IFNAR−/− and IFNAR−/− IRF-1−/− mice (Fig. 3A). Thus, IRF-1 was dispensable for systemic IFN-γ expression during acute MHV68 infection.
FIG 3.

IRF-1 and type I IFN signaling is required to induce expression of antiviral interferon-stimulated genes (ISG) in acutely infected lungs. Mice of the indicated genotypes were infected as described in the legend of Fig. 1. (A) IFN-γ levels were measured in serum collected at 9 days postinfection. (B to D) Lungs were harvested from mice of the indicated genotypes at 9 days after mock or MHV68 infection. Expression of specific interferon-stimulated genes was measured by qRT-PCR using RNA isolated from lungs. Each symbol represents an individual mouse. Data are representative of two to four independent experiments. GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
Equally elevated IFN-γ levels found in IFNAR−/− and IFNAR−/− IRF-1−/− sera did not explain differential survival of mice of these two genotypes during acute MHV68 infection (Fig. 2C). Because antiviral functions of interferon are mediated by the expression of ISGs, mRNA levels of antiviral ISGs expressed in the lungs were measured next. Of several hundred known ISGs, we focused on the genes that are known to restrict MHV68 replication, at least in vitro (30), or on antiviral genes that can be directly regulated by IRF-1, such as viperin (31). Increased mRNA levels of myeloid nuclear differentiation antigen (MNDA) and IFIH were found in the lungs of infected BL6, IRF-1−/−, and IFNAR−/− mice (Fig. 3B and D). As expected, viperin expression was increased in BL6 and IFNAR−/− but not IRF-1−/− lungs at 9 days postinfection compared to levels in mock-infected animals of the same genotypes (Fig. 3C). Surprisingly, none of the three examined ISGs were significantly induced in MHV68-infected IFNAR−/− IRF-1−/− lungs (Fig. 3B to D). Thus, increased systemic IFN-γ levels failed to generate an increase in antiviral ISG expression in MHV68-infected IRF-1−/− IFNAR−/− lungs.
IRF-1 and type I IFN receptor cooperate to induce an IFN-γ-dependent antiviral state in primary macrophages.
IRF-1 functions in a cell-autonomous fashion to potentiate IFN-γ antiviral effects in primary macrophages replicating MNV (18) and WNV (16) or in mouse embryonic fibroblasts infected with herpes simplex virus or EMCV (17). In contrast, IFN-γ suppresses VSV replication equally in wild-type and IRF-1-deficient fibroblasts (17). To determine the extent to which IRF-1 or type I IFN receptor facilitates the antiviral effects of IFN-γ in the context of MHV68 infection, primary macrophage cultures were pretreated with IFN-γ and infected with MHV68, and viral replication was assessed (Fig. 4A). As we previously demonstrated (19), replication of MHV68 in control-treated macrophages was elevated in the absence of IRF-1 and further increased in the absence of type I IFN receptor (Fig. 4B). Combined IRF-1/IFNAR deficiency resulted in titers of MHV68 similar to those observed in IFNAR−/− macrophages.
FIG 4.

IRF-1 and type I IFN receptor cooperate to induce type I or type II IFN-dependent antiviral state in primary macrophages. Primary macrophages were derived from bone marrow of mice of the indicated genotypes. (A) Following differentiation, macrophages were mock treated or treated with 1 or 10 U/ml of recombinant IFN-γ or IFN-β for 16 h. Pretreated macrophages were infected at an MOI of 1 (B and D) or 0.01 (C and E), and the same concentration of IFN or carrier was added to the medium at 1 h postinfection. Viral yields were measured at 72 h postinfection with 5 PFU as the limit of detection. Each condition was tested in triplicate within an individual experiment; data are representative of three independent experiments and are presented as means with standard errors. DKO, double knockout (IFNAR−/− IRF-1−/−).
A low concentration of IFN-γ (1 U/ml) fully suppressed MHV68 replication in wild-type and IRF-1−/− primary macrophages at 72 h postinfection (Fig. 4B). Interestingly, low levels of IFN-γ failed to fully suppress MHV68 replication in IFNAR−/− macrophages. These results indicate that the expression of type I IFN receptor was necessary for the antiviral effects of IFN-γ in the context of MHV68 infection, unlike in the context of MNV infection. Importantly, IFN-γ-treated IRF-1−/− IFNAR−/− macrophages displayed minimal attenuation of MHV68 replication compared to that of the control-treated macrophages of the same genotype (3.5-fold suppression of viral replication in IFN-γ-treated IRF-1−/− IFNAR−/− macrophages versus 22-fold suppression in BL6 macrophages and 200-fold suppression in IRF-1−/− macrophages) (Fig. 4B). Treatment of macrophages with a higher concentration of IFN-γ (10 U/ml) suppressed MHV68 replication in macrophages of all genotypes, with the exception of the IRF-1−/− IFNAR−/− cultures, which displayed MHV68 titers that were at least 150-fold higher than those of macrophages of other genotypes.
Mostly similar results were obtained when lower multiplicities of infection (MOIs) were tested (Fig. 4C, MOI of 0.01). Interestingly, under these very low-MOI conditions, 1 U/ml IFN-γ treatment failed to fully suppress MHV68 replication in IRF-1−/− macrophages. In summary, expression of type I IFN receptor and IRF-1 was required for IFN-γ-mediated induction of an optimal antiviral state in primary macrophages infected with MHV68.
IRF-1 expression potentiates type I IFN-mediated suppression of MHV68 replication in primary macrophages.
Having observed that IRF-1 expression potentiates antiviral effects of type II IFN in primary macrophages infected with MHV68 (Fig. 4B and C), the requirement of IRF-1 for antiviral effects of type I IFN was tested. Expression of type I IFN is rapidly induced in primary macrophages infected with MHV68 (26), and this induction of type I IFN and global IFN responses is independent of IRF-1 expression, regardless of the MHV68 multiplicity of infection (19; also data not shown).
In contrast to the dispensable nature of the IRF-1 expression on IFN-β-mediated suppression of MNV replication (18), suppression of MHV68 replication by exogenous IFN-β was less pronounced in IRF-1−/− macrophages (Fig. 4D and E). Specifically, when macrophages were infected at 1 PFU/cell, 1 U/ml or 10 U/ml of exogenous IFN-β treatment suppressed MHV68 replication in BL6 macrophages 7.9-fold and 13.2-fold, respectively (Fig. 4D). However, under the same experimental conditions MHV68 replication was suppressed only 2-fold and 5.6-fold, respectively, in the absence of IRF-1. Similar observations were made when lower-MOI conditions were used (Fig. 4E). Thus, IRF-1 expression by primary macrophages was necessary for optimal restriction of MHV68 replication by either type I or type II IFN.
IRF-1 and type I IFN receptor facilitate induction of antiviral ISGs in MHV68-infected macrophages.
Decreased expression of antiviral ISGs was observed in MHV68-infected lungs of IRF-1−/− IFNAR−/− mice, in spite of high levels of systemic IFN-γ (Fig. 3). Further, decreased antiviral effects of IFN-γ and IFN-β were found in MHV68-infected IRF-1−/− macrophages (Fig. 4). IFNs, whether type I or type II, mediate their antiviral effect by inducing expression of ISGs. To determine the extent to which IRF-1 and IFNAR expression are required for induction of antiviral ISGs in MHV68-infected primary macrophage cultures, macrophages were mock treated, treated with IFN-γ, infected with MHV68, or treated with IFN-γ immediately following virus absorption (Fig. 5A). Treatment with IFN-γ produced equivalently elevated levels of MNDA mRNA in macrophages of all four genotypes (Fig. 5B). Infection of primary macrophages with MHV68, which stimulates type I but not type II IFN expression in vitro (26), failed to induce MNDA expression in macrophages lacking type I IFN receptor. Interestingly, when macrophages received a combination of MHV68 and IFN-γ, MNDA expression was significantly decreased in IRF-1−/− IFNAR−/− macrophages compared to levels in BL6 and IRF-1−/− cultures, and to a lesser extent in IFNAR−/− macrophages.
FIG 5.

IRF-1 and type I IFN receptor facilitate induction of antiviral ISGs in MHV68-infected macrophages. Primary macrophages were derived from bone marrow of mice with the indicated genotypes. (A) Differentiated macrophages were mock infected or infected with MHV68 (MOI of 5). At 1 h postinfection macrophages were treated with either carrier or 10 U/ml IFN-γ (B to D) or IFN-β (E to G). Total RNA was harvested at 8 h posttreatment and subjected to qRT-PCR to determine mRNA levels of MNDA, viperin, and IFIH, as indicated. GAPDH expression was used for normalization. Within an individual experiment each condition was performed in duplicate; data are representative of two to four independent experiments and are presented as means with standard errors.
Viperin expression was induced in IFN-γ-treated BL6 macrophages and to a much lesser extent in IRF-1−/− macrophages (Fig. 5C), consistent with the known capacity of IRF-1 to directly drive viperin expression under certain conditions (31). IFN-γ treatment of IFNAR−/− macrophages increased viperin expression compared to the level in mock-treated IFNAR−/− cultures; however, these expression levels remained below those observed in BL6 and even IRF-1−/− IFN-γ-treated cells. Both MHV68 infection and MHV68/IFN-γ combined treatments efficiently induced equivalent viperin mRNA levels in BL6 and IRF-1−/− macrophages, with significantly less expression observed in IFNAR−/− and IRF-1−/− IFNAR−/− cultures (Fig. 5C).
Finally, neither IFN-γ nor MHV68 infection (separately or in combination) induced significant expression of IFIH in IFNAR−/− and IRF-1−/− IFNAR−/− macrophages compared to corresponding baseline levels of expression. Thus, similar to expression observed in acutely infected lungs in vivo, optimal IFN-γ-dependent expression of several antiviral ISGs in primary macrophages relied on expression of type I IFN receptor and IRF-1, especially in the context of IFN-γ treatment of MHV68-infected primary macrophage cultures.
When similar studies were performed using type I IFN instead of type II IFN, expression levels of MNDA, viperin, and IFIH were similar in BL6 and IRF-1−/− macrophages under all conditions tested (Fig. 5E to G), consistent with the previous observations by our group and others that IRF-1 expression is dispensable for type I IFN expression and signaling.
DISCUSSION
The discovery of IRF-1 as an inducer of IFN-β (15) established IRF-1 as a part of the type I IFN signaling network, a host response that is widely accepted as a critical innate antiviral mechanism. Our previous work has shown IRF-1 to be largely dispensable for IFN-α/β expression during MHV68 infection of cultured primary macrophages (19), with IRF-3 being the master inducer of type I IFN (26). While IRF-1 is dispensable for type I IFN expression, it is clear that IRF-1 orchestrates diverse (and poorly understood) antiviral functions, both dependent and independent of type I IFN, which help restrict viral infection. Our current study reveals the complexity of the antiviral response to MHV68 infection and highlights the contribution of IRF-1 to the control of acute gammaherpesvirus infection and host survival.
Role of IRF-1 during viral infection of an intact host.
This study demonstrated that a combined IRF-1 and IFNAR deficiency drastically impaired the ability of mice to control MHV68 replication, as indicated by a >4-log increase in lung virus titers in IRF-1−/− IFNAR−/− mice compared to titers in BL6 mice (Fig. 2). Additionally, IRF-1 promoted survival of IFNAR−/− mice, as evidenced by the significant decrease in survival of doubly deficient mice. This cooperativity between IRF-1 and type I IFN mirrors the role of IRF-1 in the control of MNV acute infection, where blockade of type I IFN signaling elevated viral burden in many organs of IRF-1−/− mice and increased mortality (18). Thus, IRF-1-type I IFN cooperation in vivo has now been demonstrated for an RNA and a DNA virus, suggesting that IRF-1 likely evolved as a broad auxiliary antiviral mechanism to augment type I IFN-mediated antiviral response.
Decreased survival of MHV68-infected IRF-1−/− IFNAR−/− mice could be a combined outcome of several factors. Our in vitro studies suggest that direct exposure of MHV68-infected macrophages to IFN-γ failed to adequately restrict MHV68 replication in IRF-1−/− IFNAR−/− macrophages along with decreased ISG expression. It is tempting to speculate that decreased sensitivity of IRF-1−/− IFNAR−/− mice to the antiviral effects of IFN-γ and subsequent increased MHV68 replication in the lungs have also contributed to the ultimate demise of these infected animals.
Interestingly, IRF-1 deficiency by itself had minimal effects on the parameters of acute MHV68 infection. This is in contrast to the sustained increase in viral titers and host mortality observed in IRF-1−/− mice infected with 100 PFU of WNV (16), a viral dose that is comparable to 500 PFU of MHV68 used to infect mice in our study. This suggests that, at least during acute MHV68 infection, IRF-1 functions are complemented by type I IFN expression and signaling. In contrast, we showed that IRF-1−/− mice have poor control of chronic MHV68 infection (23), indicating that type I IFN signaling is no longer able to compensate for IRF-1 deficiency once gammaherpesvirus infection becomes chronic.
Antiviral effects of cooperative type I IFN–IRF-1 expression.
What might be the molecular mechanism that allows cooperative IRF-1-type I IFN expression to facilitate antiviral effects of IFN-γ? The cross talk between type I and type II IFN signaling has been previously noted. Specifically, decreased expression of Stat1 in type I IFN-deficient cells contributes to the impaired responses of such cells to IFN-γ (32). Further, constitutive association between IFNAR1 and IFN-γ receptor 2 (IFNGR2) is thought to facilitate IFN-γ signaling (33). These molecular mechanisms may have contributed to the decreased antiviral effects of IFN-γ that we observed in MHV68-infected IFNAR−/− macrophages (Fig. 4). However, higher doses of IFN-γ were able to fully suppress MHV68 replication even in the absence of type I IFN receptor, suggesting that the cross talk between type I and type II IFNs is not an absolute requirement for IFN-γ antiviral effects.
In contrast, even in the absence of type I IFN signaling, IRF-1 significantly contributed to the IFN-γ-mediated suppression of MHV68 replication, as evidenced by consistently elevated viral titers in IFN-γ-treated IRF-1−/− IFNAR−/− macrophages, regardless of the MOI or IFN-γ dose (Fig. 4B and C). Further, IRF-1 expression potentiated antiviral effects of type I IFN in restricting MHV68 replication in primary macrophages (Fig. 4D and E). It is tempting to speculate that the specific IRF-1-driven genes that are induced in IFN-exposed macrophages are important antiviral effectors of the host response.
What are the IRF-1-driven genes that mediate sensitivity to the antiviral effects of type I or type II IFN? Maloney et al. have comprehensively defined IRF-1-dependent gene expression that also overlaps with STAT1-regulated gene expression in IFN-γ-treated primary macrophages (18). Further, several studies have used chromatin immunoprecipitation-sequencing approaches to catalog IRF-1 target genes in IFN-γ-treated human monocytes and mouse macrophages and explore cooperativity between IRF-1 and other IRF family members (34, 35). Interestingly, virus infection may change the repertoire of IRF-1-regulated genes. Specifically, we observed that while IFN-γ treatment alone efficiently induced MNDA mRNA in macrophages of all four genotypes, MNDA mRNA levels were significantly lower in IFN-γ-treated MHV68-infected IRF-1−/− IFNAR−/− macrophages than in similarly treated macrophages of other genotypes (Fig. 5B). Thus, one important future direction is to comprehensively define IRF-1-dependent gene expression in the context of specific virus infection and compare it to that in IFN-treated uninfected cells. However, even if such IRF-1-dependent genes are comprehensively defined in a highly physiological system, a more daunting task is to identify a precise combination of host genes/mechanisms that are responsible for IRF-1-mediated restriction of a specific virus. Further, such antiviral host systems are likely to be further modified by viral proteins that counteract intrinsic cellular immune responses. In spite of the potential complexity, defining the mechanism by which IRF-1 restricts replication of diverse viruses in vivo is likely to identify unexpected targets that could lead to the development of future broad-acting antiviral therapies.
MATERIALS AND METHODS
Animal studies.
C57BL/6J, B6.129S2-Irf1tm1Mak/J (stock 002762; referred to as IRF-1−/−), and CH25H−/− (36) mice were obtained from Jackson Laboratories (Bar Harbor, ME). IFNAR1−/− mice on the C57BL/6 genetic background were a gift from Mitchell Grayson (37); these mice were originally provided by J. Sprent (The Scripps Research Institute). IRF-1−/− IFNAR1−/− mice were generated as previously described (19). IRF-3−/− and IRF-7−/− mice were a kind gift of Michael Diamond. At 6 to 7 weeks of age mice were infected by intranasal inoculation with 500 PFU of MHV68 (WUMS strain) diluted in sterile serum-free Dulbecco's modified Eagle's medium (DMEM; 15 μl/mouse); infections were performed under light anesthesia. All experimental manipulations of mice used in this study were approved by the institutional Animal Care and Use Committee of the Medical College of Wisconsin.
Virus titers in organs.
Tissues were harvested in serum-free DMEM and disrupted by bead beating using 1-mm zirconia/silica beads (Biospec Products, Bartelsville, OK). Homogenates were cleared by brief centrifugation, and titers of supernatants were determined by plaque assay on 3T12 fibroblasts as previously described (38).
In vitro MHV68 assays.
MHV68 viral stock was prepared as previously described previously (39), and titers of stocks were determined using 3T12 fibroblast cell line (ATCC). For in vitro replication assays, primary bone marrow-derived macrophages (38) were mock treated or pretreated with recombinant IFN-β or IFN-γ (BioLegend, San Diego, CA) for 16 h and infected with MHV68, and the same IFN was added back to the culture medium at 1 h postinfection for the duration of the experiment. For IFN stimulation experiments, macrophages were mock treated or infected with MHV68, IFN was added following virus adsorption, and RNA was harvested 8 h later.
Quantitation of cellular mRNA.
Total RNA was harvested from macrophages or lungs, DNase treated, and reverse transcribed, and mRNA levels were quantified by quantitative reverse transcription-PCR (qRT-PCR) as described previously (19).
IFN-γ ELISA.
IFN-γ levels in the serum were assessed using an IFN-γ Max enzyme-linked immunosorbent assay (ELISA) kit according to the manufacturer's instructions (BioLegend, San Diego, CA).
Antiviral activity serum bioassay.
Serum was collected from infected and mock-infected mice and diluted to 30% using serum-free DMEM. The amount of antiviral activity in the serum was determined as previously described using an EMCV bioassay (26). Specifically, this bioassay compares the effectiveness of the test serum to restrict EMCV replication to that of recombinant type I IFN. Briefly, monolayers of L929 cells were overlaid with diluted serum or a known concentration of recombinant IFN-β and incubated overnight at 37°C in 5% CO2. Subsequently, monolayers were infected with EMCV (B strain) at an MOI of 5, and cell survival was determined after an additional 24 h.
Statistical analysis.
All data were analyzed using GraphPad Prism software (GraphPad Software, San Diego, CA). All P values were calculated using Student's t test.
ACKNOWLEDGMENTS
This study was supported by American Heart Association grant 15PRE22640005 (W.P.M.), ACS Research Scholar Grant RSG-12-174-01-MPC, NIH grant R01CA183593, the Wisconsin Breast Cancer Showhouse, and a Rosenberg Award (V.L.T.).
We thank Nancy Reich for helpful discussions of this study.
W.P.M. and V.L.T. designed the overall study and wrote the manuscript. W.P.M., M.M.R., M.P.L., P.T.L., K.E.S., and S.A. contributed to the design of the studies and performed the experiments.
REFERENCES
- 1.Savoie A, Perpete C, Carpentier L, Joncas J, Alfieri C. 1994. Direct correlation between the load of Epstein-Barr virus-infected lymphocytes in the peripheral blood of pediatric transplant patients and risk of lymphoproliferative disease. Blood 83:2715–2722. [PubMed] [Google Scholar]
- 2.Kenagy DN, Schlesinger Y, Weck K, Ritter JH, Gaudreault-Keener MM, Storch GA. 1995. Epstein-Barr virus DNA in peripheral blood leukocytes of patients with posttransplant lymphoproliferative disease. Transplantation 60:547–554. doi: 10.1097/00007890-199509270-00005. [DOI] [PubMed] [Google Scholar]
- 3.Cesarman E, Knowles DM. 1999. The role of Kaposi's sarcoma-associated herpesvirus (KSHV/HHV-8) in lymphoproliferative diseases. Semin Cancer Biol 9:165–174. doi: 10.1006/scbi.1998.0118. [DOI] [PubMed] [Google Scholar]
- 4.Barton ES, Lutzke ML, Rochford R, Virgin HW. 2005. Alpha/beta interferons regulate murine gammaherpesvirus latent gene expression and reactivation from latency. J Virol 79:14149–14160. doi: 10.1128/JVI.79.22.14149-14160.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chang J, Renne R, Dittmer D, Ganem D. 2000. Inflammatory cytokines and the reactivation of Kaposi's sarcoma-associated herpesvirus lytic replication. Virology 266:17–25. doi: 10.1006/viro.1999.0077. [DOI] [PubMed] [Google Scholar]
- 6.Dutia BM, Allen DJ, Dyson H, Nash AA. 1999. Type I interferons and IRF-1 play a critical role in the control of a gammaherpesvirus infection. Virology 261:173–179. doi: 10.1006/viro.1999.9834. [DOI] [PubMed] [Google Scholar]
- 7.Hwang S, Kim KS, Flano E, Wu TT, Tong LM, Park AN, Song MJ, Sanchez DJ, O'Connell RM, Cheng G, Sun R. 2009. Conserved herpesviral kinase promotes viral persistence by inhibiting the IRF-3-mediated type I interferon response. Cell Host Microbe 5:166–178. doi: 10.1016/j.chom.2008.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Krug LT, Pozharskaya VP, Yu Y, Inoue N, Offermann MK. 2004. Inhibition of infection and replication of human herpesvirus 8 in microvascular endothelial cells by alpha interferon and phosphonoformic acid. J Virol 78:8359–8371. doi: 10.1128/JVI.78.15.8359-8371.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mandal P, Krueger BE, Oldenburg D, Andry KA, Beard RS, White DW, Barton ES. 2011. A gammaherpesvirus cooperates with interferon-alpha/beta-induced IRF2 to halt viral replication, control reactivation, and minimize host lethality. PLoS Pathog 7:e1002371. doi: 10.1371/journal.ppat.1002371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Monini P, Carlini F, Sturzl M, Rimessi P, Superti F, Franco M, Melucci-Vigo G, Cafaro A, Goletti D, Sgadari C, Butto S, Leone P, Leone P, Chiozzini C, Barresi C, Tinari A, Bonaccorsi P, Capobianchi MR, Giuliani M, di Carlo A, Andreoni M, Rezza G, Ensoli B. 1999. Alpha interferon inhibits human herpesvirus 8 (HHV-8) reactivation in primary effusion lymphoma cells and reduces HHV-8 load in cultured peripheral blood mononuclear cells. J Virol 73:4029–4041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Steed A, Barton ES, Tibbetts SA, Popkin DL, Lutzke ML, Rochford R, Virgin HW. 2006. Gamma interferon blocks gammaherpesvirus reactivation from latency. J Virol 80:192–200. doi: 10.1128/JVI.80.1.192-200.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tibbetts SA, Van Dyk L, Speck SH, Virgin HW. 2002. Immune control of the number and reactivation phenotype of cells latently infected with a gamma-herpesvirus. J Virol 76:7125–7132. doi: 10.1128/JVI.76.14.7125-7132.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schoggins JW, Wilson SJ, Panis M, Murphy MY, Jones CT, Bieniasz P, Rice CM. 2011. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 472:481–485. doi: 10.1038/nature09907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schoggins JW, MacDuff DA, Imanaka N, Gainey MD, Shrestha B, Eitson JL, Mar KB, Richardson RB, Ratushny AV, Litvak V, Dabelic R, Manicassamy B, Aitchison JD, Aderem A, Elliott RM, Garcia-Sastre A, Racaniello V, Snijder EJ, Yokoyama WM, Diamond MS, Virgin HW, Rice CM. 2014. Pan-viral specificity of IFN-induced genes reveals new roles for cGAS in innate immunity. Nature 505:691–695. doi: 10.1038/nature12862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Miyamoto M, Fujita T, Kimura Y, Maruyama M, Harada H, Sudo Y, Miyata T, Taniguchi T. 1988. Related expression of a gene encoding a nuclear factor, IRF-1, that specifically binds to IFN-beta gene regulatory elements. Cell 54:903–913. doi: 10.1016/S0092-8674(88)91307-4. [DOI] [PubMed] [Google Scholar]
- 16.Brien JD, Daffis S, Lazear HM, Cho H, Suthar MS, Gale M Jr., Diamond MS. 2011. Interferon regulatory factor-1 (IRF-1) shapes both innate and CD8+ T cell immune responses against West Nile virus infection. PLoS Pathog 7:e1002230. doi: 10.1371/journal.ppat.1002230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kimura T, Nakayama K, Penninger J, Kitagawa M, Harada H, Matsuyama T, Tanaka N, Kamijo R, Vilcek J, Mak TW, Taniguchi T. 1994. Involvement of the IRF-1 transcription factor in antiviral responses to interferons. Science 264:1921–1924. doi: 10.1126/science.8009222. [DOI] [PubMed] [Google Scholar]
- 18.Maloney NS, Thackray LB, Goel G, Hwang S, Duan E, Vachharajani P, Xavier R, Virgin HW. 2012. Essential cell-autonomous role for interferon (IFN) regulatory factor 1 in IFN-γ-mediated inhibition of norovirus replication in macrophages. J Virol 86:12655–12664. doi: 10.1128/JVI.01564-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mboko WP, Mounce BC, Emmer J, Darrah E, Patel SB, Tarakanova VL. 2014. Interferon regulatory factor-1 restricts gammaherpesvirus replication in primary immune cells. J Virol 88:6993–7004. doi: 10.1128/JVI.00638-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Blanc M, Hsieh WY, Robertson KA, Kropp KA, Forster T, Shui G, Lacaze P, Watterson S, Griffiths SJ, Spann NJ, Meljon A, Talbot S, Krishnan K, Covey DF, Wenk MR, Craigon M, Ruzsics Z, Haas J, Angulo A, Griffiths WJ, Glass CK, Wang Y, Ghazal P. 2013. The transcription factor STAT-1 couples macrophage synthesis of 25-hydroxycholesterol to the interferon antiviral response. Immunity 38:106–118. doi: 10.1016/j.immuni.2012.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu SY, Aliyari R, Chikere K, Li G, Marsden MD, Smith JK, Pernet O, Guo H, Nusbaum R, Zack JA, Freiberg AN, Su L, Lee B, Cheng G. 2013. Interferon-inducible cholesterol-25-hydroxylase broadly inhibits viral entry by production of 25-hydroxycholesterol. Immunity 38:92–105. doi: 10.1016/j.immuni.2012.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nair S, Michaelsen-Preusse K, Finsterbusch K, Stegemann-Koniszewski S, Bruder D, Grashoff M, Korte M, Koster M, Kalinke U, Hauser H, Kroger A. 2014. Interferon regulatory factor-1 protects from fatal neurotropic infection with vesicular stomatitis virus by specific inhibition of viral replication in neurons. PLoS Pathog 10:e1003999. doi: 10.1371/journal.ppat.1003999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mboko WP, Olteanu H, Ray A, Xin G, Darrah EJ, Kumar SN, Kulinski JM, Cui W, Dittel BN, Gauld SB, Tarakanova VL. 2015. Tumor suppressor IRF-1 counteracts germinal center reaction driven by a cancer-associated gammaherpesvirus. J Virol 90:2818–2829. doi: 10.1128/JVI.02774-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Matsuyama T, Kimura T, Kitagawa M, Pfeffer K, Kawakami T, Watanabe N, Kundig TM, Amakawa R, Kishihara K, Wakeham A, Potter J , Furlonger CL , Narendran A , Suzuki H , Ohashi PS , Paige CJ , Taniguchi T , Mak TW. 1993. Targeted disruption of IRF-1 or IRF-2 results in abnormal type I IFN gene induction and aberrant lymphocyte development. Cell 75:83–97. doi: 10.1016/S0092-8674(05)80086-8. [DOI] [PubMed] [Google Scholar]
- 25.Sato M, Suemori H, Hata N, Asagiri M, Ogasawara K, Nakao K, Nakaya T, Katsuki M, Noguchi S, Tanaka N, Taniguchi T. 2000. Distinct and essential roles of transcription factors IRF-3 and IRF-7 in response to viruses for IFN-alpha/beta gene induction. Immunity 13:539–548. doi: 10.1016/S1074-7613(00)00053-4. [DOI] [PubMed] [Google Scholar]
- 26.Wood BM, Mboko WP, Mounce BC, Tarakanova VL. 2013. Mouse gammaherpesvirus infection acts as a rheostat to set the level of type I interferon signaling in primary macrophages. Virology 443:123–133. doi: 10.1016/j.virol.2013.04.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Salkowski CA, Thomas KE, Cody MJ, Vogel SN. 2000. Impaired IFN-gamma production in IFN regulatory factor-1 knockout mice during endotoxemia is secondary to a loss of both IL-12 and IL-12 receptor expression. J Immunol 165:3970–3977. doi: 10.4049/jimmunol.165.7.3970. [DOI] [PubMed] [Google Scholar]
- 28.Sarawar SR, Cardin RD, Brooks JW, Mehrpooya M, Tripp RA, Doherty PC. 1996. Cytokine production in the immune response to murine gammaherpesvirus 68. J Virol 70:3264–3268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tsai CY, Hu Z, Zhang W, Usherwood EJ. 2011. Strain-dependent requirement for IFN-gamma for respiratory control and immunotherapy in murine gammaherpesvirus infection. Viral Immunol 24:273–280. doi: 10.1089/vim.2011.0004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu SY, Sanchez DJ, Aliyari R, Lu S, Cheng G. 2012. Systematic identification of type I and type II interferon-induced antiviral factors. Proc Natl Acad Sci U S A 109:4239–4244. doi: 10.1073/pnas.1114981109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stirnweiss A, Ksienzyk A, Klages K, Rand U, Grashoff M, Hauser H, Kroger A. 2010. IFN regulatory factor-1 bypasses IFN-mediated antiviral effects through viperin gene induction. J Immunol 184:5179–5185. doi: 10.4049/jimmunol.0902264. [DOI] [PubMed] [Google Scholar]
- 32.Gough DJ, Messina NL, Hii L, Gould JA, Sabapathy K, Robertson AP, Trapani JA, Levy DE, Hertzog PJ, Clarke CJ, Johnstone RW. 2010. Functional crosstalk between type I and II interferon through the regulated expression of STAT1. PLoS Biol 8:e1000361. doi: 10.1371/journal.pbio.1000361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Takaoka A, Mitani Y, Suemori H, Sato M, Yokochi T, Noguchi S, Tanaka N, Taniguchi T. 2000. Cross talk between interferon-gamma and -alpha/beta signaling components in caveolar membrane domains. Science 288:2357–2360. doi: 10.1126/science.288.5475.2357. [DOI] [PubMed] [Google Scholar]
- 34.Shi L, Perin JC, Leipzig J, Zhang Z, Sullivan KE. 2011. Genome-wide analysis of interferon regulatory factor I binding in primary human monocytes. Gene 487:21–28. doi: 10.1016/j.gene.2011.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Langlais D, Barreiro LB, Gros P. 2016. The macrophage IRF8/IRF1 regulome is required for protection against infections and is associated with chronic inflammation. J Exp Med 213:585–603. doi: 10.1084/jem.20151764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bauman DR, Bitmansour AD, McDonald JG, Thompson BM, Liang G, Russell DW. 2009. 25-Hydroxycholesterol secreted by macrophages in response to Toll-like receptor activation suppresses immunoglobulin A production. Proc Natl Acad Sci U S A 106:16764–16769. doi: 10.1073/pnas.0909142106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Grayson MH, Cheung D, Rohlfing MM, Kitchens R, Spiegel DE, Tucker J, Battaile JT, Alevy Y, Yan L, Agapov E, Kim EY, Holtzman MJ. 2007. Induction of high-affinity IgE receptor on lung dendritic cells during viral infection leads to mucous cell metaplasia. J Exp Med 204:2759–2769. doi: 10.1084/jem.20070360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tarakanova VL, Leung-Pineda V, Hwang S, Yang C-W, Matatall K, Basson M, Sun R, Piwnica-Worms H, Sleckman BP, Virgin HW. 2007. Gamma-herpesvirus kinase actively initiates a DNA damage response by inducing phosphorylation of H2AX to foster viral replication. Cell Host Microbe 1:275–286. doi: 10.1016/j.chom.2007.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mounce BC, Tsan FC, Droit L, Kohler S, Cirillo LA, Tarakanova VL. 2011. Gammaherpesvirus gene expression and DNA synthesis are facilitated by viral protein kinase and histone variant H2AX. Virology 420:73–81. doi: 10.1016/j.virol.2011.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]