

ABSTRACT
Embryonic carcinoma (EC) cells are malignant counterparts of embryonic stem (ES) cells and serve as useful models for investigating cellular differentiation and human embryogenesis. Though the susceptibility of murine EC cells to retroviral infection has been extensively analyzed, few studies of retrovirus infection of human EC cells have been performed. We tested the susceptibility of human EC cells to transduction by retroviral vectors derived from three different retroviral genera. We show that human EC cells efficiently express reporter genes delivered by vectors based on human immunodeficiency virus type 1 (HIV-1) and Mason-Pfizer monkey virus (M-PMV) but not Moloney murine leukemia virus (MLV). In human EC cells, MLV integration occurs normally, but no viral gene expression is observed. The block to MLV expression of MLV genomes is relieved upon cellular differentiation. The lack of gene expression is correlated with transcriptional silencing of the MLV promoter through the deposition of repressive histone marks as well as DNA methylation. Moreover, depletion of SETDB1, a histone methyltransferase, resulted in a loss of transcriptional silencing and upregulation of MLV gene expression. Finally, we provide evidence showing that the lack of MLV gene expression may be attributed in part to the lack of MLV enhancer function in human EC cells.
IMPORTANCE Human embryonic carcinoma (EC) cells are shown to restrict the expression of murine leukemia virus genomes but not retroviral genomes of the lentiviral or betaretroviral families. The block occurs at the level of transcription and is accompanied by the deposition of repressive histone marks and methylation of the integrated proviral DNA. The host machinery required for silencing in human EC cells is distinct from that in murine EC cell lines: the histone methyltransferase SETDB1 is required, but the widely utilized corepressor TRIM28/Kap1 is not. A transcriptional enhancer element from the Mason-Pfizer monkey virus can override the silencing and promote transcription of chimeric proviral DNAs. The findings reveal novel features of human EC gene regulation not present in their murine counterparts.
KEYWORDS: DNA methylation, enhancer, chromatin immunoprecipitation, histones, repressor
INTRODUCTION
Human embryonic carcinoma (EC) cells are derived from teratocarcinomas, cancers of transformed germ cells (reviewed in reference 1). EC cells are generally considered to be malignant counterparts of embryonic stem (ES) cells and serve as useful models for investigating cell differentiation and human embryogenesis (2, 3). EC cells offer several experimental advantages over ES cells, including easy maintenance, growth without feeder layers, and resistance to spontaneous differentiation. They express useful cell surface markers, such as stage-specific embryonic antigen-3 (SSEA-3) (4) and SSEA-4 (5), that allow their undifferentiated state to be readily identified. Several lines of human EC cells are widely used, including NTERA2, whose global gene expression is highly similar to that of undifferentiated human ES cells (6). Upon exposure to retinoic acid, NTERA2 cells undergo extensive differentiation in vitro to cells of neuroectodermal lineages (7, 8), making them a useful ES cell surrogate for studying neuronal differentiation in vitro. Another commonly used cell line is the 2102Ep line, which was derived from a primary human testicular teratocarcinoma (9). Unlike NTERA2, 2102Ep cells do not differentiate in response to retinoic acid but do undergo spontaneous differentiation at low cell densities (7, 9, 10).
Retroviral transduction of human embryonic cells represents a powerful means of introducing genetic modifications in these cells. This is useful for promoting differentiation of embryonic cells along a specific developmental lineage (11) or correcting congenital mutations in a gene of interest (12). However, one obstacle with the use of retroviral vectors in embryonic cells is the transcriptional silencing of various promoter elements driving transgene expression. For example, the cytomegalovirus (CMV) promoter is potently silenced in human ES cells, while the phosphoglycerate kinase (PGK) promoter shows much higher expression (13). Why certain promoters show higher expression than others in human embryonic cells is an area of active investigation.
Much of our knowledge of ES-specific transcriptional silencing of retroviral promoters comes from studies using mouse embryonic cells. Mouse ES or EC cells are resistant to infection with Moloney murine leukemia virus (MLV) (14–16). In mouse embryonic cells, reverse transcription and proviral integration proceed normally, but viral transcription is repressed and no viral gene products can be detected. Several mechanisms are likely involved in the transcriptional silencing, including the absence of enhancer proteins recognizing binding sites in the viral long terminal repeat (LTR) (14, 15) and recruitment of trans-acting transcriptional repressors leading to chromatin modification and de novo DNA methylation of the viral promoter (16–18). One critical site for transcriptional silencing, termed the repressor binding site (RBS), shows extensive overlap (17 or 18 bp) with the primer binding site (PBSpro) of MLV (19–21), which in the context of the viral RNA genome is the site of annealing of proline tRNA used as primer for reverse transcription. The RBS by itself is sufficient to induce potent transcriptional repression of reporter constructs in mouse EC cells, irrespective of its orientation or position (22, 23). Electrophoretic mobility shift assays (EMSAs) using RBS as the probe demonstrate the presence of stem cell-specific nuclear factors. Single-base-pair mutations in the RBS are sufficient to abolish nuclear factor binding and thereby restore viral gene expression (22, 23). These findings have allowed the characterization of the stem cell-specific trans-acting transcriptional repressor complex, which contains TRIM28 (also known as KAP-1 or Tif1-beta), a known transcriptional corepressor (24–26), and Krüppel associated box (KRAB) zinc finger protein 809 (ZFP809) (27), a DNA-binding zinc finger transcription factor. The current model for PBS-mediated silencing consists of the binding of PBSpro by ZFP809 and the subsequent recruitment of TRIM28 to the provirus via the ZFP809 KRAB box domain. TRIM28 then induces the deposition of repressive histone modifications by recruiting additional chromatin modifiers such as the histone H3K9 methyltransferase SETDB1 (28), heterochromatin-associated protein 1 (HP1) (24), and the NURD histone deacetylase complex (29). Together, the ensuing histone and DNA methylation of the proviral DNA ensures transcriptional silencing. Epigenetic silencing of proviral DNA prevents expression of viral genes and blocks production of new virions that could otherwise disrupt genomic stability through insertional mutagenesis. Furthermore, deletion of Zfp809 (30), Trim28 (31), or Setdb1 (32) in mouse ES cells leads to the loss of H3K9 trimethylation (H3K9me3) marks on certain families of endogenous retroviruses (ERVs), resulting in transcriptional activation of these elements.
While much is known about retroviral silencing in mouse ES cells, retroviral infection of human embryonic cells has not been extensively characterized. Long interspersed element-1 (LINE-1 or L1) retrotransposition has been shown to be rapidly and efficiently silenced following integration in various human EC cells (33). However, the susceptibility of human EC cells to infection by exogenous retroviruses has not been examined. In the present study, we examined the susceptibility of human EC cells to infection by members of several retroviral genera. Our findings revealed that human EC cells are susceptible to transduction by retroviral vectors derived from human immunodeficiency virus type 1 (HIV-1) and Mason-Pfizer monkey virus (M-PMV), but not MLV, and that the MLV block is reversible by cellular differentiation. The block to MLV infection occurs postintegration and is associated with transcriptional silencing of the MLV promoter through the deposition of repressive histones as well as DNA methylation. Moreover, depletion of SETDB1, a histone methyltransferase responsible for the deposition of H3K9 trimethylation (H3K9me3) marks, resulted in upregulation of viral gene expression. Lastly, we present evidence showing that the lack of MLV gene expression may be attributed in part to the lack of MLV enhancer function in human EC cells. Together, our data provide insights into the susceptibility of human embryonic cells to retroviral infections and offer potential strategies to circumvent transgene silencing in human embryonic cells.
RESULTS
Characterizing retroviral transduction of human embryonic carcinoma cells.
We first assessed the ability of human EC cells to express reporter genes delivered by vectors based on HIV-1, M-PMV, and MLV. These viruses were chosen to represent lenti-, beta-, and gammaretroviruses, respectively. Vesicular stomatitis virus glycoprotein G (VSV-G)-pseudotyped single-round reporter viruses expressing green fluorescent protein (GFP) (M-PMV and MLV) or mCherry (HIV-1) were produced in 293T cells and then used to infect two different human EC cell lines, i.e., NTERA2 and 2102Ep, or HeLa cells, a differentiated human cell line. Flow cytometry analysis was carried out 48 h after infection to monitor reporter gene expression and determine transduction efficiency. As expected, HeLa cells were highly permissive to all three viruses (Fig. 1A and B). In contrast, while HIV-1 and M-PMV transduced both human EC cell lines efficiently, the MLV virus vector transduced the lines to express GFP at very low efficiency, with approximately 50-fold fewer cells scored as GFP positive, and the GFP intensity in the positive population was very low (Fig. 1A and B).
FIG 1.

Characterizing retroviral transduction of human embryonic carcinoma cells. (A) Flow cytometry analysis of HeLa or human EC cells (NTERA2 and 2102Ep) infected with VSV-G-pseudotyped single-round MLV-GFP, M-PMV–GFP, or HIV-1–mCherry reporter viruses. Data shown are from day 2 postinfection. The y axis shows side scatter (SSC); the x axis shows GFP or mCherry intensity. One representative experiment of three independent experiments is shown. (B) Quantification of the results from panel A from multiple experiments. The logarithmic graph shows the percentage of GFP+ or mCherry+ cells 2 days postinfection. Results shown are means ± standard errors of the mean (SEM) from three independent experiments. (C) Determination of viral DNA copy number. Cell lines shown were infected with VSV-G-pseudotyped MLV-GFP reporter viruses and propagated for 14 days in culture. Viral integration was determined by performing real-time quantitative PCR using primers specific for GFP and Tert on genomic DNA isolated from infected cells. The extent of viral integration (relative viral copy number) was quantified using the 2−ΔΔCT method by normalizing the GFP signal to the Tert gene. Results shown are means ± SEM from three independent experiments performed in duplicate. (D) Characterizing MLV LTR activity in HeLa and human EC cells. Individual cell lines were cotransfected with LTR firefly luciferase reporter constructs and a Renilla luciferase control plasmid for 36 h. Relative luciferase expression was calculated by dividing the firefly luciferase signal by the Renilla luciferase signal, and the resulting ratio was then normalized to the signal obtained from the same cells transfected with the M-PMV LTR luciferase construct (set to 1). Results shown are means ± SEMs from three independent experiments.
Our single-round reporter viruses monitor all the early events of infection, including reverse transcription, nuclear entry, integration, and LTR-driven reporter gene expression. To address whether the MLV block in human EC cells occurred pre- or postintegration, infected cell lines were passaged for 14 days in culture, followed by quantification of integrated proviral DNA using PCR primers against GFP. This length of passage of infected cells allows for the loss of all unintegrated viral DNAs and thus an accurate determination of the integrated proviral DNAs. The levels of integrated MLV proviral DNA were comparable between HeLa and human EC cells (Fig. 1C), indicating normal viral integration and therefore suggesting a postintegration block to MLV infection.
We next addressed whether the block to MLV transduction occurs at the level of LTR-driven reporter gene expression. HeLa or human EC cell lines were transfected with plasmid DNAs encoding firefly luciferase expressed from either MLV or M-PMV LTRs, lysates were prepared after 36 h, and levels of luciferase activity were assayed. Whereas the MLV and M-PMV LTRs directed high, comparable levels of luciferase expression in HeLa cells, the MLV LTR expressed an approximately 25- to 43-fold lower level of luciferase activity than the M-PMV LTR, in both human EC cell lines (Fig. 1D). Taken together, these results suggest that human EC cells exhibit a postintegration block to MLV infection at the level of viral gene expression.
The block to MLV infection is relieved by differentiation of human EC cells.
To test if the MLV block is relieved by differentiation, NTERA2 cells, previously shown to be sensitive to retinoic acid-induced differentiation (7), were grown in the presence or absence of retinoic acid for 14 days, followed by transduction with MLV-GFP reporter viruses and flow cytometry analysis (Fig. 2A). Retinoic acid treatment resulted in the loss of SSEA4 staining, a marker of pluripotency. In the SSEA4-negative cells, a 20-fold increase in GFP-positive (GFP+) cells was observed, indicative of increased permissiveness to MLV infection (Fig. 2B).
FIG 2.
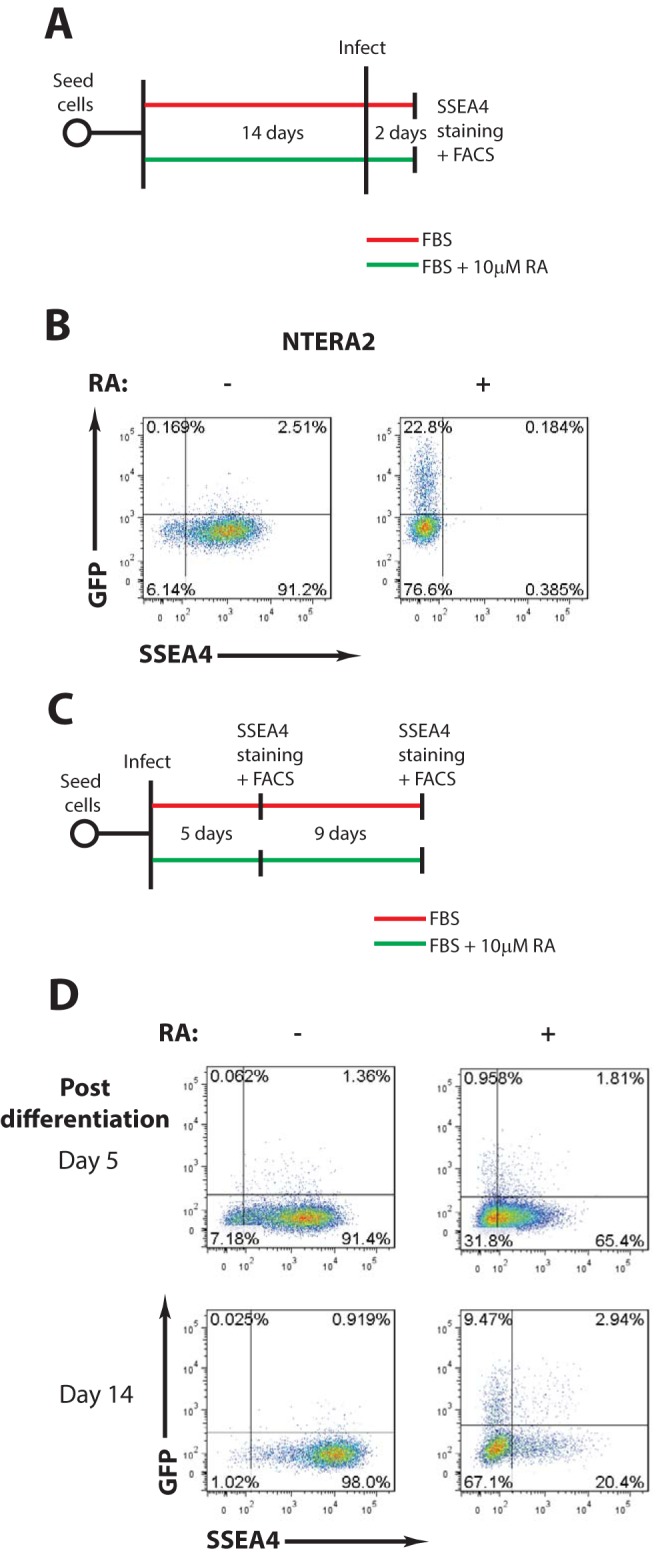
The block to MLV infection is relieved by differentiation of human EC cells (A) Scheme of experimental setup. (B) Flow cytometry analysis of undifferentiated and retinoic acid-differentiated NTERA2 cells infected with VSV-G-pseudotyped MLV-GFP reporter viruses. Results shown are at day 2 postinfection. The x axis shows SSEA4 staining (a marker of pluripotency); the y axis shows GFP intensity. One representative experiment of three independent experiments is shown. (C) Scheme of experimental setup. (D) NTERA2 cells were infected with VSV-G-pseudotyped MLV-GFP reporter viruses. At 1 day postinfection, cells were cultured with or without retinoic acid to induce differentiation. Flow cytometry analysis was carried out at the indicated times postdifferentiation to monitor MLV gene expression. The x axis shows SSEA4 staining (a marker of pluripotency); the y axis shows GFP intensity. One representative experiment of two independent experiments is shown. RA, retinoic acid; FACS, fluorescence-activated cell sorting.
The above finding raised the possibility that NTERA2 cells infected in a nonpermissive state would become permissive upon differentiation of infected cells. To test this, NTERA2 cells were infected with MLV-GFP for 24 h and then cultured for various times with or without retinoic acid. The cells were examined on days 5 and 14 by flow cytometry to monitor SSEA4 and GFP expression (Fig. 2C). At 5 days postdifferentiation, retinoic acid treatment caused a partial loss in SSEA4 staining and a minor increase in GFP+ cells, suggesting that at this time point, the cells have not yet achieved complete differentiation and that MLV gene expression is still largely repressed. By 14 days postdifferentiation, complete loss of SSEA4 staining was accompanied by a 12-fold increase in GFP+ cells, indicating the completion of differentiation and enhanced MLV gene expression (Fig. 2D). These results suggest that the block to MLV gene expression can be reversed either by infection of differentiated NTERA2 cells or by differentiation of infected NTERA2 cells soon after infection.
MLV silencing in human EC cells is associated with deposition of repressive chromatin marks and DNA methylation of the viral promoter.
We next asked whether MLV silencing in human EC cells is associated with the deposition of repressive histone marks on the proviral DNA. Chromatin immunoprecipitation (ChIP) experiments were conducted on chromatin isolated from infected NTERA2 cells using antibodies that recognize repressive histone marks (H3K9me3 and H3K27me3) or active histone marks (H3Ac) (Fig. 3A). The specificity of these antibodies for ChIP was confirmed using primers specific for promoter regions of genes encoding beta-globin (positive control for H3K9me3), myelin transcriptional factor 1 (MYT-1; positive control for H3K27me3), and glyceraldehyde-3-phosphate dehydrogenase (GAPDH; positive control for H3Ac). In MLV-infected NTERA2 cells, the LTR region of the proviral DNA showed high levels of repressive H3K9me3 and H3K27me3 marks and low levels of active H3Ac mark (Fig. 3A), consistent with the very low MLV gene expression in these cells. Importantly, retinoic acid-induced differentiation of NTERA2 cells resulted in a marked decrease in H3K9me3 and H3K27me3 marks and an increase in H3Ac marks, correlating with the markedly increased gene expression upon differentiation (Fig. 2B and D). Similar patterns of chromatin modifications on the MLV LTR were seen in another human EC cell line, 2102Ep (Fig. 3B), as well as F9 mouse EC cells (Fig. 3C). These results suggest that in both human and mouse EC cells, multiple histone methyltransferases are active in the deposition of these repressive marks. In contrast, proviral DNA from MLV-infected HeLa cells showed an abundance of the H3Ac marks but lacked both H3K9me3 and H3K27me3 marks (Fig. 3D), consistent with the high MLV gene expression in these cells. As an additional control, ChIP analysis of 2102Ep cells infected with M-PMV, which showed high gene expression in these cells (Fig. 1A and D), revealed an abundance of the active H3Ac marks and much lower levels of repressive H3K9me3 and H3K27me3 marks on the proviral DNA (Fig. 3E).
FIG 3.

MLV silencing in human EC cells is associated with deposition of repressive chromatin marks. (A) Undifferentiated NTERA2 cells or retinoic acid (RA)-induced differentiated NTERA2 cells infected with MLV-GFP for 3 days were subjected to ChIP analysis using the indicated antibodies. ChIP data are presented as the percentage of input DNA. Results shown are means ± standard deviations (SDs) from two independent experiments performed in duplicate. (B) 2102Ep cells infected with MLV-GFP for 5 days were subjected to ChIP analysis using the indicated antibodies. ChIP data are presented as the percentage of input DNA. Results shown are means ± SDs from three independent experiments performed in duplicate. (C) Experiment similar to that in panel A but performed in F9 mouse EC cells. (D) Experiment similar to that in panel A but performed in HeLa cells. (E) 2102Ep cells infected with M-PMV–GFP for 5 days were subjected to ChIP analysis as described above. qPCR, quantitative PCR.
To assess whether repressive histone marks were accompanied by DNA methylation of the proviral DNA, the various cell lines were infected and passaged for 14 days in culture, followed by DNA methylation analysis of the proviral DNA using bisulfite sequencing. Using primers targeting the MLV 5′ LTR PBS region, we observed extensive CpG methylation (80 to 90%) in both human EC and F9 cells, while little to no DNA methylation was observed in HeLa cells (Fig. 4A and B). This finding correlates with the degree of retroviral gene expression in these cells and suggests the possible involvement of de novo DNA methylation in mediating MLV silencing.
FIG 4.
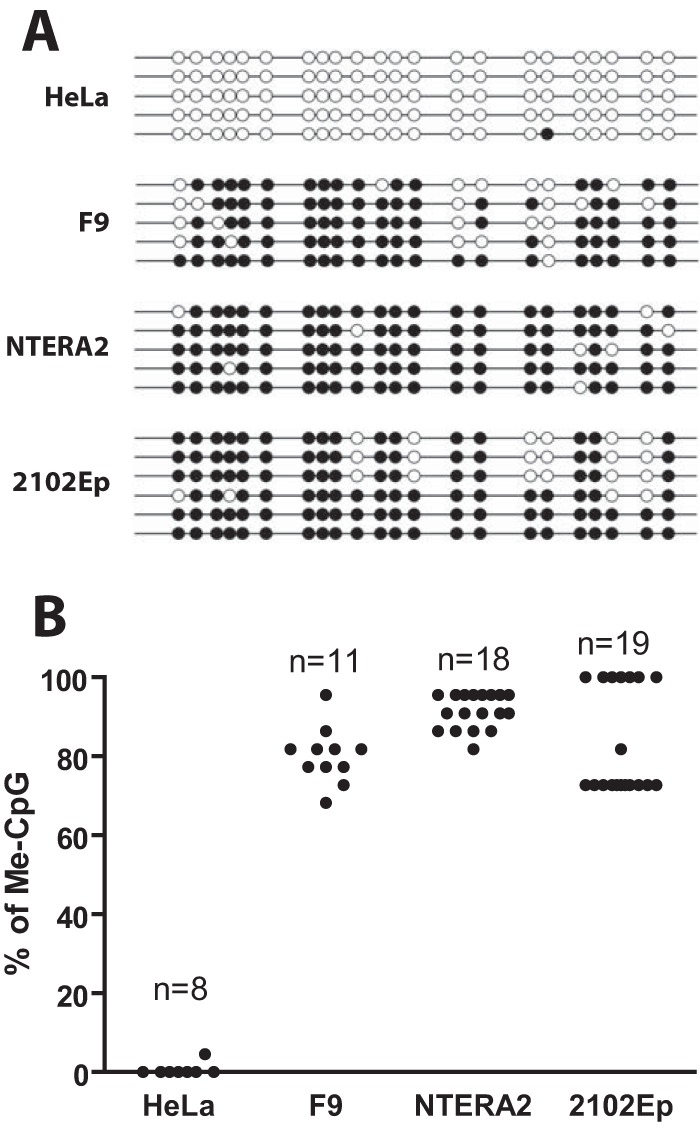
MLV silencing in human EC cells is associated with DNA methylation of the viral promoter. (A) At 14 days postinfection, bisulfite sequencing analysis of the 5′ LTR of the MLV proviral genomic DNA. Representative clones of proviral DNA genome are shown for each infected cell line. Open circles represent unmethylated CpG, filled circles represent methylated CpG (Me-CpG). (B) Quantification of the results from panel A from multiple MLV proviral genomic DNA clones for each cell line.
Depletion of SETDB1 but not TRIM28 relieves MLV transcriptional repression in human EC cells.
Trimethylation of histone H3 is associated with gene silencing and mediated by histone lysine methyltransferase SETDB1, which is recruited to target DNA via its interaction with TRIM28 (28). Given the abundance of H3K9me3 marks on the proviral DNA in human EC cells, we next determined whether TRIM28 and/or SETDB1 plays a role in transcriptional repression of MLV. 2102Ep or F9 cells were stably depleted of endogenous TRIM28 or SETDB1 expression using RNA interference (RNAi) (Fig. 5C), transduced by MLV-GFP, and analyzed by flow cytometry analysis 2 days later (Fig. 5A and B). Depletion of SETDB1 in 2102Ep cells resulted in a 6- to 10-fold increase in the number of GFP+ cells compared to that of the scrambled short hairpin RNA (shRNA) control (Fig. 5A and B). Surprisingly, in contrast, depletion of TRIM28 in 2102Ep cells had no effect. Depletion of either TRIM28 or SETDB1 in F9 cells resulted in a robust increase in the efficiency of MLV transduction, in accordance with previous reports (25, 31, 32).
FIG 5.

Depletion of SETDB1 but not TRIM28 relieves MLV transcriptional repression in human EC cells. (A) Flow cytometry analysis of indicated 2102Ep or F9 cells infected with VSV-G-pseudotyped MLV-GFP reporter viruses; this is at 2 days postinfection. The y axis shows side scatter (SSC); the x axis shows GFP intensity. Results of one representative experiment of three independent experiments are shown. (B) Quantification of the results from panel A from multiple experiments. Results shown are means ± SEM from three independent experiments. (C) Lysates prepared from 2102Ep or F9 cells stably transduced with either scrambled shRNA or TRIM28- or SETDB1-specific shRNAs were subjected to Western blotting as indicated. (D) 2102Ep cells infected with MLV-GFP for 3 days were subjected to ChIP analysis using antibodies against H3K9me3. ChIP data are presented as the percentage of input DNA. Results shown are means ± SDs from two independent experiments performed in duplicate. (E) Experiments similar to those in panel D but using antibodies against acetylated histone H3 (H3Ac).
We next examined the effect of SETDB1 knockdown on H3K9me3 of the viral promoter. As expected, ChIP experiments conducted on chromatin isolated from SETDB1 knockdown cells showed a decrease in repressive H3K9me3 marks compared to those in the scrambled shRNA control (Fig. 5D), consistent with the increase in MLV gene expression observed in these cells (Fig. 5A). Surprisingly, for reasons unclear, we did not see an increase in H3Ac marks in SETDB1-depleted cells (Fig. 5E). Nevertheless, these data indicate that SETDB1, but not TRIM28, is required for the silencing of MLV in human embryonic cells.
MLV gene expression in human EC cells is rescued by strong transcriptional enhancer sequences derived from M-PMV.
One explanation for the poor transduction of EC cells by MLV vectors would be the presence of a repressive trans-acting factor(s) in human EC cells that targets negative DNA sequences in the MLV promoter. In this case, the deletion of negative DNA sequences from the MLV LTR could restore gene expression. To test this possibility, luciferase reporter constructs containing either 5′ or 3′ deletions of the MLV promoter were introduced into HeLa or 2102Ep cells by transfection. After 36 h, cells were harvested and lysates were analyzed for luciferase activity. The wild-type MLV LTR luciferase construct showed a 60-fold reduction in luciferase activity in 2102Ep cells compared to that in HeLa cells (Fig. 6A). None of the various truncation mutants of the MLV LTR exhibited significantly higher MLV gene expression (Fig. 6A), suggesting the absence of obvious negative repressor binding sites responsible for MLV silencing in human EC cells.
FIG 6.

MLV gene expression in human EC cells is rescued by strong transcriptional enhancer sequences derived from M-PMV. (A) Functional analysis of MLV LTR sequences using mutant LTR reporter constructs. HeLa or 2102Ep cells were cotransfected with LTR firefly luciferase reporter constructs and a Renilla luciferase control plasmid for 36 h. Relative luciferase expression was calculated by dividing the firefly luciferase signal by the Renilla luciferase signal, and the resulting ratio was then normalized to the signal obtained from the same cell transfected with the M-PMV LTR luciferase construct (set to 1). Results shown are means ± SEMs from at least three independent experiments. NCR, negative-control region in MLV LTR; DR, direct repeat in MLV LTR (i.e., MLV transcriptional enhancer). Student's t test was used for statistical analysis. **, P < 0.01; *, P < 0.05 (compared to either 2102Ep or HeLa cells transfected with M-PMV LTR luciferase). (B) Assay similar to that outlined in for panel A but using chimeric LTR reporter constructs in which DNA sequences spanning the DR region of the MLV LTR were replaced with sequences from the U3 region of the M-PMV LTR. Student's t test was used for statistical analysis. **, P < 0.01; *, P < 0.05 (compared to either 2102Ep or HeLa cells transfected with M-PMV LTR luciferase). (C) Functional analysis of MLV LTR sequences using mutant LTR reporter constructs. Undifferentiated or retinoic acid (RA)-induced differentiated NTERA2 cells were cotransfected with LTR firefly luciferase reporter constructs and a Renilla luciferase control plasmid for 36 h. Relative luciferase expression was calculated as outlined above. Results shown are means ± SDs from two independent experiments. DR, direct repeat in MLV LTR (i.e., MLV transcriptional enhancer).
An alternative explanation for the low MLV gene expression in human EC cells is the lack of transcriptional activators capable of recognizing MLV enhancer sequences. Others have shown that replacement of the MLV enhancer sequence with DNA fragments from the polyomavirus enhancer resulted in enhanced MLV expression in F9 mouse embryonic cells (14). Using a similar approach, we asked whether LTR sequences derived from M-PMV, which show high gene expression in human EC cells, could rescue the low MLV gene expression. A chimeric LTR luciferase construct was generated by replacing the MLV enhancer sequence with a DNA fragment derived from the U3 region of the M-PMV LTR (position −240 to −36). This manipulation restored high levels of gene expression to the modified MLV LTR in 2102Ep cells (Fig. 6B). We further confirmed the above observations in another human EC cell line, NTERA2 cells (Fig. 6C). As with undifferentiated 2102Ep cells, the MLV LTR luciferase construct in undifferentiated NTERA2 cells also showed an ∼10-fold reduction in luciferase activity compared to that of M-PMV LTR luciferase. Differentiation of NTERA2 cells with retinoic acid caused a marked increase in MLV LTR activity. Importantly, as seen in 2102Ep cells, the very low MLV LTR activity could be rescued by replacing the MLV enhancer with a DNA fragment derived from the M-PMV U3 region (Fig. 6C).
To define the minimal M-PMV DNA sequence required to restore activity to the MLV LTR, we carried out 5′ and 3′ LTR truncation analysis of the M-PMV U3 DNA. The ability of M-PMV U3 DNA to transactivate the MLV LTR was significantly reduced upon removal of a 90-bp fragment from position −120 to −36 (Fig. 6B). Examination of this 90-bp fragment revealed two putative DNA binding sites for transcription factor E2F. Deletion of just these two E2F binding sites significantly reduced the ability of the M-PMV U3 DNA to transactivate the MLV LTR (Fig. 6B). Together, these results show that replacement of the MLV enhancer with a DNA enhancer containing E2F binding sites effectively rescues MLV LTR activity and suggest that the low MLV gene expression in human EC cells may be due at least in part to deficiency in a specific transactivator function recognizing the E2F binding site.
DISCUSSION
Retroviral transduction of human embryonic cells is a powerful means of introducing genetic changes in these cells. This technology can help expand our basic understanding of stem cell biology and can be utilized for gene therapy. In this study, we characterized the susceptibility of human EC cells to transduction by retroviruses from different families. In human EC cells, the lentivirus HIV-1 and betaretrovirus M-PMV can successfully integrate viral DNA and induce robust gene expression (Fig. 1A). In contrast, the gammaretrovirus MLV is also able to successfully integrate its viral DNA but is unable to express genes driven by the promoter sequences of the LTR (Fig. 1A to C). The lack of MLV gene expression was recapitulated upon transfection of human EC cells with plasmid DNA reporter constructs driven by the MLV LTR (Fig. 1D).
The restriction of retroviral genomes in mouse ES cells has been suggested to have been selected during evolution as a host defense mechanism against de novo infection by exogenous retroviruses as well as against activation of endogenous retroelements (34–36; for reviews, see references 37 and 38). Transcriptional silencing of MLV in mouse embryonic cells is mediated in part by the binding of stem cell-specific transcription factor ZFP809 (27) to the PBS sequence of integrated proviral DNA, leading to the downstream recruitment of transcriptional corepressor complexes containing TRIM28 (25, 31), SETDB1 (32), HP1 (24), and EBP1 (39) to induce gene silencing via the deposition of repressive histone marks such as H3K9me3 on the proviral DNA. A parallel pathway of silencing is mediated by YY1 binding to proviral DNA and also initiating histone modification and DNA methylation (40). Deletion of either TRIM28 (25, 31) or SETDB1 (32) in mouse ES cells resulted in a loss of H3K9me3 marks and upregulation of gene expression from both exogenous MLV and endogenous retroelements. In human embryonic cells, we also observed the association of the repressive histone modifications H3K9me3 and H3K27me3 with the MLV DNA, suggesting the involvement of at least two independent histone lysine methyltransferases in MLV silencing (Fig. 3). As seen in mouse ES cells, depletion of SETDB1, an enzyme involved in the deposition of H3K9me3 marks, led to a 6- to 10-fold increase in MLV gene expression in human 2102Ep cells (Fig. 5A and B). This observation correlates with a 3-fold reduction in H3K9me3 marks on the proviral DNA (Fig. 5D). Unlike the situation in mouse ES cells, however, TRIM28 depletion had no apparent effect on viral gene expression in the human cells (Fig. 5A and B). Human embryonic cells seem to lack PBS-mediated silencing, because deletion of the PBS sequence from the MLV LTR failed to upregulate MLV gene expression in human cells (Fig. 6A). Furthermore, there is no strict ZFP809 ortholog in human cells (27). It is nevertheless surprising that TRIM28 depletion had so little impact on MLV expression.
TRIM28 is clearly functional and plays an important regulatory role in human ES cells. TRIM28 has been recently implicated in the silencing of human endogenous retroviruses (HERVs) and retroelement DNAs in human ES cells (35). ChIP sequencing (ChIP-seq) analysis demonstrated the binding of TRIM28 to multiple classes of retroelements, including hominoid-restricted SINE-VNTR-Alu (SVAs), long interspersed elements (LINEs or L1s), and Alu repeats. Moreover, a fraction of TRIM28-bound endogenous retroelements also showed SETDB1 occupancy and the presence of H3K9me3 repressive marks. Importantly, depletion of TRIM28 in human ES cells resulted in the loss of H3K9me3 marks and the gain of H3K4me1 enhancer marks at some of the loci, indicating the importance of TRIM28 in maintaining silencing of these retroelements in human ES cells. These observations show that at least a subset of the players involved in the epigenetic suppression of endogenous retroelement expression are also utilized to protect human embryonic cells from de novo infection by exogenous retroviruses.
Although our studies did not reveal a significant role for TRIM28, we did observe large effects of the knockdown of SETDB1. It is not clear how SETDB1 is recruited to the MLV DNA in the absence of TRIM28. One possibility is that unidentified transcriptional corepressors might target SETDB1 to specific sequences in the MLV LTR. We tested this hypothesis using LTR truncation mutants in an attempt to identify novel repressive DNA sequences in the MLV LTR that might be recognized by human embryonic cell-specific ZFPs, but we failed to identify obvious repressive DNA sequences in the viral LTR (Fig. 6A). Instead, complete rescue of MLV gene expression was attained by replacing the MLV enhancer DNA with DNA sequences derived from the M-PMV LTR, which showed high levels of expression in two human embryonic cell lines, 2102Ep (Fig. 6B) and NTERA2 (Fig. 6C). This observation supports the notion that the block to MLV gene expression is due in part to the absence of a positive factor. In the case of the M-PMV LTR, most of the enhancer activity is contained within two E2F DNA binding sites, and deletions of these sites greatly reduced the ability of M-PMV LTR DNA to activate MLV gene expression (Fig. 6B). Based on these observations, we postulate that the inactivity of MLV LTR in human embryonic cells may be due in large part to the absence of a compatible transcriptional activator binding to the viral promoter.
How are repressive histone marks and ultimately DNA methylation induced on MLV DNA in the absence of obvious negative DNA elements? One notion is that in human embryonic cells, repressive histone marks as well as DNA methylation may be imposed on incoming viral promoters with poor transcriptional enhancer activity by default, perhaps soon after introduction of the viral DNA into the cell. Such “default” silencing is apparently overcome by viral LTRs containing strong, compatible DNA enhancer elements such as those in the M-PMV LTR (Fig. 6B). Since MLV silencing is associated with the deposition of repressive H3K9 and H3K27 trimethylation marks (Fig. 3B), we attempted to test the ability of M-PMV enhancers to decrease the repressive chromatin marks on MLV DNA in human EC cells infected with chimeric LTR viruses. During retroviral reverse transcription, DNA copies of the 5′ LTR are derived from sequences from the 3′ end of the viral RNA. Chimeric LTR viruses were generated by replacing the U3 region of the MLV 3′ LTR with M-PMV DNA sequences. This manipulation ensures that M-PMV DNA is present in both copies of the viral LTR upon integration. Chimeric LTR viruses were produced in 293T cells and used to infect human EC cells. Unfortunately, we found the titers of the chimeric LTR viruses to be 20- to 100-fold lower than those of wild-type MLV, making this approach impossible.
The refractory nature of undifferentiated human EC cells to MLV transduction raised the question of whether these cells become permissive to MLV transduction upon differentiation. Differentiation of NTERA2 cells before MLV infection indeed resulted in efficient MLV expression (Fig. 2B), much as is seen in mouse EC cells (41). Moreover, the initiation of differentiation of NTERA2 cells shortly (1 day) following infection also resulted in good MLV expression (Fig. 2D). The requirement for cellular differentiation to achieve efficient MLV gene expression is similar to the behavior seen with reversal of silencing of L1 retrotransposons in differentiated human EC cells (33). Future studies may lead to identification of the missing transcriptional activator(s) in human embryonic cells that appears upon differentiation.
In closing, we note that some human embryonic cells may exhibit other blocks to retroviral infection besides the silencing of retroviral transcription. For example, preliminary experiments using another human EC cell line, 833KE cells (42), showed a significant block at very early stages of infection, before viral DNA synthesis (data now shown). These results highlight the fact that human EC cell lines are not all alike and likely represent different stages of arrested development.
MATERIALS AND METHODS
Cell lines, transfections, and antibodies.
Cell lines, including human embryonic kidney 293T (HEK-293T; CRL-1573), HeLa (CCL-2), and F9 embryonic carcinoma (CRL-1720) cells were purchased from the American Type Culture Collection. Human EC cell lines, including NTera2D1, 2102Ep, and 833KE cells, were a gift from John Moran (University of Michigan Medical School). All cells were cultured in Dulbecco's modified Eagle's Medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum (FBS), 2 mM glutamine, 1,000 U/ml penicillin, and 100 mg/ml streptomycin. All cells were maintained in a 37°C incubator with 5% CO2. Transient-transfection studies in HeLa and HEK-293T cells were carried out using polyethylenimine (PEI), and human EC cell line transfections were carried out using Lipofectamine 2000 (Life Technologies) according to the manufacturer's instructions. Antibodies used in this study include Alexa Fluor 647-conjugated anti-human/mouse SSEA4 (BioLegend), anti-trimethyl-histone H3 (Lys9) (07-442; Millipore), anti-trimethyl-histone H3 (Lys27) (07-449; Millipore), anti-acetyl-histone H3 (06-599; Millipore), anti-TRIM28 (clone 20C1; Abcam), and rabbit IgG antibody (sc-2027; Santa Cruz).
Plasmids.
pCMV-intron expresses gag and pol from NB-tropic MLV (43). pMD.G expresses the vesicular stomatitis virus (VSV) envelope glycoprotein. pNCA-GFP is a replication-defective single-round MLV vector described previously (44). pSARM-EGFP is a replication-defective single-round M-PMV vector in which the env gene has been replaced with enhanced GFP (EGFP) as described previously (45). pNL4.3-mCherry is a replication-defective single-round HIV-1 vector described previously (46). Various LTR luciferase reporter constructs were constructed by PCR amplification of the desired LTR sequences, followed by ligation cloning into the NheI-XhoI restriction sites of pGL3-Basic vector (Promega). Cloning primers are available upon request. All constructs were verified by DNA sequencing. shRNA containing pLKO.1 lentiviral vectors was purchased from Thermo Scientific (Pittsburg, PA).
Retroviral transduction assay and flow cytometry analysis.
NB-tropic MLV-GFP reporter viruses were produced by 293T cell transfection with 8 μg of pNCA-GFP, 4 μg of pCMV-intron, and 4 μg of pMD.G DNAs using PEI. M-PMV–GFP reporter viruses were produced by transfection of 293T cells with 8 μg of pSARM-EGFP and 8 μg of pMD.G. HIV-1–mCherry reporter viruses were produced by transfection of 293T cells with 8 μg of pNL4.3-mCherry and 8 μg of pMD.G. All reporter viruses were harvested 48 h later, filtered (0.45-μm pore size), and used directly for transduction assays described previously (47). Forty-eight hours postinfection, cells were trypsinized, diluted using flow cytometry buffer (phosphate-buffered saline with 1% bovine serum albumin [BSA]), and subjected to flow cytometry using an automated cell analyzer (LSRII; BD Bioscience). For experiments involving staining with cell surface marker SSEA4, cells were detached using nonenzymatic cell dissociation solution (Sigma), stained on ice for 30 min with fluorescently labeled antibodies, washed twice, and analyzed as described above.
Chromatin immunoprecipitation.
A total of 5 × 106 cells were cross-linked with 1% formaldehyde at room temperature for 10 min, followed by quenching with 0.125 M glycine for 5 min. Cells were lysed in 0.5 ml of ChIP lysis buffer (50 mM Tris-HCl [pH 8.0], 1% SDS, 10 mM EDTA) containing protease inhibitor cocktail (Roche) and sonicated to produce an average fragment size of 200 to 800 bp. Each immunoprecipitation was performed by incubating 30 μg of sonicated chromatin together with 5 μg of the respective antibodies in ChIP dilution buffer (10 mM Tris-HCl [pH 8.0], 1% Triton X-100, 0.1% SDS, 150 mM NaCl, 2 mM EDTA) overnight at 4°C. The next day, 25 μl of protein A/G Dynabeads (Life Technologies) was added for an additional 4 h. Captured antibody-antigen complexes were washed two times each in ChIP low-salt buffer (20 mM Tris-HCl [pH 8.0], 1% Triton X-100, 0.1% SDS, 150 mM NaCl, 2 mM EDTA), ChIP high-salt buffer (20 mM Tris-HCl [pH 8.0], 1% Triton X-100, 0.1% SDS, 500 mM NaCl, 2 mM EDTA), ChIP LiCl buffer (10 mM Tris-HCl [pH 8.0], 1% NP-40, 250 mM LiCl, 1 mM EDTA), and TE buffer (10 mM Tris-HCl [pH 8.0], 1 mM EDTA). DNA was eluted from beads in 200 μl of elution buffer (TE buffer containing 1% SDS, 100 mM NaCl, 5 mM dithiothreitol [DTT]), reverse cross-linked (65°C overnight), treated with RNase A (37°C, 1 h) and proteinase K (37°C, 2 h), and purified using the QIAquick PCR purification kit according to the manufacturer's instructions (Qiagen). Quantitative real-time PCRs were performed with specific primers, and the relative enrichment for each antibody was calculated by determining the bound/input value using the 2−ΔΔCT method (48) and multiplying that value by 100. Specific primers were as follows: MLV-LTR (forward [F], 5′-AGG GTC TCC TCT GAG TGA TTG ACT-3′; reverse [R], 5′-TCG GAC AGA CAC AGA TAA GTT GCT-3′), GAPDH (F, 5′-CAA TTC CCC ATC TCA GTC GT-3′; R, 5′-TAG TAG CCG GGC CCT ACT TT-3′), MYT1 (F, 5′-AGG CAC CTT CTG TTG GCC GA-3′; R, 5′-AGG CAG CTG CCT CCC GTA CA-3′), β-globin (F, 5′-CAG AGC CAT CTA TTG CTT AC-3′; R, 5′-GCC TCA CCA CCA ACT TCA TC-3′), Mito. gene (F, 5′-AGA CAC CTG CTG CCC TAT GT-3′; R, 5′-GCT CCA TCC CAG TGC TTT AC-3′), BMP2 (F, 5′-CCG ATC ACC TCT CTT CCT CA-3′; R, 5′-CTG GGC TTC TGT TGC TTT TC-3′), and M-PMV-LTR (F, 5′-TCC TGG TTC TTC TCC GTC TTA-3′; R, 5′-CTC AGT AGC AGA GCA AGG AAA G-3′).
Bisulfite sequencing.
After infection, genomic DNA was extracted from a pool of infected cells (DNeasy blood and tissue kit; Qiagen), followed by bisulfite treatment (EZ DNA Methylation-Lightning kit; Zymo Research) and nested PCR to amplify the previously characterized CpG islands within the MLV LTR by using primers described previously (40). Following PCR amplification, the products were gel purified, ligated into the pJET1.2 vector (Life Technologies), and used to transform bacteria, and individual bacterial colonies were picked. DNAs from these colonies were sequenced using primer (5′-CGACTCACTATAGGGAGAGCGGC-3′) to determine the extent of DNA methylation. Sequence results were analyzed using QUMA (http://quma.cdb.riken.jp/). Each DNA sequence shown was derived from a single bacterial colony.
Generation of stable shRNA knockdown cell lines.
RNAi knockdown in mouse and human EC cells was performed using shRNA containing pLKO.1 lentiviral vectors purchased from Thermo Scientific (Pittsburg, PA). Briefly, VSV-G-pseudotyped shRNA lentiviral particles were generated by cotransfection of HEK-293T cells using PEI with pLKO.1-shRNA, p8.91 (encoding HIV gag-pol), and pMD.G (encoding VSV-G) plasmid DNAs at a 2:1:1 ratio of plasmids. Viral preparations were harvested 48 h posttransfection, filtered through a 0.45-μm-pore-size filter (Pall Acrodisc), and used to infect EC cells for 6 h. Infected cells were then allowed to recover in fresh medium for 36 h, after which puromycin (2 μg/ml) was added to select for stable knockdown clones.
Dual luciferase assays.
HeLa and human EC cells were seeded at 100,000 cells per well in a 12-well plate the day before transfection. The next day, cells in each well were transfected with 900 ng of various LTR firefly luciferase constructs together with 100 ng of HSV-TK Renilla luciferase control plasmid using Lipofectamine 2000 (Life Technologies) at a 1:4 ratio. Thirty-six hours later, cells were lysed in 200 μl of 1× passive lysis buffer (Promega), followed by quantification of luciferase activity using a POLARstar Omega multimode plate reader (BMG Labtech) according to the manufacturer's instructions. The relative luciferase expression was calculated by first dividing the firefly luciferase signal by the Renilla luciferase signal in a given cell, and the resulting ratio was then normalized to the ratio obtained from the same cells transfected with M-PMV LTR luciferase (set to 1).
Quantitative real-time PCR analysis of integrated proviral DNA.
Two weeks following infection, cells were washed with phosphate-buffered saline, and total DNA was isolated using a Qiagen DNeasy kit according to the manufacturer's instructions. For analysis of integrated proviral DNA copies, 100 ng of genomic DNA was combined with GFP-specific TaqMan primer/probe sets described previously (47). PCRs were performed in 96-well plates using the 7900 fast real-time PCR system (Applied Biosystems) with the following reaction conditions: 10 min at 95°C, followed by 40 cycles of 30 s at 95°C, and 1 min at 60°C. For all reactions, relative expression was quantified using the 2−ΔΔCT method (48) normalized to the human Tert housekeeping gene.
ACKNOWLEDGMENTS
This work was supported by NCI grant R01 CA 30488 from the National Cancer Institute.
S.P.G. is an investigator of the Howard Hughes Medical Institute. We thank John Moran of University of Michigan for generously providing the EC cell lines for this study.
The authors declare that they have no conflicts of interest.
REFERENCES
- 1.Andrews PW. 2002. From teratocarcinomas to embryonic stem cells. Philos Trans R Soc Lond B Biol Sci 357:405–417. doi: 10.1098/rstb.2002.1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Andrews PW, Damjanov I, Simon D, Banting GS, Carlin C, Dracopoli NC, Fogh J. 1984. Pluripotent embryonal carcinoma clones derived from the human teratocarcinoma cell line Tera-2. Differentiation in vivo and in vitro. Lab Invest 50:147–162. [PubMed] [Google Scholar]
- 3.Pera MF, Cooper S, Mills J, Parrington JM. 1989. Isolation and characterization of a multipotent clone of human embryonal carcinoma cells. Differentiation 42:10–23. doi: 10.1111/j.1432-0436.1989.tb00602.x. [DOI] [PubMed] [Google Scholar]
- 4.Shevinsky LH, Knowles BB, Damjanov I, Solter D. 1982. Monoclonal antibody to murine embryos defines a stage-specific embryonic antigen expressed on mouse embryos and human teratocarcinoma cells. Cell 30:697–705. doi: 10.1016/0092-8674(82)90274-4. [DOI] [PubMed] [Google Scholar]
- 5.Kannagi R, Cochran NA, Ishigami F, Hakomori S, Andrews PW, Knowles BB, Solter D. 1983. Stage-specific embryonic antigens (SSEA-3 and -4) are epitopes of a unique globo-series ganglioside isolated from human teratocarcinoma cells. EMBO J 2:2355–2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schwartz CM, Spivak CE, Baker SC, McDaniel TK, Loring JF, Nguyen C, Chrest FJ, Wersto R, Arenas E, Zeng X, Freed WJ, Rao MS. 2005. NTera2: a model system to study dopaminergic differentiation of human embryonic stem cells. Stem Cells Dev 14:517–534. doi: 10.1089/scd.2005.14.517. [DOI] [PubMed] [Google Scholar]
- 7.Andrews PW. 1984. Retinoic acid induces neuronal differentiation of a cloned human embryonal carcinoma cell line in vitro. Dev Biol 103:285–293. doi: 10.1016/0012-1606(84)90316-6. [DOI] [PubMed] [Google Scholar]
- 8.Pleasure SJ, Page C, Lee VM. 1992. Pure, postmitotic, polarized human neurons derived from NTera 2 cells provide a system for expressing exogenous proteins in terminally differentiated neurons. J Neurosci 12:1802–1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Andrews PW, Goodfellow PN, Shevinsky LH, Bronson DL, Knowles BB. 1982. Cell-surface antigens of a clonal human embryonal carcinoma cell line: morphological and antigenic differentiation in culture. Int J Cancer 29:523–531. doi: 10.1002/ijc.2910290507. [DOI] [PubMed] [Google Scholar]
- 10.Matthaei KI, Andrews PW, Bronson DL. 1983. Retinoic acid fails to induce differentiation in human teratocarcinoma cell lines that express high levels of a cellular receptor protein. Exp Cell Res 143:471–474. doi: 10.1016/0014-4827(83)90076-9. [DOI] [PubMed] [Google Scholar]
- 11.Reubinoff BE, Pera MF, Fong CY, Trounson A, Bongso A. 2000. Embryonic stem cell lines from human blastocysts: somatic differentiation in vitro. Nat Biotechnol 18:399–404. doi: 10.1038/74447. [DOI] [PubMed] [Google Scholar]
- 12.Chang JC, Ye L, Kan YW. 2006. Correction of the sickle cell mutation in embryonic stem cells. Proc Natl Acad Sci U S A 103:1036–1040. doi: 10.1073/pnas.0510177103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xia X, Zhang Y, Zieth CR, Zhang SC. 2007. Transgenes delivered by lentiviral vector are suppressed in human embryonic stem cells in a promoter-dependent manner. Stem Cells Dev 16:167–176. doi: 10.1089/scd.2006.0057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Linney E, Davis B, Overhauser J, Chao E, Fan H. 1984. Non-function of a Moloney murine leukaemia virus regulatory sequence in F9 embryonal carcinoma cells. Nature 308:470–472. doi: 10.1038/308470a0. [DOI] [PubMed] [Google Scholar]
- 15.Hilberg F, Stocking C, Ostertag W, Grez M. 1987. Functional analysis of a retroviral host-range mutant: altered long terminal repeat sequences allow expression in embryonal carcinoma cells. Proc Natl Acad Sci U S A 84:5232–5236. doi: 10.1073/pnas.84.15.5232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Akgun E, Ziegler M, Grez M. 1991. Determinants of retrovirus gene expression in embryonal carcinoma cells. J Virol 65:382–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Flanagan JR, Krieg AM, Max EE, Khan AS. 1989. Negative control region at the 5′ end of murine leukemia virus long terminal repeats. Mol Cell Biol 9:739–746. doi: 10.1128/MCB.9.2.739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tsukiyama T, Niwa O, Yokoro K. 1989. Mechanism of suppression of the long terminal repeat of Moloney leukemia virus in mouse embryonal carcinoma cells. Mol Cell Biol 9:4670–4676. doi: 10.1128/MCB.9.11.4670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Barklis E, Mulligan RC, Jaenisch R. 1986. Chromosomal position or virus mutation permits retrovirus expression in embryonal carcinoma cells. Cell 47:391–399. doi: 10.1016/0092-8674(86)90596-9. [DOI] [PubMed] [Google Scholar]
- 20.Feuer G, Taketo M, Hanecak RC, Fan H. 1989. Two blocks in Moloney murine leukemia virus expression in undifferentiated F9 embryonal carcinoma cells as determined by transient expression assays. J Virol 63:2317–2324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Loh TP, Sievert LL, Scott RW. 1987. Proviral sequences that restrict retroviral expression in mouse embryonal carcinoma cells. Mol Cell Biol 7:3775–3784. doi: 10.1128/MCB.7.10.3775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Loh TP, Sievert LL, Scott RW. 1990. Evidence for a stem cell-specific repressor of Moloney murine leukemia virus expression in embryonal carcinoma cells. Mol Cell Biol 10:4045–4057. doi: 10.1128/MCB.10.8.4045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Petersen R, Kempler G, Barklis E. 1991. A stem cell-specific silencer in the primer-binding site of a retrovirus. Mol Cell Biol 11:1214–1221. doi: 10.1128/MCB.11.3.1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wolf D, Cammas F, Losson R, Goff SP. 2008. Primer binding site-dependent restriction of murine leukemia virus requires HP1 binding by TRIM28. J Virol 82:4675–4679. doi: 10.1128/JVI.02445-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wolf D, Goff SP. 2007. TRIM28 mediates primer binding site-targeted silencing of murine leukemia virus in embryonic cells. Cell 131:46–57. doi: 10.1016/j.cell.2007.07.026. [DOI] [PubMed] [Google Scholar]
- 26.Wolf D, Hug K, Goff SP. 2008. TRIM28 mediates primer binding site-targeted silencing of Lys1,2 tRNA-utilizing retroviruses in embryonic cells. Proc Natl Acad Sci U S A 105:12521–12526. doi: 10.1073/pnas.0805540105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wolf D, Goff SP. 2009. Embryonic stem cells use ZFP809 to silence retroviral DNAs. Nature 458:1201–1204. doi: 10.1038/nature07844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schultz DC, Ayyanathan K, Negorev D, Maul GG, Rauscher FJ III. 2002. SETDB1: a novel KAP-1-associated histone H3, lysine 9-specific methyltransferase that contributes to HP1-mediated silencing of euchromatic genes by KRAB zinc-finger proteins. Genes Dev 16:919–932. doi: 10.1101/gad.973302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schultz DC, Friedman JR, Rauscher FJ III. 2001. Targeting histone deacetylase complexes via KRAB-zinc finger proteins: the PHD and bromodomains of KAP-1 form a cooperative unit that recruits a novel isoform of the Mi-2alpha subunit of NuRD. Genes Dev 15:428–443. doi: 10.1101/gad.869501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wolf G, Yang P, Fuchtbauer AC, Fuchtbauer EM, Silva AM, Park C, Wu W, Nielsen AL, Pedersen FS, Macfarlan TS. 2015. The KRAB zinc finger protein ZFP809 is required to initiate epigenetic silencing of endogenous retroviruses. Genes Dev 29:538–554. doi: 10.1101/gad.252767.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rowe HM, Jakobsson J, Mesnard D, Rougemont J, Reynard S, Aktas T, Maillard PV, Layard-Liesching H, Verp S, Marquis J, Spitz F, Constam DB, Trono D. 2010. KAP1 controls endogenous retroviruses in embryonic stem cells. Nature 463:237–240. doi: 10.1038/nature08674. [DOI] [PubMed] [Google Scholar]
- 32.Matsui T, Leung D, Miyashita H, Maksakova IA, Miyachi H, Kimura H, Tachibana M, Lorincz MC, Shinkai Y. 2010. Proviral silencing in embryonic stem cells requires the histone methyltransferase ESET. Nature 464:927–931. doi: 10.1038/nature08858. [DOI] [PubMed] [Google Scholar]
- 33.Garcia-Perez JL, Morell M, Scheys JO, Kulpa DA, Morell S, Carter CC, Hammer GD, Collins KL, O'Shea KS, Menendez P, Moran JV. 2010. Epigenetic silencing of engineered L1 retrotransposition events in human embryonic carcinoma cells. Nature 466:769–773. doi: 10.1038/nature09209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Friedli M, Turelli P, Kapopoulou A, Rauwel B, Castro-Diaz N, Rowe HM, Ecco G, Unzu C, Planet E, Lombardo A, Mangeat B, Wildhaber BE, Naldini L, Trono D. 2014. Loss of transcriptional control over endogenous retroelements during reprogramming to pluripotency. Genome Res 24:1251–1259. doi: 10.1101/gr.172809.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Turelli P, Castro-Diaz N, Marzetta F, Kapopoulou A, Raclot C, Duc J, Tieng V, Quenneville S, Trono D. 2014. Interplay of TRIM28 and DNA methylation in controlling human endogenous retroelements. Genome Res 24:1260–1270. doi: 10.1101/gr.172833.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ecco G, Cassano M, Kauzlaric A, Duc J, Coluccio A, Offner S, Imbeault M, Rowe HM, Turelli P, Trono D. 2016. Transposable elements and their KRAB-ZFP controllers regulate gene expression in adult tissues. Dev Cell 36:611–623. doi: 10.1016/j.devcel.2016.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Friedli M, Trono D. 2015. The developmental control of transposable elements and the evolution of higher species. Annu Rev Cell Dev Biol 31:429–451. doi: 10.1146/annurev-cellbio-100814-125514. [DOI] [PubMed] [Google Scholar]
- 38.Imbeault M, Trono D. 2014. As time goes by: KRABs evolve to KAP endogenous retroelements. Dev Cell 31:257–258. doi: 10.1016/j.devcel.2014.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang GZ, Wolf D, Goff SP. 2014. EBP1, a novel host factor involved in primer binding site-dependent restriction of Moloney murine leukemia virus in embryonic cells. J Virol 88:1825–1829. doi: 10.1128/JVI.02578-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schlesinger S, Lee AH, Wang GZ, Green L, Goff SP. 2013. Proviral silencing in embryonic cells is regulated by Yin Yang 1. Cell Rep 4:50–58. doi: 10.1016/j.celrep.2013.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Teich NM, Weiss RA, Martin GR, Lowy DR. 1977. Virus infection of murine teratocarcinoma stem cell lines. Cell 12:973–982. doi: 10.1016/0092-8674(77)90162-3. [DOI] [PubMed] [Google Scholar]
- 42.Bronson DL, Andrews PW, Solter D, Cervenka J, Lange PH, Fraley EE. 1980. Cell line derived from a metastasis of a human testicular germ cell tumor. Cancer Res 40:2500–2506. [PubMed] [Google Scholar]
- 43.Soneoka Y, Cannon PM, Ramsdale EE, Griffiths JC, Romano G, Kingsman SM, Kingsman AJ. 1995. A transient three-plasmid expression system for the production of high titer retroviral vectors. Nucleic Acids Res 23:628–633. doi: 10.1093/nar/23.4.628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ooi SK, Wolf D, Hartung O, Agarwal S, Daley GQ, Goff SP, Bestor TH. 2010. Dynamic instability of genomic methylation patterns in pluripotent stem cells. Epigenetics Chromatin 3:17. doi: 10.1186/1756-8935-3-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Newman RM, Hall L, Connole M, Chen GL, Sato S, Yuste E, Diehl W, Hunter E, Kaur A, Miller GM, Johnson WE. 2006. Balancing selection and the evolution of functional polymorphism in Old World monkey TRIM5alpha. Proc Natl Acad Sci U S A 103:19134–19139. doi: 10.1073/pnas.0605838103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sabo Y, Walsh D, Barry DS, Tinaztepe S, de Los Santos K, Goff SP, Gundersen GG, Naghavi MH. 2013. HIV-1 induces the formation of stable microtubules to enhance early infection. Cell Host Microbe 14:535–546. doi: 10.1016/j.chom.2013.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang GZ, Goff SP. 2015. Postentry restriction of Mason-Pfizer monkey virus in mouse cells. J Virol 89:2813–2819. doi: 10.1128/JVI.03051-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]